

Abstract
Two pharmacologic approaches that are currently at the forefront of treating advanced cancer are those that center on disrupting critical growth/survival signaling pathways within tumor cells (commonly referred to as “targeted therapies”) and those that center on enhancing the capacity of a patient’s immune system to mount an antitumor response (immunotherapy). Maximizing responses to both of these approaches requires an understanding of the oncogenic events present in a given patient’s tumor and the nature of the tumor-immune microenvironment. Although these 2 modalities were developed and initially used independently, combination regimens are now being tested in clinical trials, underscoring the need to understand how targeted therapies influence immunologic events. Translational studies and preclinical models have demonstrated that targeted therapies can influence immune cell trafficking, the production of and response to chemokines and cytokines, antigen presentation, and other processes relevant to antitumor immunity and immune homeostasis. Moreover, because these and other effects of targeted therapies occur in nonmalignant cells, targeted therapies are being evaluated for use in applications outside of oncology.
Keywords: Immunopharmacology, Oncology, Drug Information, Molecular Biology, Pharmaceutical R & D
The recent success of medications that enhance antitumor immune responses by disrupting “immune checkpoints” highlights the therapeutic dividends that can be paid by understanding the regulatory mechanisms of the immune system.1 Using this approach, commonly termed “immune checkpoint blockade” has yielded clinical advances across a spectrum of malignancies including melanoma, lung cancer, and renal cell carcinoma.2–4 The success of these agents illustrates the enormous translational potential of understanding and controlling the immune system and justifies additional research to identify other regulatory aspects of the immune system as candidates for new drug development.5 In addition, these clinical successes have underscored the need to define the immune effects of all forms of anticancer therapy for the following reasons: (1) a properly steered antitumor immune response can be curative even in patients with advanced cancer refractory to other therapies; (2) various anticancer agents have underappreciated immunomodulatory activity; (3) the optimal use of regimens that combine immune-based treatments with other forms of therapy will require a sophisticated and comprehensive understanding of all of the immune effects that are at play.6,7 In-depth reviews have highlighted these issues with memorable titles in which these effects have been described as “the secret ally” and in terms of what happens when “universes collide”.8,9 As the concept of cancer evolves from one that is cell-autonomous to 1 that incorporates the complex role of the tumor microenvironment on cancer growth and regression, the approach to therapy must also evolve.7,10 By understanding the numerous interactions between tumor cells and nontumor cells in the microenvironment that underpin cancer pathogenesis, progression, and the response to therapy, we can continue to build on the exciting momentum that has been generated by newer anticancer therapies. Likewise, because most anticancer treatments are not immunologically null, characterizing the occult immune effects of therapies not originally developed as immunotherapies may facilitate the development of more precise and effective combinatorial treatment paradigms.
The Immune Effects of Growth Factors
Before considering how targeted therapies influence immune responses, it is worth providing some relevant background as it relates to the underlying biology of growth factors. As described above, targeted therapies were developed to specifically block aberrantly activated signal transduction pathways within malignant cells, many of which arise from deregulated growth factor receptor tyrosine kinases (RTKs) or cellular protein kinases that can initiate oncogenic signaling.11,12 Many of these enzymes and signal transduction pathways were originally defined and conceptualized for their roles in growth factor (and growth factor receptor) biology rather than their roles in immunology.13 Thus, because many targeted therapies directly disrupt growth factor signaling (within tumor cells and potentially nontumor cells), it is important to revisit the immune effects of growth factors as a framework to understand the immune effects of targeted therapies. In the paragraphs below we use vascular endothelial growth factor (VEGF) and the epidermal growth factor receptor (EGFR) ligand amphiregulin (AREG) as model growth factors and briefly review some of their reported immune effects. These growth factors were selected because pharmacologic inhibitors of VEGF and the EGFR are currently in clinical use.14,15
VEGF is a proangiogenic growth factor that is crucial to maintaining tumor vascularity and nutrient supply.16 Its role in tumor angiogenesis fueled the development of bevacizumab, an anti-VEGF monoclonal antibody (mAb).17 In addition to its role in angiogenesis, VEGF has been shown to have multiple immunosuppressive effects including inhibition of T-cell development, inhibition of dendritic cell maturation, and an ability to promote the recruitment of immunosuppressive myeloid-derived suppressor cells (MDSCs).18–21 Moreover, VEGF has been shown to promote the induction of regulatory T cells (T regs) in the tumor microenvironment of mice as well as metastatic colorectal cancer patients.22 Recent studies have suggested VEGF expression by tumor cells promotes the expression of the inhibitory receptor programmed cell death (PD)-1 on the surface of CD8+ T cells, thereby promoting an “exhausted” phenotype of tumor-infiltrating cytotoxic T cells.23 Thus, despite being initially characterized for its effects on vasculature biology, VEGF expression can influence immune processes that may be central to the generation of an antitumor immune response.24 In support of this notion, higher pretreatment VEGF serum levels are associated with shorter overall survival times in melanoma patients treated with the immune checkpoint inhibitor ipilimumab, a mAb that blocks cytotoxic T lymphocyte antigen 4 (CTLA4).25 Moreover, regimens combining ipilimumab (anti-CTLA4) and bevacizumab (anti-VEGF) are being evaluated in patients with melanoma and glioblastoma.26–29 These studies have demonstrated that the combination of bevacizumab (anti-VEGF) and ipilimumab (anti-CTLA4) can be safely administered with predictable and manageable toxicity. Moreover, these studies illustrate that the combination of bevacizumab and ipilimumab can augment endothelial cell activation, lymphocyte infiltration into tumors, cytokine and chemokine expression, and antimelanoma humoral responses.
AREG is a member of the EGF family of ligands that includes several growth factors (additional factors include EGF, transforming growth factor-α, epigen, and several others) that bind to and activate members of the human EGF receptor (HER) family that are worth briefly reviewing.30,31 The HER family includes 4 structurally related receptors that have distinct ligand-binding abilities, intrinsic tyrosine kinase activities, and abilities to form homo- and heterodimers with other members of this family. These RTKs have sequence and structural homology with the EGFR (also referred to as ErbB1/HER1) and include ErbB2/HER2/neu, ErbB3/HER3 and ErbB4/HER4. The ErbB (or c-erb-B) designation stems from the fact that these RTKs share homology with the avian erythroblastic leukemia viral (v-erb-B) oncogene.32 The additional neu designation for ErbB2/HER2 stems from the name given to this oncogene when it was originally cloned as an oncogene isolated from the neuro/glioblastomas that developed in the offspring of rats injected with the carcinogen ethylnitrosourea.33 Both the EGFR and ErbB2 have received enormous attention as therapeutic targets for cancers of the lung, colon, and head and neck (and others) for the EGFR, and breast cancer for ErbB2.34–37
Like other ErbB ligands, AREG is known to bind to and activate the EGFR.38 AREG derives its name from its seemingly paradoxical ability to induce cell proliferation in some cell lines, whereas it induces growth arrest and differentiation in others.39 Although ErbB ligands such as AREG have been mainly studied in the context of epithelial development and epithelial cancers,40–42 their roles in the immune system have received less attention until very recently. For example, AREG is known to be expressed by epithelial cells, yet it is also expressed by cellular components of the immune system including dendritic cells, neutrophils, mast cells, and CD4+ T cells.43 Key immunologic roles for AREG have recently been uncovered by Zaiss and colleagues when they discovered that AREG modulates the activity of T regs in mouse models of colitis and melanoma.44 More recently, AREG has been implicated in the immune suppression mediated by ultraviolet radiation (UVR), which plays an important role in the development of UVR-induced skin cancers.45,46 Thus, canonical growth factor ligands have pleiotropic immune effects in part through their ability to modulate the function of immune cells such as T lymphocytes. As a result, targeted therapies that inhibit growth factor receptors and/or their downstream kinases influence processes within tumor cells and immune cells within the tumor microenvironment, and both are likely to be relevant to the generation of effective antitumor immunity.
Effects of Targeted Therapies on Tumor Cells Relevant to Antitumor Immunity
There is mounting evidence that inhibition of oncogenic signaling using targeted therapies can influence tumor:immune cell interactions. For example, the selective BRAFV600E kinase inhibitors vemurafenib and dabrafenib induce marked T lymphocyte infiltration into melanoma tumors.47 In addition, the EGFR blocking antibody cetuximab was shown to activate natural killer (NK) cells to promote dendritic cell maturation and CD8+ T-lymphocyte activation.48 Such effects likely depend on a variety of interrelated processes including those mediated by tumor-intrinsic factors, therapy-dependent factors, and host-dependent factors. Tumor-intrinsic factors will likely include the cellular origin of the tumor, its genomic and epigenetic landscape, and the activation status of signal transduction pathways within tumor cells. For example, recent studies have suggested that the load of neoantigens (altered peptides resulting from mutations, deletions, or translocations in the coding sequences of genes) expressed by a tumor can be relevant to antitumor immunity by increasing the likelihood that tumor cells are recognized as foreign (nonself) by the immune system.49
There are several therapy-dependent factors that will likely influence how these medications modulate the generation of an anticancer immune response. For example, whether a medication is a therapeutic mAb or a small molecule kinase inhibitor will likely be relevant because (as discussed in more detail below) antibodies possess unique immunologic properties. Equally important will be the medication’s mechanism(s) of action. These may include the specific pathways/enzyme(s) that are inhibited as well as the cells (tumor cells and nontumor cells) that are impacted by the medication in question. It is also worth noting the possibility of off-target effects and differences in this regard between mAbs and small molecule kinase inhibitors. The mAbs bind with high specificity to extracellular targets to disrupt growth factor receptor signaling. In contrast, small-molecule kinase inhibitors act on intracellular enzymes and may influence the activity of other targets than their intended one. This makes understanding and interpreting their immunologic impact on the tumor microenvironment more complex because both nontumor and off-target effects need to be considered.50
Host-dependent factors may include variables that influence immune responses in general such as age and comorbidities such as diabetes and obesity.51–53 It is also possible that a person’s genotype may play a role because genetic polymorphisms can influence the development of toxicity in response to targeted therapies.54 In the paragraphs below we discuss how the effects of targeted therapies on tumor cells may influence antitumor immune responses.
Antibody-Mediated Effects From Therapeutic Monoclonal Antibodies
As mentioned in the previous paragraphs, targeted therapies used in cancer can be broadly divided into small-molecule kinase inhibitors used to inhibit the activity of oncogenic enzymes and therapeutic mAbs that disrupt the activation of growth factor receptors or other receptors germane to cancer.55,56 Because antibodies are normal elements of the immune system, therapeutic mAbs not only disrupt oncogenic signaling but additionally provide a platform for interactions between tumor cells and immune cells. For example, therapeutic mAbs have been reported to enhance the ability of cells of the immune system to engulf tumor cells, a process known as opsonization.57 This occurs because many cells of the immune system express specialized antibody-binding receptors that bind to the Fc region of mAbs (Figure 1). These receptors, known as Fcγ receptors (FcγRs), are expressed by immune cells such as NK cells, monocytes, and granulocytes and can play a crucial role in the antibody-mediated recognition and killing of tumor cells.58 This antibody-mediated process is known as antibody-dependent cellular cytotoxicity (ADCC). The triggering of ADCC involves the release of cytotoxic granules containing perforin and granzyme, a theme common to other forms of antitumor immunity such as those mediated by cytotoxic T lymphocytes (CTLs). The ability to generate ADCC has been proposed as an important antitumor mechanism of action for mAbs that bind to the EGFR.59,60 Conceptually, this enables mAbs to both disrupt EGFR-mediated growth and survival signals within tumor cells while also initiating an immune-mediated attack at the surface of the tumor cell. Moreover, a better clinical response to the anti-EGFR mAb cetuximab has been linked to specific polymorphisms present within FcγRs.58,61,62 These genetic variations can influence the binding affinities of FcγRs for mAbs, which in turn can influence the potency of ADCC-mediated antitumor responses.63 This suggests that genetic variation within immunologic genes can influence the response of cancer patients to mAbs.61–64 Similar correlations of FcγR genotype and clinical outcome have been observed for trastuzumab, a HER2-specific therapeutic mAb used in treatment of breast cancer.65,66 Thus, through their ability to engage with immune cells, therapeutic mAbs are intrinsically able to mediate interactions between tumor cells and cells of the immune system to influence antitumor immunity.67
Figure 1.
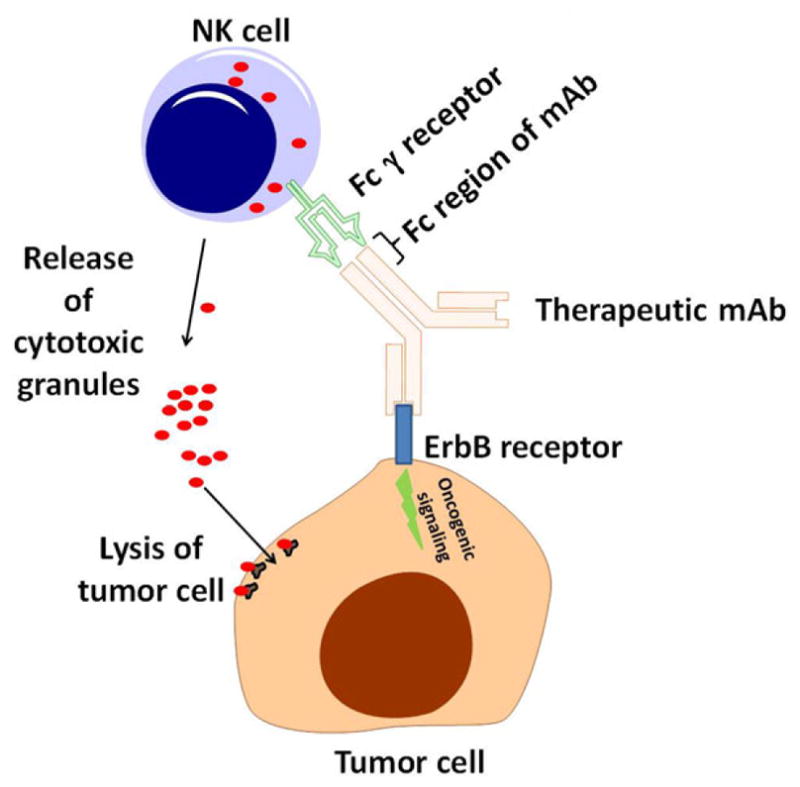
Monoclonal antibody (mAb)-based targeted therapies can facilitate interactions between tumor cells and immune cells. Therapeutic mAbs can bind to growth factor receptors (such as members of the ErbB family of receptor tyrosine kinases). In addition to disrupting oncogenic signals emanating from these receptors, mAbs mediate interactions between tumor cells and immune cells (such as natural killer [NK] cells shown above). Cells such as NK cells can recognize the Fc regions of therapeutic mAbs via their Fcγ receptors. This can lead to antibody-dependent cellular cytotoxicity and tumor cell lysis.
In line with the above concept, there is evidence that the specific isotype and/or glycosylation status of an individual therapeutic mAb is relevant in this regard because these differences influence the aforementioned interactions.59,60,68 For example, the immunoglobulin G (IgG) isotype of EGFR-specific mAbs is thought to be important in determining which lineage of cells predominates in mediating ADCC.69 FcγRIIIA (CD16) is expressed on the surface of NK cells but not neutrophils and has a higher affinity for IgG1 over IgG2 suggesting that IgG2-based therapeutic mAbs would be less able to stimulate this form of cellular killing by NK cells. There is evidence to suggest that this is indeed the case. For example, panitumumab and zalutumumab are EGFR-specific antibodies of the human IgG2 and IgG1 isotypes respectively and NK cell-mediated ADCC occurs with zalutumumab and not with panitumumab.70 In contrast, both antibodies are equally effective at stimulating ADCC by neutrophils that express different Fcγ receptors than NK cells that have different IgG isotype affinities.69
Monoclonal Antibody-Based Anticancer Therapies Can Act via Complement Activation
Another immune-related mechanism through which anticancer mAbs may be working is via the activation of complement and complement-dependent cytotoxicity (CDC). CDC can be initiated through interactions between the Fc regions of mAbs and complement proteins such as C1q.71 These interactions can promote the assembly of the membrane attack complex on the tumor cell surface, which can alter tumor cell membrane permeability and lead to tumor cell lysis.72 In vitro studies have demonstrated a complement-mediated cytotoxic effect on tumor cells for CD20-targeted mAbs rituximab and ofatumumab.73 Additional studies using EGFR-inhibiting mAbs have shown that when used alone these agents do not induce complement deposition or CDC. However, combination therapy of cetuximab and matuzimab, 2 EGFR-specific antibodies that bind to distinct parts of the receptor, elicits strong synergistic complement deposition and thus tumor cell lysis.74
Targeted Therapies Can Influence the Expression of Genes Involved in Antigen Processing, Presentation, and Tumor-Associated Antigens
The generation of an antitumor immune response mediated by CD4+ and CD8+ T lymphocytes has been and continues to be a major goal of most forms of immunotherapy. Although the generation of a T-cell-mediated antitumor immune response is a complex process, a central theme involves the presentation of peptide antigens by tumor cells to CD4+ and/or CD8+ T lymphocytes; a pivotal step in the generation of antitumor immunity. A comprehensive review of antigen processing and presentation is beyond the scope of this review, but in the subsequent paragraphs we provide a brief overview and describe how targeted therapies may influence these processes.
Antigen processing involves the cleavage of proteins into peptides that are ultimately presented by major histocompatibility complex (MHC) class I (MHCI) and class II (MHCII) molecules.75 Peptides that are presented by MHCI molecules are derived from intracellular proteins, whereas those bound for presentation by MHCII molecules are derived from exogenous sources. An important caveat to this paradigm can occur within professional antigen-presenting cells, which involves a process called “cross-presentation” whereby exogenous antigens can be presented by MHCI molecules.76 Cross-presentation aside, the typical generation of peptides for presentation by MHCI molecules that occurs within tumor cells and their normal counterparts involves 4 tasks that include peptide generation and trimming, peptide transport, assembly of the MHCI loading complex, and presentation of peptides at the cell surface.77,78 Cleavage of proteins into peptides occurs by proteolysis (via the ubiquitin-proteosome pathway), and peptide transport into the endoplasmic reticulum is mediated by the transporter associated with antigen processing (TAP) complex, which is composed of a complex of 2 proteins named TAP1 and TAP2. Importantly, targeted therapies can influence the expression of TAP proteins. For example, the anti-EGFR mAb cetuximab has been shown to increase the expression of TAP1, TAP2, and other proteins involved in antigen processing.79 In addition, the BRAFV600E kinase inhibitor vemurafenib was shown to enhance the induction (by interferon [IFN]-α2b) of calnexin and calreticulin proteins involved in antigen processing.80 Thus, although tumor cells frequently downregulate proteins involved in antigen processing and presentation as a means of immune escape, there is evidence that targeted therapies can enhance the expression of these genes.81,82
As noted above, once peptides are generated, their presentation at the cell surface ultimately involves them being “loaded” noncovalently onto MHCI or MHCII molecules.83,84 These peptide-MHC (pMHC) complexes at the cell surface are then able to engage the T-cell receptor on the surface of T lymphocytes, providing the antigen-specific signal (often referred to as “signal 1”) required for T-cell activation.85 In general, higher surface MHCI and/or MHCII expression is considered helpful for T-cell-mediated antitumor immunity and as a result is typically associated with better prognosis.86–90 This may reflect the fact that higher pMHC densities can promote T-cell activation.91,92 Not surprisingly, just as tumors downregulate proteins involved in antigen processing as a means of immune escape, they also downregulate the expression of MHC molecules via mechanisms that can be reversible or irreversible.78,93–96 As described below, in some contexts targeted therapies and the kinases they were developed to inhibit have been shown to alter the expression of MHC molecules (Figure 2).95,97–101
Figure 2.

Targeted therapies can increase the expression of MHC molecules. Before treatment (left panel), tumor cells can evade detection by CD8+ T lymphocytes due to low expression of MHC class I molecules. During treatment with a targeted therapy (right panel) the expression of MHC class I molecules may be increased either directly or by enhancing the induction of MHC molecules by cytokines present within the tumor microenvironment. This leads to enhanced recognition of tumor cells by CD8+ T lymphocytes, the activation of antitumor T lymphocytes, and enhanced antitumor immunity. Increases in MHC class II may also occur, leading to enhanced CD4+-mediated T-cell responses (not shown).
EGFR inhibitors (EGFRIs) including tyrosine kinase inhibitors (TKIs) as well as the anti-EGFR mAb cetuximab have been shown to increase surface expression of both MHC class I and class II in primary and malignant keratinocytes in vitro and in skin biopsies from cancer patients receiving anti-EGFR treatment.102 Conversely, EGFR ligands and other ErbB ligands can have the opposite effect and repress MHC induction.102–107 Several other studies have confirmed that inhibition of the EGFR or other members of the ErbB family enhances MHC expression.100,101,108,109 Likewise, vemurafenib, a selective inhibitor of the BRAFV600E mutant kinase, has been shown to augment the induction of MHC class I and II expression by interferons in some BRAFV600E-positive melanoma cells.80,110 In contrast, forced expression of BRAFV600E had the opposite effect and could repress MHC class I levels.110 This finding has been further supported by a study demonstrating that the BRAFV600E mutant kinase can directly target MHC class I molecules for internalization and degradation, thereby promoting tumor growth and immune evasion.111 These effects are consistent with in vivo studies demonstrating that in response to BRAFV600E inhibitors, tumor infiltration by CD8+ T cells is increased.112 Moreover, increases in tumor-infiltrating CD8+ T cells during treatment with the BRAFV600E inhibitors vemurafenib and dabrafenib were associated with a reduction in tumor size.47 It is worth noting that although an increase in MHCI expression on tumor cells could potentially enhance the recognition and killing of tumor cells by CD8+ T cells, it could have the opposite effect on antitumor NK cells, which express inhibitory receptors for MHCI molecules known as killer immunoglobulin-like receptors.113 Further complicating this picture is the fact that ligands for activating NK-cell receptors such as MHC class I–related chain A and B can be regulated by growth factor signaling.114 These findings illustrate the intimate yet complex relationship between canonical oncogenic signaling pathways and the cellular machinery that influences interactions between tumor cells and cells of the immune system. In addition to altering the expression of proteins involved in antigen processing and presentation, targeted therapies have also been shown to alter the expression of proteins and the peptides derived from them that can function as tumor-associated antigens (TAAs).
TAAs are usually defined as peptides that are expressed, processed, and presented by tumor cells.115 While they may not be entirely tumor specific and may be expressed by nontumor cells, TAAs have the potential to facilitate the recognition of tumor cells by T lymphocytes.116 Importantly, targeted therapies have been shown to influence antitumor immunity by altering the expression of TAAs. For example, melanocyte differentiation antigens (MDAs) are TAAs derived from gene products (proteins) that play a role in melanocyte differentiation and as such can be expressed by melanoma cells. In response to treatment with a selective BRAFV600E kinase inhibitor (named PLX4720), the expression of MDAs was found to be augmented with resulting increases in antigen-specific immune responses against MDA-derived peptides.117 Thus, targeted therapies can influence antitumor immunity by changing the expression of proteins, and thus the pool of presented peptides, in a manner that can facilitate the generation of an antigen-specific antitumor immune response (Figure 3).
Figure 3.

Targeted therapies can increase expression of tumor-associated antigens. Targeted therapies may influence antitumor immunity by increasing the expression of tumor-associated antigens that can be recognized by host T lymphocytes. Before treatment (left panel) the tumor cell is not recognized by host T lymphocytes. In response to treatment with a BRAFV600E inhibitor (right panel) there is a change in the expression of tumor-associated antigens derived from melanocyte differentiation antigens that can promote the recognition of tumor cells by T lymphocytes. The recognition of tumor-associated antigens by T lymphocytes facilitates their activation and the generation of an antitumor immune response.
Recent studies have further highlighted the importance of the antigens presented by tumors by demonstrating that the presentation of neoantigens is pivotal for clinical responses to ipilimumab (anti-CTLA4 antibody).118 It is worth noting that neoantigen expression is influenced by the mutational burden of a tumor. Some tumors are associated with high mutational burdens (such as ultraviolet radiation–induced skin cancers) and therefore high neoantigen expression, whereas other tumors with lower mutational burdens may express few such neoantigens.49 Although the impact of targeted therapies on neoantigen expression is unknown, it is conceivable that through their ability to influence antigen processing, presentation, and gene expression in general, they may influence the expression of neoantigens.
Targeted Therapies Can Influence PD-L1 Expression
In settings of chronic antigen exposure, the normal development of activated CD8+ T cells into memory T cells can become disrupted. In such settings, CD8+ T cells can enter a state where they lose important functions such as the ability to secrete cytokines and kill (virally infected or malignant) cells.119 In addition to these deficits, another important hallmark of this “exhausted” state is an increased expression of inhibitory receptors such as PD-1.120 When PD-1 binds its ligands programmed death ligand-1 and 2 (PD-L1 and PD-L2), T-cell activation is repressed.121 This “immune checkpoint” limits immune responses including those against tumors and chronic viral infections.1 Fortunately, the functional deficits exhibited by exhausted CD8+ T cells can be reversed by blocking the interactions between inhibitory receptors and their ligands. This is highlighted by the success and expanding use of anti-PD-1 therapies (such as nivolumab and pembrolizumab) to treat advanced-stage cancer.122–124 It also highlights the need to understand how oncogenic signaling and targeted therapies influence the expression of regulatory proteins such as PD-L1/L2 because such effects could potentially influence antitumor immune responses and/or the response to immunotherapies. This is particularly relevant when one considers that treatment regimens utilizing both targeted and anti-PD-1 therapies are currently undergoing clinical trials (phases 1 to 3).
There is evidence that oncogenic signaling and targeted therapies influence the expression of PD-L1 in some cellular contexts. For example, in non-small-cell lung cancer (NSCLC), treatment of cells with a preclinical mitogen-activated protein kinase (MAPK)/ERK kinase (MEK) inhibitor (U0126) was shown to decrease PD-L1 expression.125 In another study on NSCLC, increased expression of PD-L1 was noted in tumors with activating mutations in the EGFR and gefitinib, a small molecule inhibitor of the EGFR, attenuated the expression of PD-L1.126,127 Another study using NSCLC cell lines demonstrated that small-molecule kinase inhibitors of the EGFR, MEK, PI3 kinase, and anaplastic lymphoma kinase could repress PD-L1 expression.128 In the context of melanoma, the MAPK pathway has been implicated in regulating PD-L1 expression, although cellular context is important because inhibitors of BRAF, MEK, and phosphoinositide 3-kinase (PI3K) have shown variable effects with both reduction and induction of PD-L1 being reported.129,130 In triple-negative breast cancer, the phosphatase and tensin homologue (PTEN)/PI3K/protein kinase B (Akt) pathway has been linked to regulating PD-L1 expression as both an Akt inhibitor (MK-2206) and rapamycin decreased PD-L1 expression.131 Although the modulation of inhibitory receptor ligands such as PD-L1 on tumor cells by targeted therapies is incompletely understood, such effects have the potential to influence the response to immunotherapy and warrant further investigation.
Alterations in Chemokine/Cytokine Expression by Targeted Therapies
In addition to altering the expression of molecules on the tumor cell surface (such as MHC molecules and PD-L1/2), oncogenic signaling and the targeted therapies developed to disrupt it can affect cytokine secretion within the tumor microenvironment. For example, the Ras oncoprotein has been shown to upregulate the expression of interleukin (IL)-1β, IL-6, and IL-8 in hematologic and epithelial cancers.132–134 The secretion of cytokines has been shown to be indispensable for tumorigenesis, and neutralization of both IL-6 and IL-8 has been shown to hinder angiogenesis and tumor growth and progression.132,133 Increased cytokine production is also elicited by the BRAFV600E mutation within melanoma cells, which has been shown to upregulate expression of IL-1α, IL-1β, IL-6, IL-8, IL-10, and VEGF.135,136 Treatment with the BRAFV600E kinase inhibitor vemurafenib is able to attenuate the production of these cytokines.135,136 It is worth noting that IL-10 and VEGF are immunosuppressive cytokines compared to the proinflammatory IL-1α, IL-1β, IL-6, and IL-8, which further obscures the impact of small-molecule inhibitors such as the BRAFV600E inhibitor vemurafenib because the expression of both proinflammatory and immunosuppressive cytokines may be altered. The mechanism by which interleukins contribute to the immunosuppressive tumor microenvironment has been reported to depend heavily on cells known as tumor-associated fibroblasts (TAFs), which interact closely with tumor-infiltrating lymphocytes.136 TAFs isolated from melanoma patients were treated with IL-1 and were able to reduce the functional response of tumor-infiltrating lymphocytes in vitro by 4- to 5-fold compared to TAFs that did not receive IL-1 pretreatment. The mechanism of this response was shown to be dependent on the increased expression of COX-2, PD-L1, and PD-L2 by the TAF.136 Importantly, the BRAFV600E inhibitor was able to relieve this TAF-mediated immune suppression.136 The ability of targeted therapies to reduce the immunosuppressive ability of TAFs within the tumor microenvironment provides further evidence supporting the use of targeted therapies with immunotherapies that stimulate a tumor-specific T-cell response. These findings illustrate that targeted therapies influence the expression of soluble factors released by tumor cells that in turn can influence the recruitment and/or behavior of nontumor cells within the tumor microenvironment.
Effects of Targeted Therapies on Immune Cells Relevant to Antitumor Immunity
It has become clear that targeted therapies act on many cell types within the tumor microenvironment including those of the immune system. This is relevant because the tumor microenvironment contains immune cells (such as CD8+ and CD4+ T cells) that can recognize and eradicate tumor cells and other immune cells such as T regs and MDSCs that hinder antitumor immunity.137 Recent studies have demonstrated a role for some targeted therapies in combating the immunosuppressive tumor microenvironment by downregulating T regs and/or MDSC function, thus permitting the antitumor lymphocyte response to predominate (Figure 4). As previously mentioned, Zaiss et al showed in a murine model of melanoma that tumor-resident T regs expressed the EGFR on their surface, and EGFR ligands were able to stimulate the suppressive capacity of T regs.44 Further, treatment with the EGFR inhibitor gefitinib was able to abrogate the response seen by ligand addition.44 Elegant in vivo experiments using treatment with EGFRIs in mice bearing B16-F10 melanoma tumors (which lack EGFR expression) showed significant reduction in tumor size compared to peptide immunization alone, supporting an indirect antitumor activity of the EGFRI mediated through its action on the tumor microenvironment.44 This may explain EGFRI efficacy in patients with EGFR-negative tumors.44,138–140 Similar effects have also been observed with sunitinib, an inhibitor of multiple tyrosine kinases including VEGF and platelet-derived growth factor receptors.141 In a murine model of colon carcinoma, sunitinib treatment was shown to decrease both the number and suppressive capacity of T regs from peripheral blood, and a 2-week interruption of therapy was accompanied by a rebound of suppressive activity mediated by MDSCs.141 A similar study in metastatic renal cell carcinoma patients showed a decrease in MDSCs and T regs in the peripheral blood in response to sunitinib therapy and a subsequent increase in effector cytokine production by circulating T lymphocytes.142 In mice, sunitinib administration prior to tumor-antigen vaccination also augmented tumor-specific T-lymphocyte responses, suggesting that targeted-therapy-mediated inhibition of existing T-reg responses is important for the priming and development of tumorlytic T-cell responses.141 Sunitinib has been shown to decrease frequencies of circulating T regs and MDSCs in patients.142,143 This effect was perhaps potentiated by the increase in frequency of IFN-γ-producing type 1 helper T cells leading to an increased T-helper 1:T-helper 2 ratio and an overall augmented antitumor effect.143 Sunitinib has also been shown to lead to a decrease in expression of immunosuppressive cytokines including TGF-β and IL-10 and of negative regulatory immune checkpoint receptors, CTLA4 and PD-1 on tumor-infiltrating lymphocytes.144
Figure 4.

Targeted therapies can enhance antitumor immunity by disrupting the function of suppressive immune cells. Cells such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (T regs) block antitumor immune responses by inhibiting the function of antitumor T lymphocytes. By disrupting the function of MDSCs and/or T regs, targeted therapies can enhance the function of antitumor T lymphocytes, thereby augmenting antitumor immunity.
In addition to acting on T regs, a role for the EGFR in the recruitment and response of innate immune cells was demonstrated in a study examining the role of the EGFR in hepatocellular carcinoma (HCC). In a series of conditional knockout experiments, it was shown that intact EGFR signaling leads to a substantial increase of serum CCL2, which directs the recruitment of liver macrophages or Kupffer cells.145 This study further demonstrated opposing roles for EGFR signaling in hepatocytes and Kupffer cells with EGFR inhibition in hepatocytes contributing to the deregulated growth observed in HCC while EGFR inhibition in Kupffer cells attenuated tumor growth by reducing their secretion of the hepatocyte growth factor IL-6. The polarizing effect EGFRI treatment has on these 2 cell types in the tumor microenvironment provides an explanation of why, despite the overexpression of the EGFR in 40% to 70% of human HCCs, EGFRI therapies have yielded disappointing results in clinical trials.146,147
Targeted therapies have also been noted for their unintended deleterious effects on immune cells and immune responses. In vitro studies have shown that imatinib inhibits T-cell proliferation and results in reduced expression of activation markers CD25 and CD69, leading some to posit its immunosuppressive activity.148–150 Likewise, sorafenib, a small-molecule tyrosine kinase inhibitor targeting RAF as well as the VEGF receptor, the platelet-derived growth factor receptor, Flt-3, and c-KIT, has been shown to inhibit the maturation and costimulatory activity of dendritic cells, which results in decreased ability to stimulate T cells.151 This effect was reversible on discontinuation of the drug. Thus, having a detailed understanding of the effects of targeted therapies on immune cells will be crucial to allow their optimal use alone and in combination with immune-based therapies.152 Indeed, the hepatotoxicity seen in patients receiving the BRAFV600E inhibitor vemurafenib with the anti-CTLA4 ipilimumab underscores the challenges that can arise when targeted therapies are combined with immunotherapy.153
Effects of Targeted Therapies on Normal Tissue Homeostasis: The Skin as a Model
In addition to acting within the tumor microenvironment, the immune effects of targeted therapies are also at play within normal cells and tissues remote from the area of a tumor. This is evidenced by the common immune-mediated side effects of these medications such as those affecting the skin and gastrointestinal tract.154–158 These effects are not trivial because they can interfere with anticancer therapy and quality of life. Changes induced within the skin by treatment with EGFRIs have received the most attention and, as outlined below, have shed light on how EGFR signaling influences immune homeostasis within the context of a normal tissue.
The effects of EGFRIs in patients were predicted by murine knockout studies that illustrated the role of the EGFR in the maintenance of healthy follicular structures and epidermal barrier function and have recapitulated the effects of EGFRIs in patients.159 Additional studies have highlighted the profound impact of EGFR inhibition on immune cell trafficking into the skin (Figure 5). For example, dense immune infiltrates can be found in the affected skin of erlotinib (EGFR TKI)-treated patients consisting of macrophages, Langerhans cells, and both CD4+ and CD8+ lymphocytes.160,161 These changes in immune cell recruitment are likely mediated by alterations in the expression of chemokines such as CCL2, CCL5, CCL27, and CXCL14 within normal keratinocytes, which are crucial to the recruitment of monocytes, T cells, and other innate and adaptive effector immune cells to the skin.161,162 In addition, treatment of primary epidermal keratinocytes with EGFR inhibitor erlotinib decreased the expression of antimicrobial peptides such as human β-defensin 3, cathelicidin, and ribonuclease 7, suggesting that EGFR TKIs also alter host defenses against bacteria.161 In studies using mice with a conditional knockout of the EGFR, it was shown that, despite their recruitment to the skin, neither T cells nor Langerhans cells were solely responsible for the cutaneous inflammatory phenotype, but rather, it was a combination of innate immune cells (macrophages, granulocytes, and mast cells) that when trafficked to the skin could elicit a phenotype comparable to the EGFRI-related rash observed in patients.161,163 The immune alterations described above further illustrate the immune impact of targeted therapies and shed light on observations from clinical trials using EGFRIs that indicated a relationship between rash severity and efficacy of therapy.164–166 This relationship is in line with the notion that EGFRIs and other targeted therapies influence immune events within normal tissues as well as within the tumor microenvironment (Figure 6). Understanding the impact of targeted therapies on the immune system will help to (1) facilitate their optimal use in the treatment of cancer and (2) promote the development of approaches to mitigate their immune-mediated side effects.
Figure 5.
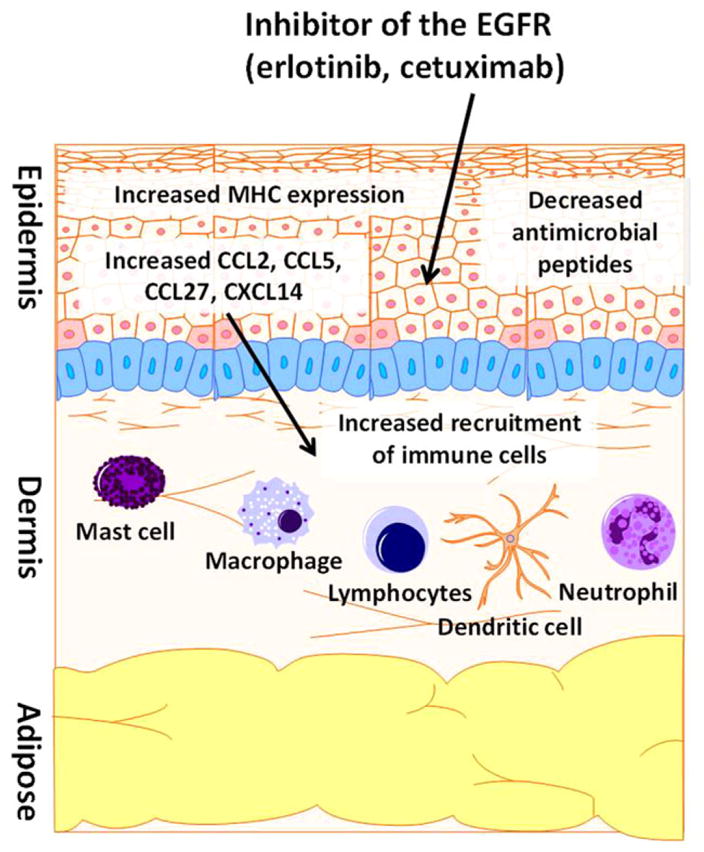
EGFR inhibitors alter immune homeostasis within the skin via numerous mechanisms. Studies in humans and mice have shown that disruption of the EGFR genetically or pharmacologically can influence numerous immune-related processes. These include changes in the expression of chemoattractant chemokines within epidermal keratinocytes such as CCL2, CCL5, CXCL14, and CCL27. These changes likely influence the alterations in immune-cell recruitment seen within the skin in patients treated with EGFR inhibitors. Other reported effects induced by EGFR inhibition on epidermal keratinocytes include alterations in MHC expression and the expression of antimicrobial peptides. EGFR indicates epidermal growth factor receptor.
Figure 6.
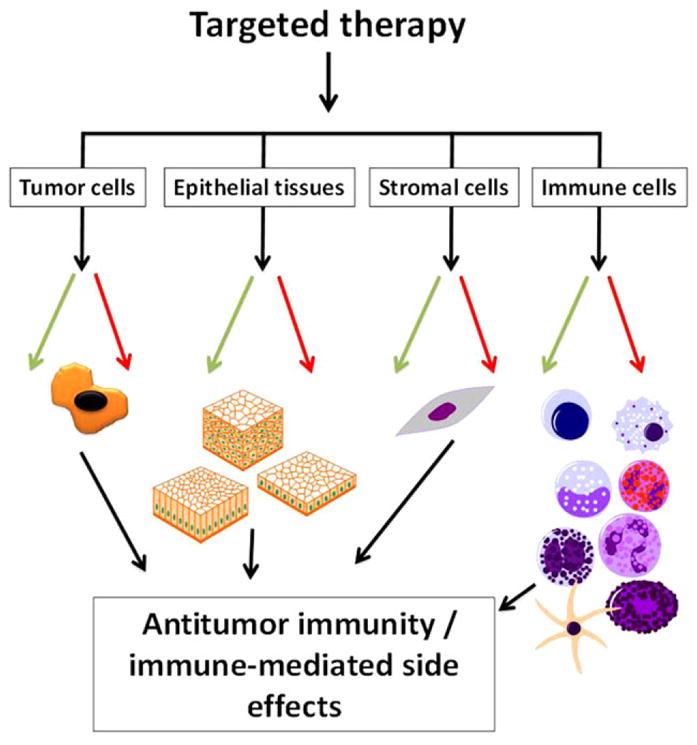
Targeted therapies influence immune-related events by acting on diverse cell types within the tumor microenvironment and remotely. Targeted therapies can influence immune-related processes within numerous cell types. In doing so, they can influence immune responses against tumor cells and immune homeostasis within normal tissues. These changes may potentially enhance (green arrows) and/or hinder (red arrows) antitumor immunity and the response to immunotherapy. In addition, these effects may contribute to the immune-related side effects of targeted therapies.
The Use of Targeted Therapies Outside of Advanced Cancer
At present, the medications discussed above are used exclusively for the treatment of advanced cancer. However, it is important to consider that the ability to pharmacologically disrupt specific signaling pathways may provide additional therapeutic opportunities. The repurposing of medications is a well-recognized approach to develop new treatments for diseases with limited therapeutic options.167–169 In the subsequent paragraphs we seek to provide a broad overview of how targeted therapies (such as EGFR inhibitors) have been applied in settings outside the treatment of advanced cancer (Figure 7).
Figure 7.

The use of targeted therapies in settings other than the treatment of cancer. The EGFR and its downstream signaling pathways play complex roles in numerous processes in addition to their roles in cancer. As a result, inhibitors of the EGFR and other targeted therapies are being tested systemically and locally for diverse applications. For example, targeted therapies are being evaluated as antiviral agents, vaccine adjuvants, to enhance wound healing, to promote nerve regrowth following nerve injury, and in other settings. EGFR indicates epidermal growth factor receptor.
Use of EGFR Inhibitors as Antiviral Agents
As described in earlier sections of this article, the EGFR plays diverse roles in physiologic processes and in the pathogenesis of several forms of cancer. In addition, the EGFR has also been shown to play important roles in the pathogenesis of viral infections. The EGFR has been implicated as a host factor that is relevant to infections caused by poxviruses, human cytomegalovirus (CMV), influenza A virus (IAV), and hepatitis C virus (HCV).170–173
Many viruses exploit the EGFR as a means to internalize by using it as a coreceptor for entry into host cells. For example, the principal envelope glycoprotein of human CMV preferentially binds to an EGFR monomer and oligomerizes with ErbB3, which then allows the virus to infect human fibroblast HEL cells.172 Recently, Kim et al demonstrated that human CMV binds to the EGFR and induces signaling via PI3K in CD34+ human progenitor cells, which mediate HCMV entry and viral trafficking as well as the establishment of viral latency.174 IAV also uses the EGFR as a coreceptor for binding to host cells. IAV utilizes the EGFR in a sialic acid–dependent manner for internalization.170 In regard to HCV, a screen using siRNAs identified the EGFR as a key mediator of its entry.171 Last, the EGFR has been shown to act as a coreceptor for adeno-associated virus serotype 6 entry, an adeno-associated virus vector used for gene therapy.175 These findings illustrate that the EGFR is an important growth factor receptor that facilitates viral infection.
In addition to mediating viral entry into cells, EGFR activation by respiratory viruses inhibits antiviral defense mechanisms. IFN-λ, a type III interferon, provides an antiviral mucosal response to viral infection via IRF1 signaling.176,177 IAV and rhinovirus were able to activate the EGFR, and its activation suppressed IRF1 induction of IFN-λ.178 In a related way, EGFR activation by respiratory viruses including IAV, rhinovirus, and respiratory syncytial virus decreased the production of CXCL10, a lymphocyte-recruiting chemokine, in an IRF1-dependent manner in airway epithelial cells.179 Given that the EGFR plays an important role in viral entry and host antiviral suppression, it is not surprising that EGFR inhibitors have been shown to have antiviral activity as described in more detail below.
Blockade of EGFR kinase activity utilizing the EGFR TKI erlotinib has been investigated in the context of HCV infection. Lupberger et al showed that erlotinib could effectively block viral transmission in vitro utilizing cocultures involving HCV-producing and uninfected Huh-7.5 human hepatocytes.171 They also demonstrated that treatment with erlotinib could delay the kinetics of HCV infection and decrease viral load in a human-liver chimeric mouse model.171 Erlotinib has since been found to synergistically enhance the anti-HCV effects of IFN-α as well as the viral protease inhibitor telaprevir and the cyclophilin A inhibitor alisporivir.180,181 Utilizing erlotinib with sofosbuvir, the recently FDA-approved HCV NS5B polymerase inhibitor, reduced the IC50 of sofosbuvir by 210-fold in in vitro HCV-infection assays.181 The use of EGFR inhibitors in HCV-infected patients showed that treatment with erlotinib could reduce HCV viral load in sporadic case reports.182 These data prompted the initiation of phase 1/2a clinical trials investigating the safety and efficacy of erlotinib in HCV infections (NCT01835938).183 In addition to HCV, treatment with the preclinical EGFR tyrosine kinase inhibitor AG1478 prevented CMV-induced PI3K activation and subsequent expression of viral proteins in host target cells.172 In IAV models, blockade of EGFR signaling by anti-EGFR antibodies or treatment with EGFR tyrosine kinase inhibitors diminished virion uptake into human lung epithelial cells in vitro.170 The EGFR TKI gefitinib was found to decrease respiratory viral burden in C57Bl/6 mice infected with IAV in both prophylactic and therapeutic models, a mechanism dependent on higher levels of IFN-λ produced by respiratory epithelial cells in gefitinib-treated animals.178,179 EGFR blockade has also been found to inhibit the pathogenesis of poxvirus,173,185 dengue virus,186 and viral hemorrhagic fevers187 in in vitro studies. Due to its importance in maintaining cellular homeostasis, it is unsurprising that many viruses have evolved mechanisms to co-opt the EGFR signaling pathway for their gain. The use of EGFR inhibitors as antiviral agents will likely grow as additional interactions between clinically relevant viruses and this critical receptor complex are uncovered. It is worth noting that EGFR inhibition may not always be beneficial in the setting of viral infection because treatment with the EGFR kinase inhibitor AG1478 or gefitinib was shown to disrupt EGFR signaling in CMV-infected primary bone-marrow–derived CD34+ hematopoietic progenitor cells resulting in viral reactivation.184
EGF and EGFR Inhibitors in Models of Sepsis
The effects of EGF and inhibition of the EGFR have been studied in different animal models of sepsis. Early studies looking at the effect of EGF on the cecal ligation and puncture (CLP) polymicrobial model of sepsis in mice showed increased expression levels of EGFR and EGF within the gut tissue after CLP.188 In septic mice, exogenous EGF treatment was able to attenuate expression of proapoptotic proteins Bid, Fas-associated death domain, and p21 as well as decrease sepsis-mediated mortality by 30%.188 More recently, EGF was shown to improve intestinal integrity and mortality following sepsis in a CLP model incorporating chronic alcohol ingestion.189 Alternatively, studies using LPS treatment to model endotoxemia demonstrated that the EGFR is crucial to the production of TNF-α within the heart.190 Treatment with an irreversible EGFR inhibitor (PD168393) or the EGFR inhibitor erlotinib was able to decrease TNF-α production, improve cardiac ejection fraction, and improve survival rates in endotoxemic mice.190 Likewise, treatment with the EGFR inhibitor gefitinib was also shown to protect mice from LPS-mediated septic shock by blocking aspects of toll-like receptor 4 signaling.191 The results of these studies, using different models, indicate a role for the EGFR in regulating the response to sepsis. The nature of the influence of the EGFR in sepsis, whether regenerative or pathologic, may depend on the presence and location of injury among other factors. Taken together, the aforementioned studies clearly imply that EGFR signaling can influence the response to sepsis and underscore the importance of further elucidating the role of EGFR signaling in this context.
Neurobiology and Targeted Therapies
Growth factors play vital roles within the nervous system, and the signaling pathways at play within tumor cells (and thus inhibited by targeted therapies) are also important to the biology of many cell types within the central and peripheral nervous systems. It is therefore not surprising that targeted therapies have shown activity in several models relevant to neurobiology. For example, early studies seeking to identify pathways involved in inhibiting nerve regrowth following injury revealed that EGFR kinase inhibitors can promote nerve regrowth after injury.192 Similarly, EGFR inhibitors enhanced recovery in models of spinal cord injury.193 In line with these neurologic effects, there is intriguing evidence that EGFRIs can alleviate neuropathic pain in humans.194,195 More recently, ErbB2 blockade with trastuzumab has been reported to enhance peripheral nerve regeneration after nerve injury.196 With regard to the central nervous system, a MEK inhibitor (PD325901) has been recently reported to reduce cocaine-mediated behaviors in a murine model of cocaine addiction.197 These studies further illustrate how the development of targeted therapies for cancer can potentially be used to address unmet needs in other areas of medicine.
Local Application of Targeted Therapies
Although targeted therapies are currently used systemically, there is preclinical evidence that small-molecule TKIs can be locally applied to manipulate immune responses. The recent development of topical formulations of Janus kinase inhibitors illustrates how locally applied kinase inhibitors can be used to treat autoimmune skin diseases and inflammation of the eye.198–201 In addition, inhaled kinase inhibitors are also being explored for the treatment of lung diseases.202,203 Several studies have reported the immunologic impact of topically applied EGFR inhibitors. For example, topical EGFR inhibitors have been shown to enhance the effector phase of contact hypersensitivity.162 Moreover, topical application of an EGFR inhibitor has been shown to prevent UVR-induced skin tumors and block the immunosuppressive effects of UVR.204,205 Our group has shown that the topical application of an EGFR inhibitor can enhance the immune response to cutaneous vaccination.206 More recently, the topical application of the BRAFV600E-selective inhibitor vemurafenib has shown promise as a treatment to accelerate wound healing through its paradoxical ability to activate the MAPK pathway.207 Thus, in addition to their use as systemic agents, locally applied targeted therapies may harbor potential for the treatment of diseases affecting accessible epithelial tissues.
Summary and Conclusions
There is ample evidence that targeted therapies influence immunologic processes through varied mechanisms. These effects are at play within malignant cells, nontumor cells within the tumor microenvironment, and normal tissues remote from the site of a tumor (Figure 6). Although many of these effects likely contribute to the adverse effects of targeted therapies, there is a growing appreciation that targeted therapies modulate antitumor immunity and that the immune system plays a role in the response to targeted therapies. Understanding these immunologic effects will likely inform how to optimally use these medications (alone and in combination) for the treatment of cancer. Some of these effects seen with small-molecule TKIs may represent off-target effects, but they may still be clinically relevant. In addition, there is growing evidence that because of their ability to influence immune responses and/or to disrupt key pathogenic signaling events, targeted therapies have the potential to be repurposed for use in the treatment of other human diseases.
Acknowledgments
Sources of Support
A.E.K., S.N., and Y.M.C. are supported by the Medical Scientist Training Program at Emory University School of Medicine. S.N.T. is supported by National Institutes of Health (NIH) Grant R01CA207619 and Department of Defense Grant CA150523. H.K. is supported by NIH grant K99CA197801. G.B.L. is supported by NIH Grant R01CA208253-01. E. K.W. is supported by NIH Grant R01CA74364. B.P.P. is supported by a Merit Review Award (5I01BX001922) from the Department of Veterans Affairs, a Career Development Award from the Melanoma Research Foundation, funding from the Skin Cancer and Melanoma Fund of the Winship Cancer Institute, and the Melanoma Innovation Fund of the Winship Cancer Institute.
Footnotes
Declaration of Conflicting Interests
B.P.P. is an inventor on patent number US 2016/0228533. R.R.K receives research funding from Merck.
References
- 1.Lee L, Gupta M, Sahasranaman S. Immune checkpoint inhibitors: an introduction to the next-generation cancer immunotherapy. J Clin Pharmacol. 2016;56(2):157–169. doi: 10.1002/jcph.591. [DOI] [PubMed] [Google Scholar]
- 2.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koster BD, de Gruijl TD, van den Eertwegh AJ. Recent developments and future challenges in immune checkpoint inhibitory cancer treatment. Curr Opin Oncol. 2015;27(6):482–488. doi: 10.1097/CCO.0000000000000221. [DOI] [PubMed] [Google Scholar]
- 6.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714. doi: 10.1016/j.ccell.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L, Zitvogel L, Kroemer G. Immunological mechanisms underneath the efficacy of cancer therapy. Cancer Immunol Res. 2016;4(11):895–902. doi: 10.1158/2326-6066.CIR-16-0197. [DOI] [PubMed] [Google Scholar]
- 8.Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11(3):215–233. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- 9.Wargo JA, Cooper ZA, Flaherty KT. Universes collide: combining immunotherapy with targeted therapy for cancer. Cancer Discov. 2014;4(12):1377–1386. doi: 10.1158/2159-8290.CD-14-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palucka AK, Coussens LM. The basis of oncoimmunology. Cell. 2016;164(6):1233–1247. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shepard HM, Lewis GD, Sarup JC, et al. Monoclonal antibody therapy of human cancer: taking the HER2 protooncogene to the clinic. J Clin Immunol. 1991;11(3):117–127. doi: 10.1007/BF00918679. [DOI] [PubMed] [Google Scholar]
- 12.Mauro MJ, O’Dwyer M, Heinrich MC, Druker BJ. STI571: a paradigm of new agents for cancer therapeutics. J Clin Oncol. 2002;20(1):325–334. doi: 10.1200/JCO.2002.20.1.325. [DOI] [PubMed] [Google Scholar]
- 13.Prendergast GC, Jaffee EM. Cancer immunologists and cancer biologists: why we didn’t talk then but need to now. Cancer Res. 2007;67(8):3500–3504. doi: 10.1158/0008-5472.CAN-06-4626. [DOI] [PubMed] [Google Scholar]
- 14.Okines A, Cunningham D, Chau I. Targeting the human EGFR family in esophagogastric cancer. Nat Rev Clin Oncol. 2011;8(8):492–503. doi: 10.1038/nrclinonc.2011.45. [DOI] [PubMed] [Google Scholar]
- 15.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 16.Arbiser JL, Moses MA, Fernandez CA, et al. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc Natl Acad Sci U S A. 1997;94(3):861–866. doi: 10.1073/pnas.94.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8(4):210–221. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabrilovich DI, Chen HL, Girgis KR, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–1103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 19.Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res. 1999;5(10):2963–2970. [PubMed] [Google Scholar]
- 20.Ko JS, Bukowski RM, Fincke JH. Myeloid-derived suppressor cells: a novel therapeutic target. Curr Oncol Rep. 2009;11(2):87–93. doi: 10.1007/s11912-009-0014-6. [DOI] [PubMed] [Google Scholar]
- 21.Ohm JE, Gabrilovich DI, Sempowski GD, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101(12):4878–4886. doi: 10.1182/blood-2002-07-1956. [DOI] [PubMed] [Google Scholar]
- 22.Terme M, Tartour E, Taieb J. VEGFA/VEGFR2-targeted therapies prevent the VEGFA-induced proliferation of regulatory T cells in cancer. Oncoimmunology. 2013;2(8):e25156. doi: 10.4161/onci.25156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voron T, Colussi O, Marcheteau E, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–148. doi: 10.1084/jem.20140559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voron T, Marcheteau E, Pernot S, et al. Control of the immune response by pro-angiogenic factors. Front Oncol. 2014;4:70. doi: 10.3389/fonc.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan J, Zhou J, Dong Z, et al. Pretreatment serum VEGF is associated with clinical response and overall survival in advanced melanoma patients treated with ipilimumab. Cancer Immunol Res. 2014;2(2):127–132. doi: 10.1158/2326-6066.CIR-13-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carter T, Shaw H, Cohn-Brown D, Chester K, Mulholland P. Ipilimumab and bevacizumab in glioblastoma. Clin Oncol (R Coll Radiol) 2016;28(10):622–626. doi: 10.1016/j.clon.2016.04.042. [DOI] [PubMed] [Google Scholar]
- 27.Hodi FS, Lawrence D, Lezcano C, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2(7):632–642. doi: 10.1158/2326-6066.CIR-14-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishino M, Giobbie-Hurder A, Ramaiya NH, Hodi FS. Response assessment in metastatic melanoma treated with ipilimumab and bevacizumab: CT tumor size and density as markers for response and outcome. J Immunother Cancer. 2014;2(1):40. doi: 10.1186/s40425-014-0040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu X, Giobbie-Hurder A, Liao X, et al. VEGF neutralization plus CTLA-4 blockade alters soluble and cellular factors associated with enhancing lymphocyte infiltration and humoral recognition in melanoma. Cancer Immunol Res. 2016;4(10):858–868. doi: 10.1158/2326-6066.CIR-16-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsujioka H, Yotsumoto F, Shirota K, et al. Emerging strategies for ErbB ligand-based targeted therapy for cancer. Anticancer Res. 2010;30(8):3107–3112. [PubMed] [Google Scholar]
- 31.Roskoski R., Jr The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Downward J, Yarden Y, Mayes E, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307(5951):521–527. doi: 10.1038/307521a0. [DOI] [PubMed] [Google Scholar]
- 33.Schechter AL, Stern DF, Vaidyanathan L, et al. The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature. 1984;312(5994):513–516. doi: 10.1038/312513a0. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Z, Stiegler AL, Boggon TJ, Kobayashi S, Halmos B. EGFR-mutated lung cancer: a paradigm of molecular oncology. Oncotarget. 2010;1(7):497–514. doi: 10.18632/oncotarget.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saif MW. Colorectal cancer in review: the role of the EGFR pathway. Expert Opin Investig Drugs. 2010;19(3):357–369. doi: 10.1517/13543781003593962. [DOI] [PubMed] [Google Scholar]
- 36.Egloff AM, Grandis JR. Improving response rates to EGFR-targeted therapies for head and neck squamous cell carcinoma: candidate predictive biomarkers and combination treatment with Src inhibitors. J Oncol. 2009;2009:896407. doi: 10.1155/2009/896407. https://doi.org/10.1155/2009/896407. Epub 2009 Jul 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nahta R, Shabaya S, Ozbay T, Rowe DL. Personalizing HER2-targeted therapy in metastatic breast cancer beyond HER2 status: what we have learned from clinical specimens. Curr Pharmacogenomics Person Med. 2009;7(4):263–274. doi: 10.2174/187569209790112337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson GR, Kannan B, Shoyab M, Stromberg K. Amphiregulin induces tyrosine phosphorylation of the epidermal growth factor receptor and p185erbB2. Evidence that amphiregulin acts exclusively through the epidermal growth factor receptor at the surface of human epithelial cells. J Biol Chem. 1993;268(4):2924–2931. [PubMed] [Google Scholar]
- 39.Shoyab M, McDonald VL, Bradley JG, Todaro GJ. Amphiregulin: a bifunctional growth-modulating glycoprotein produced by the phorbol 12-myristate 13-acetate-treated human breast adenocarcinoma cell line MCF-7. Proc Natl Acad Sci U S A. 1988;85(17):6528–6532. doi: 10.1073/pnas.85.17.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schelfhout VR, Coene ED, Delaey B, et al. The role of heregulin-alpha as a motility factor and amphiregulin as a growth factor in wound healing. J Pathol. 2002;198(4):523–533. doi: 10.1002/path.1240. [DOI] [PubMed] [Google Scholar]
- 41.Schuger L, Johnson GR, Gilbride K, Plowman GD, Mandel R. Amphiregulin in lung branching morphogenesis: interaction with heparan sulfate proteoglycan modulates cell proliferation. Development. 1996;122(6):1759–1767. doi: 10.1242/dev.122.6.1759. [DOI] [PubMed] [Google Scholar]
- 42.Luetteke NC, Qiu TH, Fenton SE, et al. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development. 1999;126(12):2739–2750. doi: 10.1242/dev.126.12.2739. [DOI] [PubMed] [Google Scholar]
- 43.Zaiss DM, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. 2015;42(2):216–226. doi: 10.1016/j.immuni.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaiss DM, van Loosdregt J, Gorlani A, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. 2013;38(2):275–284. doi: 10.1016/j.immuni.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Gruijl FR. UV-induced immunosuppression in the balance. Photochem Photobiol. 2008;84(1):2–9. doi: 10.1111/j.1751-1097.2007.00211.x. [DOI] [PubMed] [Google Scholar]
- 46.Meulenbroeks C, van Weelden H, Schwartz C, et al. Basophil-derived amphiregulin is essential for UVB irradiation-induced immune suppression. J Invest Dermatol. 2015;135(1):222–228. doi: 10.1038/jid.2014.329. [DOI] [PubMed] [Google Scholar]
- 47.Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18(5):1386–1394. doi: 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- 48.Srivastava RM, Lee SC, Andrade Filho PA, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19(7):1858–1872. doi: 10.1158/1078-0432.CCR-12-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto N, Honma M, Suzuki H. Off-target serine/threonine kinase 10 inhibition by erlotinib enhances lymphocytic activity leading to severe skin disorders. Mol Pharmacol. 2011;80(3):466–475. doi: 10.1124/mol.110.070862. [DOI] [PubMed] [Google Scholar]
- 51.Derhovanessian E, Solana R, Larbi A, Pawelec G. Immunity, ageing and cancer. Immun Ageing. 2008;5:11. doi: 10.1186/1742-4933-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia-Jimenez C, Garcia-Martinez JM, Chocarro-Calvo A, De la Vieja A. A new link between diabetes and cancer: enhanced WNT/beta-catenin signaling by high glucose. J Mol Endocrinol. 2014;52(1):R51–66. doi: 10.1530/JME-13-0152. [DOI] [PubMed] [Google Scholar]
- 53.Catalan V, Gomez-Ambrosi J, Rodriguez A, Fruhbeck G. Adipose tissue immunity and cancer. Front Physiol. 2013;4:275. doi: 10.3389/fphys.2013.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu S, Kurzrock R. Toxicity of targeted therapy: implications for response and impact of genetic polymorphisms. Cancer Treat Rev. 2014;40(7):883–891. doi: 10.1016/j.ctrv.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 55.You B, Chen EX. Anti-EGFR monoclonal antibodies for treatment of colorectal cancers: development of cetuximab and panitumumab. J Clin Pharmacol. 2012;52(2):128–155. doi: 10.1177/0091270010395940. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt KT, Chau CH, Price DK, Figg WD. Precision oncology medicine: the clinical relevance of patient-specific biomarkers used to optimize cancer treatment. J Clin Pharmacol. 2016;56(12):1484–1499. doi: 10.1002/jcph.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee SC, Lopez-Albaitero A, Ferris RL. Immunotherapy of head and neck cancer using tumor antigen-specific monoclonal antibodies. Curr Oncol Rep. 2009;11(2):156–162. doi: 10.1007/s11912-009-0023-5. [DOI] [PubMed] [Google Scholar]
- 58.Roda JM, Joshi T, Butchar JP, et al. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin Cancer Res. 2007;13(21):6419–6428. doi: 10.1158/1078-0432.CCR-07-0865. [DOI] [PubMed] [Google Scholar]
- 59.Patel D, Guo X, Ng S, et al. IgG isotype, glycosylation, and EGFR expression determine the induction of antibody-dependent cellular cytotoxicity in vitro by cetuximab. Hum Antibodies. 2010;19(4):89–99. doi: 10.3233/HAB-2010-0232. [DOI] [PubMed] [Google Scholar]
- 60.Trivedi S, Srivastava RM, Concha-Benavente F, et al. Anti-EGFR targeted monoclonal antibody isotype influences anti-tumor cellular immunity in head and neck cancer patients. Clin Cancer Res. 2016;22(21):5229–5237. doi: 10.1158/1078-0432.CCR-15-2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bibeau F, Lopez-Crapez E, Di Fiore F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27(7):1122–1129. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 62.Zhang W, Gordon M, Schultheis AM, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol. 2007;25(24):3712–3718. doi: 10.1200/JCO.2006.08.8021. [DOI] [PubMed] [Google Scholar]
- 63.Lopez-Albaitero A, Lee SC, Morgan S, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58(11):1853–1864. doi: 10.1007/s00262-009-0697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: clinical response, cellular immunity, and immunoescape. J Clin Oncol. 2010;28(28):4390–4399. doi: 10.1200/JCO.2009.27.6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Musolino A, Naldi N, Bortesi B, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26(11):1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 66.Tamura K, Shimizu C, Hojo T, et al. FcgammaR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol. 2011;22(6):1302–1307. doi: 10.1093/annonc/mdq585. [DOI] [PubMed] [Google Scholar]
- 67.Trivedi S, Concha-Benavente F, Srivastava RM, et al. Immune biomarkers of anti-EGFR monoclonal antibody therapy. Ann Oncol. 2015;26(1):40–47. doi: 10.1093/annonc/mdu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kubach J, Hubo M, Amendt C, Stroh C, Jonuleit H. IgG1 anti-epidermal growth factor receptor antibodies induce CD8-dependent antitumor activity. Int J Cancer. 2015;136(4):821–830. doi: 10.1002/ijc.29037. [DOI] [PubMed] [Google Scholar]
- 69.Barnhart BC, Quigley M. Role of Fc-FcgammaR interactions in the antitumor activity of therapeutic antibodies. Immunol Cell Biol. 2017;95(4):340–346. doi: 10.1038/icb.2016.121. [DOI] [PubMed] [Google Scholar]
- 70.Schneider-Merck T, Lammerts van Bueren JJ, Berger S, et al. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol. 2010;184(1):512–520. doi: 10.4049/jimmunol.0900847. [DOI] [PubMed] [Google Scholar]
- 71.Lee CH, Romain G, Yan W, et al. IgG Fc domains that bind C1q but not effector Fcgamma receptors delineate the importance of complement-mediated effector functions. Nat Immunol. 2017 doi: 10.1038/ni.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu XY, Wang XY, Li RY, et al. Recent progress in the understanding of complement activation and its role in tumor growth and anti-tumor therapy. Biomed Pharmacother. 2017;91:446–456. doi: 10.1016/j.biopha.2017.04.101. [DOI] [PubMed] [Google Scholar]
- 73.Pawluczkowycz AW, Beurskens FJ, Beum PV, et al. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J Immunol. 2009;183(1):749–758. doi: 10.4049/jimmunol.0900632. [DOI] [PubMed] [Google Scholar]
- 74.Dechant M, Weisner W, Berger S, et al. Complement-dependent tumor cell lysis triggered by combinations of epidermal growth factor receptor antibodies. Cancer Res. 2008;68(13):4998–5003. doi: 10.1158/0008-5472.CAN-07-6226. [DOI] [PubMed] [Google Scholar]
- 75.Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 76.Lopez-Albaitero A, Mailliard R, Hackman T, et al. Maturation pathways of dendritic cells determine TAP1 and TAP2 levels and cross-presenting function. J Immunother. 2009;32(5):465–473. doi: 10.1097/CJI.0b013e3181a1c24e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst. 2013;105(16):1172–1187. doi: 10.1093/jnci/djt184. [DOI] [PubMed] [Google Scholar]
- 78.Bukur J, Jasinski S, Seliger B. The role of classical and non-classical HLA class I antigens in human tumors. Semin Cancer Biol. 2012;22(4):350–358. doi: 10.1016/j.semcancer.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 79.Srivastava RM, Trivedi S, Concha-Benavente F, et al. STAT1-induced HLA class I upregulation enhances immunogenicity and clinical response to anti-EGFR mAb cetuximab therapy in HNC patients. Cancer Immunol Res. 2015;3(8):936–945. doi: 10.1158/2326-6066.CIR-15-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sabbatino F, Wang Y, Scognamiglio G, et al. Antitumor activity of BRAF inhibitor and IFNalpha combination in BRAF-mutant melanoma. J Natl Cancer Inst. 2016;108(7) doi: 10.1093/jnci/djv435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today. 2000;21(9):455–464. doi: 10.1016/s0167-5699(00)01692-3. [DOI] [PubMed] [Google Scholar]
- 82.Leibowitz MS, Andrade Filho PA, Ferrone S, Ferris RL. Deficiency of activated STAT1 in head and neck cancer cells mediates TAP1-dependent escape from cytotoxic T lymphocytes. Cancer Immunol Immunother. 2011;60(4):525–535. doi: 10.1007/s00262-010-0961-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boss JM, Jensen PE. Transcriptional regulation of the MHC class II antigen presentation pathway. Curr Opin Immunol. 2003;15(1):105–111. doi: 10.1016/s0952-7915(02)00015-8. [DOI] [PubMed] [Google Scholar]
- 84.Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol. 2009;9(7):503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 85.Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. 2010;22(3):333–340. doi: 10.1016/j.coi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Houdt IS, Sluijter BJ, Moesbergen LM, et al. Favorable outcome in clinically stage II melanoma patients is associated with the presence of activated tumor infiltrating T-lymphocytes and preserved MHC class I antigen expression. Int J Cancer. 2008;123(3):609–615. doi: 10.1002/ijc.23543. [DOI] [PubMed] [Google Scholar]
- 87.Leffers N, Lambeck AJ, de Graeff P, et al. Survival of ovarian cancer patients overexpressing the tumour antigen p53 is diminished in case of MHC class I down-regulation. Gynecol Oncol. 2008;110(3):365–373. doi: 10.1016/j.ygyno.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 88.Zia A, Schildberg FW, Funke I. MHC class I negative phenotype of disseminated tumor cells in bone marrow is associated with poor survival in R0M0 breast cancer patients. Int J Cancer. 2001;93(4):566–570. doi: 10.1002/ijc.1362. [DOI] [PubMed] [Google Scholar]
- 89.Weidanz JA, Nguyen T, Woodburn T, et al. Levels of specific peptide-HLA class I complex predicts tumor cell susceptibility to CTL killing. J Immunol. 2006;177(8):5088–5097. doi: 10.4049/jimmunol.177.8.5088. [DOI] [PubMed] [Google Scholar]
- 90.Matsushita H, Sato Y, Karasaki T, et al. Neoantigen load, antigen presentation machinery, and immune signatures determine prognosis in clear cell renal cell carcinoma. Cancer Immunol Res. 2016;4(5):463–471. doi: 10.1158/2326-6066.CIR-15-0225. [DOI] [PubMed] [Google Scholar]
- 91.Gonzalez PA, Carreno LJ, Coombs D, et al. T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell. Proc Natl Acad Sci U S A. 2005;102(13):4824–4829. doi: 10.1073/pnas.0500922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vanguri V, Govern CC, Smith R, Huseby ES. Viral antigen density and confinement time regulate the reactivity pattern of CD4 T-cell responses to vaccinia virus infection. Proc Natl Acad Sci U S A. 2013;110(1):288–293. doi: 10.1073/pnas.1208328110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Melief CJ, Kast WM. Cytotoxic T lymphocyte therapy of cancer and tumor escape mechanisms. Semin Cancer Biol. 1991;2(5):347–354. [PubMed] [Google Scholar]
- 94.Kono K, Halapi E, Hising C, et al. Mechanisms of escape from CD8+ T-cell clones specific for the HER-2/neu proto-oncogene expressed in ovarian carcinomas: related and unrelated to decreased MHC class 1 expression. Int J Cancer. 1997;70(1):112–119. doi: 10.1002/(sici)1097-0215(19970106)70:1<112::aid-ijc17>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 95.Seliger B, Ritz U, Abele R, et al. Immune escape of melanoma: first evidence of structural alterations in two distinct components of the MHC class I antigen processing pathway. Cancer Res. 2001;61(24):8647–8650. [PubMed] [Google Scholar]
- 96.Ferris RL, Hunt JL, Ferrone S. Human leukocyte antigen (HLA) class I defects in head and neck cancer: molecular mechanisms and clinical significance. Immunol Res. 2005;33(2):113–133. doi: 10.1385/IR:33:2:113. [DOI] [PubMed] [Google Scholar]
- 97.Seliger B, Pfizenmaier K. Post-transcriptional downregulation of MHC class I expression in oncogene-transformed cells is reverted by IFN-gamma and TNF-alpha. J Immunogenet. 1989;16(4–5):315–320. doi: 10.1111/j.1744-313x.1989.tb00477.x. [DOI] [PubMed] [Google Scholar]
- 98.Lohmann S, Wollscheid U, Huber C, Seliger B. Multiple levels of MHC class I down-regulation by ras oncogenes. Scand J Immunol. 1996;43(5):537–544. doi: 10.1046/j.1365-3083.1996.d01-73.x. [DOI] [PubMed] [Google Scholar]
- 99.Herrmann F, Lehr HA, Drexler I, et al. HER-2/neu-mediated regulation of components of the MHC class I antigen-processing pathway. Cancer Res. 2004;64(1):215–220. doi: 10.1158/0008-5472.can-2522-2. [DOI] [PubMed] [Google Scholar]
- 100.Kumai T, Matsuda Y, Oikawa K, et al. EGFR inhibitors augment antitumour helper T-cell responses of HER family-specific immunotherapy. Br J Cancer. 2013;109(8):2155–2166. doi: 10.1038/bjc.2013.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumai T, Ohkuri T, Nagato T, et al. Targeting HER-3 to elicit antitumor helper T cells against head and neck squamous cell carcinoma. Sci Rep. 2015;5:16280. doi: 10.1038/srep16280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pollack BP, Sapkota B, Cartee TV. Epidermal growth factor receptor inhibition augments the expression of MHC class I and II genes. Clin Cancer Res. 2011;17(13):4400–4413. doi: 10.1158/1078-0432.CCR-10-3283. [DOI] [PubMed] [Google Scholar]
- 103.Mitra RS, Nickoloff BJ. Epidermal growth factor and transforming growth factor-alpha decrease gamma interferon receptors and induction of intercellular adhesion molecule (ICAM-1) on cultured keratinocytes. J Cell Physiol. 1992;150(2):264–268. doi: 10.1002/jcp.1041500207. [DOI] [PubMed] [Google Scholar]
- 104.Schreiber AB, Schlessinger J, Edidin M. Interaction between major histocompatibility complex antigens and epidermal growth factor receptors on human cells. J Cell Biol. 1984;98(2):725–731. doi: 10.1083/jcb.98.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lahat N, Sheinfeld M, Sobel E, Kinarty A, Kraiem Z. Divergent effects of cytokines on human leukocyte antigen-DR antigen expression of neoplastic and non-neoplastic human thyroid cells. Cancer. 1992;69(7):1799–1807. doi: 10.1002/1097-0142(19920401)69:7<1799::aid-cncr2820690723>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 106.Mimura K, Shiraishi K, Mueller A, et al. The MAPK pathway is a predominant regulator of HLA-A expression in esophageal and gastric cancer. J Immunol. 2013;191(12):6261–6272. doi: 10.4049/jimmunol.1301597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen XH, Liu ZC, Zhang G, et al. TGF-beta and EGF induced HLA-I downregulation is associated with epithelial-mesenchymal transition (EMT) through upregulation of snail in prostate cancer cells. Mol Immunol. 2015;65(1):34–42. doi: 10.1016/j.molimm.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 108.Okita R, Wolf D, Yasuda K, et al. Contrasting effects of the cytotoxic anticancer drug gemcitabine and the EGFR tyrosine kinase inhibitor gefitinib on NK cell-mediated cytotoxicity via regulation of NKG2D ligand in non-small-cell lung cancer cells. PLoS One. 2015;10(10):e0139809. doi: 10.1371/journal.pone.0139809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mortenson ED, Park S, Jiang Z, Wang S, Fu YX. Effective anti-neu-initiated antitumor responses require the complex role of CD4+ T cells. Clin Cancer Res. 2013;19(6):1476–1486. doi: 10.1158/1078-0432.CCR-12-2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAFV600E homozygous melanoma cells. Oncoimmunology. 2013;2(1):e22890. doi: 10.4161/onci.22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bradley SD, Chen Z, Melendez B, et al. BRAFV600E coopts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol Res. 2015;3(6):602–609. doi: 10.1158/2326-6066.CIR-15-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025–1036. doi: 10.1038/ni.3518. [DOI] [PubMed] [Google Scholar]
- 114.Vantourout P, Willcox C, Turner A, et al. Immunological visibility: posttranscriptional regulation of human NKG2D ligands by the EGF receptor pathway. Sci Transl Med. 2014;6(231):231ra249. doi: 10.1126/scitranslmed.3007579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125(9):3413–3421. doi: 10.1172/JCI80008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pitcovski J, Shahar E, Aizenshtein E, Gorodetsky R. Melanoma antigens and related immunological markers. Crit Rev Oncol Hematol. 2017;115:36–49. doi: 10.1016/j.critrevonc.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 117.Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70(13):5213–5219. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 118.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee J, Ahn E, Kissick HT, Ahmed R. Reinvigorating exhausted T cells by blockade of the PD-1 pathway. Forum Immunopathol Dis Ther. 2015;6(1–2):7–17. doi: 10.1615/ForumImmunDisTher.2015014188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med. 2015;21(1):24–33. doi: 10.1016/j.molmed.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ferris RL, Blumenschein G, Jr, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sumimoto H, Takano A, Teramoto K, Daigo Y. RAS-mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PLoS One. 2016;11(11):e0166626. doi: 10.1371/journal.pone.0166626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Akbay EA, Koyama S, Carretero J, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3(12):1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen N, Fang W, Zhan J, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015;10(6):910–923. doi: 10.1097/JTO.0000000000000500. [DOI] [PubMed] [Google Scholar]
- 128.Ota K, Azuma K, Kawahara A, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. 2015;21(17):4014–4021. doi: 10.1158/1078-0432.CCR-15-0016. [DOI] [PubMed] [Google Scholar]
- 129.Atefi M, Avramis E, Lassen A, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20(13):3446–3457. doi: 10.1158/1078-0432.CCR-13-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19(3):598–609. doi: 10.1158/1078-0432.CCR-12-2731. [DOI] [PubMed] [Google Scholar]
- 131.Mittendorf EA, Philips AV, Meric-Bernstam F, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2(4):361–370. doi: 10.1158/2326-6066.CIR-13-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6(5):447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 133.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21(14):1714–1719. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Beaupre DM, Talpaz M, Marini FC, 3rd, et al. Autocrine interleukin-1beta production in leukemia: evidence for the involvement of mutated RAS. Cancer Res. 1999;59(12):2971–2980. [PubMed] [Google Scholar]
- 135.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203(7):1651–1656. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Khalili JS, Liu S, Rodriguez-Cruz TG, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18(19):5329–5340. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gajewski TF, Meng Y, Harlin H. Immune suppression in the tumor microenvironment. J Immunother. 2006;29(3):233–240. doi: 10.1097/01.cji.0000199193.29048.56. [DOI] [PubMed] [Google Scholar]
- 138.Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23(9):1803–1810. doi: 10.1200/JCO.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 139.Chan G, Pilichowska M. Complete remission in a patient with acute myelogenous leukemia treated with erlotinib for non small-cell lung cancer. Blood. 2007;110(3):1079–1080. doi: 10.1182/blood-2007-01-069856. [DOI] [PubMed] [Google Scholar]
- 140.Pitini V, Arrigo C, Altavilla G. Erlotinib in a patient with acute myelogenous leukemia and concomitant non-small-cell lung cancer. J Clin Oncol. 2008;26(21):3645–3646. doi: 10.1200/JCO.2008.17.0357. [DOI] [PubMed] [Google Scholar]
- 141.Farsaci B, Higgins JP, Hodge JW. Consequence of dose scheduling of sunitinib on host immune response elements and vaccine combination therapy. Int J Cancer. 2012;130(8):1948–1959. doi: 10.1002/ijc.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 143.Finke JH, Rini B, Ireland J, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14(20):6674–6682. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- 144.Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69(6):2514–2522. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lanaya H, Natarajan A, Komposch K, et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat Cell Biol. 2014;16(10):972–977. doi: 10.1038/ncb3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Buckley AF, Burgart LJ, Sahai V, Kakar S. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. Am J Clin Pathol. 2008;129(2):245–251. doi: 10.1309/WF10QAAED3PP93BH. [DOI] [PubMed] [Google Scholar]
- 147.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29(36):4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 148.Dietz AB, Souan L, Knutson GJ, Bulur PA, Litzow MR, Vuk-Pavlovic S. Imatinib mesylate inhibits T-cell proliferation in vitro and delayed-type hypersensitivity in vivo. Blood. 2004;104(4):1094–1099. doi: 10.1182/blood-2003-12-4266. [DOI] [PubMed] [Google Scholar]
- 149.Cwynarski K, Laylor R, Macchiarulo E, et al. Imatinib inhibits the activation and proliferation of normal T lymphocytes in vitro. Leukemia. 2004;18(8):1332–1339. doi: 10.1038/sj.leu.2403401. [DOI] [PubMed] [Google Scholar]
- 150.Wolf D, Tilg H, Rumpold H, Gastl G, Wolf AM. The kinase inhibitor imatinib—an immunosuppressive drug? Curr Cancer Drug Targets. 2007;7(3):251–258. doi: 10.2174/156800907780618293. [DOI] [PubMed] [Google Scholar]
- 151.Hipp MM, Hilf N, Walter S, et al. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood. 2008;111(12):5610–5620. doi: 10.1182/blood-2007-02-075945. [DOI] [PubMed] [Google Scholar]
- 152.Hu-Lieskovan S, Robert L, Homet Moreno B, Ribas A. Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challenges. J Clin Oncol. 2014;32(21):2248–2254. doi: 10.1200/JCO.2013.52.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–1366. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 154.Chu D, Lacouture ME, Fillos T, Wu S. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol. 2008;47(2):176–186. doi: 10.1080/02841860701765675. [DOI] [PubMed] [Google Scholar]
- 155.Lynch TJ, Jr, Kim ES, Eaby B, Garey J, West DP, Lacouture ME. Epidermal growth factor receptor inhibitor-associated cutaneous toxicities: an evolving paradigm in clinical management. Oncologist. 2007;12(5):610–621. doi: 10.1634/theoncologist.12-5-610. [DOI] [PubMed] [Google Scholar]
- 156.Rosenbaum SE, Wu S, Newman MA, West DP, Kuzel T, Lacouture ME. Dermatological reactions to the multitargeted tyrosine kinase inhibitor sunitinib. Support Care Cancer. 2008;16(6):557–566. doi: 10.1007/s00520-008-0409-1. [DOI] [PubMed] [Google Scholar]
- 157.Davila M, Bresalier RS. Gastrointestinal complications of oncologic therapy. Nat Clin Pract Gastroenterol Hepatol. 2008;5(12):682–696. doi: 10.1038/ncpgasthep1277. [DOI] [PubMed] [Google Scholar]
- 158.Pessi MA, Zilembo N, Haspinger ER, et al. Targeted therapy-induced diarrhea: a review of the literature. Crit Rev Oncol Hematol. 2014;90(2):165–179. doi: 10.1016/j.critrevonc.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 159.Roberts RB, Arteaga CL, Threadgill DW. Modeling the cancer patient with genetically engineered mice: prediction of toxicity from molecule-targeted therapies. Cancer Cell. 2004;5(2):115–120. doi: 10.1016/s1535-6108(04)00032-7. [DOI] [PubMed] [Google Scholar]
- 160.Guttman-Yassky E, Mita A, De Jonge M, et al. Characterisation of the cutaneous pathology in non-small cell lung cancer (NSCLC) patients treated with the EGFR tyrosine kinase inhibitor erlotinib. Eur J Cancer. 2010;46(11):2010–2019. doi: 10.1016/j.ejca.2010.04.028. [DOI] [PubMed] [Google Scholar]
- 161.Lichtenberger BM, Gerber PA, Holcmann M, et al. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Sci Transl Med. 2013;5(199):199ra111. doi: 10.1126/scitranslmed.3005886. [DOI] [PubMed] [Google Scholar]
- 162.Mascia F, Mariani V, Girolomoni G, Pastore S. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163(1):303–312. doi: 10.1016/S0002-9440(10)63654-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mascia F, Lam G, Keith C, et al. Genetic ablation of epidermal EGFR reveals the dynamic origin of adverse effects of anti-EGFR therapy. Sci Transl Med. 2013;5(199):199ra110. doi: 10.1126/scitranslmed.3005773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 165.Wacker B, Nagrani T, Weinberg J, Witt K, Clark G, Cagnoni PJ. Correlation between development of rash and efficacy in patients treated with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in two large phase III studies. Clin Cancer Res. 2007;13(13):3913–3921. doi: 10.1158/1078-0432.CCR-06-2610. [DOI] [PubMed] [Google Scholar]
- 166.Perez-Soler R, Saltz L. Cutaneous adverse effects with HER1/EGFR-targeted agents: is there a silver lining? J Clin Oncol. 2005;23(22):5235–5246. doi: 10.1200/JCO.2005.00.6916. [DOI] [PubMed] [Google Scholar]
- 167.Langedijk J, Mantel-Teeuwisse AK, Slijkerman DS, Schutjens MH. Drug repositioning and repurposing: terminology and definitions in literature. Drug Discov Today. 2015;20(8):1027–1034. doi: 10.1016/j.drudis.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 168.Papanagnou P, Stivarou T, Tsironi M. Unexploited antineo-plastic effects of commercially available anti-diabetic drugs. Pharmaceuticals (Basel) 2016;9(2) doi: 10.3390/ph9020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rumore MM. Medication repurposing in pediatric patients: teaching old drugs new tricks. J Pediatr Pharmacol Ther. 2016;21(1):36–53. doi: 10.5863/1551-6776-21.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Eierhoff T, Hrincius ER, Rescher U, Ludwig S, Ehrhardt C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathogens. 2010;6(9):e1001099. doi: 10.1371/journal.ppat.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lupberger J, Zeisel MB, Xiao F, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17(5):589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature. 2003;424(6947):456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 173.Yang H, Kim SK, Kim M, et al. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J Clin Invest. 2005;115(2):379–387. doi: 10.1172/JCI23220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kim JH, Collins-McMillen D, Buehler JC, Goodrum FD, Yurochko AD. HCMV requires EGFR signaling to enter and initiate the early steps in the establishment of latency in CD34+ human progenitor cells. J Virol. 2016;91(5):e01206–e01216. doi: 10.1128/JVI.01206-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Weller ML, Amornphimoltham P, Schmidt M, Wilson PA, Gutkind JS, Chiorini JA. Epidermal growth factor receptor is a co-receptor for adeno-associated virus serotype 6. Nat Med. 2010;16(6):662–664. doi: 10.1038/nm.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Khaitov MR, Laza-Stanca V, Edwards MR, et al. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy. 2009;64(3):375–386. doi: 10.1111/j.1398-9995.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 177.Mordstein M, Neugebauer E, Ditt V, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol. 2010;84(11):5670–5677. doi: 10.1128/JVI.00272-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ueki IF, Min-Oo G, Kalinowski A, et al. Respiratory virus-induced EGFR activation suppresses IRF1-dependent interferon lambda and antiviral defense in airway epithelium. J Exp Med. 2013;210(10):1929–1936. doi: 10.1084/jem.20121401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kalinowski A, Ueki I, Min-Oo G, et al. EGFR activation suppresses respiratory virus-induced IRF1-dependent CXCL10 production. Am J Physiol Lung Cell Mol Physiol. 2014;307(2):L186–L196. doi: 10.1152/ajplung.00368.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lupberger J, Duong FH, Fofana I, et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology. 2013;58(4):1225–1235. doi: 10.1002/hep.26404. [DOI] [PubMed] [Google Scholar]
- 181.Xiao F, Fofana I, Thumann C, et al. Synergy of entry inhibitors with direct-acting antivirals uncovers novel combinations for prevention and treatment of hepatitis C. Gut. 2015;64(3):483–494. doi: 10.1136/gutjnl-2013-306155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bardou-Jacquet E, Lorho R, Guyader D. Kinase inhibitors in the treatment of chronic hepatitis C virus. Gut. 2011;60(6):879–880. doi: 10.1136/gut.2010.212068. [DOI] [PubMed] [Google Scholar]
- 183.Qian XJ, Zhu YZ, Zhao P, Qi ZT. Entry inhibitors: new advances in HCV treatment. Emerg Microbes Infect. 2016;5:e3. doi: 10.1038/emi.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Buehler J, Zeltzer S, Reitsma J, et al. Opposing regulation of the EGF receptor: a molecular switch controlling cytomegalovirus latency and replication. PLoS Pathogens. 2016;12(5):e1005655. doi: 10.1371/journal.ppat.1005655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Langhammer S, Koban R, Yue C, Ellerbrok H. Inhibition of poxvirus spreading by the anti-tumor drug gefitinib (Iressa) Antiviral Res. 2011;89(1):64–70. doi: 10.1016/j.antiviral.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 186.Limjindaporn T, Panaampon J, Malakar S, Noisakran S, Yenchitsomanus PT. Tyrosine kinase/phosphatase inhibitors decrease dengue virus production in HepG2 cells. Biochem Biophys Res Commun. 2017;483(1):58–63. doi: 10.1016/j.bbrc.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 187.Mohr EL, McMullan LK, Lo MK, et al. Inhibitors of cellular kinases with broad-spectrum antiviral activity for hemorrhagic fever viruses. Antiviral Res. 2015;120:40–47. doi: 10.1016/j.antiviral.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 188.Clark JA, Clark AT, Hotchkiss RS, Buchman TG, Coopersmith CM. Epidermal growth factor treatment decreases mortality and is associated with improved gut integrity in sepsis. Shock (Augusta, Ga) 2008;30(1):36–42. doi: 10.1097/shk.0b013e31815D0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Klingensmith NJ, Yoseph BP, Liang Z, et al. Epidermal growth factor improves intestinal integrity and survival in murine sepsis following chronic alcohol ingestion. Shock (Augusta, Ga) 2017;47(2):184–192. doi: 10.1097/SHK.0000000000000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sun X, Liang J, Yao X, et al. The activation of EGFR promotes myocardial tumor necrosis factor-alpha production and cardiac failure in endotoxemia. Oncotarget. 2015;6(34):35478–35495. doi: 10.18632/oncotarget.6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chattopadhyay S, Veleeparambil M, Poddar D, et al. EGFR kinase activity is required for TLR4 signaling and the septic shock response. EMBO Rep. 2015;16(11):1535–1547. doi: 10.15252/embr.201540337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Koprivica V, Cho KS, Park JB, et al. EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science. 2005;310(5745):106–110. doi: 10.1126/science.1115462. [DOI] [PubMed] [Google Scholar]
- 193.Erschbamer M, Pernold K, Olson L. Inhibiting epidermal growth factor receptor improves structural, locomotor, sensory, and bladder recovery from experimental spinal cord injury. J Neurosci. 2007;27(24):6428–6435. doi: 10.1523/JNEUROSCI.1037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kersten C, Cameron MG. Cetuximab alleviates neuropathic pain despite tumour progression. BMJ Case Rep. 2012;2012 doi: 10.1136/bcr.12.2011.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kersten C, Cameron MG, Laird B, Mjaland S. Epidermal growth factor receptor-inhibition (EGFR-I) in the treatment of neuropathic pain. Br J Anaesthesia. 2015;115(5):761–767. doi: 10.1093/bja/aev326. [DOI] [PubMed] [Google Scholar]
- 196.Hendry JM, Alvarez-Veronesi MC, Placheta E, Zhang JJ, Gordon T, Borschel GH. ErbB2 blockade with Herceptin (trastuzumab) enhances peripheral nerve regeneration after repair of acute or chronic peripheral nerve injury. Ann Neurol. 2016;80(1):112–126. doi: 10.1002/ana.24688. [DOI] [PubMed] [Google Scholar]
- 197.Papale A, Morella IM, Indrigo MT, et al. Impairment of cocaine-mediated behaviours in mice by clinically relevant Ras-ERK inhibitors. eLife. 2016;5:e17111. doi: 10.7554/eLife.17111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fridman JS, Scherle PA, Collins R, et al. Preclinical evaluation of local JAK1 and JAK2 inhibition in cutaneous inflammation. J Invest Dermatol. 2011;131(9):1838–1844. doi: 10.1038/jid.2011.140. [DOI] [PubMed] [Google Scholar]
- 199.Punwani N, Scherle P, Flores R, et al. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J Am Acad Dermatol. 2012;67(4):658–664. doi: 10.1016/j.jaad.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 200.Bissonnette R, Papp KA, Poulin Y, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. 2016;175(5):902–911. doi: 10.1111/bjd.14871. [DOI] [PubMed] [Google Scholar]
- 201.Sakimoto T, Ishimori A. Anti-inflammatory effect of topical administration of tofacitinib on corneal inflammation. Exp Eye Res. 2016;145:110–117. doi: 10.1016/j.exer.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 202.Pitsiou G, Zarogoulidis P, Petridis D, et al. Inhaled tyrosine kinase inhibitors for pulmonary hypertension: a possible future treatment. Drug Design Dev Ther. 2014;8:1753–1763. doi: 10.2147/DDDT.S70277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jones P, Storer RI, Sabnis YA, et al. Design and synthesis of a pan-Janus kinase inhibitor clinical candidate (PF-06263276) suitable for inhaled and topical delivery for the treatment of inflammatory diseases of the lungs and skin. J Med Chem. 2017;60(2):767–786. doi: 10.1021/acs.jmedchem.6b01634. [DOI] [PubMed] [Google Scholar]
- 204.El-Abaseri TB, Fuhrman J, Trempus C, Shendrik I, Tennant RW, Hansen LA. Chemoprevention of UV light-induced skin tumorigenesis by inhibition of the epidermal growth factor receptor. Cancer Res. 2005;65(9):3958–3965. doi: 10.1158/0008-5472.CAN-04-2204. [DOI] [PubMed] [Google Scholar]
- 205.Yao Y, Wolverton JE, Zhang Q, et al. Ultraviolet B radiation generated platelet-activating factor receptor agonist formation involves EGF-R-mediated reactive oxygen species. J Immunol. 2009;182(5):2842–2848. doi: 10.4049/jimmunol.0802689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pulit-Penaloza JA, Sapkota B, Stein Esser E, Compans RW, Pollack BP, Skountzou I. Modulation of influenza vaccine immune responses using an epidermal growth factor receptor kinase inhibitor. Sci Rep. 2015;5:12321. doi: 10.1038/srep12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Escuin-Ordinas H, Li S, Xie MW, et al. Cutaneous wound healing through paradoxical MAPK activation by BRAF inhibitors. Nat Commun. 2016;7:12348. doi: 10.1038/ncomms12348. [DOI] [PMC free article] [PubMed] [Google Scholar]