

Abstract
Epstein-Barr virus (EBV) is an oncogenic virus that infects more than 90% of the world’s population1. EBV predominantly infects human B cells and epithelial cells, which is initiated by fusion of the viral envelope with a host cellular membrane2. The mechanism of EBV entry into B cells has been well characterized3. However, the mechanism for epithelial cell entry remains elusive. Here, we show that the integrins (αvβ5, αvβ6, and αvβ8) do not function as an entry and fusion receptor for epithelial cells whereas ephrin receptor tyrosine kinase A2 (EphA2) functions well for both. EphA2 overexpression significantly increased EBV infection of HEK 293 cells. Using a virus-free cell-cell fusion assay, we found that EphA2 dramatically promoted EBV but not HSV fusion with HEK293 cells. EphA2 silencing using shRNA or knockout by CRISPR/Cas9 blocked fusion with epithelial cells. This inhibitory effect was rescued by the expression of EphA2. Antibody against EphA2 blocked epithelial cell infection. Using label-free Surface Plasmon Resonance (SPR) binding studies, we confirmed that EphA2 but not EphA4 specifically bound to EBV gHgL and this interaction is through the EphA2 extracellular domain (EphA2-ECD). The discovery of EphA2 as an EBV epithelial cell receptor has important implications for EBV pathogenesis and may uncover new potential targets that can be used for the development of novel interventional strategies.
Epstein-Barr virus (EBV) is a member of the gammaherpesvirus family, which was discovered in 1964 and was the first human virus associated with cancer4. EBV is the causative agent of infectious mononucleosis and is associated with Burkitt lymphoma, Hodgkin disease, nasopharyngeal carcinoma, and gastric carcinoma, indicating the EBV tropism for B cells and epithelial cells. EBV infects more than 90% of the world’s population1; however, there is a lack of therapies and vaccines. EBV entry into target cells is an essential step for EBV to cause disease and requires the fusion of viral and host membranes mediated by viral glycoproteins and cellular receptors2. The viral glycoproteins important for EBV entry include gp350, gHgL, gB, and gp422. Among these glycoproteins, gp350 is important for virus attachment by binding to complement receptor type 2 (CR2/CD21), which is abundantly expressed on B cells and expressed on tonsilar epithelial cells5. gHgL and gB are the core fusion machinery and are both required for B cell and epithelial cell fusion. However, gp42 is the tropism determinant required only for B cell fusion and inhibits epithelial cell fusion, indicating different infection mechanisms for these two cell types6. The mechanism for B cell infection is better understood than the mechanism of epithelial cell infection. The B cell receptor HLA-DR was identified to bind to gp42 by a gp42 ligand binding screen in 19967. In 1997, it was found that HLA-DR functions as a cofactor for infection of B lymphocytes8. Since that time, we have worked extensively on EBV entry determining the structures of unbound gp42, the gp42:HLA complex, the gHgL complex, and gB in the post-fusion form9, 10, 11, 12. Recently, we assembled and analyzed the reconstituted B cell entry complex comprised of gHgL, gp42, and HLA class II and the crystal structure of the gHgL/gp42 complex bound to an anti-gHgL antibody (E1D1), providing an overall structural basis for Epstein-Barr virus host cell tropism3, 13.
To characterize the EBV epithelial cell entry complex similarly to what we have done for the B cell entry complex3, we first wanted to verify the receptor used for epithelial cell entry. We chose the AGS cell line that has been extensively used as a model of EBV epithelial cell entry and HEK293 cells, which we use in our cell based fusion assay. Previous studies had indicated that the integrins (αvβ5, αvβ6, and, αvβ8 but not αvβ3) functioned as receptors for epithelial cell entry14, 15. It was also found that blocking antibodies to integrins and siRNA targeting of integrin αv did not completely abolish epithelial cell fusion or infection14. In addition, three anti-gHgL monoclonal antibodies (CL40, CL59 and E1D1) targeting different epitopes can all inhibit epithelial cell infection, indicating that multiple regions on gHgL may participate in EBV infection16. To determine if integrins are the primary epithelial cell receptor, we chose to knock out the integrin αv gene using the CRISPR/Cas9 system in HEK293-T14 cells. The integrins αvβ5, αvβ6, and αvβ8 are heterodimeric complexes composed of the αv subunit and a β subunit. The crystal structure of EBV gHgL with an exposed KGD motif (RGD motif mimic) within gH domain II (D-II) suggested it might bind to the αv subunit of the heterodimeric integrins reported to function as entry receptors14, 17. By knocking out αv, we would block the expression of the integrins previously shown to mediate EBV entry of epithelial cells. Integrin αv knockout clonal cell lines were readily obtained and were first analyzed by flow cytometry verifying the absence of αv (Fig. 1a). We next used our cell-cell fusion assay to monitor syncytia formation. We found that cells lacking integrin αv still fused with CHO-K1 cells that expressed gHgL and gB (Fig. 1b) indicating that αvβ5, αvβ6, and αvβ8 integrins are not the primary receptor(s) in HEK293 cells.
Figure 1. Identification of EphA2 as the potential EBV epithelial cell receptor.
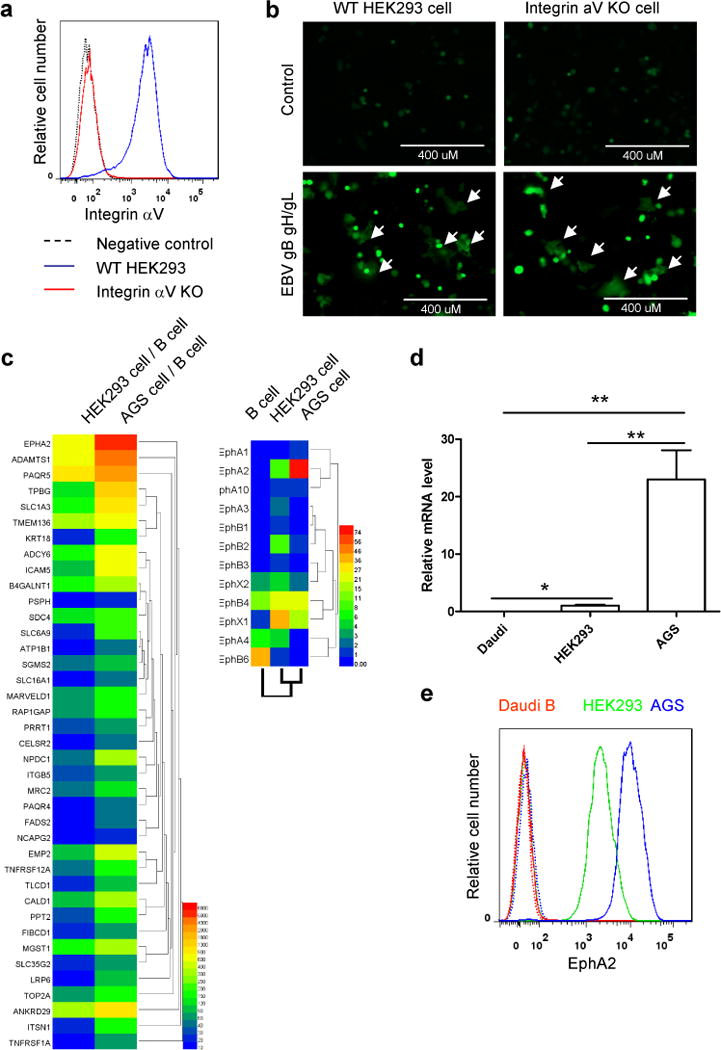
a, Integrin aV cell surface expression in integrin aV WT and KO HEK293 cells by flow cytometry. b, CHO-K1 cells transfected with GFP plasmid plus either control plasmid or EBV gHgL and gB were overlaid with integrin aV WT and KO HEK293 cells. Syncytia formation was visualized and captured with a EVOS fluorescence microscope (representative data from three independent experiments). c, Heat map of the normalized expression of potential epithelial cell receptors compared to B cells for HEK293 and AGS cells (left). Heat map of Ephs for B cells, HEK293 cells, and AGS cells (right). d, Comparison of EphA2 expression in different cell types. Total RNA from each cell type was assayed for EphA2 and GAPDH mRNA levels by quantitative RT-PCR. The bars represent the relative EphA2 mRNA expression that was normalized to GAPDH. Data are means plus standard errors of the means for three independent experiments. e, Flow cytometry histograms of EphA2 in Daudi B, HEK293 and AGS cells, with the dotted line as a negative control. One representative flow cytometry data of at least two independent experiments is shown in a and e. *P<0.05 and **P<0.01 (ANOVA followed by post-hoc Tukey’s multiple comparison test).
To identify essential epithelial cell receptors in HEK293 cell fusion, wecompared RNA seq data of human B cells and HEK293 cells. We chose this comparison because it had been previously shown that EBVcontaining only gB and gHgL but lacking gp42 is unable to infect B cells18. In addition, previous data had shown that CHO-K1 cells that expressgHgL and gB but not gp42 can fuse with HEK293 cells but not B cells, further indicating that B cells do not express epithelial cell receptors19. To identify candidate epithelial cell receptors, wescreened B cell and HEK293 cell RNA seq data sets. In our analysis, we screened the data sets such that the mRNA of HEK293candidates versus the mRNA of B cell candidates was 10-fold greater andthat the HEK293 cell gene reads were over 5 FPKM (Fragment PerKilobase per Million mapped reads) threshold as the lower boundary for reliabledetection of gene expression. From this analysis, we selected 2,039genes out of 18,400 genes that were compared. We then selectedmembrane proteins and excluded genes that had B cell reads of morethan 5 FPKM. This analysis resulted in 245 genes (Supplementary Table 1). To further reduce the number of potential genes, we used RNA seq data from AGS cells. AGScells are a human gastric adenocarcinoma and have been usedextensively as a model of EBV epithelial cell entry14, 20. This cell line formslarger syncytia and are more readily infected when compared to HEK293 cells (data not shown) and gastric epithelial cells are a target of EBV infection21. Whencomparing the expression of the 245 genes (EphA2 ranked No. 14, fold difference 864, Supplementary Table 1) identified in the HEK293 cellRNA seq data and selecting only those genes that are expressed twofold greater in AGS cells, we identified 39 genes. After ranking these genes by the ratio of AGS to B cell expression from high to low, EphA2 was ranked as the No. 1 candidate (Fig. 1c and Supplementary Table 2). Interestingly, if we first compared AGS cells with B cells using the similar parameters (B cell expression less than 5 FPKM, AGS cell expression more than 5 FPKM, AGS cell/B cell > 10, membrane protein), this resulted 278 genes, Supplementary Table 3 and EphA2 ranked No. 9 (fold difference 6798). If we select only those genes that are expressed twofold greater in AGS cells, we identified 65 genes (Supplemenarty Fig 1 and Supplementary Table 4). If we compared the first 15 genes of membrane proteins selected out by two methods (Supplementary Table 1 and 3), EphA2 and PAQR5 are the only genes shared by the two methods. EphA2 is also the receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV), another human gammaherpesvirus22, 23.
EphA2 belongs to the Eph receptor family, which constitutes the largest family of tyrosine kinase receptors. The function of EphA2 includes cell migration, adhesion, proliferation and differentiation. Eph receptors are divided into two classes, A and B, based upon sequence similarity and ligand affinity24. Since we had found that EphA2 had the greatest differential expression profile among B cells, HEK293 cells, and AGS cells (Fig. 1c), we wanted to verify expression using quantitative RT-PCR and flow cytometry. As expected, we confirmed that Daudi B cells do not express EphA2 (Fig. 1d) and the expression level of EphA2 in AGS cells is about 10-fold more than that of HEK293 cells (Fig. 1d and 1e). It has been shown that EphA2 is expressed on tongue mucosa and salivary gland25, 26. Additionally, EphA2 has also been shown to be expressed on normal gastric mucosa and EphA2 expression is upregulated in the carcinogenesis of gastric mucosa epithelial cells27, 28.
To determine whether EphA2 serves as a receptor for EBV infection of epithelial cells, we first transiently transfected control vector, EphA2 or EphA4 into HEK293 cells to see if EphA2 increased EBV infection or fusion. We used expression vectors for EphA2 or EphA4. Over-expression of EphA2 increased the cell surface expression of EphA2 by about 10-fold in HEK293 cells (Fig 2b). These cells were also infected with EBfaV-GFP (recombinant EBV reporter virus expressing GFP29) with the resulting infection increasing the detection of green cells in control transfected HEK293 cells (Fig. 2a and 2b). The number of GFP-expressingcells also increased in EphA2 but not EphA4 transfected HEK293 cells(Fig. 2b). Flow cytometry data and quantification confirmedthat EphA2 but not EphA4 increased EBV infection in HEK293 cells(Fig. 2c). Furthermore, EphA2 expression in HEK293 cellsled to significantly increased fusion with CHO-K1 cells expressing theEBV glycoproteins gB, gHgL but not HSV glycoproteins. EphA4 over-expression had no effect on fusion of either EBV or HSV (Fig 2d).
Figure 2. EphA2 can promote both EBV infection and virus-free cell-cell fusion.
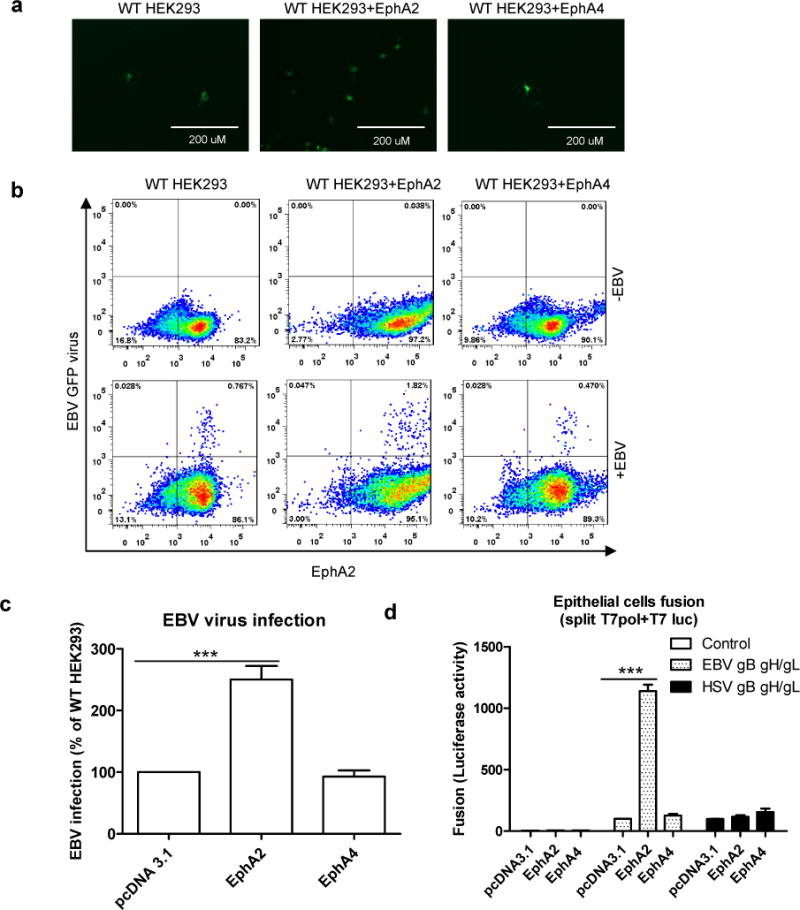
a, HEK293 cells were transfected with pcDNA 3.1, EphA2 or EphA4. 24h post transfection, 5×104 cells were seeded onto a 48 well plate. 24 hours later, the cells were infected with 100 mL EBfaV-GFP virus concentrated from 1 mL virus-containing supernatant. 72 hours later, the infected GFP cells were visualized and captured with a EVOS fluorescence microscope (a) or analyzed by flow cytometry (b). Data are representative data from three independent experiments. c, Quantification of the flow cytomery data from three independent experiments, the bars represent the percentage of infection with infection of pcDNA 3.1 transfected HEK293 cells set to 100%. d, CHO-K1 cells transfected with T7 luciferase plasmid together with either control plasmid, EBV gHgL and gB, or HSV gHgL, gB and gD were overlaid with HEK293 cells transfected with pcDNA 3.1, EphA2 or EphA4. EBV fusion with HEK293 cells transfected with pcDNA 3.1 was set to 100%. HSV fusion was normalized to EBV fusion and standardized with pcDNA 3.1 transfected cells set to 100%. Data are means plus standard errors of the means for three independent experiments for c and d. ***P<0.001 (ANOVA followed by post-hoc Tukey’s multiple comparison test).
To further confirm that EphA2 can serve as a cellular receptor for EBV infection, we knocked out EphA2 using the CRISPR/Cas9 system. Following knockout, the EphA2 cell surface expression was decreased to the level of the negative control (Fig 3a). Both EBV fusion and infection were dramatically decreased in EphA2 knockout cells (Fig. 3b–e). This marked reduction was rescued by over-expression of EphA2 (Fig. 3b–e) but not EphA4. HSV fusion activity was not affected in all the cells tested, indicating the specificity of EphA2 as an EBV receptor (Fig. 3b). We performed additional control experiments using shRNA to knock down EphA2 in HEK293-T14 and AGS cells and found similar reduction in fusion (Supplementary Fig. 2–3). As a further confirmation of EphA2 EBV receptor activity, we found that an antibody against EphA2 but not EphA4 can block the infection of nasopharyngeal carcinoma cell line Detroit 562 (Supplementary Fig. 4a). Interestingly, murine EphA2 which shares 93% identity with human EphA2, can also increase fusion and infection with EBV (Supplementary Fig. 4b and 4c). When we mixed the EphA2 KO cells and WT cells at a 1:1 ratio, the infection ability of EphA2 KO cells by EBV was further decreased (Supplementary Fig. 5a and b), indicating there may be another low affinity receptor that unable to compete with the EphA2 when EphA2 is present.
Figure 3. EphA2 is essential for EBV infection and virus-free cell-cell fusion.
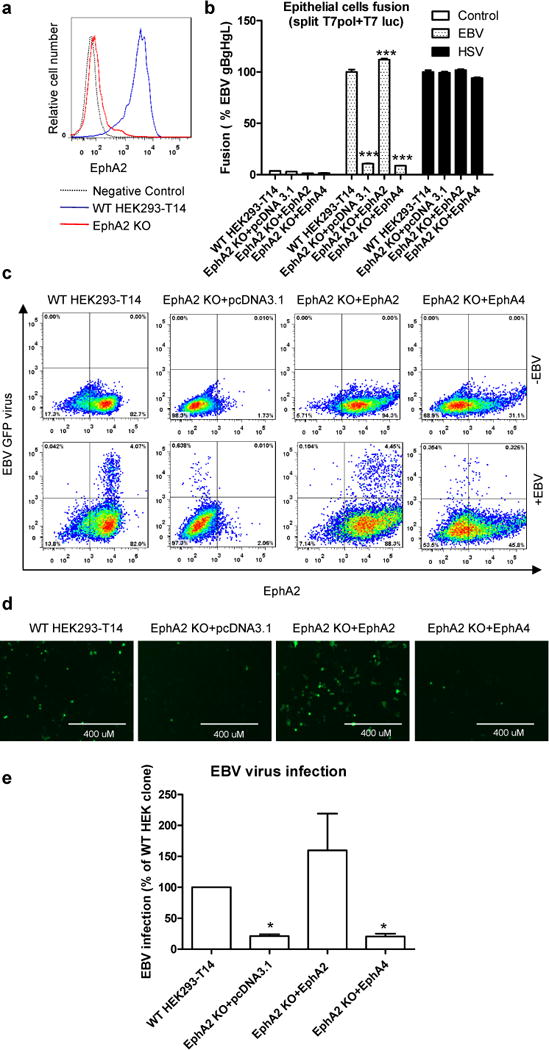
a, EphA2 cell surface expression in EphA2 WT and KO HEK293-T14 cells by flow cytometry. One representative flow cytometry data of three independent experiments is shown. b, Virus-free EBV or HSV fusion with EphA2 WT and KO HEK293-T14 cells or EphA2 KO cells that overexpress EphA2 or EphA4. The bars represent the fusion activity and data are means plus standard errors of the means for three independent experiments. c-e, EphA2 WT and KO HEK293-T14 cells or EphA2 KO cells that overexpress EphA2 or EphA4 were infected with EBfaV-GFP virus. 72 h post infection, the infected GFP cells were visualized and captured with a EVOS fluorescence microscope (d) or GFP-positive cells were analyzed by flow cytometry (c). Data are representative data from three independent experiments. e, Quantification of the flow cytometry data from three independent experiments, the bars represent the percentage of infection and infection of WT HEK293-T14 cells was set to 100. Data are means plus standard errors of the means. *P<0.05 and ***P<0.001 vs WT HEK293-T14 (ANOVA followed by post-hoc Tukey’s multiple comparison test).
EphA2 is also the receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV), another human gammaherpesvirus22, 23. Previous data showed that KSHV gHgL binds to the ligand binding domain of EphA230. To examine whether the function of EphA2 in EBV fusion is through the ligand binding domain, we swapped the ligand binding domain of EphA2 and EphA4 to generate EphA2A4 or EphA4A2 chimeras. We found replacing the EphA4 ligand binding domain with the EphA2 ligand binding domain (EphA2A4 chimera) increased fusion activity 3-fold, suggesting that like KSHV, EBV gHgL also binds the ligand binding domain of EphA2 (Fig. 4a). To examine whether the EphA2 kinase activity is important for fusion, we also tested three EphA2 kinase dead mutants that were mutated in the EphA2 kinase domain and found that kinase activity of EphA2 is not important for EBV fusion (Fig. 4b). It has been shown that the kinase activity of EphA2 is important for KSHV infection23. Depending upon cell type, EBV can enter cells by fusion at the plasma membrane (epithelial cells) or fusion with an endocytic membrane after endocytosis (B cells)2. KSHV enters human B cells, fibroblast, epithelial, and endothelial cells by endocytosis and EphA2 regulates clathrin mediated KSHV endocytosis31, 32. The different route of entry may explain the differing requirements of EphA2 kinase function in EBV and KSHV infection. To explore the region of gHgL that binds EphA2, we took advantage of previous observations indicating that the gHgL domain for gp42 binding and epithelial cell binding overlaps17, 33. When an expression vector expressing gp42 or purified soluble gp42 is included in our fusion assay, the level of fusion is dramatically reduced (Fig. 4c) indicating that gp42 binding to gHgL blocks EphA2-mediated membrane fusion, in line with previous observations. To further confirm the direct binding of EphA2-ECD and EBV gHgL, we used label-free Surface Plasmon Resonance (SPR) binding studies and found that purified EBV gHgL bound to EphA2 with a KD of 5 μM in three independently repeated binding kinetics experiments but EBV gHgL did not bind to EphA4 under the same conditions (Fig. 4d and 4e and Supplementary Table 5). The 200 nM concentration of EBV gHgL was injected in duplicates each time with randomized order of injections where the second 200 nM injection occurred after the 1.8 μM EBV gHgL injection to serve as internal control. Fig. 4d show that both injections of the same concentration (200 nM) overlap each other. Previous data from a flow cytometry binding assay showed that KSHV gHgL can bind tightly with EphA2 particularly but only weakly with other Ephrins including EphA434. This suggests that gammaherpesviruses share a common entry receptor. Taken together, we identify EphA2 as an important epithelial cell entry receptor for EBV infection. This result provides important insight into the mechanism EBV utilizes to infect epithelial cells and may provide a better understanding of the development of epithelial pathologies and cancers associated with EBV infection and strategies to lessen EBV-associated epithelial cancers. Finally, our studies using bioinformatics to implicate EphA2 as an EBV epithelial receptor highlight the benefit of using readily obtainable sequencing data to aid in the discovery of pathogen receptors.
Figure 4. EphA2 binds to EBV gHgL with low affinity.
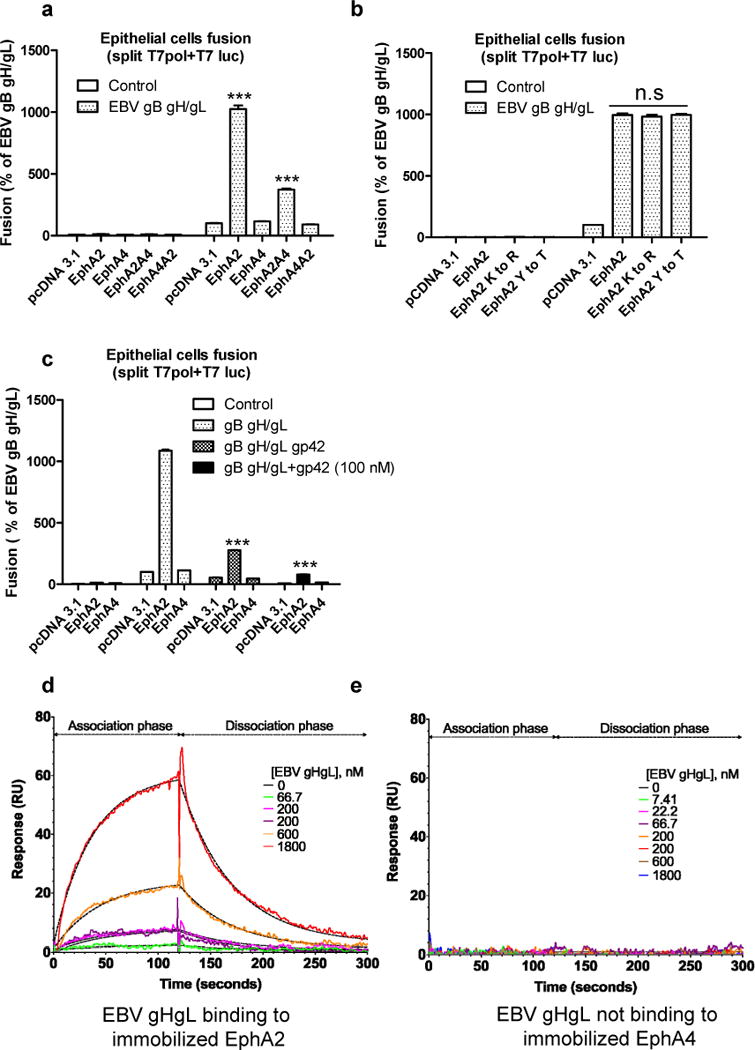
a, virus-free EBV fusion with HEK293 cells transfected with pcDNA 3.1, EphA2, EphA4 or EphA2 and EphA4 chimeras as indicated. b, virus-free EBV fusion with HEK293 cells transfected with control, EphA2, EphA4 or EphA2 kinase-dead mutants as indicated. Fusion activity of HEK293 cells transfected with pcDNA 3.1 was set to 100 (a) and (b). c, HEK293 cells transfected with T7 polymerase together with pcDNA 3.1, EphA2, or EphA4 were overlaid with CHO-K1 cells transfected with T7 luciferase plasmid together with EBV gB, gHgL in the presence or absence of gp42 plasmid or 100 nM soluble gp42. EBV fusion with HEK293 cells transfected with pcDNA 3.1 was set to 100. The bars represent the fusion activity (a, b and c) and data are means plus standard errors of the means for three independent experiments. d and e, EphA2 and EphA4 were immobilized on two separate channels of a CMD biosensor chip surface. Label-free SPR binding kinetics of EBV gHgL with EphA2 (d) or EphA4 (e) are depicted with model fits represented by dashed line. Global curve fitting with a 1:1 interaction model (dashed lines) closely matches the experimental data for EphA2 binding to EBV gHgL. EphA4 does not bind to EBV gHgL under identical conditions (representative data from three independent experiments). ***P<0.001 vs EBV gHgL, pcDNA 3.1 in a, vs gB gHgL, EphA2 in c (ANOVA followed by post-hoc Tukey’s multiple comparison test).
Methods
Cell culture
Chinese Hamster Ovary (CHO-K1) cells (ATCC CCL-61 or CRL-9618) were grown in Ham’s F-12 medium (Corning) containing 10% heat inactivated fetal bovine serum complex (FBS) (Corning) and 1% penicillin-streptomycin (100 U penicillin/mL, 100 μg streptomycin/mL; Sigma). Human embryonic kidney 293 (HEK293) cells (ATCC CRL-1573) or HEK293-T14 cells (HEK293T cells stably expressing T7 RNA polymerase, ATCC CRL-3216) were grown DMEM with 100 μg/mL zeocin (Invitrogen, for HEK293T cells expressing T7 RNA polymerase only), respectively containing 10% heat inactivated FBS and 1% penicillin-streptomycin. Detroit 562 cells (ATCC CCL138) were grown in MEM medium (Corning) containing 10% heat inactivated fetal bovine serum complex (FBS) (Corning) and 1% penicillin-streptomycin (100 U penicillin/mL, 100 μg streptomycin/mL; Sigma). CHO-K1, HEK283, HEK293T, AGS, and Detroit 562 cells were obtained from ATCC and routinely tested for mycoplasma using LookOut® Mycoplasma PCR Detection Kit from Sigma. The cell lines were authenticated by ATCC and not authenticated in our laboratory.
Constructs
The EphA2, EphA2 kinase-dead mutants, and EphA4 constructs are a gift from Dr. Spiro Getsios. The construction of the EphA2 and EphA4 chimeras (EphA2A4 or EphA4A2) was done using overlapping PCR with the primers listed. EphA4A2 HindIII F: 5-TTAAGCTTATGGCTGGGATTTTCTATTTC-3, EphA4A2 F: 5-CGTGTGTTCTATAAAAAGTGTCCCGAGCTGCTGCAGGGCCTG-3, EphA4A2 R: 5-CAGGCCCTGCAGCAGCTCGGGACACTTTTTATAGAACACACG-3, EphA4A2 HindIII F: 5-TTAAGCTTATGGCTGGGATTTTCTATTTC-3, EphA4A2 BsRGI R: 5-ACCGGCACCTCCTTCTTCCCC-3; EphA2A4 F: AfeI 5-ACGTGAAGCTGAACGTGGAGG-3, EphA2A4 F: 5-CGTGTCTACTACAAGAAGTGCCCACTCACAGTCCGCAATCTG-3, EphA2A4 R: 5-CAGATTGCGGACTGTGAGTGGGCACTTCTTGTAGTAGACACG-3, EphA4 pCDNA 3.1 AflII F: 5-AACTTAAGCTTATGGCTGGGATTTTCTATTTC-3. mouse EphA2 was cloned into pCDNA 3.1 vector using primers mEphA2 AflII F: 5- AACTTAAGCTTATGGAGCTCCGGGCAGTCGGT-3 and m EphA2 Xho I R 5-GACTCGAGTCACTTGTCATCGTCGTCCTTGTAATCGATAGGAATCCCCACTGTGTTGAC-3. Integrin αV sgRNA constructs: oligos 5-CACCGCCTGTGCCCCATTGTACCAT-3 and 5-AAACATGGTACAATGGGGCACAGGC-3 were annealed and ligated into a plasmid pU6-(BbsI)_CBh-Cas9-T2A-BFP (Addgene, # 64323) bearing both Cas9 and the remainder of the sgRNA as an invariant scaffold immediately following the oligo cloning sites. EphA2 sgRNA constructs: oligos 5-AAACGTGTGCGCTACTCGGAGCCTC-3 and 5-CACCGGAAGCGCGGCATGGAGCTCC-3 were annealed and ligated into a lentiGuide-Puro plasmid (Addgene, # 52963). Sequencing was done for all the constructs to confirm the correct sequence.
Generation of Integrin and EphA2 KO cells
For Integrin αV KO cells, 5 × 105 HEK293-T14 cells per well in a 6 well plate were transfected with 3 μg Integrin αV sgRNA/Cas9 or sgRNA/Cas9 control plasmids. 48 hours later, the cells were dissociated and resuspended in 10 mL 10% FBS-DMEM and passed through a 100 μm-cell strainer. The cells were counted and seeded at a concentration of 0.5 cell per 100 μL. After 3 weeks, single cell colonies were picked and expanded for functional assays. The knockout of integrin αV in these cells was confirmed by flow cytometry. For EphA2 KO cells, Cas9-expressing stable HEK293-T14 cells were established by infecting with lentivirus that had Cas9 for 24 h. 24 hours later, the cells were changed to fresh medium with 5 ug/mL blasticidin for selection. After one week, the cells were colonized as previously described and expanded for 2-3 weeks. The Cas9 expression in these single cell colonies was analyzed using western blotting against Flag. 2.5 × 105 HEK293-T14-Cas9 cells per well in a 12-well plate were infected with lentivirus that has control sgRNA or EphA2 sgRNA. The cells were selected with 2 ug/mL puromycin and colonized as previously described. After 2-3 weeks of expansion, the knockout of EphA2 was confirmed by flow cytometry.
Generation of EphA2 Knockdown cells in HEK and AGS cells
EphA2 knockdown cells were created by lentiviral transduction of a pLKO.1 vector containing EPHA2-specific shRNA (sequence: CCGGCGGACAGACATATAGGATATTCTCGAGAATATCCTATATGTCTGTCCGTTTTT, underlined sequence is the hairpin loop) in HEK 293 and AGS cells. The AGS and HEK293-T14 cells were under puromycin (2.5 ug/mL and 2ug/mL, respectively) selection for two weeks and the surface expression of EphA2 were analyzed by flow cytometry before using in the fusion experiments.
Fusion assay
The virus-free cell-based fusion assay was performed as described previously. Briefly, CHO-K1 cells grown to approximately 80% confluency were transiently transfected with T7 luciferase reporter plasmid with a T7 promoter (1.5 μg) and other essential glycoproteins for EBV fusion including gB (0.8 μg), gH (0.5 μg), gL (0.5 μg) or for HSV fusion including HSV gB (0.8μg), gH (0.5μg) gL (0.5μg), gD (0.5μg) by using Lipofectamine 2000 transfection reagent (Invitrogen) in Opti-MEM (Gibco-life technology) as previously described. HEK293 cells or HEK293-T14 WT and EphA2 knockout (KO) cells were transfected with T7 polymerase (1.5 μg) plus 1.5 μg pcDNA 3.1, EphA2 or EphA4 for the fusion assay. Twenty-four hours post transfection, the cells were detached, counted, and mixed 1:1 with target cells (HEK293 cells, 2.0 × 105 per sample) into a 48-well plate in 0.5 mL Ham’s F-12 medium with 10% heat-inactivated FBS. Twenty-four hours later, the cells were washed once with PBS and lysed with 50 μL of passive lysis buffer (Promega). Luciferase activity was quantified by transferring 20 μL of lysed cells to a 96-well plate and adding 50 μL of luciferase assay reagent (Promega). Luminescence was measured on a Perkin-Elmer Victor plate reader.
Cell surface expression
Surface expression of EphA2 or Integrin αV was performed by flow cytometry analysis. 1× 106 cells were harvested and washed in PBS containing 1% bovine serum albumin (BSA) and incubated with 5 μL of PE-conjugated EphA2 Antibody (Biolegend, SHM16) or PE-conjugated Integrin αV (R&D systems, FAB1219P) in 50 μL PBS containing 1% BSA for 30 mintues at 4 °C. Cells were then washed and diluted in 300 μl PBS containing 1% BSA. Data were acquired using a BD LSR Fortessa instrument and FlowJo software was used for analysis.
EBV Virus infection
7.5×105 mL of cells infected with EBfaV-GFP (recombinant EBV reporter virus expressing GFP29) were grown in 100 mL RPMI with 10% FBS, Pen/Strep, and 30 ng/mL tiglian 12-O-tetradecanoylphorbol-13-acetate (TPA) at 37°C, 5% CO2 for 4 days. The supernatant was collected by spin down at 1500 RPM for 10 minutes, aliquoted in 1 mL, and frozen at −80 °C, or it was centrifuged at 13000 RPM for 30 minutes at 4 °C. The pellets were resuspended in 100 μL of 10% FBS DMEM. 5×104 HEK293 cells/well were seeded on a 48-well plate and infected on the second day with 100 μL EBV virus in 10% FBS DMEM. For blocking assay, the Detroit 562 cells growing in trans-well were pretreated with anti-EphA2 (2ug/uL, R&D system, AF3035) or anti-EphA4 (2ug/uL, Life technologies, 371600) for one hour and infected with EBV virus concentrated from 5 mL supernatant35.
Bioinformatics
The Sequence Read Archive (SRA) data of RNA-seq for HEK293 (SRR5011298, SRR5011302 and SRR5011303), AGS (SRR2084426, SRR2084600 and SRR2084602) and B cells (SRR5048161, SRR5048160, SRR5048157 and SRR5048158) were downloaded from the SRA database (https://www.ncbi.nlm.nih.gov/sra). The SRA data were transformed into the original FASTQ documents using NCBI SRA Toolkit fastq-dump. The original documents were trimmed using FASTX and aligned to the reference genome using TopHat. The differential expression analysis was performed using Cuffdiff software. The genes encoding membrane proteins which had a 10-fold increase in HEK293 cells compared with B cells were chosen as candidates, and further filtered according to original reads (more than 5 FPKM in HEK293 cells and less than 5 FPKM in B cells). Among the candidate genes from comparison of B and HEK293 cells, the final top candidate genes were chosen if they had the maximum reads in AGS cells. The heatmap was generated using HemI software.
qRT-PCR
Expression of EphA2 and EphA4 mRNA in HEK293 cells was examined by quantitative RT-PCR (qRT-PCR) analysis, using the following primers: EphA2 F 5-AAGGAAGTGGTACTGCTGGA-3 and R: ACGTTGCACACGGAGTACAT ; and control GAPDH primers: F: 5-TGGTATCGTGGAAGGACTCATGAC-3, R: 5-ATGCCAGTGAGCTTCCCGTTCAGC-3. Real-time PCR was performed using a StepOnePlus Real-Time PCR System from Applied Biosciences with iQ SYBR supermix from BioRad. We used a 2-step amplification (40 cycles of 95°C, 15 seconds; 60°C, 30 seconds; followed by melting temperature determination stage) and quantified relative changes in gene expression using the ΔΔCt method as per manufacturer’s instructions.
Cloning, Expression and Purification of EBV proteins
EBV gH residues 18-679 and EBV gL 22-137 representing the soluble EBV gHgL protein were cloned by Gibson assembly into pTTVH8G expression vector (National Research Council Canada, NRC license file 11266). Either Vascular Endothelial Growth Factor (VEGF) as the signal sequence (SS) as part of the pTTVH8G vector or the native gH or gL signal sequence (SS) were used to make 4 different plasmids (two for EBV gH and two for EBV gL, Supplementary Fig. 6). Empty pTTVH8G served as the ‘vector’, while pSG5-gH and gL (insert reference) expressing full length EBV proteins were used as the ‘fragment’ templates in the Gibson assembly reaction (New England Biolabs). Following are the different primers used in Gibson assembly reaction: (1) EBV gH, VEGF SS resulting in a 8012 bp plasmid used Fragment-Forward: 5′-atgccaagtggtcccaggctGCCAGCCTCAGC-3′; Fragment-Reverse: 5′-gtcgaggtcgggggatctcaGTGTGCTCTTTCTTCATACAGGCCC-3′; Vector-Forward: 5′-TGTATGAAGAAAGAGCACACtgagatcccccgacctcgac-3′; Vector-Reverse: 5′-TTAACCTCGCTGAGGCTGGCagcctgggaccact-3′, (2) EBV gH, native SS plasmid resulting in a 7985 bp plasmids used Fragment-Forward: 5′- aacggatctctagcgaattcATGCAGTTGCTCTGTGTTTTTTGC-3′; Fragment-Reverse: 5′- gtcgaggtcgggggatctcaGTGTGCTCTTTCTTCATACAGGCCC-3′; Vector-Forward: 5′- TGTATGAAGAAAGAGCACACtgagatcccccgacctcgac-3′; Vector-Reverse: 5′- AAAACACAGAGCAACTGCATgaattcgctagagatccgtttaaact-3′, (3) EBV gL, VEGF SS resulting in a 6374 bp plasmid used Fragment-Forward: 5′- ccaagtggtcccaggctgcaTGGGCATACCCATGTTGTCAC-3′; Fragment-Reverse: 5′- gtcgaggtcgggggatctcaCTAGCCCCCGCGATG-3′; Vector-Forward: 5′-CATGGCATCGCGGGGGCTAGtgagatcccccgacctcgacctct-3′; Vector-Reverse: 5′- TGACAACATGGGTATGCCCAtgcagcctgggaccacttg-3′, (4) EBV gL, native SS resulting in a 6362 bp plasmid used Fragment-Forward: 5′- aacggatctctagcgaattcATGCGTGCTGTTGGTGTATTTCT-3′; Fragment-Reverse: 5′- gtcgaggtcgggggatctcaCTAGCCCCCGCGATG-3′; Vector-Forward: 5′- CATGGCATCGCGGGGGCTAGtgagatcccccgacctcgacctct-3′; Vector-Reverse: 5′- AATACACCAACAGCACGCATgaattcgctagagatccgtttaaact-3′. The sequence of the final 4 assembled plasmids including the entire gH or gL insert was verified by sanger sequencing using vector specific and custom sequencing primers. Using the pair of either VEGF SS or native SS assembled EBV pTTVH8G gH and gL plasmids in transient transfections when the cell density was between 1.5-2.0 ×106 cells/mL in a 1:1 ratio of gH:gL plasmids in HEK 293EBNA1-6E suspension cells (National Research Council Canada, NRC license file 11565) with PEI:plasmid DNA at 3:1 weight ratio resulted in a similar protein yield. The cells were grown with HEK 293 serum-free growth media (Freestyle 293, Gibco) with 0.1 % Kolliphor P188 and 25 μg/mL G418 and shaking at 135 rpm at 37°C with 5% CO2. 24 hours after transfection with PEI, 0.5% v/v final Tryptone-N1 (TN1) supplement (from a 20% w/v sterile filtered TN1 stock made in Freestyle 293) was added. The supernatant was collected after 4 days by centrifugation and passed through E1D1 antibody affinity column as previously described (insert PPathogens reference), eluted with Pierce Gentle Ag/Ab elution buffer pH 6.6 (Thermo Fisher Scientific) and purified by final Superdex 200 gel filtration column (GE Life Sciences) in 1× Phosphate Buffered Saline pH 7.4. Gp42 was expressed and purified as previously described (insert PPathogens reference).
SPR-binding kinetics
The binding kinetics assay to determine on-rate (ka), off-rate (kd), and affinity (KD) between EBV gHgL with EphA2 or EphA4 was performed using a Bioptix 404pi biosensor instrument (BiOptix, CO). 1× Phosphate Buffered Saline (PBS) pH 7.4 with 0.05% (v/v) Tween-20 was used as the running buffer. EphA2 (R&D systems 3035-A2-100) or EphA4 (R&D systems, 6827-A4-050) (as the ‘Ligand’) were immobilized onto separate channels on a carboxy-methyl dextrose (CMD-200m; BiOptix, CO) biosensor chip by the amine-coupling method using EDC/NHS chemicals (Sigma). Sensorgrams with different serial dilution, i.e., concentration series of EBV gHgL as the mobile analyte were flown over the ligand and the sensorgram data fit globally to a 1:1 interaction model using GraphPad Prism 7. Kinetic parameters from the model fit are collected in Supplementary Table 5. Sensorgram traces with the model fit overlaid on the data are shown in Fig. 4d and 4e.
Statistical analysis
The data were collected from three independent experiments. Statistical differences between multiple groups were determined by one-way ANOVA with post-hoc Tukey’s multiple comparison test. Two-group comparisons were analyzed by the two-tailed unpaired Student’s t test. P < 0.05 denotes the presence of a statistically significant difference. Data are expressed as mean ± SE. The analysis was performed using GraphPad Prism, version 6.0c for Mac (GraphPad Software, San Diego, California, USA). Flow cytometry histograms and microscopy images are representative of at least two independent experiments.
Data availability
For the comparative transcriptomics analysis, we used the following RNA-seq datasets from the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra): HEK293 (SRR5011298, SRR5011302 and SRR5011303), AGS (SRR2084426, SRR2084600 and SRR2084602) and B cells (SRR5048161, SRR5048160, SRR5048157 and SRR5048158). The rest of the data that support the findings of this study are available within this Letter and its supplementary information files or upon requesting the relevant information from the corresponding author.
Supplementary Material
Acknowledgments
We appreciate the help and advice from members of the Longnecker and Jardetzky laboratories, especially Nanette Susmarski. We also thank Dr. Mark Lingen and Terri Li from the Human Tissue Resource Center (HTRC) at the University of Chicago for performing experiments during the revision. This research was supported by AI076183 (R.L. and T.J.) from the National Institute of Allergy and Infectious Diseases, by CA117794 (R.L. and T.J.) from the National Cancer Institute as well as by Chicago Biomedical Consortium (Fall 14-0121 and 15-0257) to J.C.
Footnotes
Contributions
J.C. and R.L. designed the overall study with input from the co-authors. J.C. performed the key experiments. K.S. performed the initial microarray analysis, cloning gH and gL expression plasmids, purified protein preparations and EphA2 binding experiments. X.Z. performed the RNA seq analysis and helped with the sgRNA constructs and statistical analysis. S.S. helped with cell cultures. B.E.P.W contributed key reagents. J.C. and R.L. wrote the manuscript. X.Z., K.S. and T.J. contributed expertise and helped write the paper. All authors analyzed the results, read and approved the manuscript for submission.
Competing interests
The authors declare no competing financial interests.
References
- 1.Longnecker R, Kieff E, Cohen JI. Fields Viorology. 6. Lippincott Williams & Wilkins; Philadelphia, PA: 2013. [Google Scholar]
- 2.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nature reviews Microbiology. 2011;9(5):369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sathiyamoorthy K, Jiang J, Hu YX, Rowe CL, Möhl BS, Chen J, et al. Assembly and architecture of the EBV B cell entry triggering complex. 2014 doi: 10.1371/journal.ppat.1004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Epstein MA, Achong BG, Barr YM. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet. 1964;1(7335):702–703. doi: 10.1016/s0140-6736(64)91524-7. [DOI] [PubMed] [Google Scholar]
- 5.Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearon DT. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(14):4510–4514. doi: 10.1073/pnas.81.14.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Kenyon WJ, Li Q, Mullberg J, Hutt-Fletcher LM. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. Journal of virology. 1998;72(7):5552–5558. doi: 10.1128/jvi.72.7.5552-5558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spriggs MK, Armitage RJ, Comeau MR, Strockbine L, Farrah T, Macduff B, et al. The extracellular domain of the Epstein-Barr virus BZLF2 protein binds the HLA-DR beta chain and inhibits antigen presentation. Journal of virology. 1996;70(8):5557–5563. doi: 10.1128/jvi.70.8.5557-5563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, et al. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. Journal of virology. 1997;71(6):4657–4662. doi: 10.1128/jvi.71.6.4657-4662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullen MM, Haan KM, Longnecker R, Jardetzky TS. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Molecular cell. 2002;9(2):375–385. doi: 10.1016/s1097-2765(02)00465-3. [DOI] [PubMed] [Google Scholar]
- 10.Backovic M, Longnecker R, Jardetzky TS. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(8):2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirschner AN, Sorem J, Longnecker R, Jardetzky TS. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure. 2009;17(2):223–233. doi: 10.1016/j.str.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(52):22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sathiyamoorthy K, Hu YX, Mohl BS, Chen J, Longnecker R, Jardetzky TS. Structural basis for Epstein-Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nature communications. 2016;7:13557. doi: 10.1038/ncomms13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(48):20464–20469. doi: 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chesnokova LS, Hutt-Fletcher LM. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. Journal of virology. 2011;85(24):13214–13223. doi: 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. Journal of virology. 2000;74(14):6324–6332. doi: 10.1128/jvi.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Rowe CL, Jardetzky TS, Longnecker R. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. mBio. 2012;3(1) doi: 10.1128/mBio.00290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Hutt-Fletcher LM. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. Journal of virology. 1998;72(1):158–163. doi: 10.1128/jvi.72.1.158-163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, Jardetzky TS, Longnecker R. The large groove found in the gH/gL structure is an important functional domain for Epstein-Barr virus fusion. Journal of virology. 2013;87(7):3620–3627. doi: 10.1128/JVI.03245-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borza CM, Hutt-Fletcher LM. Epstein-Barr virus recombinant lacking expression of glycoprotein gp150 infects B cells normally but is enhanced for infection of epithelial cells. Journal of virology. 1998;72(9):7577–7582. doi: 10.1128/jvi.72.9.7577-7582.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasahara Y, Yachie A. Cell type specific infection of Epstein-Barr virus (EBV) in EBV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Critical reviews in oncology/hematology. 2002;44(3):283–294. doi: 10.1016/s1040-8428(02)00119-1. [DOI] [PubMed] [Google Scholar]
- 22.Boshoff C. Ephrin receptor: a door to KSHV infection. Nature medicine. 2012;18(6):861–863. doi: 10.1038/nm.2803. [DOI] [PubMed] [Google Scholar]
- 23.Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nature medicine. 2012;18(6):961–966. doi: 10.1038/nm.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kania A, Klein R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nature reviews Molecular cell biology. 2016;17(4):240–256. doi: 10.1038/nrm.2015.16. [DOI] [PubMed] [Google Scholar]
- 25.Shao Z, Zhang WF, Chen XM, Shang ZJ. Expression of EphA2 and VEGF in squamous cell carcinoma of the tongue: correlation with the angiogenesis and clinical outcome. Oral oncology. 2008;44(12):1110–1117. doi: 10.1016/j.oraloncology.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 26.Shao Z, Zhu F, Song K, Zhang H, Liu K, Shang Z. EphA2/ephrinA1 mRNA expression and protein production in adenoid cystic carcinoma of salivary gland. J Oral Maxillofac Surg. 2013;71(5):869–878. doi: 10.1016/j.joms.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 27.Wang DH, Zhang YJ, Zhang SB, Liu H, Liu L, Liu FL, et al. Geldanamycin mediates the apoptosis of gastric carcinoma cells through inhibition of EphA2 protein expression. Oncology reports. 2014;32(6):2429–2436. doi: 10.3892/or.2014.3542. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura R, Kataoka H, Sato N, Kanamori M, Ihara M, Igarashi H, et al. EPHA2/EFNA1 expression in human gastric cancer. Cancer science. 2005;96(1):42–47. doi: 10.1111/j.1349-7006.2005.00007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Speck P, Longnecker R. Epstein-Barr virus (EBV) infection visualized by EGFP expression demonstrates dependence on known mediators of EBV entry. Archives of virology. 1999;144(6):1123–1137. doi: 10.1007/s007050050574. [DOI] [PubMed] [Google Scholar]
- 30.Hahn AS, Desrosiers RC. Binding of the Kaposi’s sarcoma-associated herpesvirus to the ephrin binding surface of the EphA2 receptor and its inhibition by a small molecule. Journal of virology. 2014;88(16):8724–8734. doi: 10.1128/JVI.01392-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar B, Chandran B. KSHV Entry and Trafficking in Target Cells-Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics. Viruses. 2016;8(11) doi: 10.3390/v8110305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutta D, Chakraborty S, Bandyopadhyay C, Valiya Veettil M, Ansari MA, Singh VV, et al. EphrinA2 regulates clathrin mediated KSHV endocytosis in fibroblast cells by coordinating integrin-associated signaling and c-Cbl directed polyubiquitination. PLoS pathogens. 2013;9(7):e1003510. doi: 10.1371/journal.ppat.1003510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirschner AN, Lowrey AS, Longnecker R, Jardetzky TS. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. Journal of virology. 2007;81(17):9216–9229. doi: 10.1128/JVI.00575-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hahn AS, Desrosiers RC. Rhesus monkey rhadinovirus uses eph family receptors for entry into B cells and endothelial cells but not fibroblasts. PLoS pathogens. 2013;9(5):e1003360. doi: 10.1371/journal.ppat.1003360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tugizov SM, Berline JW, Palefsky JM. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nature medicine. 2003;9(3):307–314. doi: 10.1038/nm830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For the comparative transcriptomics analysis, we used the following RNA-seq datasets from the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra): HEK293 (SRR5011298, SRR5011302 and SRR5011303), AGS (SRR2084426, SRR2084600 and SRR2084602) and B cells (SRR5048161, SRR5048160, SRR5048157 and SRR5048158). The rest of the data that support the findings of this study are available within this Letter and its supplementary information files or upon requesting the relevant information from the corresponding author.