

Introduction
Despite suppressive combination antiretroviral therapy (ART), latent HIV-1 proviruses persist. This latent reservoir is established within 48-72 hours after infection, has a long half-life1,2, enables viral rebound when ART is interrupted, and is the major barrier to HIV-1 cure3. Latent cells are exceedingly rare in the blood (≈ 1/106 CD4+ T cells) and typically enumerated by indirect means such as viral outgrowth assays4,5. We report a novel strategy to purify and characterize single reactivated latent cells from HIV-1 infected individuals on suppressive antiretroviral therapy. Surface expression of viral Envelope protein was used to enrich reactivated latent T cells producing HIV-RNA, and single cell analysis was performed to identify intact virus. Reactivated latent cells produce full length viruses that are identical to those found in viral outgrowth cultures, and represent clones of in vivo expanded T cells as determined by the sequence of their T cell receptors. Gene expression analysis revealed that these cells share a transcriptional profile that includes expression of genes implicated in silencing the virus. We conclude that reactivated latent T cells isolated from the blood can share a gene expression program that allows for cell division without activation of the cell death pathways that are normally triggered by HIV-1 replication.
To investigate the cells that contribute to the latent reservoir, we developed a method to enrich and isolate reactivated latent cells by combining antibody staining, magnetic enrichment, and flow cytometry6 (latent cell capture, or LURE). Purified CD4+ T cells from ART suppressed donors were activated with PHA, a robust in vitro latency reactivation agent,5,7 for 36h in the presence of 5 potent antiretroviral drugs, and a pan-caspase inhibitor. Cells expressing surface HIV-1 Envelope (Env) protein were labeled with a cocktail of biotinylated anti-Env broadly neutralizing antibodies (bNAbs, 3BNC117,8 10-1074,9 and PG16,10), and enriched with magnetic beads.
Relative enrichment of the magnetically isolated, Env+ cellular fraction was measured by comparison to unfractionated control cells from the same culture by flow cytometry (Fig. 1a and Supplemental Data Fig. 1a) and by quantitative PCR for HIV-1 gag mRNA (Fig. 1c). Enrichment of cell associated HIV-1 RNA was entirely dependent on cellular activation with PHA (Supplemental Data Fig. 1b). Enrichment was measured in samples from 10 individuals and was found to be dependent in part (r2 = 0.5609, p = 0.0127) on the size of the latent reservoir as measured by viral outgrowth assays in infectious units per million (IUPM) (Fig. 1d). We conclude that reactivated latently infected cells can be enriched based on HIV-1 Env surface expression.
Figure 1. Latency capture enriches for HIV-RNA producing cells.
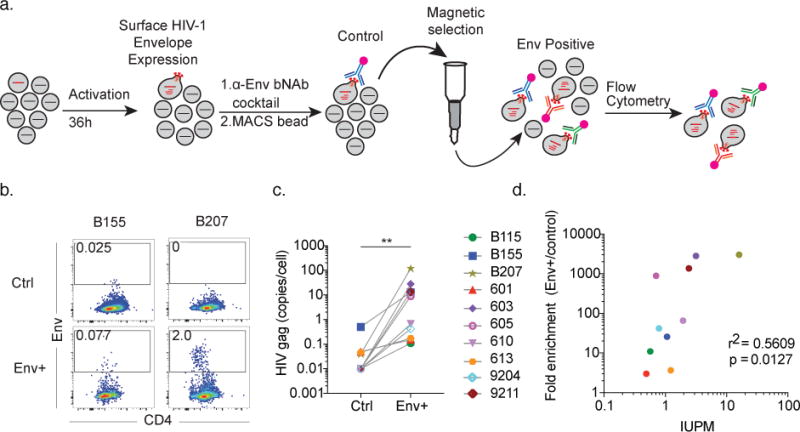
a) Diagrammatic representation of latency capture (LURE) protocol. CD4+ T cells from ART suppressed donors are cultured in conditioned media with PHA, IL-2, antiretroviral drug cocktail and pan-caspase inhibitor for 36h. Cells are labeled with a biotinylated bNAb cocktail, followed by Streptavidin PE and anti-PE magnetic beads, passed over a magnetic column, and FACS analysis. b) Envelope-expressing cell enrichment. Dot plots show Env vs. CD4 staining on pre-enrichment control (top row), and positively selected cells (bottom row) for donors B155 and B207. Gate shows frequency of Env+ cells in each population. Shown is two representative experiments of 15 independent experiments. c) HIV-gag mRNA was measured in equivalent numbers of Env+ and control cells. Graph shows results of qPCR (12.8-copy limit of detection) for HIV-gag mRNA, normalized to the number of sorted cells. p = 0.002, Wilcoxon matched-pairs signed rank two-tailed test. Shown is representative data from 10 individuals from more than 30 independent experiments. d) Fold-enrichment (Env+/control) in (c) compared to IUPM. Shown is representative data from 10 individuals from more than 30 independent experiments.
To further purify the reactivated latent cells, we used flow cytometry to sort single cells from the magnetically enriched fraction based on Env staining. Individual cells expressing both env and gag were identified by the combination of surface Env staining and single cell HIV-1 gag mRNA expression. The frequency of gag mRNA expressing single cells in patients with high IUPMs ranged from 10-50% of sorted cells (Supplemental Table 1). In individuals with relatively lower IUPMs (0.49-2.43), the percent of Env+gag+ single cells isolated varied from 0-4% (Supplemental Table 1).
We performed single cell RNA sequencing (scRNASeq) on gag+Env+ single cells captured by LURE and control unfractionated single cells from the exact same PHA activated culture from donors 603, 605 and B207. In addition, we performed scRNASeq on activated CD4+ T cells that were productively infected with HIV-1YU2 (YU2) in vitro and purified by cell sorting using anti-Env antibodies (Supplemental Data Fig. 2). Overall 249 cells were characterized, of which 22 cells (8.8%) were removed by quality metrics11. Of the 227 cells retained, 33 were YU2 infected cells, 85 were cells captured by LURE, and 109 were unfractionated control cells from the same cultures (Fig. 2A). On average, we obtained ~1500 expressed genes per cell (Supplemental Data Fig. 3).
Figure 2. Full length virus sequences recovered by scRNASeq.
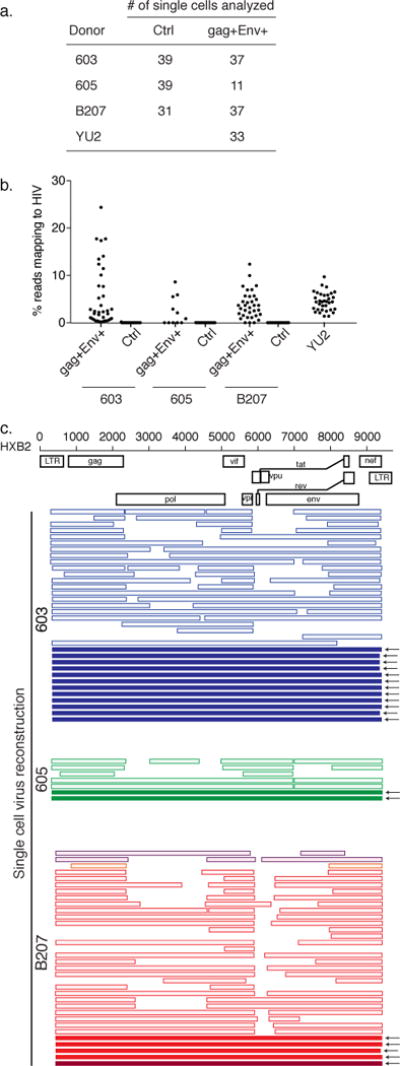
a) Number of single cells analyzed by RNASeq. b) Fraction of reads mapping to HIV-1 in unfractionated control, LURE purified gag+Env+, and YU2 infected scRNASeq libraries. c) Map of individual viruses reconstructed from scRNASeq. Each horizontal bar represents a single virus from an individual cell. Solid bars indicate that the entire virus was reconstructed from scRNASeq reads. Outlined, lighter colored bars indicate incomplete genome reconstruction. Different colors indicate different sequences. For participants 603 and 605, every virus identified was identical. For B207, we identified 4 unique viruses, with one clone (in red) predominating.
As expected, HIV reads were not detectable in the unfractionated, activated control cells (Fig. 2b). In contrast, cells captured by LURE and YU2 infected cells showed similar percentages of total mRNA reads mapping to the HIV-1 genome (3.8 and 4.5% respectively12) (Fig. 2b). We conclude that scRNASeq performed on reactivated latent cells captured by LURE contains RNA sequences mapping to the human genome and HIV-1.
We used Iterative Virus Assembler software to reconstruct the virus from scRNASeq reads in each individual CD4+ T cell13. HIV RNA recovered by scRNASeq was dependent on proviral transcription as determined by analysis of HIV-1 splice variants (Supplemental Data Fig. 4a). Fully reconstructed viruses were obtained from 26 cells infected with YU2, and 19 cells captured by LURE (Fig. 2c and Supplemental Data Fig. 4b). All viruses obtained from 603 and 605 belonged to a single expanded viral clone (Fig. 2c). We identified 4 different viruses in B207: 2 were fully reconstructed, and 2 others partially reconstructed (Fig. 2c). All of the fully reconstructed viruses were completely intact when analyzed by Gene Cutter software. Thus, the combination of LURE and scRNASeq can be used to recover full length, intact HIV-1 from single reactivated latent cells.
To determine whether the full-length viruses expressed in the purified single cells obtained by LURE correspond to the intact latent viruses that emerge in viral outgrowth assays, we compared their Env sequences (Fig. 3a). To do so, we performed quantitative and qualitative viral outgrowth assays (Q2VOA)14, Env SGA on DNA isolated from CD4+ T cells, and compared these sequences to those found in LURE cells.
Figure 3. Captured cells express Env that is identical to latent virus emerging in Q2VOA and represent expanded clones.
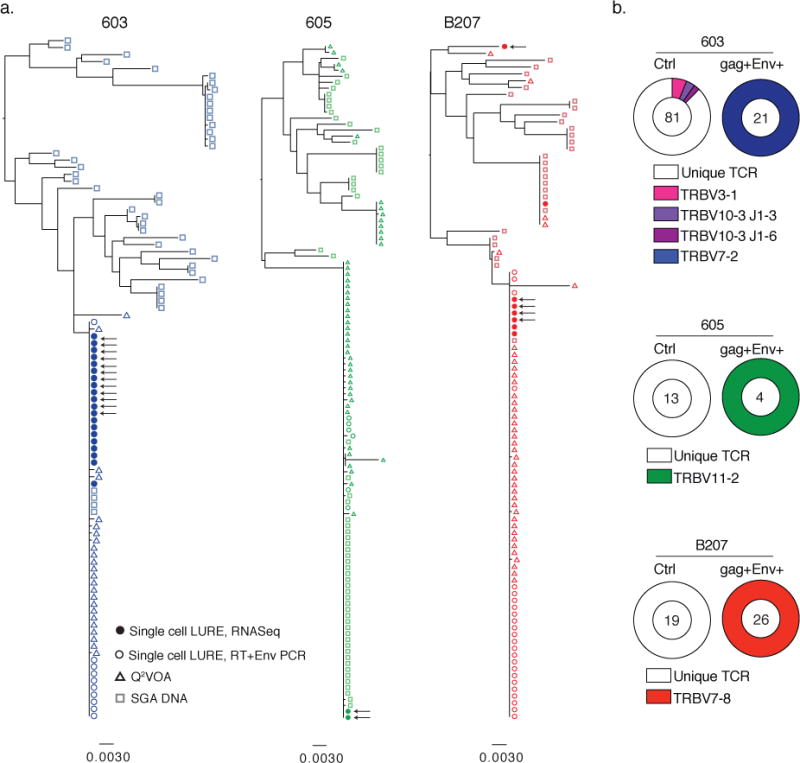
a) Maximum likelihood phylogenetic trees compare full length Env sequences derived from single cells capture by LURE (solid and open circles), DNA proviruses (open squares) and replication-competent single cell viral outgrowth cultures (Q2VOA) (open triangles) from participants 603, 605, and B207. Sequences from LURE cells were obtained either by recovery and assembly from RNASeq reads (closed circles) or from reverse transcription of RNA in single cells followed by specific Env PCR from single gag+Env+ LURE cells (open circles). Arrows indicate confirmed full-length sequences. b) TCR sequences recovered from scRNASeq or amplified by PCR, for control (unfractionated pre-enrichment) and gag+Env+ LURE purified cells. The number in the center of the pie denotes the number of cells sequenced; slices are proportional to clone size showing unique TCRs (white slices) and clonal TCRs (colored slices). Clones were identified by their shared TCR alpha and beta sequences.
Phylogenetic analysis of Env sequences revealed that in donors 603 and B207 the Env sequences obtained by LURE and Q2VOA generally clustered together, were part of an expanded clone, and did not overlap significantly with sequences obtained by proviral DNA SGA (Fig. 3a). Participant 605 has an unusual distribution of DNA SGA proviral sequences in that there is a significant overlap with the Env sequences found in viral outgrowth cultures. Nevertheless, the majority of LURE derived Env sequences belong to the major viral outgrowth clone found in Q2VOA (Fig. 3a) in all three individuals. We conclude that the Env sequences expressed by cells purified by LURE are typically identical to those found in viruses that emerge from latent cells in viral outgrowth cultures and therefore are replication competent.
Latent cells harboring identical replication competent viruses may arise by T cell clonal expansion14–22 or during a viral replicative burst when identical viruses infect a diverse group of T cells. To definitively distinguish between these possibilities, we analyzed the T cell receptor (TCR) sequences obtained from single latent cells captured by LURE. CD4+ T cells express unique antigen receptors produced by random TCR variable, diversity and joining gene segment (VDJ) recombination. T cells with identical TCRs are only produced by clonal expansion. As a control, we obtained TCR sequences from nearly 600 single CD4+ T cells from 3 healthy and 3 ART treated HIV-1 infected donors. We found that 99.9% of all control TCR sequences were unique, with only a single 2-member clone identified in 1 of the 6 individuals (Supplemental Data Fig. 5). In contrast, the TCR sequences derived from the latent cells with identical proviruses captured by LURE (Fig. 2c and 3a) were entirely clonal in all 3 donors (Fig. 3b and Supplemental Data Fig. 6). The clonality was not due to T cell division in vitro, since there was no measurable T cell division in 36h under our culture conditions (Supplemental Data Fig. 7). Our data demonstrates that groups of latent cells containing identical replication competent viruses are products of CD4+ T cell clonal expansion in vivo.
To further characterize the reactivated latent cells captured by LURE, we performed single-cell transcriptome analysis, and compared the results to unfractionated, PHA stimulated control cells from the same cultures, and to activated CD4+ T cells productively infected with YU2. We performed hierarchical clustering using a principal-component analysis (PCA) called Seurat23 using gene expression data from the 227 cells. This unbiased analysis identified three unique groups of genes that segregated the cells into three separate clusters. Each of these clusters was found to correspond to one of the three input groups: control, LURE, and YU2 infected cells (Fig. 4a, Supplemental Data Fig. 8, and Supplemental Table 2). Additional analysis which employs unsupervised clustering using all gene expression data (Single-cell Consensus Clustering, or SC3), confirmed these results comparing control to LURE cells (Supplemental data Figure 9). Thus, reactivated latent cells captured by LURE cluster separately from uninfected (control) and actively infected CD4+ T cells by PCA and unsupervised clustering.
Figure 4. Distinct gene signature defines reactivated latent cells.
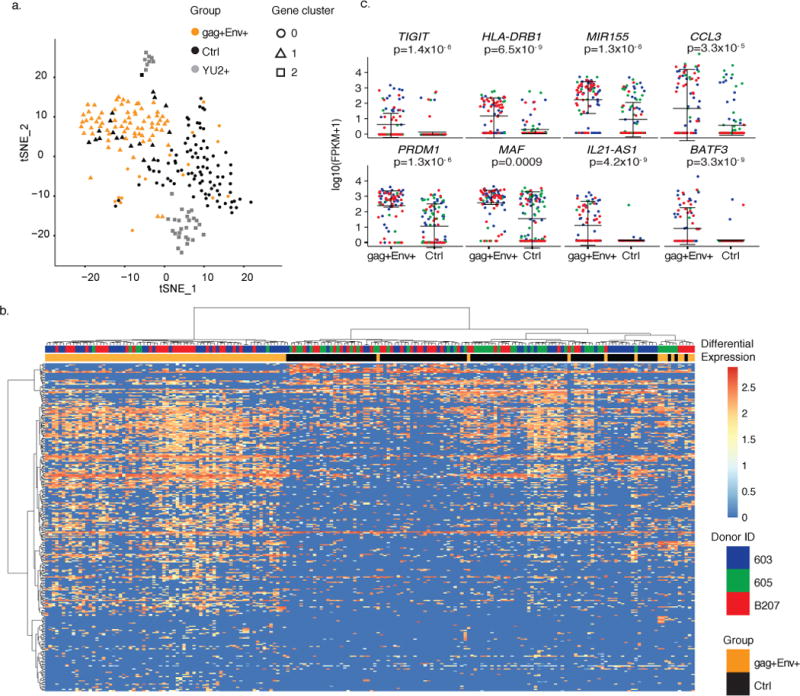
a) Principal components analysis (PCA) clusters cells by group. Shown is the Seurat t-SNE displayed output for the three groups. Plot shows single cells (Control (black), Env+ LURE (orange) and YU2 (gray)). Seurat analysis identified 3 distinct clusters of genes which define three groups of cells (circles (gene cluster 0), triangles (gene cluster 1) and squares (gene cluster 2)) by performing graph-based clustering over 6 principal components. Shown is all data obtained from individuals 603, 605, and B207 (control and LURE cells) and HIV-1YU2 infected healthy donor cells (109 control cells, 85 LURE cells and 33 HIV-1YU2 infected cells). b) Heat-map shows unsupervised clustering of differentially expressed genes between the gag+Env+ LURE purified group (orange bars) and control unfractionated group (black bars). Cells from donor 603 are indicated in blue, 605 in green, and B207 in red. Color indicates the normalized level of expression. c) Graphs show expression of selected significantly differentially expressed genes in individual gag+Env+ LURE purified and control unfractionated cells as determined by MAST software in participants 603 (blue), 605 (green), B207(red). Shown is all data obtained from individuals 603, 605, and B207 (109 control cells and 85 LURE cells). Error bars show mean and standard deviation. Significant differential expression was determined using the likelihood ratio test embedded in the MAST software.
To further understand the transcriptional differences between the three groups of cells, we identified differentially expressed genes (DEG) (p < 0.01) between reactivated latent cells and PHA activated control cells. Using unsupervised clustering, we grouped the cells based on the expression of all significantly differentially expressed genes between LURE and control cell groups (p<0.01, 778 genes Supplemental Table 3). Irrespective of donor, reactivated cells purified by LURE generally segregate from unfractionated, activated control cells in 2 of 3 individuals (Fig. 4b), with cells from the third individual split between the LURE group and control group. Similar results were also obtained by comparison with YU2 infected cells (Supplemental Data Fig. 10). We conclude that cells captured by LURE segregate from activated control cells and productively infected cells by three different methods of analysis.
Among the 240 genes which overlapped between the PCA and DEG (p < 0.01), we find a number of genes highly expressed in the isolated LURE cells which have been shown by independent analyses to be associated with HIV-1 latency (Fig. 4d). For example, Tigit24,25 and HLA-DR26 were 140 and 76-fold up-regulated in cells purified by LURE, and CD32a27 was not (Fig. 4c, and Supplemental Data Fig. 11). MiR-155, which inhibits TRIM32, prevents its interaction with HIV tat and reinforces viral latency28, was 368 times more highly expressed in LURE cells compared to controls. Chemokine CCL3, which is reported to have HIV-1 suppressive effects29,30, is expressed 795 times higher in LURE cells compared to controls. Finally, a number of transcription factors were among the top 15 differentially expressed genes, including the top differentially expressed gene, PRDM1 (1365x). PRDM1 represses HIV-1 proviral transcription in memory CD4+ T cells by inhibition of HIV tat31, and its overexpression is associated with lower levels of HIV-1 transcription in elite controllers32.
To further examine the differences between LURE and control cells, we performed enrichment analysis using the Gene Ontology database with the 240 genes that overlapped between the DEG and PCA analyses. Among the top ten most significantly enriched biological processes, eight are related to immune system function, suggesting that PHA stimulated LURE and control cells differ in their expression of genes related to responses to pathogens. For example, LURE and control cells differ markedly in response to type I interferon and regulation of type I interferon production (Supplemental Tables 4 and 5) with control cells having higher expression of type I interferon responsive genes such as IFIT3, ISG20, IRF1, IFI6, RSAD2, STAT1, XAF1, CTNNB1 and UBE2L6. Consequently, the control cells also show a higher overall expression of genes involved in response to viruses such as CCL5, IFIT3, ISG20, IRF1, SERINC5, IL2RA, RSAD2, DDIT4, STAT1, and PIM2. Consistent with the altered gene expression program in reactivated latent cells, LURE and control cells show significant differences in the expression of genes that regulate transcription (Supplemental Tables 4 and 5). For example, reactivated latent cells have higher levels of expression of transcriptional regulators PRDM1, MAF, IRF4, MTDH, IKZF3, and BATF3, whereas control cells have higher expression of PIM2, STAT1, HNRNPA2B, EZR, IRF1, CTNNB1 and NFKBIZ (Supplemental Tables 4 and 5). We conclude that reactivated latent cells differ from control cells in a number of ways, many of which are related to the suppression of cellular anti-viral immunity.
Our analysis is limited to 3 individuals and to a single reactivation agent, PHA. Examination of additional individuals and methods of latent cell reactivation may reveal additional and or different genes and pathways involved in maintaining latency. LURE purification of reactivated latent cells requires proviral activation to induce Env protein expression on the cell surface. Therefore, LURE captures a subset of latent cells with proviruses that can be reactivated in a single round of potent T cell stimulation33,34. Due to the relative resistance of some latent cells to reactivation,7 LURE mirrors the viral outgrowth assay and is unable to capture the entirety of the latent reservoir. Furthermore, our analysis is limited to circulating CD4+ T cells that express Env on the cell surface that are recognized by our antibody cocktail. Finally, some reactivated latent cells are certainly lost during the multiple processing stages involved in the LURE protocol. Thus, the cells captured by LURE represent a fraction of the circulating latent reservoir that is closely related to and overlapping with the latent cells that emerge in traditional viral outgrowth assays. Further experiments will be required to determine whether tissue resident latent cells have a similar gene program upon reactivation.
T cell division in response to antigen or mitogens like PHA and HIV-1 reactivation from latency are stimulated by shared metabolic and transcriptional pathways including NFκB35. Once activated, productive HIV-1 infection typically leads to CD4+ T cell death by apoptosis or pyroptosis36. However, cell death after latency reactivation in vitro appears to be stochastic with some cells being able to divide and survive after strong stimulation19. Our finding that latent cells can survive upon cell division in vivo confirms in vitro experiments19 and is also consistent with the observation that the latent compartment contains groups of CD4+ T cells that harbor proviruses with identical Env sequences14,19. Purification of reactivated latent cells by LURE and subsequent TCR sequencing provides definitive evidence that these cells arise by clonal expansion in vivo. The data is consistent with the idea that the protracted longevity of the latent compartment is due at least in part to cell division14–22. Finally, because the reservoir is stable over time1,2, the finding that latent cells divide implies that they are also dying at similar rate, and that the reservoir is a dynamic compartment.
Antibody binding to Env expressing cells in vivo leads to their accelerated clearance37,38. Should latent cells undergoing clonal expansion in vivo also express viral proteins, they too could be targeted for clearance by HIV-1 specific cytotoxic T cells, NK cells or by antibody dependent cellular cytotoxicity.
How does a subset of latent cells divide and still survive despite expression of HIV-1? Our single cell transcriptomic analysis of purified primary CD4+ T cells demonstrates that reactivated latent cells can express a distinct transcriptional program that includes muted responses to type I interferon and factors such as MiR-155 and PRDM1 that can suppress HIV-1 transcription28,31,32. We speculate that active HIV-1 suppression during CD4+ T cell division could be one of the mechanisms that maintains the latent reservoir. Further studies will be required to determine whether interference with these cellular safeguards could contribute to accelerating latent HIV-1 clearance.
Online Methods
Study subjects
All study participants were recruited by the Rockefeller University Hospital, New York, USA. Informed consent was obtained from all subjects, all relevant ethical regulations were followed, and leukapheresis was performed according to protocols approved at the Rockefeller University by the Rockefeller Internal Review Board. PBMCs were isolated by Ficoll separation and frozen in aliquots. In all cases, HIV-1 infected patients on therapy were confirmed to be aviremic at the time of sample collection.
Latency capture protocol
CD4+ T cells were isolated from ~1×109 PBMCs by negative selection using the Miltenyi CD4+ T isolation kit. Cells were cultured at 2×106/mL in R10 (RPMI supplemented with 10% heat inactivated FCS, 10mM HEPES, 100U/mL PenStrep), and 25% volume conditioned media. Conditioned media was made by culturing healthy PBMCs in R10 with PHA and IL-2 for 2 days, followed by a wash and 5 days in culture with IL-2 alone. The conditioned media was then collected and frozen at −80C until use. 100U/mL IL-2 (Peprotech), 1ug/mL PHA (Sigma), 10uM Z-VAD-FKM (R&D), 10uM Ritonavir, 10uM Dolutegravir, 10uM Emtricitabine, 5uM Tenofovir, and 10uM Maraviroc (all Selleckchem) were added to the media. 36h later, cells were labeled with 5ug/mL each of biotinylated 3BNC117, 10-1074, PG16, followed by Streptavidin PE (1:500, BD) and anti-PE magnetic beads (Miltenyi Biotech). Cells were then passed over a magnetic column and bound cells were eluted for downstream analysis. For FACS sorting, cells were labeled with the following antibodies, all Biolegend: CD1c (cat. no. 331510), CD3 (cat. no. 300430), CD4 (cat. no. 317444), CD8 (cat. no. 344726), CD14 (cat. no. 301812), CD20 (cat. no. 302318), CD32a (cat. no. 303204), and CD56 (cat. no. 318314).
Gag bulk qPCR
RNA was extracted from equivalent numbers of cells irrespective of enrichment. Gag qPCR was performed using RNA-to-CT one-step RT-PCR mix (ThermoFisher) and previously described primers39.
Single Cell sorting
All sorts were performed on BD FACS Aria into 96-well plates containing guanidine thiocyanate buffer (Qiagen) supplemented with 1% β-mercaptoethanol. Plates were immediately frozen on dry ice and transferred to long-term storage at −80C. LURE cells were gated on live, CD1c, CD8, CD14, CD20, and CD56 negative, CD3 positive and sorted based on Env staining. Control cells were gated on live, CD1c, CD8, CD14, CD20, and CD56 negative and sorted CD3 positive cells.
Single Cell gag qPCR and ENV PCR
Nucleic acids were isolated by SPRI bead cleanup as described40. RNA was reverse-transcribed into cDNA using an oligo(dT) primer. Gag qPCR was performed on one-fifth of the cDNA39. Gag+Env+ cells were selected based on the presence of cell-associated gag RNA measured by qPCR. Control cells were assayed for gag RNA and none was detected. Nested Env PCR was performed on one-fifth of the cDNA14.
Env DNA SGA and Q2VOA
DNA was extracted from isolated CD4+ T cells as previously described16 and Env SGA was performed as previously described14. Qualitative and quantitative viral outgrowth assays and downstream analysis were performed and processed as previously described14. For quality control, Q2VOA assays were performed more than once, and for donor B207, on samples taken at two different time-points. IUPM calculations were performed using the data from all independent experiments using the calculator IUPMStats44.
Clustering Env Sequences
Env nucleotide sequences were translation-aligned using ClustalW 2.1 with the BLOSUM cost matrix in Geneious v10.0.3. A maximum-likelihood tree was then inferred using PhyML 3.1 under the GTR model with 1,000 bootstrap replicates.
YU2 infection and sorting
CD4+ T cells were activated and infected with YU2 and labeled as previously described37. CD4lo, Envelope positive cells were sorted.
Single Cell RNASeq
RNASeq libraries were constructed based on Trombetta et al.41 using primers from Islam et al.42 Briefly, RNA was converted to full-length cDNA using oligo(dT) priming (Bio-AATGATACGGCGACCACCGATCGTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT) and SMART template switching technology (all RNA oligo: Bio-AAUGAUACGGCGACCACCGAUNNNNNGGG) followed by 24 cycles of PCR preamplification of cDNA (primer Bio-GAATGATACGGCGACCACCGAT). We used the amplified cDNA to construct standard Illumina sequencing libraries Nextera XT library preparation kit. Samples were sequenced by Illumina NextSeq.
RNASeq Analysis
The quality of the RNASeq libraries was evaluated using the fastQC143. We used STAR (2.4.1d)44 aligner to map the raw paired-end reads to the reference genome GRCh37/hg19. The gene-level counts were obtained using HTSEQ43. We performed a saturation analysis to detect the number of detected genes and filtered out the outlier cells as in Gaublomme et al11. Briefly, we excluded cells with number of aligned reads <25,000 and percentage of identified genes <20% of the group maximum. Normalized expression values were calculated using the scran package45. Heatmaps and dotplots were generated in R. The gene counts were used to infer the differentially expressed genes (DEG) in the data by MAST (v1.2.1)50.
HIV Splice variant analysis
We recovered the reads which failed to map to the human genome and mapped these reads to annotated junctions between HIV splice donors and acceptors to reconstruct the splice variants present in the scRNASeq data.
HIV reads alignment and reconstruction
We carried out HIV assembly analysis on the all reads which failed to map to the human genome by the IVA de novo assembler (v1.0.7)13.
TCR identification
TraceR46 was used to reconstruct full-length, paired T cell receptor (TCR) sequences. TCR sequences unable to be recovered from RNASeq reads were amplified as previously described47.
PCA Seurat
We used the Seurat package (v1.4.0.16) to identify variable genes, principal components (PCs), clusters and gene markers as described23. Briefly, the software identifies highly variably expressed genes using a normalized z-score, performs linear dimensional reduction (PCA) on the filtered genes, obtains additional transcriptome PCA loading genes using projection of these principal components to the entire dataset, determines groups by density clustering of the t-SNE significant principal component scores and performs gene marker discovery. We also used the Improved Stochastic Ranking Evolution Strategy algorithm53, implemented by NLopt, to find the optimal set of PCs and parameters, and to find the optimal set of clusters that best correlate with each group of cells.
Single Cell Consensus Clustering
Single-Cell Consensus Clustering (SC3) tool48 (default settings) was used for unsupervised clustering of single cells in this study. SC3 consistently integrates different clustering solutions through a consensus approach and identifies marker genes which are highly expressed in only one of the clusters and are able to distinguish it from all the remaining ones.
We have tested combinations of clustering settings (k=2, 3 and 4) and used a quantitative measure of the diagonality of the consensus matrix to select the k in which the measure is closest to 1 (k=3). We then used SC3 (AUROC>0.6 and FDR < 0.1) to identify marker genes which are highly expressed in only one of the clusters and are able to distinguish it from all the remaining clusters.
Data availability and Acession Code Availability Statement
The data reported in this paper is archived at the following databases: Single cell RNASeq data (Fig 2 and 4) is available at NCBI GEO (GSM2801437); Envelope sequences (Fig 3) are available in the Genebank database (MG196359 - MG196639); TCR sequences (Supplemental Data Fig 5) are available in the Genebank database (MG192535-MG193127).
Supplementary Material
Acknowledgments
We thank all participants who contributed to this study; members of the Nussenzweig laboratory for helpful discussions, particularly Ervin Kara and Thiago Oliveira; our lab manager Zoran Jankovic; Luka Mesin and Moshe Biton for advice on single cell RNASeq; Anna Gazumyan for bNAb production; Gaelle Breton for help with FACS; Kristie Gordon and Neena Thomas for performing all FACSorting experiments; Arnold Han and Mark Davis for TCR sequencing advice; Daniel Mucida, Charles Rice and Paul Bieniasz for helpful discussion; Katrina Millard for recruitment of study subjects, and Michael Deal for assistance with figures. This work was supported by the Bill and Melinda Gates Foundation Collaboration for AIDS Vaccine Discovery (OPP1033115 and OPP1124068) the NIH Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery (CHAVI-ID) (1UM1 AI100663), BEAT-HIV Delaney Collaboratory (UM1 AI126620), the National Institute of Allergy and Infectious Diseases of the NIH (AI100148, AI037526), the Robertson Foundation, and the Rockefeller University. Marina Caskey is supported by NIH grant U01AI118536. M.C.N. is an HHMI investigator.
Footnotes
Author Contributions
L.B.C., M.J., and M.C.N. wrote the manuscript; L.B.C., M.J., and M.C.N. designed and analyzed experiments; L.B.C and M.J. performed LURE experiments, RNA sequencing, Q2VOA, TCR sequencing, and virus SGA. R.V., I.T.d.S performed bioinformatics analysis of RNASeq data. A.S.H. performed TCR sequencing and virus SGA. J.C.C.L and Y.Z.C. performed Q2VOA; J.A.P. performed phylogenetic analysis of Env sequencing and gene enrichment analysis; A.L.B and M.C. performed study subject recruitment and oversaw sample collection.
References
- 1.Crooks AM, et al. Precise Quantitation of the Latent HIV-1 Reservoir: Implications for Eradication Strategies. The Journal of infectious diseases. 2015;212:1361–1365. doi: 10.1093/infdis/jiv218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siliciano JD, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nature medicine. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 3.Murray AJ, Kwon KJ, Farber DL, Siliciano RF. The Latent Reservoir for HIV-1: How Immunologic Memory and Clonal Expansion Contribute to HIV-1 Persistence. Journal of immunology. 2016;197:407–417. doi: 10.4049/jimmunol.1600343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henrich TJ, Deeks SG, Pillai SK. Measuring the Size of the Latent Human Immunodeficiency Virus Reservoir: The Present and Future of Evaluating Eradication Strategies. The Journal of infectious diseases. 2017;215:S134–S141. doi: 10.1093/infdis/jiw648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spina CA, et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS pathogens. 2013;9:e1003834. doi: 10.1371/journal.ppat.1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laird GM, et al. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS pathogens. 2013;9:e1003398. doi: 10.1371/journal.ppat.1003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheid JF, et al. Sequence and Structural Convergence of Broad and Potent HIV Antibodies That Mimic CD4 Binding. Science. 2011;333:1633–1637. doi: 10.1126/science.1207227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mouquet H, et al. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E3268–E3277. doi: 10.1073/pnas.1217207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker LM, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaublomme JT, et al. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell. 2015;163:1400–1412. doi: 10.1016/j.cell.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherrill-Mix S, Ocwieja KE, Bushman FD. Gene activity in primary T cells infected with HIV89.6: intron retention and induction of genomic repeats. Retrovirology. 2015;12:79. doi: 10.1186/s12977-015-0205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunt M, et al. IVA: accurate de novo assembly of RNA virus genomes. Bioinformatics. 2015;31:2374–2376. doi: 10.1093/bioinformatics/btv120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lorenzi JC, et al. Paired quantitative and qualitative assessment of the replication-competent HIV-1 reservoir and comparison with integrated proviral DNA. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E7908–E7916. doi: 10.1073/pnas.1617789113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullins JI, Frenkel LM. Clonal Expansion of Human Immunodeficiency Virus-Infected Cells and Human Immunodeficiency Virus Persistence During Antiretroviral Therapy. The Journal of infectious diseases. 2017;215:S119–S127. doi: 10.1093/infdis/jiw636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohn LB, et al. HIV-1 integration landscape during latent and active infection. Cell. 2015;160:420–432. doi: 10.1016/j.cell.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner TA, et al. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 2014 doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maldarelli F, et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014;345:179–183. doi: 10.1126/science.1254194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hosmane NN, et al. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J Exp Med. 2017;214:959–972. doi: 10.1084/jem.20170193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bui JK, et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS pathogens. 2017;13:e1006283. doi: 10.1371/journal.ppat.1006283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee GQ, et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. The Journal of clinical investigation. 2017;127:2689–2696. doi: 10.1172/JCI93289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simonetti FR, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:1883–1888. doi: 10.1073/pnas.1522675113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fromentin R, et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS pathogens. 2016;12:e1005761. doi: 10.1371/journal.ppat.1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baxter AE, et al. Single-Cell Characterization of Viral Translation-Competent Reservoirs in HIV-Infected Individuals. Cell Host Microbe. 2016;20:368–380. doi: 10.1016/j.chom.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cockerham LR, et al. CD4+ and CD8+ T cell activation are associated with HIV DNA in resting CD4+ T cells. PloS one. 2014;9:e110731. doi: 10.1371/journal.pone.0110731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Descours B, et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature. 2017;543:564–567. doi: 10.1038/nature21710. [DOI] [PubMed] [Google Scholar]
- 28.Ruelas DS, et al. MicroRNA-155 Reinforces HIV Latency. J Biol Chem. 2015;290:13736–13748. doi: 10.1074/jbc.M115.641837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hudspeth K, et al. Engagement of NKp30 on Vdelta1 T cells induces the production of CCL3, CCL4, and CCL5 and suppresses HIV-1 replication. Blood. 2012;119:4013–4016. doi: 10.1182/blood-2011-11-390153. [DOI] [PubMed] [Google Scholar]
- 30.Abdelwahab SF, et al. HIV-1-suppressive factors are secreted by CD4+ T cells during primary immune responses. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15006–15010. doi: 10.1073/pnas.2035075100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaczmarek Michaels K, et al. Blimp-1, an intrinsic factor that represses HIV-1 proviral transcription in memory CD4+ T cells. Journal of immunology. 2015;194:3267–3274. doi: 10.4049/jimmunol.1402581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Masson A, et al. Blimp-1 overexpression is associated with low HIV-1 reservoir and transcription levels in central memory CD4+ T cells from elite controllers. Aids. 2014;28:1567–1577. doi: 10.1097/QAD.0000000000000295. [DOI] [PubMed] [Google Scholar]
- 33.Bruner KM, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nature medicine. 2016;22:1043–1049. doi: 10.1038/nm.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho YC, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siliciano RF, Greene WC. HIV latency. Cold Spring Harbor perspectives in medicine. 2011;1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doitsh G, Greene WC. Dissecting How CD4 T Cells Are Lost During HIV Infection. Cell Host Microbe. 2016;19:280–291. doi: 10.1016/j.chom.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu CL, et al. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science. 2016;352:1001–1004. doi: 10.1126/science.aaf1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horwitz JA, et al. Non-neutralizing Antibodies Alter the Course of HIV-1 Infection In Vivo. Cell. 2017;170:637–648 e610. doi: 10.1016/j.cell.2017.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer S, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 2003;41:4531–4536. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tas JM, et al. Visualizing antibody affinity maturation in germinal centers. Science. 2016;351:1048–1054. doi: 10.1126/science.aad3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trombetta JJ, et al. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr Protoc Mol Biol. 2014;107(4):22, 21–17. doi: 10.1002/0471142727.mb0422s107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Islam S, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014;11:163–166. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 43.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lun AT, McCarthy DJ, Marioni JC. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 2016;5:2122. doi: 10.12688/f1000research.9501.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stubbington MJT, et al. T cell fate and clonality inference from single-cell transcriptomes. Nat Methods. 2016;13:329–332. doi: 10.1038/nmeth.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han A, Glanville J, Hansmann L, Davis MM. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat Biotechnol. 2014;32:684–692. doi: 10.1038/nbt.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Runarsson T, Yao X. Search biases in constrained evolutionary optimization. IEEE Trans on Systems, Man, and Cybernetics Part C: Applications and Reviews. 2005;35(3):233–243. [Google Scholar]
- 49.Kiselev VY, et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods. 2017;14:483–486. doi: 10.1038/nmeth.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data reported in this paper is archived at the following databases: Single cell RNASeq data (Fig 2 and 4) is available at NCBI GEO (GSM2801437); Envelope sequences (Fig 3) are available in the Genebank database (MG196359 - MG196639); TCR sequences (Supplemental Data Fig 5) are available in the Genebank database (MG192535-MG193127).