

Abstract
We report on a 5-year-old white girl with Prader-Willi syndrome (PWS) and a submicroscopic deletion of 15q11q13 of approximately 100–200 kb in size. High resolution chromosome analysis was normal but fluorescence in situ hybridization (FISH), Southern hybridization, and microsatellite data from the 15q11q13 region demonstrated that the deletion was paternal in origin and included the SNRPN, PAR-5, and PAR-7 genes from the proximal to distal boundaries of the deletion segment. SNRPN and PW71B methylation studies showed an abnormal pattern consistent with the diagnosis of PWS and supported the presence of a paternal deletion of 15q11q13 or an imprinting mutation. Biparental (normal) inheritance of PW71B (D15S63 locus) and a deletion of the SNRPN gene were observed by microsatellite, quantitative Southern hybridization, and/or FISH analyses. Our patient met the diagnostic criteria for PWS, but has no reported behavior problems, hyperphagia, or hypopigmentation. Our patient further supports SNRPN and possibly other genomic sequences which are deleted as the cause of the phenotype recognized in PWS patients.
Keywords: Prader-Willi syndrome, submicroscopic deletion, FISH, genotype/phenotype correlations
INTRODUCTION
Prader-Willi Syndrome (PWS) is characterized by infantile hypotonia with feeding problems, hypogonadism, early childhood onset of obesity, small hands and feet, short stature, minor facial anomalies including almond-shaped palpebral fissures, downturned corners of the mouth, and a narrow bifrontal diameter, psychomotor retardation, and hypopigmentation. While about 70% of PWS patients have an interstitial deletion of the proximal long arm of chromosome 15, between 20 and 25% have maternal disomy of chromosome 15, and 5 to 10% have biparental inheritance with normal appearing chromosome 15s [Mascari et al., 1992]. The clinically typical PWS patient either has a deletion or maternal disomy of chromosome 15 [Lai et al., 1993]. Interestingly, a similar deletion of the maternal chromosome 15 (15q11q13) produces an entirely different clinical condition, Angelman syndrome, characterized by severe mental retardation, absent speech, brachycepahly, ataxia, large mouth and bouts of inappropriate laughter. Thus, Prader-Willi and Angelman syndromes are considered the first examples of genetic imprinting in man characterized by differential expression of genetic material from the mother versus the father. Clinical differences have been reported between those PWS subjects with or without the cytogenetic deletion, the former having lighter hair, eye and skin color, and higher IQ scores than those with apparently normal chromosomes [Butler et al., 1986; Butler, 1990].
To better characterize the cytogenetic/molecular genetic status of a relatively large number of individuals presenting with findings consistent with PWS, a molecular and cytogenetic study was undertaken in a large group of patients [Butler et al., 1994]. During this analysis a PWS patient with a small submicroscopic deletion of the paternal chromosome 15 was found. Because of a paucity of clinical reports of PWS patients with submicroscopic deletions of chromosome 15, we would like to report the clinical, molecular, and cytogenetic findings of this patient.
CLINICAL REPORT
EE is a 5-year-old white girl who was referred for genetic evaluation at age 3 days because of a history of severe hypotonia and a poor suck reflex. She weighed 3,334 g (30th centile) at birth and was 54.6 cm (90th centile) long. She was born to a 19-year-old G2P1 mother following a 42 week pregnancy. The pregnancy was complicated by morning sickness requiring medication. Decreased fetal vigor was noted. A C-section was performed secondary to failure of labor progression. Apgar scores were 6 and 7 at one and five minutes, respectively. The family history was unremarkable. The patient has one younger normal brother. During the neonatal period, hypotonia, a weak cry, poor suck reflex, poor head control, and a poor Moro reflex were noted as well as an abnormal resting posture. At age 4 weeks, the infant was considered to have failure to thrive, although she was normocephalic and had a narrow forehead, short upturned nose, downturned corners of the mouth, sticky saliva and micrognathia. A rapid weight gain was observed at 2½ years of age, although the parents have not observed hyperphagia. She has borderline normal intelligence and currently attends normal kindergarten at age 6 years. Her weight at age 6 years was 23.3 kg (>95th centile), height 104.2 cm (10th centile), head circumference 52 cm (75th centile), hand length 12 cm (10th centile), mid-finger length 5.2 cm (25th centile), foot length 15.9 cm (3rd centile), ear length 5.9 cm (75th centile), and outer canthal distance 6.5 cm (<3rd centile). By history the patient has not experienced any unusual behavior problems. Photographs of patient EE taken at 3 months and 5 years, respectively, are shown in Figure 1.
Fig. 1.

Frontal photographs of patient EE with a submicroscopic deletion of proximal 15q at age 3 months and 5 years.
At age 4 weeks, a normal EEG, EMG, and head MRI were noted. Urine for organic acids showed trace adipic, suberic, and sebacic acids (but normal when repeated); normal electrolytes, serology, amino acids, and hematologic findings were observed. The urine analysis showed no reducing substances or other abnormalities. A muscle biopsy during the neonatal period was not diagnostic for any specific neuromuscular disorder. Auditory evaluation by BAER was interpreted as normal at 4 weeks of age. An ophthalmology consultation also showed a healthy optic nerve and a normal retinal exam.
CYTOGENETIC STUDIES
High resolution cytogenetic analysis (≥550 bands) using routine G-banding techniques with Actinomycin D [Butler et al., 1986] were normal and fluorescence in situ hybridization (FISH) using SNRPN, D15S10, D15S11, and GABRB3 probes from Oncor, Inc. (Gaithersburg, MD) following manufacturers’ protocols revealed a submicroscopic deletion with the SNRPN probe only. We have used successfully FISH analysis of cosmids from a contig developed for YAC 457B4 [Sutcliffe et al., 1994] in patient EE. Cosmids 93 and 102 which contain the SNRPN gene were both deleted in patient EE as well as cosmid 125 (Fig. 2). Cosmid 102 contains the SNRPN gene plus the 5′ flanking region [Sutcliffe et al., 1994]. Cosmid 106, located just proximal to cosmid 102, is not deleted and establishes the proximal breakpoint. Cosmid 107, which contains PAR-7, located distal to cosmid 125 also contains the MN1 microsatellite. PAR-5 is located between the SNRPN and PAR-7 genes [Sutcliffe et al., 1994].
Fig. 2.
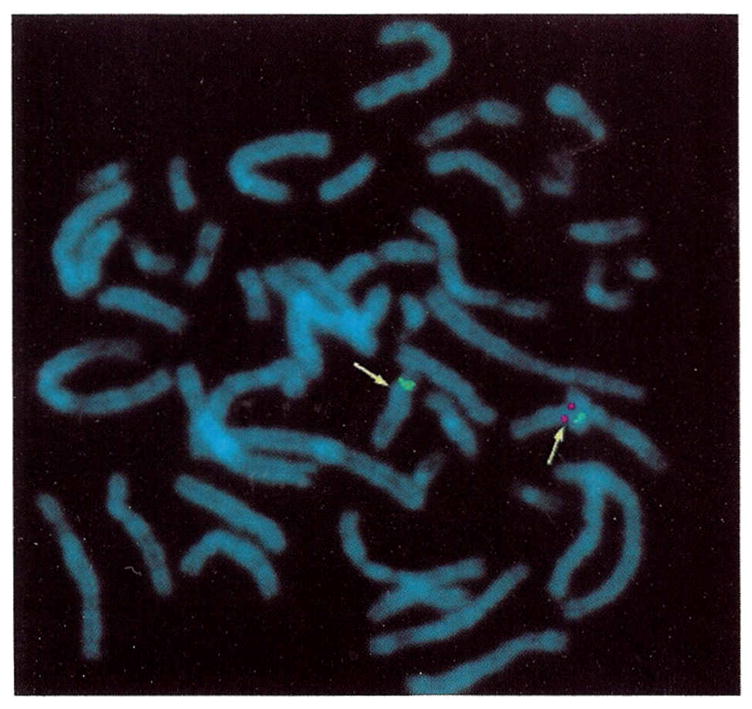
Fluorescence in situ hybridization (FISH) using cosmid 102, containing most of the SNRPN gene, from the proximal 15q11q13 region (red signal; yellow arrow) and a chromosome 15 alpha satellite control probe (green) from patient EE. Only one chromosome 15 (normal) shows both the alpha satellite and cosmid 102 hybridization signals, while the other chromosome 15 (deleted) shows only the control alpha satellite hybridization signal and no cosmid 102 signal.
MOLECULAR STUDIES
Quantitative hybridization using a radio-labeled PCR product generated from primers of exons 4 and 6 of the SNRPN gene using protocols described by Ozcelik et al. [1992] with genomic DNA digested with Bgl II enzyme supported a submicroscopic deletion (Fig. 3). The SNRPN gene is a paternally expressed candidate gene for PWS [Ozcelik et al., 1992]. This analysis produces a 7.5 kb upper fragment representing the SNRPN pseudogene from chromosome 6 (used as a control) and a 5.8 kb lower fragment representing the SNRPN gene from the 15q11q13 region. An autoradiograph was analyzed by densitometry to calculate the copy number of the SNRPN gene comparing the control pseudogene upper band with the lower SNRPN band from the autoradiograph following the methodology described by Ozcelik et al. [1992] (Fig. 3). The results indicated a copy number of 0.7 or a deletion of one of the SNEPN alleles.
Fig. 3.
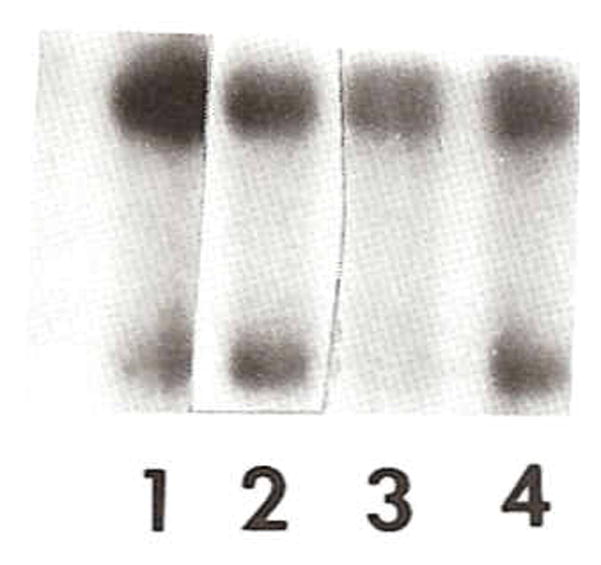
Quantitative hybridization using a radio-labeled PCR product generated using primers of exons 4 and 6 of the SNRPN gene [Ozcelik et al., 1992] with genomic DNA digested with Bgl II. The 7.5 kb (upper band) fragment represents a pseudogene (used as control) from chromosome 6 and the 5.8 kb (lower band) fragment represents the SNRPN gene from 15q11q13 region. Lanes 1 and 3 are from patient EE and show a submicroscopic deletion (e.g., calculated copy number of 0.7 or a deletion for SNRPN) of 15q11q13 and confirmed by FISH analysis. Lanes 2 and 4 are a normal control female.
In addition, the PW71B probe (recognizing D15S63, a differentially methylated locus) was hybridized to genomic DNA isolated from peripheral blood digested with Hind III/Hpa II enzymes which produces two different size fragments (upper 6.6 kb band from the maternal chromosome 15 and a lower 4.7 kb band from the paternal chromosome 15) following protocols reported by Dittrich et al. [1992]. If both fragments are present then a biparental (normal) inheritance or normal methylation of D15S63 loci is indicated. If only the upper allele is present then a paternal 15q11q13 deletion or maternal disomy of chromosome 15 would be indicated. The results indicated the presence of only the upper maternal band consistent with either a paternal deletion or loss of imprinting at this locus (Fig. 4). Methylation was also analyzed at the SNRPN CpG island using the methylation sensitive enzyme, Not I, and digested with Xba I [Sutcliffe et al., 1994] from genomic DNA isolated from peripheral blood. An abnormal result was obtained with the presence of the 4.2 kb maternal band and absence of the 0.9 kb paternal band (Fig. 5). The methylation analysis using the SNRPN CpG island probe was normal in the father. Quantitative Southern hybridization of several DNA probes from 15q11q13 (e.g., IR10, IR39, 189-1, 34, 4-3R, and 3-21) and control probes from 13q (e.g., H2-42 and H2-26) were also studied as previously described [Mbikay et al., 1988; Roback et al., 1991; Butler et al., 1993; Tantravahi et al., 1989] and a normal copy number of 2 was calculated indicating a nondeletion status for these loci.
Fig. 4.
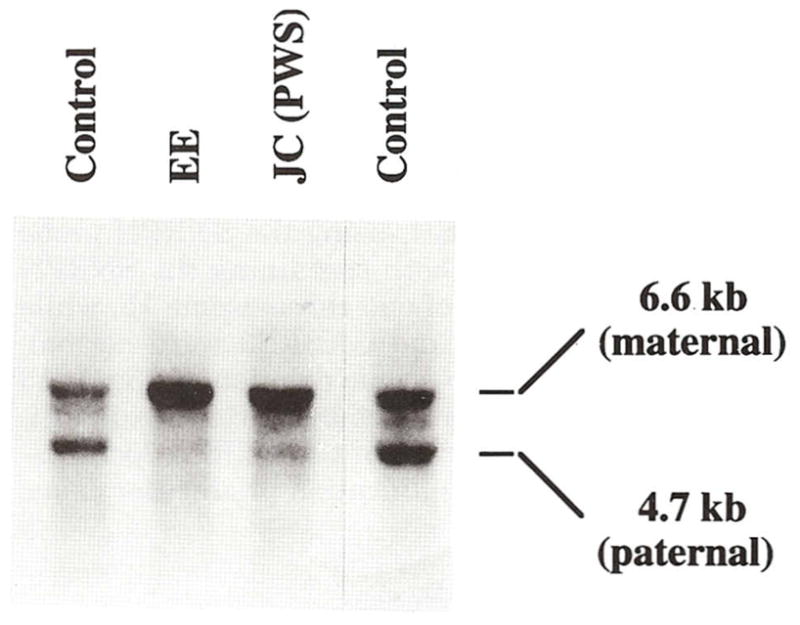
Southern hybridization using a radio-labeled PW71B probe (recognizing D15S63, a differentially methylated locus) hybridized to 5 μg of genomic DNA isolated from peripheral blood and digested with Hind III/Hpa II enzymes which produces two different size fragments (upper 6.6 kb band from maternal chromosome 15 and a lower 4.7 kb band from the paternal chromosome 15). Four individuals were studied. Our patient (EE) and a PWS patient (JC) both show only the upper band (maternal) indicating the PWS diagnosis or absence of a lower band (paternal) while the two controls have both upper and lower bands.
Fig. 5.
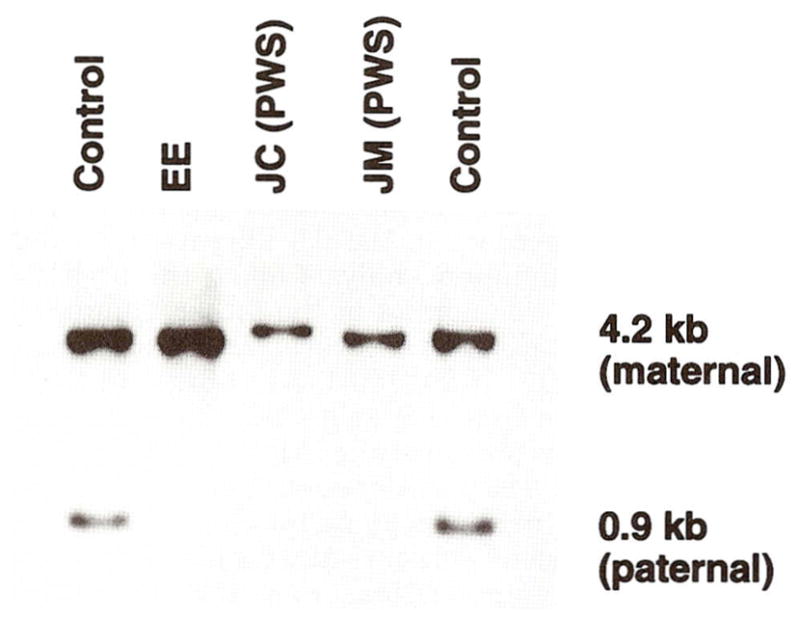
Southern hybridization using radio-labeled SNRPN CpG island probe (0.9 kb) [Sutcliffe et al., 1994] hybridized to 5 μg genomic DNA isolated from peripheral blood and digested with Xba I and Not I enzymes, produces two different size fragments (upper 4.2 kb band from the maternal chromosome 15 and lower 0.9 kb band from the paternal chromosome 15). Five individuals were studied. Our patient (EE) and two PWS patients (JC and JM) showed only the upper band (maternal) indicating the PWS diagnosis or absence of the lower band (paternal) while the two controls have both the upper and lower bands.
Standard PCR amplifications were undertaken on patient EE and other PWS patients using 18 short tandem repeats or microsatellites from the 15q11q13 region (D15S541, D15S542, D15S543, D15S11, D15S63, SNRPN, D15S128, MN1, D15S210, D15S10, D15S122, D15S113, GABRB3, D15S97, GABRA5, D15S156, D15S219, and D15S165—represented in linear order) following established protocols [Mutirangura et al., 1992a, b; 1993a, b]. The PCR amplification and polyacrylamide gel electrophoresis of 32P end-labeled amplification products were analyzed according to modifications of the method described by Weber and May [1989] and Mutirangura et al. [1992a, b]. Oligonucleotide sequences were obtained from the NIH-DOE Genome Data Base (Johns Hopkins University) and from the report of the Second International Chromosome 15 Workshop [Malcom and Donlon, 1994]. PCR amplification showed biparental inheritance for D15S63 (Fig. 6) and heterozygous results for D15S10, D15S11, D15S113, GABRB3, D15S122, GABRA5, D15S165, D15S210, and D15S542. Single, uninformative alleles were seen for D15S97, D15S156, D15S128, D15S543, D15S541, and D15S219. The only informative marker detecting a deletion was MN1 identified in the 3′ UTR region of the PAR-7 gene (Nakao, M and Sutcliffe J, unpublished) (Fig. 7).
Fig. 6.

PCR amplification with radio-labeled primers from D15S63 locus from PWS patient EE (middle lane), her mother (right lane), and her father (left lane) generating two fragment sizes [167 bp (bottom band) and 171 bp (upper band)]. PCR amplification of D15S63 produces 38% heterozygosity. Biparental (normal) inheritance of D15S63 was seen for patient EE.
Fig. 7.
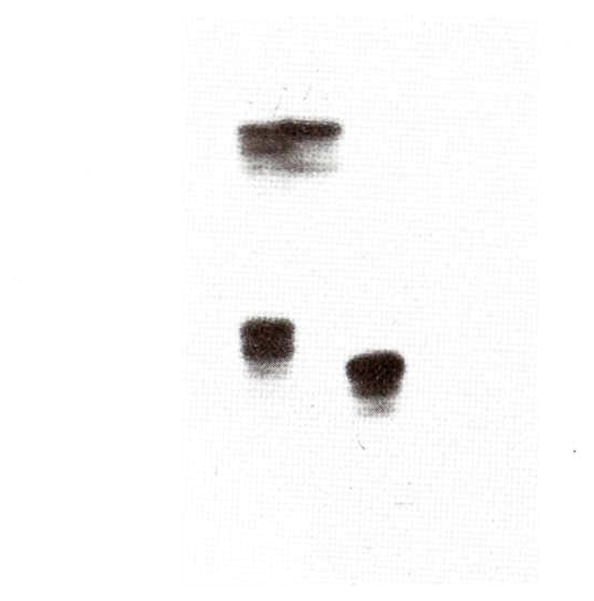
PCR amplification with radio-labeled primers from MN1 locus (identified in the 3′ UTR region of the PAR-7 gene) from patient EE (middle lane), her mother (left lane) and her father (right lane). The mother showed two alleles (heterozygous) and the father showed one allele (homozygous) but for a different size allele. Patient EE shows only one allele from the mother and no allele from the father, thus a paternal deletion of the MN1 locus.
DISCUSSION
Our patient clearly has the Prader-Willi syndrome [Holm et al., 1993]. However, high resolution chromosome analysis (≥550 band level) was performed with patient EE and no deletion of 15q11q13 region was observed cytogenetically. FISH, using the SNRPN probe and cosmids 93 and 102 (both containing the SNRPN gene), detected a deletion of one chromosome 15, while probes D15S10, D15S11, and GABRB3 were normal, thus indicating a submicroscopic deletion of 15q11q13 region. These findings were also supported by quantitative Southern hybridization of the SNRPN locus using a radiolabeled PCR amplification product for axons 4 through 6. Microsatellite analysis using 18 polymorphic markers within 15q11q13 detected a deletion in only one marker, MN1, located in the 3′ UTR of the PAR 7 gene. Methylation analysis at both the PW71 (D15S63) and SNRPN loci detected an abnormal pattern consistent with PWS. Thus, using FISH and microsatellite analyses indicated a paternal submicroscopic deletion of the SNRPN, PAR-5 and PAR-7 genes in patient EE. The full extent of the deletion has not been completely defined, but does not extend to either D15S63 or D15S10. It is not known, definitively, whether the deletion includes the imprinting control region [Buiting et al., 1995], but the abnormal methylation pattern at PW71, which is not deleted by microsatellite analysis, provides strong evidence that the imprinting control region is deleted. This patient showed biparental (normal) inheritance of chromosome 15 and not uniparental maternal disomy and clearly shows a paternal deletion of MN1 while other loci in the region were intact (non-deleted) or were uninformative. There was no evidence of non-paternity when loci of other chromosomes (e.g., chromosome 2 —data not shown) were evaluated from this family.
Our patient is one of the first patients clinically described with such a small submicroscopic deletion, which is estimated to be approximately 100–200 kb in size as determined by chromosome 15q11q13 map region reported by Mutirangura et al. [1993] and Sutcliffe et al. [1994], yet the patient has PWS. The patient does not show the hypopigmentation which is frequently seen in PWS patients [Butler, 1989] and thought due to deletions of locus D15S12 which is intact in our patient. In addition, at age 6 years there have been no reported behavior problems or hyperphagia reported by the parents or observed during regular medical follow-up since the neonatal period. A rapid weight gain was noted at 2½ years of age without documented hyperphagia. Her weight is maintained by caloric reduction (approximately 1,000 kcal/day) and regular exercise (e.g., riding a bicycle and walking daily).
We present this patient with hopes that other investigators will report their typical as well as atypical PWS patients with submicroscopic deletions in order to identify genotype/phenotype correlations and to better characterize the size, location, and type of genetic lesion found in the 15q11q13 region. Our patient further supports SNRPN and other genomic DNA sequences (e.g., PAR genes) which are deleted in our patient as the cause of the phenotype recognized in PWS patients.
Acknowledgments
We thank Peggy Perkinson and Verna Wyatt for expert preparation of the manuscript. This research was partially supported by NICHD grants (PO1 HD 30329-01A2 and P30 HD15052).
References
- Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, Horsthemke B. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet. 1995;9:395–400. doi: 10.1038/ng0495-395. [DOI] [PubMed] [Google Scholar]
- Butler MG. Hypopigmentation: A common feature of Prader-Willi syndrome. Am J Hum Genet. 1989;45:140–146. [PMC free article] [PubMed] [Google Scholar]
- Butler MG. Prader-Willi syndrome: Current understanding of cause and diagnosis. Am J Med Genet. 1990;35:319–332. doi: 10.1002/ajmg.1320350306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dahir GA, Schwartz HS. Molecular analysis of transforming growth factor beta in giant cell tumor of bone. Cancer Genet Cytogenet. 1993;66:108–112. doi: 10.1016/0165-4608(93)90237-g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Forrest KB, Miller LK. Cytogenetic and molecular characterization of 57 individuals with the Prader-Willi syndrome. Am J Hum Genet. 1994;55:A100. [Google Scholar]
- Butler MG, Meaney FJ, Palmer CG. Clinical and cytogenetic survey of 39 individuals with Prader-Labhart-Willi syndrome. Am J Med Genet. 1986;23:793–809. doi: 10.1002/ajmg.1320230307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich B, Robinson WP, Knoblauch H, Buiting K, Schmidt K, Gillessen-Kaesbach G, Horsthemke B. Molecular diagnosis of the Prader-Willi and Angelman syndromes by detection of parent-of-origin specific DNA methylation in 15q11-13. Hum Genet. 1992;90:313–315. doi: 10.1007/BF00220089. [DOI] [PubMed] [Google Scholar]
- Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F. Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics. 1993;91:398–402. [PMC free article] [PubMed] [Google Scholar]
- Lai LW, Erickson RP, Cassidy SB. Clinical correlates of chromosome 15 deletions and maternal disomy in Prader-Willi syndrome. Am J Dis Child. 1993;147:1217–1223. doi: 10.1001/archpedi.1993.02160350091014. [DOI] [PubMed] [Google Scholar]
- Malcolm S, Donlon TA. Report of the second international workshop on human chromosome 15 mapping. Cytogenet Cell Genet. 1994;67:2–14. doi: 10.1159/000133717. [DOI] [PubMed] [Google Scholar]
- Mascari MJ, Gottlieb W, Rogan PK, Butler MG, Waller DA, Armour JAL, Jeffreys Ladda RL, Nicholls RD. The frequency of uniparental disomy in Prader-Willi syndrome. New Engl J Med. 1992;326:1599–1607. doi: 10.1056/NEJM199206113262404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbikay M, Linard CG, Sirois F, Lazure C, Seidah NG, Chrétien M. Tissue-specific expression of the prostatic secretory protein PSP94 in cynomolgus monkey (Macaca fascicularis) Cell Mol Bio. 1988;34:387–398. [PubMed] [Google Scholar]
- Mutirangura A, Jayakumar A, Sutcliffe JS, Nakao M, McKinney MJ, Buiting K, Horsthemke B, Beaudet AL, Chinault AC, Ledbetter DH. A complete YAC contig of the Prader-Willi/Angelman chromosome region (15q11q13) and refined localization of the SNRPN gene. Genomics. 1993b;18:546–552. doi: 10.1016/s0888-7543(11)80011-x. [DOI] [PubMed] [Google Scholar]
- Mutirangura A, Greenberg F, Butler MG, Malcolm S, Nicholls RD, Chakravarti A, Ledbetter DH. Multiplex PCR of three dinucleotide repeats in the Prader-Willi/Angelman critical region (15q11q13): Molecular diagnosis and mechanism of uniparental disomy. Hum Mol Genet. 1993a;2:143–151. doi: 10.1093/hmg/2.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutirangura A, Kuwano A, Ledbetter SA, Chinault AC, Ledbetter DH. Dinucleotide repeat polymorphism at the D15S11 locus in the Angelman/Prader-Willi region (AS/PWS) of chromosome 15. Hum Mol Genet. 1992a;1:139. doi: 10.1093/hmg/1.2.139-a. [DOI] [PubMed] [Google Scholar]
- Mutirangura A, Ledbetter SA, Kuwano A, Chinault AC, Ledbetter DH. Dinucleotide repeat polymorphism at the GABAa receptor B3(GABRB3) locus in the Angelman/Prader-Willi region (AS/PWS) of chromosome 15. Hum Mol Genet. 1992b;1:67. doi: 10.1093/hmg/1.1.67. [DOI] [PubMed] [Google Scholar]
- Ozcelik T, Leff S, Robinson W, Donlon T, Lalande M, Sanjines E, Schinzel A, Francke U. Small nuclear ribonucleoprotein polypeptide N (SNRPN), an expressed gene in the Prader-Willi syndrome critical region. Nature Genet. 1992;2:265–269. doi: 10.1038/ng1292-265. [DOI] [PubMed] [Google Scholar]
- Roback EW, Barakat AY, Dev VG, Mbikay M, Chretien M, Butler MG. An infant with deletion of the distal long arm of chromosome 15 (q26.1→qter) and loss of insulin-like growth factor 1 receptor gene. Am J Med Genet. 1991;38:74–79. doi: 10.1002/ajmg.1320380117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JS, Nakao M, Christian S, Orstavik KH, Tommerup N, Ledbetter DH, Baudet AL. Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat Genet. 1994;8:52–58. doi: 10.1038/ng0994-52. [DOI] [PubMed] [Google Scholar]
- Tantravahi U, Nicholls RD, Stroh H, Ringer S, Neve RL, Kaplan L, Wharton R, Wurster-Hill D, Graham JM, Cantu ES, Frias JL, Kousseff B, Latt SA. Quantitative calibration and use of DNA probes for investigating chromosome abnormalities in Prader-Willi syndrome. Am J Med Genet. 1989;33:78–87. doi: 10.1002/ajmg.1320330110. [DOI] [PubMed] [Google Scholar]
- Weber JL, May PE. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet. 1989;44:388–396. [PMC free article] [PubMed] [Google Scholar]