

Abstract
We evaluated the in vivo efficacy of humanized exposures of cefiderocol, a novel siderophore cephalosporin, against a test panel of P. aeruginosa (PSA) previously shown to develop resistance to 2 preclinical candidate siderophores (MB-1 and SMC-3176). In the thigh infection model, the PSA bacterial density in untreated controls grew from 5.54 ± 0.23 to 8.68 ± 0.57 log10 CFU over 24 h. The humanized cefiderocol exposure resulted in >1 log10 CFU reduction in all 8 isolates, while MB-1 and SMC-3176 exhibited variable activity similar to that previously reported. Humanized exposures of cefepime and levofloxacin, acting as positive antimicrobial controls displayed activity consistent with that of the bacterial phenotypic susceptibility profiles. Cefiderocol manifested in vivo efficacy against all PSA isolates including those resistant to cefepime and levofloxacin in contrast to its predecessor siderophore compounds. These preclinical data are supportive of further evaluation of cefiderocol in the treatment of P. aeruginosa.
Keywords: In vivo activity, Cefiderocol, Siderophore, Humanized exposure, Pseudomonas aeruginosa
Introduction
The evolution of human bacterial pathogens and their resistance to antibiotics has been an ongoing process since the introduction of these therapeutic entities. In the current era of multidrug-resistant organisms that defy most, if not all, available antibiotics, the search for effective agents with novel mechanisms of action is of paramount importance. To this end, the development of compounds that exploit bacterial iron assimilation may be a viable area of investigation [1, 2].
Iron is a fundamental element to every living organism, and bacteria have developed pathways to acquire iron especially under deficient conditions, which occurs with infection. The primary route for iron acquisition is siderophores [3], which are sequestering molecules with powerful iron-binding affinity. Bacterial cells retrieve secreted siderophores through outer membrane receptors [4, 5, 6]. Therefore, the coupling of antibiotics to siderophores presents an opportunity to evade conventional bacterial mechanisms that may limit sufficiently high antibiotic concentrations at the target site [7, 8].
At present, this approach has been utilized to design antibiotics capable of overcoming resistance due to membrane penetration by delivering the lethal moiety to the site of action [9]. For example, the conjugation of artemisinin to mycobactin siderophore yielded a compound with potent antimycobacterial activity [10]. When considering Gram-negative pathogens, the siderophore conjugated monobactam (MB-1) [11] and monocarbam (SMC-3176) [12] displayed potent in vitro activity; however, in preclinical trials, the in vivo development of adaptive type resistance, which manifested itself as regrowth of the organism in the face of drug exposures that far exceeded the minimum inhibitory concentrations (MICs) of the test isolates halted their transition to clinical candidate evaluation.
Cefiderocol (formerly known as S-649266) is a catechol substituted cephalosporin conjugated siderophore developed by Shionogi & Co., Ltd. (Osaka, Japan). The compound has shown a favorable iron-binding profile [13]. In vitro data demonstrate activity against Enterobacteriaceae, Acinetobacter baumannii, and Pseudomonas aeruginosa. Moreover, this compound is active against multidrug-resistant organisms including those producing carbapenem destroying serine and metallo-β-lactamases [14, 15]. Additionally, a recently conducted in vivo murine-based study has demonstrated an incremental exposure-response profile over a dose range without the appearance of adaptive resistance [16].
This study was conducted using a neutropenic murine thigh infection model to demonstrate the effect of the studied drugs against the isolates tested in a biological system with minimum interference from the immune system. The objective was to investigate the efficacy of humanized exposures of cefiderocol against the same isolates utilized previously with MB-1 and SMC-3176 to establish a robust comparison with respect to efficacy. In addition, comparison with the commercially available antibiotics cefepime and levofloxacin were conducted to further assess the clinical utility of cefiderocol.
Materials and Methods
Compounds Tested
Test compounds cefiderocol, MB-1, and SMC-3176 were synthesized and provided by Shionogi & Co. Ltd. Cefepime (Sagent, Schaumberg, IL, USA), and levofloxacin (Akorn Inc. Lake Forest, IL, USA) commercial vials were acquired from the Hartford Hospital Pharmacy Department (Hartford, CT, USA) for in vivo experimentation. Standard analytical powders of cefepime and levofloxacin were purchased from Sigma-Aldrich (St. Louis, MO, USA) for MIC determination in the Center for Anti-Infective Research and Development at the Hartford Hospital.
In vitro Susceptibility
MIC determinations for the 3 siderophores were performed using iron-depleted cation adjusted Mueller Hinton broth (ID-CAMHB; as recommended by CLSI for this class of compounds), while cefepime and levofloxacin MICs were determined in conventional CAMHB in accordance with the CLSI methodology [17]. MIC studies were conducted in triplicate and the modal values are reported.
Bacterial Isolates
The same 8 clinical P. aeruginosa (PSA) isolates (PSA 1403, PSA 1401, PSA JJ8–16, PSA JJ5–35, PSA AZ8–18, PSA AZ32-13, PSA JJ4–36, and PSA JJ11–54) used in the previous siderophore investigations [11, 12] were tested in the current study. Isolates were maintained in double-strength skim milk (Remel Inc., Lenexa, KS, USA) at −80°C. Each isolate was transferred twice on Trypticase soy agar with 5% sheep blood (Becton, Dickinson & Co.; Sparks, MD, USA) prior to use in animal infection.
Animal Model
The protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Hartford Hospital. Animals were maintained and used in accordance with National Research Council recommendations and provided food and water ad libitum. As done previously [18], the animal model was constructed by using pathogen-free ICR female mice weighing 20–22 grams and 7–8 weeks at the time of delivery (Envigo RMS, Inc. Indianapolis, IN, USA) that were rendered transiently neutropenic by intraperitoneal injection of cyclophosphamide (Sigma-Aldrich Co., St. Louis, MO, USA) 150 and 100 mg/kg on day 4 and 1 respectively, prior to inoculation. Mice also received a single intraperitoneal injection of 5 mg/kg of uranyl nitrate inducing a predictable degree of renal impairment to assist with humanizing the target exposures of these renally eliminated compounds. Groups of 3 animals (n = 6 thighs) were inoculated intramuscularly in each thigh 2 h before the start of the experiment with 0.1 mL of a solution containing the test isolate at approximately 107 CFU/mL. To minimize animal distress after inoculation with bacteria, animals are monitored throughout the 24-h study period for any abnormal signs or behavior and euthanized if necessary as per protocol used at the Hartford Hospital.
Pharmacokinetic Study
Determination of human-simulated dose of cefiderocol mimicking the profile observed in humans given a 2,000 mg cefiderocol every 8 h (q8h) as a 3-h infusion (inf) was undertaken by simulation (Phoenix version 6.3, Pharsight Corp., Mountain View, CA, USA) of pharmacokinetic (PK) data derived from infected mice receiving the compound in our previously conducted study [16]. Corrections for free-drug concentrations were incorporated into the calculation of simulated regimen using a protein-binding value of 31.6% [19].
Confirmatory PK studies for cefiderocol, cefepime, and levofloxacin were carried out to ensure that the regimens utilized provided humanized exposures. Infected animals received incremental doses 0.2 mL subcutaneous injections as required. Groups of 6 mice were euthanized at predefined time points. Terminal blood samples from CO2-asphyxiated mice were collected via cardiac puncture and placed in sodium heparin Vacutainer® tubes (BD, Franklin Lakes, NJ, USA). Plasma was separated by centrifugation for 10 min at 4°C at 10,000 relative centrifugal force and transferred into polypropylene tubes. These tubes were stored at −80°C until analyzed. The plasma concentrations of cefiderocol were determined by Shionogi using a validated liquid chromatography double mass spectrometry assay (LC/MS/MS). Mouse plasma samples were protein precipitated using 0.1 % trifluoroacetic acid- methanol. The mobile phase A was water/heptafluorobutyric acid and mobile phase B was acetonitrile/heptafluorobutyric acid. The internal standard used was S-649266-d12 sodium. The lower limit of detection was 0.1 mg/L. The assays of cefepime [20] and levofloxacin [21] were undertaken in our center.
Pharmacodynamic Study
Cefiderocol, cefepime, and levofloxacin were administered to mice using dosing regimens that mimicked human exposures of the following doses: 2g q8h (3 h inf), 1g q8 h (30 min inf) [22], and 750 mg q24h [23] respectively. The MB-1 dose equivalent to 2g q8h (4 h inf) [11] and SMC-3176 [12] dose of 50 mg/kg q3h were used as in the previously cited studies to facilitate siderophore comparisons and validate the performance of the PSA isolates in this study [11, 12]. While MB-1 and SMC-3176 were included in the current study to act as inter-study controls, SMC-3176 was tested only against 2 isolates, one with expected growth and the other was killed due to limitations in the availability of the compound and minimizing the number of animals used in this protocol. MB-1 and SMC-3176 were administered in renally intact mice in a fashion identical to that of the previous studies [11, 12]. The mice used in the study were grouped in cages of 3 animals and all received subcutaneous injections of study agents. The sample size for this portion of the study was calculated as follows: (1) for typical antimicrobial agents, the optimal dosing regimen usually produces an approximately 2–3 log decrease in bacterial density with a %CV of 40 in order to have an observed mean which deviates from the true mean by no more than 1 SD using a 2-sided 95% CI with 80% probability; n = 6 data points are required. Using our proposed methodology, each animal will produce 2 data points, since both thighs will be infected and cultured for bacterial count. At the initiation of dosing, 1 group was sacrificed and harvested to serve as the 0-h control group. All other mice were harvested after 24 h of treatment. In addition to the mice that received the study medications, 1 group of mice received sterile normal saline injections at the same frequency, route, and volume. After sacrifice, thighs were removed and individually homogenized in normal saline. Serial dilutions of the thigh homogenate were plated on Trypticase soy agar with 5% sheep blood for CFU determination. Efficacy, defined as the change in bacterial density, was calculated as the log10 change in numbers of bacterial CFU/mL obtained for treated mice after 24 h from the pre-antibiotic counts measured from the 0-h control animals.
Results
In vitro Susceptibility
The range of MICs (mg/L) for cefiderocol, MB-1, SMC-3176, cefepime, and levofloxacin were 0.063–0.5, 0.063–0.5, 0.063–1, 2–64, and 1—32 respectively. Individual MICs for all compounds are displayed in Table 1.
Table 1.
Minimum inhibitory concentrations (MICs, mg/L) of the study isolates
| Cefiderocol | MB-1 | SMC-3176 | Cefepime | Levofloxacin | |
|---|---|---|---|---|---|
| PSA 1403 | 0.063 | 0.063 | 0.063 | 2 | 1 |
| PSA 1401 | 0.25 | 0.125 | 0.25 | 2 | 1 |
| PSA JJ8–16 | 0.125 | 0.5 | 0.125 | 32 | 2 |
| PSA JJ5–35 | 0.25 | 0.25 | 0.25 | 32 | 4 |
| PSA AZ8–18 | 0.5 | 0.5 | 1 | 4 | 32 |
| PSA AZ32-13 | 0.25 | 0.25 | 0.25 | 8 | 16 |
| PSA JJ4–36 | 0.125 | 0.25 | 0.063 | 32 | 16 |
| PSA JJ11–54 | 0.25 | 0.5 | 0.125 | 64 | 16 |
MICs were determined in ID-CAMHB for cefiderocol, MB-1, and SMC-3176 in conventional CAMHB for cefepime and levofloxacin.
PKs Data
Simulation of cefiderocol PK data in mice resulted in a humanized regimen that consisted of the following doses of 15, 20, 25, 10, and 5 mg/kg administered at 0, 1, 2, 4, and 6 h, respectively, every 8 h. Confirmatory PK studies of cefiderocol revealed that the delivered dosing regimen provided a similar profile to that of the human regimen. Moreover, this murine regimen provides a similar fT>MIC profile to that in man (Fig. 1). Additional studies confirmed the adequacy of cefepime and levofloxacin humanized regimens (data not shown).
Fig. 1.
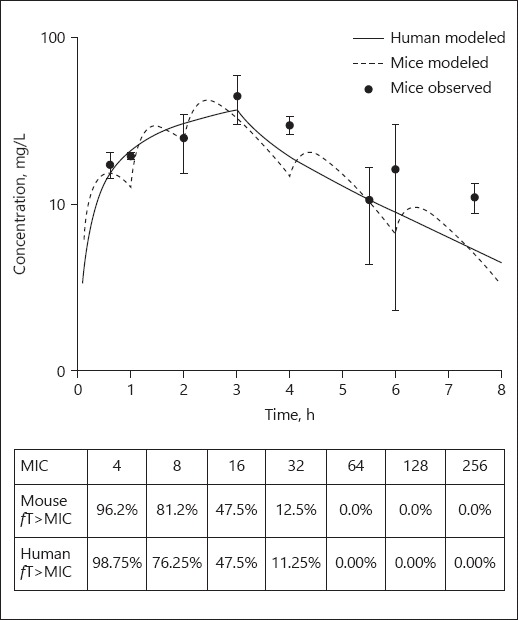
Comparative total drug concentration profile and fT>MIC of cefiderocol in mouse and man. Dot, mean; whiskers, SD.
Pharmacodynamics Data
The 0-h control groups showed log10 CFU (mean ± SD) 5.5 ± 0.12 and unopposed 24 h growth was 8.6 ± 0.17. Cefiderocol was efficacious, defined ≥1 log10 reduction in CFU, against all the isolates and produced ≥2 log10 reduction in 7 of 8 PSA tested. In contrast, MB-1 demonstrated activity only against 3 of 8, as no meaningful activity was observed against the other 5 isolates. SMC-3176 was tested against 2 isolates for comparison purposes. The compound displayed activity against PSA 1403, while PSA JJ4–36 regrew in a similar fashion to untreated controls. A comparison of activity for the 3 siderophore compounds is provided in Figure 2. Cefepime and levofloxacin display in vivo activity concordant with their phenotypic profiles, while cefiderocol displayed activity against all the PSA isolates independent of fluoroquinolone and/or β-lactam susceptibility (Fig. 3).
Fig. 2.
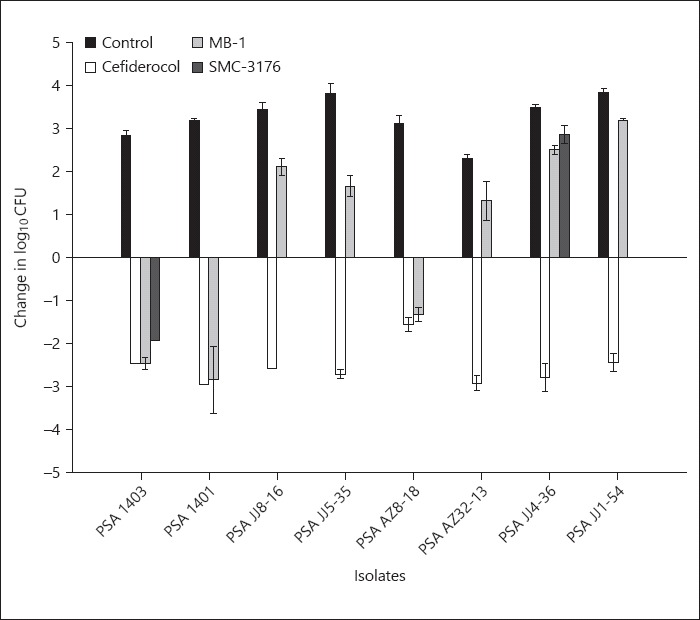
Comparative efficacy of cefiderocol versus MB-1 and SMC-3176. Bars, mean; whiskers, SD.
Fig. 3.
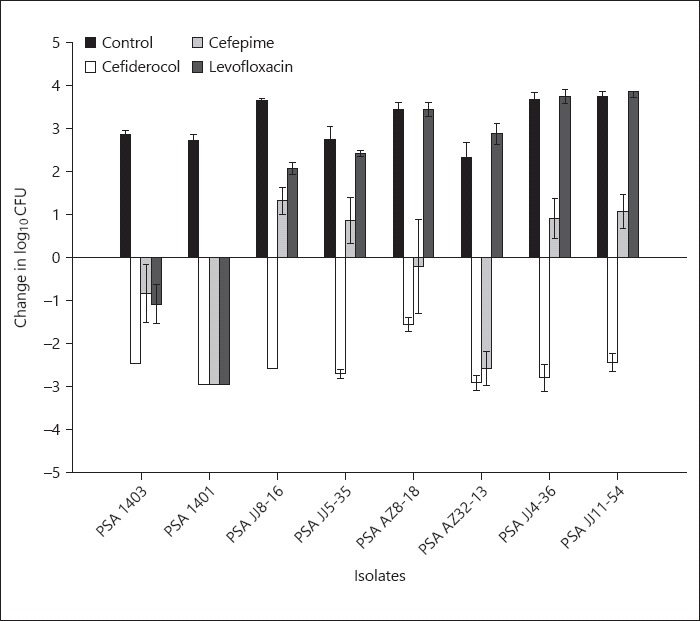
Comparative efficacy of cefiderocol versus cefepime and levofloxacin. Bars, mean; whiskers, SD.
Discussion
While siderophore conjugated antibiotics have been investigated as a means of overcoming bacterial resistance, compounds such as MB-1 and SMC-3176 failed to sustain their antibacterial effects during pre-clinical in vivo testing, despite drug concentrations that exceeded their in vitro MICs [11, 12]. Subsequent studies revealed that this lack of efficacy was due to adaptive mechanisms [11]. This phenomenon is not new and has historically been described as the ability of bacterial cells to adjust the concentrations of various cellular enzymes and components in response to change in the surrounding medium in order to sustain survival and growth. In the case of MB-1, the compound was able only to produce CFU reduction against 3 of 8 PSA isolates, despite the fact that MICs of all isolates were low and the dose regimen given would achieve >90% fT>MIC. The authors explained this in vitro-in vivo disconnect by the existence of subpopulation hyper-producing high iron affinity native siderophore pyoverdine sufficient to confer protection to neighboring cells. Under extremely low iron conditions in vivo, bacterial cells will not depend on less efficient siderophore for iron acquisition, thus diminishing the MB-1 activity [11]. A subsequent study demonstrated that using an inhibitor of the efflux pump that exports pyoverdine out of cells potentiated the in vivo activity of MB-1; thus, these data substantiate the hypothesis of importance of iron-binding affinity [24]. Similarly, SMC-3176 was vulnerable to the development of the same type of resistance that is reversible with drug exposure cessation. The authors found that the resistance was mediated by genes responsible for siderophore uptake, but no colonies with stable mutation were isolated. The study illustrated through dose ranging that increasing the fT>MIC can improve the efficacy but could not establish a threshold exposure [12].
In the current study, we investigated the in vitro and in vivo potency of cefiderocol in comparison to the previously studied siderophores, MB-1 and SMC-3176 against the same PSA isolates to which these latter compounds failed in the in vivo setting. In an effort to create a robust testing environment for cefiderocol, we repeated the previous experiments conducted with MB-1 and SMC-3176 [11, 12]. As such, we reconfirmed not only a similar in vitro potency profile for MB-1 and SMC-3176 against all 8 P. aeruginosa but also established the same in vivo efficacy profile for both compounds in different studies conducted months apart. In contrast to the observations of poor efficacy for MB-1 and SMC-3176 over the 24-h dosing scheme, the humanized regimen of cefiderocol displayed potent in vivo activity as evidenced by sustained efficacy over the treatment period. The sustained activity of cefiderocol is believed to be due in part to the compound's catechol moiety substitution, which enhances its iron-chelating affinity compared to that of the other siderophores. Moreover, the faster uptake of cefiderocol in P. aeruginosa when compared to native pyoverdine appears to contribute to the enhanced killing of the compound [13].
In addition, to the sustained antibacterial effects displayed by cefiderocol relative to the other siderophores tested, it should also be noted that cefiderocol displayed potent in vivo activity against all the P. aeruginosa irrespective of their susceptibility to cefepime and levofloxacin. Thus, our data support consideration of this compound in the context of new therapeutic entities for the multidrug resistant phenotype of this problematic nosocomial pathogen.
In this comparative study, the cefiderocol siderophore conjugated cephalosporin appeared to elude the rapidly adaptive mechanisms in this group of P. aeruginosa test isolates that defeated 2 predecessors in the siderophore class. Moreover, the observed concordance between in vitro and in vivo potency of the compound against this challenging cache of P. aeruginosa renders cefiderocol a good candidate for more extensive pharmacodynamic profile of this novel class of agents and subsequently in clinical trials.
Ethical Approval
Ethical approval was obtained from the Institutional Animal Care and Use Committee before the start of the study.
Disclosure Statement
I.M.G. and M.L.M. have no conflicts of interest. M.T. is an employee of Shionogi & Co., Ltd. Osaka, Japan. D.P.N received a research grant from Shionogi & Co., Ltd., Osaka, Japan.
Acknowledgments
This work was supported by a research grant from Shionogi & Co., Ltd., Osaka, Japan. The sponsor had no involvement in the conduct of the study, data collection, and analysis. We acknowledge Kimelyn Greenwood, Jennifer Tabor-Rennie, Lucinda Lamb, Sara Robinson, Debora Santini, and Abrar Thabit (Center for Anti-Infective Research and Development, Hartford Hospital) for their assistance with animal experiments and MIC determination.
References
- 1.Baym M, Stone LK, Kishony R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science. 2016;351 doi: 10.1126/science.aad3292. aad3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmes AH, Moore LS, Sundsfjord A, Steinbakk M, Regmi S, Karkey A, et al. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet. 2016;387:176–187. doi: 10.1016/S0140-6736(15)00473-0. [DOI] [PubMed] [Google Scholar]
- 3.Foley TL, Simeonov A. Targeting iron assimilation to develop new antibacterials. Expert Opin Drug Discov. 2012;7:831–847. doi: 10.1517/17460441.2012.708335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neilands JB. Iron absorption and transport in microorganisms. Annu Rev Nutr. 1981;1:27–46. doi: 10.1146/annurev.nu.01.070181.000331. [DOI] [PubMed] [Google Scholar]
- 5.Ratledge C. Iron metabolism and infection. Food Nutr Bull. 2007;28:S515–S523. doi: 10.1177/15648265070284S405. [DOI] [PubMed] [Google Scholar]
- 6.Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 7.Hancock RE, et al. The bacterial outer membrane as a drug barrier. Trends Microbiol. 1997;5:37–42. doi: 10.1016/S0966-842X(97)81773-8. [DOI] [PubMed] [Google Scholar]
- 8.Braun V, Pramanik A, Gwinner T, Köberle M, Bohn E. Sideromycins: tools and antibiotics. Biometals. 2009;22:3–13. doi: 10.1007/s10534-008-9199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller MJ, Zhu H, Xu Y, Wu C, Walz AJ, Vergne A, et al. Utilization of microbial iron assimilation processes for the development of new antibiotics and inspiration for the design of new anticancer agents. Biometals. 2009;22:61–75. doi: 10.1007/s10534-008-9185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller MJ, Walz AJ, Zhu H, Wu C, Moraski G, Möllmann U, et al. Design, synthesis, and study of a mycobactin-artemisinin conjugate that has selective and potent activity against tuberculosis and malaria. J Am Chem Soc. 2011;133:2076–2079. doi: 10.1021/ja109665t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomaras AP, Crandon JL, McPherson CJ, Banevicius MA, Finegan SM, Irvine RL, et al. Adaptation-based resistance to siderophore-conjugated antibacterial agents by Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2013;57:4197–4207. doi: 10.1128/AAC.00629-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim A, Kutschke A, Ehmann DE, Patey SA, Crandon JL, Gorseth E, et al. Pharmacodynamic profiling of a siderophore-conjugated monocarbam in Pseudomonas aeruginosa: assessing the risk for resistance and attenuated efficacy. Antimicrob Agents Chemother. 2015;59:7743–7752. doi: 10.1128/AAC.00831-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuji M, Ito A, Horiyama T, et al. S-649266, a Novel Siderophore: Mechanism of Enhanced Activity and β-Lactamase Stability. Poster number 256. ID week 2014.
- 14.Kohira N, West J, Ito A, Ito-Horiyama T, Nakamura R, Sato T, et al. In vitro antimicrobial activity of a siderophore cephalosporin, S-649266, against Enterobacteriaceae clinical isolates, including carbapenem-resistant strains. Antimicrob Agents Chemother. 2015;60:729–734. doi: 10.1128/AAC.01695-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito A, Kohira N, Bouchillon SK, West J, Rittenhouse S, Sader HS, et al. In vitro antimicrobial activity of S-649266, a catechol-substituted siderophore cephalosporin, when tested against non-fermenting Gram-negative bacteria. J Antimicrob Chemother. 2016;71:670–677. doi: 10.1093/jac/dkv402. [DOI] [PubMed] [Google Scholar]
- 16.Ghazi IM, Monogue ML, Tsuji M, Nicolau DP. Pharmacodynamics of S-649266, a Novel Siderophore Conjugated Cephalosporin, Explored in a Pseudomonas aeruginosa Infected Murine Thigh Model. Poster number 516. ASM Microbe. 2016 [Google Scholar]
- 17.Clinical and Laboratory Standards Institute . Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Sixth Informational Supplement (Document M07-A10). Wayne: National Committee for Clinical Laboratory Standards; 2016. [Google Scholar]
- 18.Crandon JL, Nicolau DP. In Vivo activities of simulated human doses of cefepime and cefepime-AAI101 against multidrug-resistant gram-negative Enterobacteriaceae. Antimicrob Agents Chemother. 2015;59:2688–2694. doi: 10.1128/AAC.00033-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura R, Toba S, Ito A, Tsuji M, Yamano Y, Shimada J. S-649266, a Novel Siderophore Cephalosporin; V. Pharmacodynamic Assessment in Murine Thigh Infection Model. Poster number F1559. 54th Interscience Conference Antimicrobial Agents and Chemotherapy, 2014.
- 20.Barbhaiya RH, Forgue ST, Shyu WC, Papp EA, Pittman KA. High-pressure liquid chromatographic analysis of BMY-28142 in plasma and urine. Antimicrob Agents Chemother. 1987;31:55–59. doi: 10.1128/aac.31.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tobin CM, Sunderland J, White LO, MacGowan AP. A reverse-phase, isocratic high-performance liquid chromatography assay for levofloxacin. J Antimicrob Chemother. 1999;43:434–435. doi: 10.1093/jac/43.3.434. [DOI] [PubMed] [Google Scholar]
- 22.Tam VH, McKinnon PS, Akins RL, Drusano GL, Rybak MJ. Pharmacokinetics and pharmacodynamics of cefepime in patients with various degrees of renal function. Antimicrob Agents Chemother. 2003;47:1853–1861. doi: 10.1128/AAC.47.6.1853-1861.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rebuck JA, Fish DN, Abraham E. Pharmacokinetics of intravenous and oral levofloxacin in critically ill adults in a medical intensive care unit. Pharmacotherapy. 2002;22:1216–1225. doi: 10.1592/phco.22.15.1216.33484. [DOI] [PubMed] [Google Scholar]
- 24.Tomaras AP, Crandon JL, McPherson CJ, Nicolau DP. Potentiation of antibacterial activity of the MB-1 siderophore-monobactam conjugate using an efflux pump inhibitor. Antimicrob Agents Chemother. 2015;59:2439–2442. doi: 10.1128/AAC.04172-14. [DOI] [PMC free article] [PubMed] [Google Scholar]