

Abstract
This manuscript reports on the first two-photon, label-free, metabolic imaging of biological tissues in vivo at histological resolution on an extremely compact, fiber-optic endomicroscopy platform. This system provides new opportunities for performing non-invasive and functional histological imaging of internal organs in vivo, in situ and in real time. As a routine clinical procedure, traditional histology has made significant impacts on medicine. However, the procedure is invasive and time consuming, suffers random sampling errors, and cannot provide in vivo functional information. The technology reported here features an extremely compact and flexible fiber-optic probe ~2 mm in diameter, enabling direct access to internal organs. Unprecedented two-photon imaging quality comparable to a large bench-top laser scanning microscope was achieved through technological innovations in double-clad fiber optics and miniature objective lenses (among many others). In addition to real-time label-free visualization of biological tissues in situ with subcellular histological detail, we demonstrated for the first time in vivo two-photon endomicroscopic metabolic imaging on a functioning mouse kidney model. Such breakthroughs in nonlinear endoscopic imaging capability present numerous promising opportunities for paradigm-shifting applications in both clinical diagnosis and basic research.
Keywords: functional histology in vivo, label-free imaging, nonlinear endomicroscopy, two-photon metabolic imaging
Introduction
Optical microscopy has played an indispensable role in conventional histopathological analyses of biopsy specimens, which serves as the current gold standard for disease diagnosis. The entire process, which typically entails endoscopy examination, tissue extraction, pathological preparation and microscopy interrogation, is time consuming (typically a few days) and prone to sampling error. In particular, static images cannot reveal any in vivo dynamic functional information. An endoscopic imaging technology that allows direct imaging with subcellular resolution in vivo and in situ would eliminate the need for tissue removal and histopathological processing and promote the accuracy of targeted biopsies and the overall diagnostic yield. One recent technical innovation toward this goal is confocal laser endomicroscopy (CLE)1; however, CLE still relies on the use of extrinsic fluorescent agents applied either topically or intravenously to attain sufficient imaging contrast2. Meanwhile, despite its promising label-free, structural and metabolic imaging potential3, 4, 5 and minimal regulatory hurdles in in vivo applications, ubiquitous tissue autofluorescence is under-exploited owing to the phototoxicity associated with the high-power ultraviolet excitation needed for CLE. One strategy to circumvent this difficulty is to integrate two-photon excitation (2PE) into an endoscopy device6, 7. In addition to dramatically reducing photodamage, other merits of 2PE, for example, deeper tissue penetration8 and intrinsic depth-sectioning capability9, are readily available to the endomicroscope. Moreover, both two-photon fluorescence (2PF) from endogenous fluorophores (such as reduced nicotinamide adenine dinucleotide (NADH) and oxidized flavin adenine dinucleotide (FAD)) and second-harmonic generation (SHG) signals from collagen fibers can be acquired simultaneously with single-wavelength excitation, permitting label-free, in vivo, high-resolution and functional histological assessment of internal organs that was previously impossible.
To develop a nonlinear endomicroscope that lends itself to practical label-free functional histological imaging in vivo, one central challenge in addition to the miniaturization difficulty involves achieving high detection sensitivity. Here we use the term ‘detection sensitivity’ to represent the achievable signal-to-noise ratio (SNR) with a given excitation power and fluorophore concentration. High detection sensitivity is of paramount importance for label-free functional imaging in vivo based on intrinsic 2PF and SHG signal given that (1) both endogenous fluorophores and collagen fibers generally exist in relatively low abundance, and (2) the two-photon action cross-sections of endogenous fluorophores (<0.1 GM for NADH and FAD3) are much smaller than common exogenous dyes or fluorescent proteins (~10–100 GM10, 11). Several innovative two-photon endomicroscope designs have been reported in past decades12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23. However, most of the early prototypes lacked detection sensitivity and thus were incapable of performing in vivo label-free, dynamic and functional two-photon imaging in real time. Here we present the first histological and functional visualization of unstained/unprocessed biological tissues in vivo and in real time on an ultracompact, flexible two-photon endomicroscopy setting. Unprecedented two-photon imaging quality was achieved on various tissue structures. Moreover, we also demonstrated that this nonlinear endomicroscope is capable of capturing and quantifying dynamic, cellular metabolic information on internal organs (mouse kidney as an example) in vivo, in situ and in real time. Such an in vivo dynamic functional histological imaging modality can be highly valuable for translational applications (for example, early disease diagnosis) and for basic research.
Materials and methods
Scanning fiber-optic endomicroscope and system set-up
A piezoelectric (PZT) tube with its outer surface divided into two orthogonal pairs of electrodes functions as an actuator to sweep a fiber-optic cantilever to perform imaging beam scans. The working principle of a fiber-optic scanner has been detailed in previous reports12, 24. Briefly, the double-clad fiber (DCF) is threaded through and anchored to the PZT tube, and its freely standing end (~11 mm long) serves as a cantilever. By driving the two orthogonal pairs of electrodes with two amplitude-modulated sinusoidal waveforms that are 90° out of phase, the fiber tip can be resonantly (~1.4 kHz) vibrated into a desired open–close spiral scanning pattern. Throughout this paper, 512 spirals were acquired to construct one frame, corresponding to a frame acquisition time of ~0.38 s. The spatial resolution of the endomicroscope was ~0.7 μm laterally and ~6.5 μm axially, and was estimated by the full width at half maximum of the point-spread functions when imaging across 0.1 μm diameter fluorescent beads and a 1 μm thick fluorescent thin film, respectively, using the 200-μm working distance (WD) miniature objective. A schematic of the basic system is provided in Supplementary Note S1 and Supplementary Fig. S1. Moreover, the entire imaging system (including a home-built 20 × 20 inch tunable femtosecond Ti:Sapphire laser; not shown here) can be made compact and portable.
Customized DCF
The pure silica-core DCF was fabricated using outside vapor deposition technology. A core NA of ~0.12 was realized by tuning the fluorine-doping concentration in the inner clad, and single-mode operation was achieved by precisely controlling the core size (~5.5 μm). Similarly, the high inner clad NA (~0.35) was realized by further tuning the fluorine-doping concentration in the outer clad.
Fiber background characterization
The measurement set-up was similar to the system schematic except that the emission signal was sent to a grating-based monochromator (Acton SP2300i, Princeton Instruments, Inc., Trenton, NJ, USA) and detected by a photon-counting module (SPC-150, Becker & Hickl GmbH, Berlin, Germany) along with a photomultiplier tube (PMT; H7422P-50, Hamamatsu Photonics, Hamamatsu City, Japan). We coupled femtosecond laser pulses (λcenter~890 nm) into the DCF core and detected the backward-propagating nonlinear emission exiting the proximal end of the DCF. The photomultiplier tube was cooled to ensure a stable dark count rate (~120 photons s−1). The emission spectra were characterized at a 1-nm interval, and the total emission was averaged over 12 s to estimate the emission rate. The fiber length (~75 cm) and laser power propagated within the fiber (40 mW) were maintained the same for the three pieces of DCFs under testing, with the distal end of the DCF exposed to air. Physical parameters of the two commercial DCFs are listed below: (1) SM-GDF-5/130 from NuFern, Inc. (East Granby, CT, USA), which has a single-mode core 5 μm in diameter and 0.12 NA and an inner clad 130 μm in diameter and 0.46 NA; (2) SMM900 from Fibercore Ltd. (Chilworth, Southampton, UK), which has a mode-field diameter of 6.5–8.2 μm at 1550 nm and a core NA of 0.18–0.20 and an inner clad 100 μm in diameter and 0.24–0.28 NA.
Tissue preparation for ex vivo imaging
The liver was dissected from a euthanized mouse (BALB/c strain, 16–18 weeks age, Taconic Biosciences, Hudson, NY, USA) and pinned to a wax block. The endomicroscope was positioned gently against the liver surface (without breaking the liver surface), and phosphate-buffered saline (PBS) was added to maintain water immersion for the endomicroscope during imaging. Intact cervices were dissected from normal pregnant mice (C57Bl6/129S) at gestation day 15 and mifepristone-treated Ru486 preterm-birth (PTB) mouse models25, 26, 27. The tissue was then snap frozen in liquid nitrogen in the optimal cutting temperature compound (Tissue-Tek, Sakura Finetek USA, Inc., Torrance, CA, USA). Before imaging, the entire tissue-embedded optimal cutting temperature block was first thawed at room temperature. Then, the intact cervix was rehydrated with PBS and fixed on a wax block with the ectocervix facing upwards for imaging.
Animal preparation for in vivo imaging
A mouse (BALB/c strain, 16–18 weeks age, Taconic Biosciences) was first sedated in an induction chamber with 3% isoflurane-oxygen mixture gas until adequate anesthesia was assured. The mouse was then moved onto a temperature-controlled heating pad (36 °C) and fitted to a rodent mask to maintain anesthesia with ~1%–2% isoflurane-oxygen mixture. For small intestine imaging, a small abdominal incision was created, through which a loop of small intestine (jejunum or ileum) was identified, carefully lifted out and fixed onto a home-built supporting frame without damaging blood vessels in the mesentery. To access the intestinal mucosa, a short intestine segment (~1 cm) was cut open along the dorsal side with respect to the intestinal artery arcades to minimize bleeding. The exposed mucosa surface was flushed using PBS to remove all visible intestinal contents. Afterwards, a No. 0 coverslip (~100 μm thick) was gently placed on the mucosa to minimize potential motion artifacts. Then, the endomicroscope was placed on top of the coverslip under water immersion during imaging. For kidney imaging, the mouse was positioned to lie on its right side, and a small dorsal incision was made, through which the left kidney was exteriorized. After carefully separating the kidney from its surrounding fatty tissue using cotton swabs, the kidney was mounted onto a custom-built tiny rack, which not only helped isolate breathing-induced motion, but also exposed the renal pedicle without stretching the blood vessels. The renal artery and vein, which accompany each other, were identified and separated from the ureter and other surrounding tissues of the renal pedicle. A vascular clamp was deployed using a specially designed clamp applier to allow blood vessel clamping and releasing without inducing large global kidney movement, such that the same field of view could be tracked throughout the ischemia–reperfusion process. The endomicroscope was gently placed against the kidney surface to image the renal cortex. All animal housing and experimentation procedures were performed in accordance with the standards of humane animal care described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Protocols were approved by the institutional animal care and use committee of Johns Hopkins University.
Results and discussion
Two-photon endomicroscopy with superb detection sensitivity
A flexible endomicroscope relies on optical fibers to deliver the excitation beam (through the single-mode fiber core). We identified that one major source of background noise is the in-fiber nonlinear luminescence background emitted from the germanium-doped core of most commercial fibers. The dopant, which was originally used to increase the refractive index, also generates multiphoton fluorescence and SHG emission when subject to strong femtosecond laser pulses that propagate through the fiber core28, 29, 30. As shown in Figure 1b, such background luminescence exhibits a broad spectrum that overlaps with the intrinsic 2PF and SHG signals from biological tissues3, 9, making it impossible to separate and eliminate the background signal from the sample-generated nonlinear signals within the overlapping spectrum if the same fiber is used for both excitation laser delivery and emission signal collection. One work-around is to employ two optical fibers with one single-mode fiber for excitation light delivery and another large diameter multimode fiber for two-photon signal collection16, 31. However, to make the endomicroscope as compact and flexible as possible, a single DCF-based design is preferable12. To suppress the in-fiber background in this context, we designed and customized a DCF featuring a pure silica single-mode core, aiming to curtail the in-fiber nonlinear luminescence and the chance of quasi-phase matching for in-fiber SHG generation. The inner clad of the customized DCF is doped with fluorine to reduce the refractive index to guarantee a single-mode operation within the core. In addition, the DCF was designed to have an inner clad (185 μm in diameter) much larger than the commercial DCF inner clad (typically 100–130 μm in diameter; see ‘Methods’ section for details), representing a 2.0- to 3.4-fold increase in the collection area for two-photon signals.
Figure 1.

Fiber-optic scanning two-photon endomicroscope. The endomicroscope involved a single piece of DCF, and the mechanical assembly is illustrated in a; the inset is a photograph of the endomicroscope head (along with a ruler with 1-mm graduations) depicting the physical dimensions of the endomicroscope, which has an outer diameter of ~2.1 mm and a rigid length of ~35 mm. (b) Spectra of nonlinear in-fiber background emission from our customized DCF and two common types of commercial DCFs (SM-GDF-5/130 from NuFern and SMM900 from Fibercore), demonstrating the significantly reduced in-fiber background noise of our customized DCF.
The background spectrum of the customized DCF was measured and compared with two common commercial DCFs. Qualitatively, all three spectra exhibit a broad (~350–650 nm) nonlinear luminescence bandwidth plus a prominent SHG peak. Quantitatively, the total background emission rates from the two commercial DCFs are ~40- and ~15-fold increased, respectively, compared with the customized DCF (Figure 1b). Assuming the in-fiber background emission follows a Poisson distribution, the final imaging SNR (defined as the ratio of the average signal level to the standard deviation of the background) can be described as 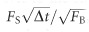 , where FS and FB denote the average emission rate of signal photons from the sample and the in-fiber background, respectively, and Δt the pixel dwell time. Therefore, suppressing the background signal FB by N-fold promotes the system detection sensitivity to the intrinsic 2PF or SHG signal by
, where FS and FB denote the average emission rate of signal photons from the sample and the in-fiber background, respectively, and Δt the pixel dwell time. Therefore, suppressing the background signal FB by N-fold promotes the system detection sensitivity to the intrinsic 2PF or SHG signal by 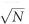 times. Quantitatively, considering the SHG emission band (435–455 nm with excitation at 890 nm), the pure silica-core DCF boosts the SNR by
times. Quantitatively, considering the SHG emission band (435–455 nm with excitation at 890 nm), the pure silica-core DCF boosts the SNR by 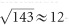 - and
- and 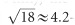 -fold compared with the commercial DCF SMM900 and NuFern 5/130, respectively. Regarding the NADH emission band used in this manuscript (417–477 nm), the corresponding SNR is enhanced by ~10- and ~3.8-fold, respectively (Supplementary Table S1).
-fold compared with the commercial DCF SMM900 and NuFern 5/130, respectively. Regarding the NADH emission band used in this manuscript (417–477 nm), the corresponding SNR is enhanced by ~10- and ~3.8-fold, respectively (Supplementary Table S1).
In addition to the customized DCF and effective temporal pulse-width management (Supplementary Note S1), a miniature objective is indispensable for focusing the excitation light to the sample and for epi-collecting 2PF (or SHG) photons from the sample back into the same DCF (primarily into the inner clad). Commonly used gradient-index (GRIN) lenses suffer from severe chromatic aberration, which leads to a substantial longitudinal focal shift (up to 1 mm) between the excitation (λ~750–900 nm) and emission (λ~400–550 nm) wavelengths19. The severe focal shift will cause emission light to be focused far away from the DCF end surface, substantially impairing the collection efficiency even with DCFs of a large diameter inner clad19. We therefore customized an achromatic miniature objective that integrated a diffractive mask. The mask adopted here, basically a circular phase diffractive grating32, bends longer wavelength light by a relatively larger angle and provides chromatic dispersion complementary to the GRIN elements in the miniature objective (Figure 2a). We measured the focal length at several wavelengths spanning a range of 480–900 nm. We observed that the sample-side longitudinal focal shift is well maintained below 10 μm (Figure 2b). Given the magnification (~4.8 ×) and the fiber-side NA (~0.175) of the miniature objective, one can estimate that the fiber-side focal shift is <230 μm. Ray-tracing computation indicates that ballistic signal photons emitted within the focal volume, once collected by the miniature objective (of an NA ~0.8, water immersion), will strike the DCF end surface over an area of 82 μm in diameter with a divergence NA of ~0.175. Thus, in principle, all ballistic signal photons can be readily collected by the DCF inner clad, which has a 185-μm diameter and a 0.35 NA. The extra allowance in the inner clad diameter and acceptance NA was designed to facilitate the collection of non-ballistic emission photons that experience multiple scattering in tissues. As revealed by ray-tracing simulation (Supplementary Note S3 and Supplementary Fig. S3), when imaging scattering tissues, the combination of our customized DCF (with a large inner clad diameter of 185 μm and high NA of 0.35) and the achromatic miniature objective can enhance the overall signal collection efficiency by at least ~2.5-fold compared with an endomicroscope employing a commercial DCF (Nufern 5/130 with a 130-μm inner clad diameter and a 0.46 NA) and a state-of-the-art GRIN objective without chromatic correction (GT-MO-080–018–810, GRINTECH GmbH, Jena, Germany). Combining this approximately 2.5 × enhancement of collection efficiency, the 4–10 × reduction of in-fiber background noise and the 2 × increase of 2PE efficiency provided by the innovative pulse-width management strategy (Supplementary Note S1), our endomicroscope affords an overall imaging SNR at least approximately 20–50 × as high as the prior art.
Figure 2.

Miniature objective and longitudinal focal shift. (a) Illustration of the customized miniature objective. A phase diffractive grating is sandwiched between two GRIN elements. The first GRIN element (~1/4 pitch) collimates the light from the DCF core, and the second GRIN element (<1/4 pitch) pre-focuses the light before it enters a high NA plano-convex lens for tight focusing. All the glass elements are encapsulated and fixed within a protective stainless-steel tube (1.0 mm inner diameter and 1.4 mm outer diameter). The fiber- and sample-side WDs are designed to be 200 μm in air and 200 μm in water, respectively. (b) Sample-side longitudinal focal shift measured over the wavelength range commonly involved in two-photon microscopy (see Supplementary Note S2 and Supplementary Fig. S2 for details of the measurement method).
A prototype flexible endomicroscope of an extremely compact form (that is, 2.1 mm in diameter) was successfully developed with the customized DCF and the achromatic miniature objective of a 200-μm sample-side WD and 0.8 NA (along with a PZT-based fiber-optic scanner) (Figure 1a). To meet the need of certain applications for a longer WD, we also developed a highly customized miniature objective of 300-μm sample-side WD while maintaining a similar sample-side NA (0.75 in water), achromaticity and a compact footprint (1.8-mm optics diameter). A prototype flexible endomicroscope equipped with this long WD miniature objective was also engineered accordingly. The two prototypes (compared in Supplementary Table S2) offered similar SNR improvement and image quality, as demonstrated in tissue imaging (see Figures 3 and 4, and Supplementary Figs. S4–S6).
Figure 3.

Endomicroscopy 2PF and SHG label-free structural imaging. (a, b) Overlay of intrinsic 2PF and SHG images acquired from mouse liver ex vivo. The emission signal was detected through two spectral channels: 496–665 nm (green, 2PF signal) and 435–455 nm (red, SHG signal). (c, d) Two-photon autofluorescence images of the mucosa of mouse small intestine in vivo. The two detection channels are 417–477 nm for NADH (green) and 496–665 nm for FAD (red). (e, f) SHG images of cervical collagen fiber network acquired through intact ectocervical epithelium of cervices dissected from preterm-birth mouse models (e) and normal pregnant mice (f) at gestation day 15. All images were acquired using the 200-μm WD miniature objective. The excitation conditions include ~30 mW at 890 nm (a, b), ~30 mW at 750 nm (c, d) and ~40 mW at 890 nm (e, f). Four raw frames are averaged in a–d and 10 in e, f, corresponding to an effective frame acquisition time of ~1.5 s (a–d) and ~3.8 s (e, f), respectively. Scale bar=10 μm.
Figure 4.

Endomicroscopy 2PF redox imaging of a mouse kidney ischemia–reperfusion model in vivo. Top row (a–c): 2PF intensity images from the NADH detection channel (417–477 nm); middle row (d–f): 2PF intensity images from the FAD detection channel (496–665 nm); bottom row (g–i): 2PF intensity images color-coded by the measured optical redox ratio defined as FAD/(FAD+NADH), where more reddish (greenish) color corresponds to an increased (reduced) concentration of NADH and thus a reduced (increased) redox ratio. The dark round-to-elliptical spots scattered along the renal tubule wall (dashed squares) correspond to the nuclei of renal tubular cells. Each column corresponds to one specific time point: normal (left), 2 min 30 s post ischemia (center) and 3 min 05 s post reperfusion (right). All images shown are averaged over five raw frames acquired using the 200-μm WD miniature objective with an incident power of ~33 mW at 750 nm, corresponding to an effective frame acquisition time of ~1.9 s. Scale bar=10 μm.
Label-free histological imaging of biological tissues
In light of the dramatically enhanced detection sensitivity, we assessed the performance of our fiber-optic endomicroscope for label-free imaging of various organs on ex vivo and in vivo mouse models. Figures 3a and 3b present overlaid intrinsic 2PF (green) and SHG (red) images acquired from the same lateral position on the surface of a freshly resected mouse liver. With the focal plane in Figure 3b positioned ~20 μm deeper compared with that in Figure 3a, the SHG signal from collagen fibers decreases, whereas cytoplasm 2PF increases, reflecting three-dimensional interlacing of individual hepatocytes and the supporting collagen fiber network. Notable in both figures are bright granules scattered around in the cytoplasm (arrows); these granules are believed to be from vitamin A, which is mainly stored in liver and known to emit bright autofluorescence33.
Figures 3c and 3d present superposed dual-channel intrinsic 2PF images at subcellular level acquired from intestinal villi of mouse small intestine mucosa in vivo with the NADH (FAD) detection channel pseudo-colored in green (red). Epithelial cells (mostly enterocytes) lining the villus wall are arranged in a mosaic pattern on the villus tip (Figure 3c), and the green-yellowish pseudo-color reflects the abundance of both NADH and FAD in the enterocytes34. A mucus-secreting goblet cell can be identified as a dark patch (dash box in Figure 3c) surrounded by enterocytes34. Moving the focal plane toward the basal side, we obtained a cross-section view of the villus wall (Figure 3d), where individual enterocytes and their nuclei (dark regions) are clearly delineated. Pseudo-yellowish punctate granules visible in both Figures 3c and 3d (arrows) are attributed to lysosomes34, which are critical for the absorption function of enterocytes and preferably cluster around the apical cytoplasm (arrows in Figure 3d). Bright reddish granules appearing within the villus lamina propria (arrowheads in Figure 3d) stem from the antigen-presenting cells, which also contain a large quantity of lysosomes34.
SHG images of cervical tissues from the Ru486 PTB mouse model25 (Figure 3e) and normal pregnant mice at the same gestation time (Figure 3f) are compared, revealing dramatic changes in the cervical collagen fiber architecture. The images of subepithelial collagen fibers were acquired through the intact ectocervical epithelium by gently pressing the endomicroscope against the ectocervix of dissected mouse cervices and imaging. The collagen fiber architecture from the PTB group is more porous, suggesting the presence of pre-mature cervical ripening as reported in previous studies performed using bench-top SHG microscopy26. Although the images presented in Figure 3 are averaged over multiple raw frames, the corresponding single-frame images (Supplementary Fig. S4) can generally capture very similar structural details, demonstrating the excellent detection sensitivity of the endomicroscope.
Redox ratio/metabolic function imaging of the ischemia–reperfusion process in vivo
In addition to structural information, the endomicroscope can also assess the spatially resolved metabolic status of biological tissues in vivo by quantitatively imaging the ratio of FAD and NADH fluorescence; this optical redox ratio is sensitive to the vascular oxygen supply35. For demonstration, we chose an in vivo acute mouse kidney ischemia–reperfusion model, where we clamped the left renal artery and vein of an anesthetized mouse to induce ischemia (~4 min) and then released the clamp to allow reperfusion. Changes in the redox ratio of renal cortical tubules were monitored on top of cellular structures in vivo with our endomicroscope.
First, the nuclei of tubular cells are discernible as dark round circles scattered along renal tubule walls (dashed squares in Figure 4). Notably, both NADH and FAD fluorescence concentrate predominantly along the basolateral side of the tubular cells (arrowheads), whereas the fluorescence signal along the apical side is comparatively much dimmer (arrows). This observation is consistent with the known basolateral distribution of mitochondria in renal tubular cells36. Second, in response to ischemia, the NADH fluorescence intensity increased (Figure 4b and Supplementary Fig. S5b), whereas the redox ratio defined as FAD/(FAD+NADH) decreased markedly (Figure 4h and Supplementary Fig. S5h). Both changes were subsequently reversed upon reperfusion (Figures 4c and 4i;Supplementary Figs. S5c and S5i). The reduced NADH fluorescence and increased redox ratio observed under normal (Figures 4a and 4g) and post-reperfusion (Figures 4c and 4i) conditions reflect the relatively reduced amount of NADH (compared to ischemia), which is consistent with previous findings that the mitochondrial NAD pool of renal tubular cells resides in a more oxidized state (that is, containing more NAD+ than NADH) under resting conditions36, 37. For quantitative analysis, we repeated this acute ischemia–reperfusion process at nine randomly selected imaging locations (all on renal tubules). For each location, NADH and FAD images were acquired at five different time points, including one time point under normal conditions (before ischemia), two under ischemia and another two following reperfusion. For each pair of NADH and FAD images, we calculated the mean NADH intensity and mean redox ratio (which were averaged over the entire imaging field of view). Using the three data sets of mean values (9 data points under normal conditions, 18 under ischemia and 18 post reperfusion), an unpaired two-sample Welch’s t-test was performed in MATLAB (MATLAB 2013b, The MathWorks, Inc., Natick, MA, USA) to compare the normal (or reperfusion) against the ischemia conditions, and the resultant P-values corroborated the statistical significance of ischemia-induced fluctuations in both NADH intensity and redox ratio (Figures 5a and 5b). We also observed that the tubular cells after reperfusion exhibited a reduced mean NADH intensity (Figure 5a, P=0.001) and increased mean redox ratio (Figure 5b, P<0.001) compared with normal conditions. This finding is also reflected by the overall more greenish tone of Figure 4i compared with Figure 4g. Such phenomena suggest incomplete recovery of tissue metabolic states post reperfusion, which might result from either acute ischemia-induced tissue damage or insufficient reperfusion time. More importantly, all the image features and redox ratio changes were also discernible even from the non-averaged single-frame images (Supplementary Fig. S6), demonstrating the superb detection sensitivity of the endomicroscopy system that enables real-time visualization of both structural and functional information on label-free tissues in vivo.
Figure 5.

Comparison of mean NADH intensity and mean redox ratio between normal (or reperfusion) and ischemia conditions. By repeating the ischemia–reperfusion process for nine random imaging locations on renal tubules and five different time points for each location, we calculated 45 data points of mean NADH intensity (or redox ratio) values. Error bars represent the standard deviation of those mean values across 9 (normal), 18 (ischemia) and 18 (reperfusion) data points. Unpaired two-sample Welch’s t-tests were performed between normal (or reperfusion) and ischemia conditions, and the resultant P-values reveal that the increase (decrease) of NADH intensity (redox ratio) upon ischemia and the reverse trends following reperfusion are statistically significant. See Supplementary Note S4 and Supplementary Figs. S7–S8 for details on redox ratio validation using our two-photon endomicroscope.
Conclusions
In summary, our newly developed fiber-optic two-photon endomicroscope has achieved unprecedented high detection sensitivity and is capable of capturing label-free sub-cellular structural and dynamic functional/metabolic information in vivo with spatial resolution and image quality approaching a standard bench-top two-photon microscope. Notably, only a moderate level of incident power (~30–40 mW) was adequate for all experiments reported above, and no visual signs of photodamage were observed throughout the entire imaging process. The flexible and extremely compact outfit of this endomicroscopy technology enables its delivery to internal luminal organs for label-free, biopsy-free, histological imaging in vivo, in situ and in real time, which opens up new possibilities to a wide range of translational preclinical and clinical applications, such as guidance of biopsy in internal organs with improved diagnostic yield and assessment of abnormal cervical collagen remodeling to predict the risk of preterm birth26. Furthermore, this cost-effective, lightweight (<1 g) and compact two-photon endomicroscope also serves as an enabling tool for basic research, for example, for brain imaging on freely behaving animals with a head-mount configuration to gain new insights into dynamic neurological processes with high spatial resolution and for imaging studies of infectious disease for which expensive bench-top scanning two-photon microscope might not be an economically viable choice. The imaging speed of the current endomicroscope is ~3 frames per second, and increased speed represents one future direction for further development of this enabling technology.
Acknowledgments
The authors gratefully thank Mala Mahendroo and Kate Phelps for providing the mouse cervical tissues and Jiefeng Xi for helpful discussions. This research was partially supported by the National Institutes of Health under a Grant R01CA153023 (XL), the National Science Foundation under Grant CBET-1430040 (XL) and the Individual Biomedical Research Award (XL) from The Hartwell Foundation.
Footnotes
Note: Supplementary Information for this article can be found on the Light: Science & Applications’ website(http://www.nature.com/lsa).
The authors declare no conflict of interest.
Supplementary Material
References
- Kiesslich R, Goetz M, Vieth M, Galle PR, Neurath MF. Technology insight: confocal laser endoscopy for in vivo diagnosis of colorectal cancer. Nat Clin Pract Oncol 2007; 4: 480–490. [DOI] [PubMed] [Google Scholar]
- Mondal SB, Gao SK, Zhu N, Liang RG, Gruev V et al. Real-time fluorescence image-guided oncologic surgery. Adv Cancer Res 2014; 124: 171–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SH, Heikal AA, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J 2002; 82: 2811–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnola PJ, Loew LM. Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. Nat Biotechnol 2003; 21: 1356–1360. [DOI] [PubMed] [Google Scholar]
- Quinn KP, Sridharan GV, Hayden RS, Kaplan DL, Lee K et al. Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci Rep 2013; 3: 3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science 1990; 248: 73–76. [DOI] [PubMed] [Google Scholar]
- Flusberg BA, Cocker ED, Piyawattanametha W, Jung JC, Cheung ELM et al. Fiber-optic fluorescence imaging. Nat Methods 2005; 2: 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods 2005; 2: 932–940. [DOI] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol 2003; 21: 1369–1377. [DOI] [PubMed] [Google Scholar]
- Xu C, Zipfel W, Shear JB, Williams RM, Webb WW. Multiphoton fluorescence excitation: new spectral windows for biological nonlinear microscopy. Proc Natl Acad Sci USA 1996; 93: 10763–10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobizhev M, Makarov NS, Tillo SE, Hughes TE, Rebane A. Two-photon absorption properties of fluorescent proteins. Nat Methods 2011; 8: 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myaing MT, MacDonald DJ, Li XD. Fiber-optic scanning two-photon fluorescence endoscope. Opt Lett 2006; 31: 1076–1078. [DOI] [PubMed] [Google Scholar]
- Fu L, Jain A, Xie HK, Cranfield C, Gu M. Nonlinear optical endoscopy based on a double-clad photonic crystal fiber and a MEMS mirror. Opt Express 2006; 14: 1027–1032. [DOI] [PubMed] [Google Scholar]
- Fu L, Jain A, Cranfield C, Xie HK, Gu M. Three-dimensional nonlinear optical endoscopy. J Biomed Opt 2007; 12: 040501. [DOI] [PubMed] [Google Scholar]
- Fu L, Gu M. Fibre-optic nonlinear optical microscopy and endoscopy. J Microsc 2007; 226: 195–206. [DOI] [PubMed] [Google Scholar]
- Le Harzic R, Riemann I, Weinigel M, König K, Messerschmidt B. Rigid and high-numerical-aperture two-photon fluorescence endoscope. Appl Opt 2009; 48: 3396–3400. [DOI] [PubMed] [Google Scholar]
- Piyawattanametha W, Cocker ED, Burns LD, Barretto RPJ, Jung JC et al. In vivo brain imaging using a portable 2.9g two-photon microscope based on a microelectromechanical systems scanning mirror. Opt Lett 2009; 34: 2309–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Leng YX, Xi JF, Li XD. Scanning all-fiber-optic endomicroscopy system for 3D nonlinear optical imaging of biological tissues. Opt Express 2009; 17: 7907–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Xi JF, Cobb MJ, Li XD. Scanning fiber-optic nonlinear endomicroscopy with miniature aspherical compound lens and multimode fiber collector. Opt Lett 2009; 34: 953–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YB, Nakamura H, Gordon RJ. Development of a versatile two-photon endoscope for biological imaging. Biomed Opt Express 2010; 1: 1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera DR, Brown CM, Ouzounov DG, Pavlova I, Kobat D et al. Compact and flexible raster scanning multiphoton endoscope capable of imaging unstained tissue. Proc Natl Acad Sci USA 2011; 108: 17598–17603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CM, Rivera DR, Pavlova I, Ouzounov DG, Williams WO et al. In vivo imaging of unstained tissues using a compact and flexible multiphoton microendoscope. J Biomed Opt 2012; 17: 040505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducourthial G, Leclerc P, Mansuryan T, Fabert M, Brevier J et al. Development of a real-time flexible multiphoton microendoscope for label-free imaging in a live animal. Sci Rep 2015; 5: 18303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WX, Murari K, Zhang YY, Chen YP, Li MJ et al. Increased illumination uniformity and reduced photodamage offered by the Lissajous scanning in fiber-optic two-photon endomicroscopy. J Biomed Opt 2012; 17: 021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DJ, Branch DW, Edwin SS, Mitchell MD. Induction of preterm birth in mice by RU486. Biol Reprod 1996; 55: 992–995. [DOI] [PubMed] [Google Scholar]
- Akins ML, Luby-Phelps K, Mahendroo M. Second harmonic generation imaging as a potential tool for staging pregnancy and predicting preterm birth. J Biomed Opt 2010; 15: 026020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Akins ML, Murari K, Xi J, Li M-J et al. A compact fiber-optic SHG scanning endomicroscope and its application to visualize cervical remodeling during pregnancy. Proc Natl Acad Sci USA 2012; 109: 12878–12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonyuk BP, Antonyuk VB. Second-harmonic generation in Ge-doped silica fibres: experiment and theory. J Mod Opt 1998; 45: 257–268. [Google Scholar]
- Udovich JA, Kirkpatrick ND, Kano A, Tanbakuchi A, Utzinger U et al. Spectral background and transmission characteristics of fiber optic imaging bundles. Appl Opt 2008; 47: 4560–4568. [DOI] [PubMed] [Google Scholar]
- Chang Y-C, Ye JY, Thomas T, Chen Y, Baker JR et al. Two-photon fluorescence correlation spectroscopy through a dual-clad optical fiber. Opt Express 2008; 16: 12640–12649. [DOI] [PubMed] [Google Scholar]
- Engelbrecht CJ, Johnston RS, Seibel EJ, Helmchen F. Ultra-compact fiber-optic two-photon microscope for functional fluorescence imaging in vivo. Opt Express 2008; 16: 5556–5564. [DOI] [PubMed] [Google Scholar]
- Stone T, George N. Hybrid diffractive-refractive lenses and achromats. Appl Opt 1988; 27: 2960–2971. [DOI] [PubMed] [Google Scholar]
- Suematsu M, Oda M, Suzuki H, Kaneko H, Watanabe N et al. Intravital and electron microscopic observation of Ito cells in rat hepatic microcirculation. Microvasc Res 1993; 46: 28–42. [DOI] [PubMed] [Google Scholar]
- Orzekowsky-Schroeder R, Klinger A, Martensen B, Blessenohl M, Gebert A et al. In vivo spectral imaging of different cell types in the small intestine by two-photon excited autofluorescence. J Biomed Opt 2011; 16: 116025. [DOI] [PubMed] [Google Scholar]
- Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci USA 2007; 104: 19494–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AM, Rhodes GJ, Sandoval RM, Corridon PR, Molitoris BA. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int 2013; 83: 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AM, Unwin RJ, Parker N, Duchen MR. Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J Am Soc Nephrol 2009; 20: 1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.