

Experimental studies indicate that, following focal or global ischaemia, progressive recruitment of neocortical neurons into death begins when spreading depolarization gives way to a negative ultraslow potential. By monitoring patients with subarachnoid haemorrhage, Lückl et al. identify this process as the electrophysiological correlate of infarction and a neuromonitoring-detected medical emergency.
Keywords: cytotoxic oedema, ischaemia, spreading depression, stroke, subarachnoid haemorrhage
Abstract
Spreading depolarizations are characterized by abrupt, near-complete breakdown of the transmembrane ion gradients, neuronal oedema, mitochondrial depolarization, glutamate excitotoxicity and activity loss (depression). Spreading depolarization induces either transient hyperperfusion in normal tissue; or hypoperfusion (inverse coupling = spreading ischaemia) in tissue at risk for progressive injury. The concept of the spreading depolarization continuum is critical since many spreading depolarizations have intermediate characteristics, as opposed to the two extremes of spreading depolarization in either severely ischaemic or normal tissue. In animals, the spreading depolarization extreme in ischaemic tissue is characterized by prolonged depolarization durations, in addition to a slow baseline variation termed the negative ultraslow potential. The negative ultraslow potential is initiated by spreading depolarization and similar to the negative direct current (DC) shift of prolonged spreading depolarization, but specifically refers to a negative potential component during progressive recruitment of neurons into cell death in the wake of spreading depolarization. We here first quantified the spreading depolarization-initiated negative ultraslow potential in the electrocorticographic DC range and the activity depression in the alternate current range after middle cerebral artery occlusion in rats. Relevance of these variables to the injury was supported by significant correlations with the cortical infarct volume and neurological outcome after 72 h of survival. We then identified negative ultraslow potential-containing clusters of spreading depolarizations in 11 patients with aneurysmal subarachnoid haemorrhage. The human platinum/iridium-recorded negative ultraslow potential showed a tent-like shape. Its amplitude of 45.0 (39.0, 69.4) mV [median (first, third quartile)] was 6.6 times larger and its duration of 3.7 (3.3, 5.3) h was 34.9 times longer than the negative DC shift of spreading depolarizations in less compromised tissue. Using Generalized Estimating Equations applied to a logistic regression model, we found that negative ultraslow potential displaying electrodes were significantly more likely to overlie a developing ischaemic lesion (90.0%, 27/30) than those not displaying a negative ultraslow potential (0.0%, 0/20) (P = 0.004). Based on serial neuroimages, the lesions under the electrodes developed within a time window of 72 (56, 134) h. The negative ultraslow potential occurred in this time window in 9/10 patients. It was often preceded by a spreading depolarization cluster with increasingly persistent spreading depressions and progressively prolonged DC shifts and spreading ischaemias. During the negative ultraslow potential, spreading ischaemia lasted for 40.0 (28.0, 76.5) min, cerebral blood flow fell from 57 (53, 65) % to 26 (16, 42) % (n = 4) and tissue partial pressure of oxygen from 12.5 (9.2, 15.2) to 3.3 (2.4, 7.4) mmHg (n = 5). Our data suggest that the negative ultraslow potential is the electrophysiological correlate of infarction in human cerebral cortex and a neuromonitoring-detected medical emergency.
Introduction
The evolution of parenchymal lesions after aneurysmal subarachnoid haemorrhage (aSAH) shows a bimodal distribution with an early and a delayed peak. Infarcts developing between Days 0 and 4 are usually subsumed under the term ‘early cerebral ischaemia’ (Sarrafzadeh et al., 2002) in contrast to those developing between Days 5 and 14 termed ‘delayed cerebral ischaemia’ (Vergouwen et al., 2010). In addition to aSAH, aneurysmal rupture may also lead to large intracerebral haemorrhages contributing to parenchymal damage. In our recent study on 23 patients with anterior communicating artery aneurysms, intracerebral haemorrhages with surrounding compression-induced parenchymal damage were already visible on the initial CT scan, performed at a median of 2.5 h after the initial haemorrhage. In contrast, early cerebral ischaemia-related infarcts were observed only by an early MRI scan 24–48 h after aneurysm treatment (Hartings et al., 2017c).
Cerebral ischaemia after aSAH is of particular interest because it is a model disease of hypoxic-ischaemic injury for two reasons (Dreier, 2011). First, it often occurs while the patient is already in the neurocritical care unit. In animal experiments, neuroprotective interventions have been found to be most effective either when given within minutes after the onset of ischaemia or when they have been pre-administered. Thus, cerebral ischaemia after aSAH allows the treatment of patients with a neuroprotective intervention before the possible insult or very shortly thereafter to prove or disprove the neuroprotective concept. Second, neurosurgical procedures are indicated in patients early after the initial haemorrhage, allowing implantation of invasive probes. This provides the unique option to invasively monitor the whole period of ischaemic stroke development, to perform early treatment stratification according to changes in diagnostic summary measures sensed in real time by neuromonitoring devices, and then to record the responses of the diagnostic summary measures to the neuroprotective intervention.
Direct current (DC)/alternate current (AC)-electrocorticography (ECoG), tissue partial pressure of oxygen (ptiO2) and regional cerebral blood flow (CBF) represent diagnostic summary measures indicating disturbances of brain energy metabolism that allow continuous bedside monitoring with high temporal resolution and a good safety profile (Dreier et al., 2009; Waziri et al., 2009; Bosche et al., 2010; Hinzman et al., 2014; Citerio et al., 2015; Drenckhahn et al., 2016; Winkler et al., 2017). A future goal is that these measures should be subject to an automated real-time analysis to minimize human workload and allow prespecified diagnostic criteria to trigger an alarm (Dreier et al., 2017). Spreading depolarizations (SDs) play an important role in this concept because they are indicators of disturbed energy metabolism and propagate widely from ischaemic or metabolically stressed zones in both animals and humans, thereby affording even remote detection of newly developing injury (Hartings et al., 2017c). Previous animal experiments and human case reports moreover suggest that their local characteristics inform whether a given electrode is positioned either (i) directly at; (ii) in the neighbourhood of; or (iii) remote from a newly developing ischaemic zone (Oliveira-Ferreira et al., 2010; Dreier et al., 2017; Hartings et al., 2017b; Milakara et al., 2017; Winkler et al., 2017).
Of particular interest is the zone of developing injury. In animals, it is characterized by an SD that initiates the so-called negative ultraslow potential (NUP) (Dreier and Reiffurth, 2015; Hartings et al., 2017b). Here we sought to better characterize this phenomenon as a hypothesized electrophysiological correlate of a newly developing infarct. We first recognized and quantified the NUP during filament occlusion of the middle cerebral artery in rats and correlated it with the cortical and striatal infarct volumes as well as the neurological outcome. Clinical recordings are often subject to artefacts. Therefore, we additionally characterized two complementary electrophysiological biomarkers indicating cerebral ischaemia/infarction in order to increase the diagnostic accuracy: (i) the persistent depression of spontaneous activity; and (ii) the rundown in amplitudes between subsequent SDs during the NUP. These variables were also correlated with the cortical and striatal infarct volumes as well as the neurological outcome. We then identified and quantified characteristic NUP-containing clusters of SDs in 11 patients with aSAH using simultaneous DC/AC-ECoG, ptiO2, CBF as well as intracranial and arterial pressure recordings. Finally, we used serial neuroimaging scans to determine whether electrodes displaying the NUP are more likely to overlie a newly developing ischaemic lesion than electrodes not displaying the NUP.
Materials and methods
Animals
The reporting of animal experiments complies with the ARRIVE guidelines. All animal experiments were approved by the Landesamt für Arbeitsschutz, Gesundheitsschutz und technische Sicherheit Berlin (LAGetSi, G0152/11). Naïve, male, wild-type Sprague-Dawley rats (n = 64; 250–400 g, ∼10–16 weeks old; Charles River) were anaesthetized with isoflurane 4% for induction and 1.2–1.6% for maintenance dissolved in 70% N2O and 30% O2. The tail artery was cannulated with a polyethylene catheter. Body temperature was maintained at 37.5 ± 0.5°C using a rectal probe, a heating pad and a homoeothermic control unit. Systemic arterial pressure (Pressure Monitor BP-1, World Precision Instruments) was monitored. Arterial pO2 (paO2), partial pressure of CO2 (paCO2) and pH were serially measured using a Compact 1 Blood Gas Analyzer (AVL Medizintechnik). Adequacy of the level of anaesthesia was assessed with testing motor and blood pressure responses to tail pinch. Head surgery, filament occlusion and electrical stimulation are explained in the Fig. 1 and the online Supplementary material. After 72 h of survival, all animals were neurologically examined. Thereafter, they underwent cardiac perfusion fixation and histological assessment, as explained in the Supplementary material. The neurological exam followed the protocol of Belayev et al. (1996). Postural reflex, visual placing in forward and sideways directions, tactile placing of the dorsal and lateral paw surfaces and proprioceptive placing were tested and scored on a 12-point scale ranging from 0 (no deficit) to 12 (maximum deficit) (Belayev et al., 1996). The statistical analysis of the animal data is explained in the Supplementary material.
Figure 1.

Experimental set-ups and paradigms. (A) The top left panel shows a schematic of a single, linear, six-contact (platinum/iridium) ECoG recording strip placed on human cortex. The other five panels show the animal experimental set-ups, which are further explained in the Supplementary material. (B) The top left panel explains the intraluminal filament occlusion. After head surgery and placement of laser-Doppler flowmetry probe and electrodes, the filament was later advanced during the experiment until the laser-Doppler flowmetry probe indicated adequate MCAO by a sharp decrease in CBF. After either 15 min (Group 1B) or 90 min (Groups 1A and 1C) of occlusion the filament was gently withdrawn and the reperfusion was monitored. ACA = anterior cerebral artery; AChA = anterior choroidal artery; BA = basilar artery; CCA = common carotid artery; HA = hypothalamic artery; ICA = internal carotid artery; PCA = posterior cerebral artery; SCA = superior cerebellar artery. In the other five panels, representative example traces and histological outcomes of the animal-experimental groups are shown. Infarcts are macroscopically identified as pale areas using haematoxylin staining (cf. right hemispheres in Groups 1A and 1C). SDs are observed as slow potential changes in the DC/AC-ECoG (bandpass: 0–45 Hz) and depression of activity in the AC-ECoG (bandpass: 0.5–45 Hz) (Dreier et al., 2017). Whereas the first SD initiated the NUP in Group 1A, subsequent SDs typically occurred superimposed on the NUP. In the two control Groups 2A and 2B, no filament occlusion was performed but three SDs were electrically triggered at the same time intervals as measured on average in Group 1A between first, second and third SD during filament occlusion (second SD: 20.6 ± 14.9 min, third SD: 36.0 ± 15.8 min). Also in the scalp EEG recordings of Groups 1C and 2B, DC drifts were seen, as exemplified in the figure, but they were not consistent within or between the groups and may thus represent complex summary potentials of different generators. For example, changes in sympathetic tone can produce scalp DC-EEG drifts through activation/deactivation of cutaneous sweat glands. Additional contributors could be DC potential deflections generated at the blood–brain barrier, e.g. by changes in partial pressures of CO2 or O2 (Lehmenkühler et al., 1999; Voipio et al., 2003).
Patients
For the clinical study we selected patients from a prospectively collected database using pre-specified criteria and endpoints, as described below. The protocol was approved by the ethics committee of the Charité – Universitätsmedizin Berlin. Either informed consent or surrogate informed consent was obtained for all patients, and research was conducted in accordance with the Declaration of Helsinki. Results are reported in accordance with the STrengthening the Reporting of OBservational studies in Epidemiology (STROBE) guidelines.
Patient selection and characterization
Patients with aSAH consecutively enrolled in the Co-Operative Studies on Brain Injury Depolarizations (COSBID) at two centres (Campus Virchow Klinikum and Campus Benjamin Franklin, Berlin, Germany) were screened for study inclusion. Prospective inclusion criteria for COSBID have been described previously (Winkler et al., 2017). For this study, patients were further screened retrospectively for (i) the availability of DC/AC recordings (bandpass: 0–45 Hz) of sufficient quality; (ii) a NUP-containing cluster (of at least 4 SDs in a row with inter-SD intervals of <1 h); and (iii) the availability of serial neuroimages performed before and after the occurrence of the NUP. Eleven patients enrolled between December 2007 and February 2014 met criteria, including seven at one site and four at the other. Seven cases were previously reported (Dreier et al., 2012; Winkler et al., 2017).
Aneurysms were assessed using four-vessel digital subtraction angiography, or more restricted studies when indicated. Haemorrhage was graded according to the original Fisher scale (Fisher et al., 1980). After aneurysm treatment, patients were transferred to intensive care where continuous neuromonitoring commenced. Glasgow coma scale, blood gases, glucose and electrolytes were documented at least every 6 h. Thorough neurological examinations and transcranial Doppler sonography were performed daily and oral nimodipine was given prophylactically.
Neuroimaging
The MRI exam protocol included a T2-weighted fluid-attenuated inversion recovery (FLAIR) sequence, a T1-weighted sequence pre- and post-gadolinium, a T1-weighted 3D high resolution sequence [i.e. magnetization prepared rapid gradient echo (MPRAGE)], and a diffusion weighted imaging (DWI) sequence. The first MRI was performed 24–48 h after aneurysm treatment to assess the initial structural brain injury, the second MRI around Day 7, and the third one on the day of electrode withdrawal (∼Day 15) to assess the occurrence of early ischaemic strokes and delayed ischaemic strokes during the ECoG monitoring period. For analysis, the region of interest was classified into normal, ischaemic or haemorrhagic for each patient using all sequences including DWI and apparent diffusion coefficient (ADC) mapping. In addition to MRI scans, serial CT scans were performed at times of clinical deterioration. CT scans were used in addition to MRI scans to identify the time period of infarct evolution. In addition, they provided visualization of the six subdural electrode positions. The serial images were analysed by V.K. and V.H. blinded to the clinical findings and the ECoG recordings.
Neuromonitoring
For continuous ECoG recordings, a 6-contact, collinear platinum/iridium electrode strip (10-mm spacing; Wyler, Ad-Tech Medical) was placed on cerebral cortex accessible either through craniotomy during aneurysm surgery or via an extended burr hole (Dreier et al., 2009). The near-DC/AC-ECoG (0.01–45 Hz) was recorded in five bipolar channels with a GT205 amplifier (ADInstruments). A subdermal platinum/iridium needle electrode was used as reference for additional monopolar recordings. The full-band DC/AC-ECoG (0–45 Hz) was measured using a DC-coupled amplifier (BrainAmp MR plus, Brain Products) and BrainVision Recorder software (Brain Products). Data were sampled at 200 Hz and recorded and analysed with a Powerlab 16/SP and Chart-7 software (ADInstruments).
Analyses of the bipolar near-DC/AC-ECoG data of the full recording period were performed by M.W. with blinding to the clinical courses and neuroimaging findings, and were discussed with J.D. The analyses followed the recently published COSBID recommendations (Dreier et al., 2017). SDs were identified by the consecutive occurrence of slow potential changes (<0.05 Hz) of neighbouring ECoG channels. In electrically active tissue, SD typically causes spreading depression of spontaneous activity in the AC-ECoG range (0.5–45 Hz). Such SDs received the epithet ‘spreading depression’. By contrast, SDs measured in a zone of electrically-inactive tissue were denoted with the adjective ‘isoelectric’ (Dreier et al., 2017). For each SD, the depression durations were calculated in minutes for each channel. Only depressions observed to be induced by SD were included. The duration of the depression period was scored beginning at the initial decrease in the integral of power and ending at the start of the recovery phase (Dreier et al., 2006). Among all depression durations in individual channels, the longest was then determined for each SD. Subsequently, the total depression duration of each 24-h period following the initial insult was calculated as the sum of the longest depression durations of all individual SDs during that 24-h period. If there was temporal overlap between depression periods of successive SDs on different electrodes, the overlapping period was only counted once. The first 24-h period was denoted as ‘Day 0’, the second 24-h period as ‘Day 1’ and so on. Subsequently, the total SD-induced depression duration of each day (abbreviated as TDDD in Dreier et al., 2017) was defined as the value after normalization to the total time of valid recordings during that 24-h period. For instance, if 240 min (4 h) of total depression duration were recorded in 22 h of a 24-h period, the normalized total SD-induced depression duration of a recording day was 262 min [(4/22) × 24 × 60]. The peak total SD-induced depression duration of a recording day (abbreviated as PTDDD in Dreier et al., 2017) was then defined as the longest total SD-induced depression duration among all recording days in a given patient.
NUP-containing clusters of at least 4 SDs in a row with inter-SD intervals of <1 h were analysed by C.L. using monopolar DC/AC-ECoG, ptiO2 and CBF recordings as further described in the results section. C.L. was blinded to the clinical courses and neuroimaging findings, and discussed the data with J.D. PtiO2 was recorded in eight patients using a Clark-type intraparenchymal sensor (Licox CC1P1, Integra) (Dreier et al., 2009; Bosche et al., 2010; Winkler et al., 2017). In four patients, a subdural opto-electrode strip allowed the simultaneous measurement of ECoG and CBF using LDF (Perimed AB) (Dreier et al., 2009; Oliveira-Ferreira et al., 2010; Drenckhahn et al., 2016). Intracranial pressure was monitored via ventricular drainage catheter or intracranial pressure transducer (Codman or Camino systems). Arterial pressure was recorded from the radial artery. At the conclusion of the monitoring period, the subdural (opto-)electrode strip and the ptiO2 probe were removed at the bedside by gentle traction.
Statistics
The primary hypothesis in the analysis of patient data was that NUPs predicted infarcts, where both variables were measured for the regions defined by the locations of multiple electrodes per patient. Dependency of data was adjusted for by the method of Generalized Estimating Equations (GEE, specification Independent Estimation Equations, IEE) applied to a logistic regression model with NUP (yes/no) as single predictor, infarct (yes/no) as outcome and patient as cluster variable (Zeger and Liang, 1986). All other patient-related analyses are purely exploratory and were not corrected for multiple testing. The data in the text and figures are given as median (first quartile, third quartile). The statistical tests are stated in the ‘Results’ section and were two-sided. P < 0.05 was accepted as statistically significant.
Results
Middle cerebral artery occlusion in rats induces characteristic changes in cerebral blood flow and electrophysiology
CBF significantly dropped in response to filament occlusion (Fig. 1B). Thereafter, four characteristic parameter changes were typically observed: depression of spontaneous activity that simultaneously occurred at the rostral and the caudal electrode (non-spreading depression) (Leão, 1947; Dijkhuizen et al., 1999; Dreier, 2011; Dreier et al., 2017), rise in mean arterial pressure, SD and the SD-initiated NUP consistent with previous literature (Dijkhuizen et al., 1999; Higuchi et al., 2002; Dreier et al., 2017; Hartings et al., 2017b). Supplementary Table 1 provides the changes in CBF, brain surface AC-ECoG power, scalp AC-EEG power and mean arterial pressure from baseline during middle cerebral artery occlusion (MCAO), which were statistically highly significant (P < 0.001).
In Groups 1A (epidural ECoG, n = 28) and 1C (scalp EEG, n = 12), filament occlusion lasted for 90 min, and in Group 1B (epidural ECoG, n = 8) for 15 min (Fig. 1B). In the two groups with 90 min of filament occlusion, 36 of 40 animals showed an infarct of the striatum with a median (first, third quartile) size of 44 (27, 49) mm3 (Fig. 2A) and 32 of 40 had a cortical infarct with a median size of 84 (45, 108) mm3 (Fig. 2B). Linear regression found that cortical and striatal infarct sizes significantly correlated (adjusted R2 = 0.537, P < 0.001, n = 40).
Figure 2.


Histological outcome and electrophysiology. Necrotic neurons were found in the infarcted area of striatum (A) and cortex (B) of the ipsilateral hemisphere 72 h after 90 min of MCAO (Group 1A). By contrast, neurons appeared to be normal, having well-defined nuclei, prominent nucleoli, and clearly visible cytoplasm in the striatum (C) and cortex (D) of the ipsilateral hemisphere 72 h after 15 min of MCAO (Group 1B). Likewise, neurons were normal in striatum (E) and cortex (F) in the control group (Group 2A) 72 h after three SDs had been triggered by electrical stimulation. All pictures were taken at ×40 magnification after haematoxylin staining. (G) Representative recording site of an animal with smaller infarct, fewer SDs, less pronounced NUP and less depression of spontaneous activity. First trace: to quantify the NUP, the integral of the DC potential change was calculated after correction for the electrode drift; second trace: DC/AC-ECoG with SDs and NUPs; third trace: bandpass filtered (AC-ECoG) data with initial non-spreading depression of activity and two SD-induced spreading depressions of activity; fourth trace: total AC-ECoG power; fifth trace: it is convention in neurocritical care to review the raw signal alongside a leaky integral of the total power of the AC-ECoG data (Dreier et al., 2017). (H) Representative recording site of an animal with larger infarct, more SDs and more pronounced NUP than in G. (I–L) Comparison of the electrophysiological biomarkers at 10-min intervals from the onset to the end of filament occlusion between Group 1A (90 min filament occlusion, solid line), Group 1B (15 min filament occlusion, dashed line) and control Group 2A (electrical stimulation of three SDs instead of filament occlusion, dotted line). (I) Pooled data of rostral and caudal mean values of the DC integral. The DC integral over the 90 min recording period was significantly larger in Group 1A (90 min filament occlusion) (rostral: n = 27; caudal: n = 28) compared to both Group 1B (15 min filament occlusion, n = 8) and Group 2A (control, n = 8) at both recording sites (Kruskal-Wallis one-way ANOVA on Ranks with Dunn’s post hoc test, P < 0.05). By contrast, the DC integral over only the first 15 min was significantly larger both in Group 1A (rostral: n = 27; caudal: n = 28) and 1B (n = 8) compared to Group 2A (n = 8) (Kruskal-Wallis one-way ANOVA on Ranks with Dunn’s post hoc test, P < 0.05). (J) Pooled data of rostral and caudal mean values of the AC-ECoG power. The mean AC-ECoG power over the full 90-min period was significantly smaller in Group 1A (n = 27/n = 28) compared to both Group 1B (n = 8) and Group 2A (n = 8) at both recording sites (one-way ANOVA with Fisher’s LSD post hoc test, rostral: Group 1A versus 1B, P = 0.021; Group 1A versus 2A, P < 0.001; caudal: Group 1A versus 1B, P = 0.039; Group 1A versus 2A, P < 0.001). By contrast, the mean AC-ECoG power over the first 15 min was significantly smaller both in Group 1A (n = 27/n = 28) and Group 1B (n = 8) compared to Group 2A (n = 8) (one-way ANOVA with Fisher’s LSD post hoc test, rostral: Group 1A versus 2A, P < 0.001; Group 1B versus 2A, P = 0.024; caudal: Group 1A versus 2A, P < 0.002; Group 1B versus 2A, P = 0.029). (K) Pooled data of rostral and caudal mean values of the integral of AC-ECoG power. The findings were similar to the AC-ECoG power. (L) Pooled data of rostral and caudal mean values of the scalp AC-EEG power. The mean scalp AC-EEG power values over the first 15 min and the full 90-min period were significantly smaller in Group 1C (90 min filament occlusion, n = 12) compared to Group 2B (control, n = 8) at both recording sites (15 min: rostral: t-test, P = 0.005, caudal: t-test, P < 0.001; 90 min: rostral: t-test, P < 0.001, caudal: Mann-Whitney U-test, P < 0.008).
Spreading depolarization and negative ultraslow potentials as biomarkers of cerebral ischaemia/infarction
SD, as indicated by the large negative DC shift, started only 80 (65, 116) s after the drop in CBF (Fig. 2G and H) consistent with previous literature (Leão, 1947; Dijkhuizen et al., 1999). To quantify the NUP over the 90-min period of filament occlusion in Group 1A, we calculated the integral of the DC potential change after correction for the electrode drift as explained in the Supplementary material (Fig. 2I). Linear regression found that the DC integral significantly correlated with the infarct volumes (Table 1).
Table 1.
Correlation of electrophysiological variables and CBF with infarct volumes and neurological outcome after MCAO in rats
| Variable | Group/animals, n | Linear regression with cortical infarct | Linear regression with striatal infarct | Linear regression with neurological outcome | Receiver operating characteristic | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Adjusted R2 | P | Adjusted R2 | P | Adjusted R2 | P | AUCs and cut-off values | Sensitivity of the cut-off value | Specificity of the cut-off value | |||
| Rostral AC-ECoG power | |||||||||||
| First 15 min | 1A/27 | 0.128 | 0.038 | 0.026 | n.s. | 0.047 | n.s. | Group 1A (90 min ischaemia) versus control group 2A (electrical stimulation of SDs) |
|
85% | 63% |
| First 30 min | 1A/27 | 0.145 | 0.028 | 0.047 | n.s. | 0.075 | n.s. | 89% | 88% | ||
| Full 90 min | 1A/27 | 0.198 | 0.012 | 0.099 | n.s. | 0.113 | 0.048 | 89% | 100% | ||
| Caudal AC-ECoG power | |||||||||||
| First 15 min | 1A/28 | 0.318 | <0.001 | 0.055 | n.s. | 0.184 | 0.013 | 86% | 63% | ||
| First 30 min | 1A/28 | 0.342 | <0.001 | 0.091 | n.s. | 0.209 | 0.008 | 82% | 75% | ||
| Full 90 min | 1A/28 | 0.267 | 0.003 | 0.104 | n.s. | 0.179 | 0.014 | 79% | 75% | ||
| Rostral scalp AC-EEG power | |||||||||||
| First 15 min | 1C/12 | 0.359 | 0.023 | 0.280 | 0.044 | 0.128 | n.s. | Group 1C (90 min ischaemia) versus control group 2B (electrical stimulation of SDs) |
|
83% | 75% |
| First 30 min | 1C/12 | 0.352 | 0.025 | 0.278 | 0.045 | 0.156 | n.s. | 83% | 100% | ||
| Full 90 min | 1C/12 | 0.684 | <0.001 | 0.712 | <0.001 | 0.430 | 0.012 | 92% | 100% | ||
| Caudal scalp AC-EEG power | |||||||||||
| First 15 min | 1C/12 | 0.278 | 0.045 | 0.294 | 0.040 | 0.225 | n.s. | 83% | 88% | ||
| First 30 min | 1C/12 | 0.565 | 0.003 | 0.654 | 0.001 | 0.299 | 0.038 | 83% | 88% | ||
| Full 90 min | 1C/12 | 0.567 | 0.003 | 0.468 | 0.008 | 0.461 | 0.009 | 83% | 88% | ||
| Rostral DC-ECoG amplitude ratio 2nd/1st SD | 1A/22 | 0.274 | 0.007 | 0.180 | 0.028 | 0.230 | 0.014 | Group 1A versus control group 2A |
|
94% | 75% |
| Caudal DC-ECoG amplitude ratio 2nd/1st SD | 1A/23 | 0.224 | 0.013 | 0.061 | n.s. | 0.140 | <0.019 | 83% | 88% | ||
| Rostral near-DC-ECoG amplitude ratio 2nd/1st SD | 1A/20 | 0.023 | n.s. | 0.000 | n.s. | 0.000 | n.s. | Group 1A versus control group 2A |
|
83% | 100% |
| Caudal near-DC-ECoG amplitude ratio 2nd/1st SD | 1A/21 | 0.168 | 0.037 | 0.033 | n.s. | 0.354 | <0.003 | 83% | 63% | ||
| Rostral DC-ECoG integral | 1A/28 | 0.323 | <0.001 | 0.222 | <0.007 | 0.262 | <0.003 | ||||
| Caudal DC-ECoG integral | 1A/27 | 0.326 | <0.001 | 0.179 | 0.016 | 0.279 | <0.003 | ||||
| CBF decrease | 1A, 1C/40 | 0.229 | <0.001 | 0.205 | <0.001 | 0.259 | <0.001 | ||||
| MAP increase | 1A, 1C/40 | 0.000 | n.s. | 0.000 | n.s. | 0.000 | n.s. | ||||
| Number of SDs | 1A/28 | 0.361 | <0.001 | 0.332 | <0.001 | 0.291 | 0.002 | ||||
| Number of CBF responses to SD | 1A/28 | 0.351 | <0.001 | 0.313 | 0.002 | 0.240 | 0.005 | ||||
| Neurological score after 72 h | 1A, 1C/40 | 0.693 | <0.001 | 0.624 | <0.001 | ||||||
The left part of the table shows linear regression analyses of the different variables in the left column with the volumes of cortical and striatal infarcts and neurological outcome after 72 h. The right part of the table gives cut-off values for the variables in the left column that provide the best separation of the ischaemia and control groups. Note that 68 statistical tests are reported in the left part of the table without correction for multiple testing. Using a strict Bonferroni correction, only P-values below 0.05/68 = 0.00074 indicate significance. However, with 68 tests, only three or four uncorrected significant results are expected by chance. We observe 49 such significances. Taking into account that many of the statistical hypotheses tested are related to the same fundamental hypothesis—electrophysiological variables monitored during transient ischaemia correlate with the histological and neurological outcome after 72 h—we think that the Bonferroni correction is not adequate in this case. AUC = area under the curve; n.s. = not significant.
During the 90 min of filament occlusion in Group 1A, three (2, 4) SDs were recorded. Whereas the first SD initiated the NUP, subsequent SDs typically occurred superimposed on the NUP. Their amplitudes were significantly smaller than that of the first SD (Fig. 3A). Linear regression found that the ratio between the amplitudes of first and second SD significantly correlated with the cortical infarct volume (Table 1). To determine a cut-off value for this amplitude ratio a receiver operating characteristic (ROC) analysis was performed using the empirical data of Groups 1A (90 min filament occlusion) and 2A (control). Because of the small sample sizes, a subdivision into learning and validation samples was not performed. The rostral and caudal areas under the curve (AUC) were 0.944 and 0.972, respectively, indicating that it was possible to separate Groups 1A and 2A. As best cut-off value between the two groups, we chose a value of 70% (cf. Table 1 for sensitivities and specificities of the cut-off value).
Figure 3.
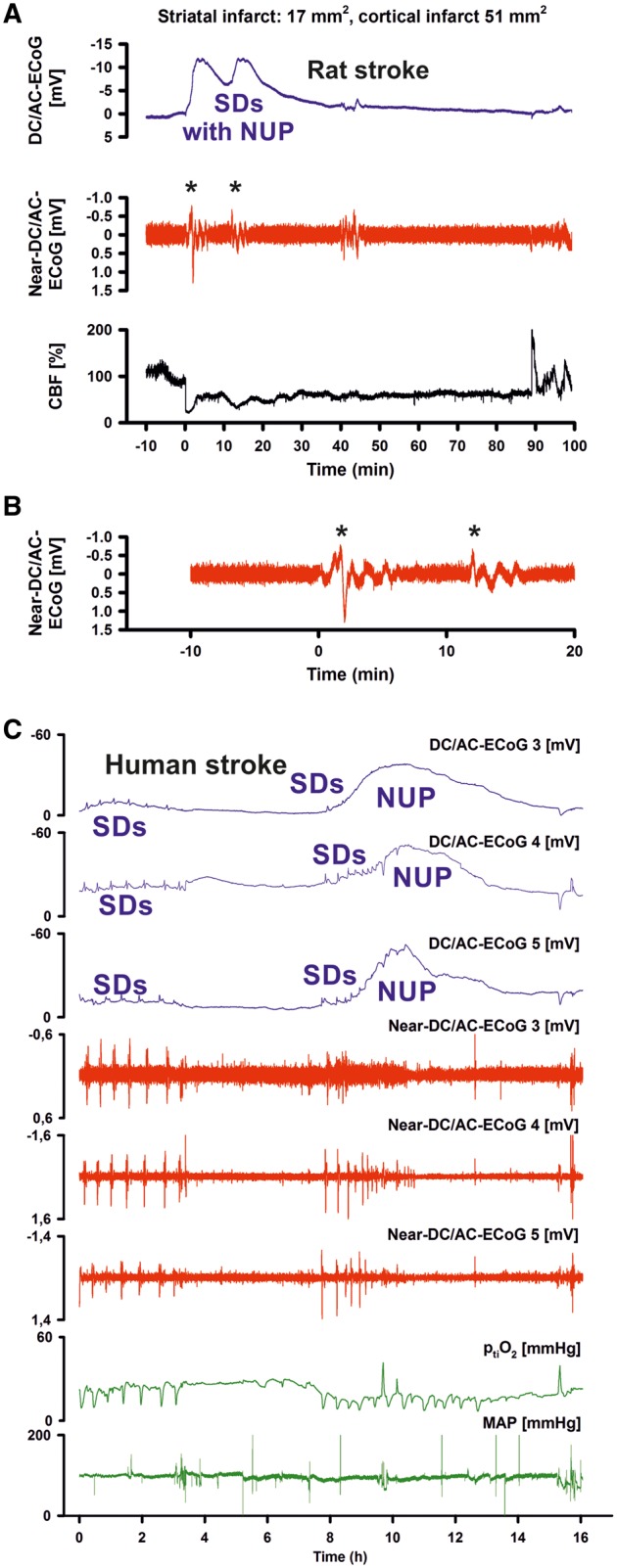
Predictive value of the amplitude ratio between first and second SD. (A) In Group 1A (90 min filament occlusion), the amplitudes of subsequent SDs were significantly smaller than that of the first SD (Kruskal-Wallis one-way ANOVA on Ranks with Dunn’s post hoc test, first SD versus second SD: P < 0.05, first SD versus third SD: P < 0.05). The median amplitude of the first SD was 8.5 (6.5, 10.3) mV (n = 28), the second one 2.2 (1.6, 4.4) mV (n = 22) and the third one 1.9 (1.6, 2.6) mV (n = 15) at the rostral recording electrode. At the caudal recording site, the results were similar [first SD: 8.4 (7.1, 10.7) (n = 28), second SD: 2.4 (1.7, 3.1) mV (n = 23), third SD: 2.0 (1.6, 2.6) mV (n = 14)]. When the raw data were filtered by a high-pass filter with a lower frequency limit of 0.01Hz (second trace), the mean amplitude of the first SD was 1.8 (1.4, 2.3) mV (n = 25), of the second one 0.7 (0.4, 1.3) mV (n = 22) and of the third one 0.8 (0.6, 1.1) mV (n = 14) at the rostral recording electrode. At the caudal recording site, the results were similar [first SD: 1.5 (1.1, 2.0) mV (n = 28), second SD: 0.7 (0.4, 1.0) mV (n = 22), third SD: 0.6 (0.4, 1.1) mV (n = 13)]. (B) The same near-DC/AC-ECoG data from A on an expanded time scale to visualize the difference between the amplitudes of the first and second SD (asterisks). Such high-pass filtered near-DC recordings are relevant because they are often used in neurocritical care (Dreier et al., 2006; Fabricius et al., 2006; Dohmen et al., 2008; Hartings et al., 2011). (C) A 68-year-old female (Table 1, Patient 4) developed a Fisher grade 3 haemorrhage due to rupture of a posterior communicating artery aneurysm. An external ventricular drain was established and the aneurysm was coiled on Day 1. The NUP occurred at electrodes 3–5 (traces 1–3, DC/AC-ECoG) on Day 4 (onset: 101 h 0 min) after the initial haemorrhage. In addition, it involved electrode 6 (not shown). MRI on Day 3 had already shown an infarct in the posterior territory of the left middle cerebral artery. An MRI on Day 7 revealed an additional new infarct in the left anterior middle cerebral artery territory that involved the cortex underlying electrodes 3–6 (Supplementary Fig. 1). Similar to the rodent recordings, the amplitudes of the SD-indicating near-DC/AC shifts showed a marked rundown between subsequent SDs during the NUP (traces 4–6). This amplitude reduction between subsequent SDs may be used as an important criterion to differentiate the NUP from large negative DC shifts of other nature. For a simple quantitative comparison of amplitude ratios between subsequent SDs, near-DC/AC recordings show the advantage over DC/AC recordings when platinum/iridium electrodes are used that the subjacent continuously rising NUP is filtered out of the signal. Trace 7 shows a persistent decrease of ptiO2 during the NUP. Note the recurrent decreases of ptiO2 in response to the SDs. Mean arterial pressure (MAP) and intracranial pressure were constant during the NUP. Notably, the daily performed transcranial Doppler sonography examination showed a normal mean velocity below 120 cm/s until Day 5.
We also filtered the DC/AC-ECoG with a high-pass filter (lower frequency limit: 0.01 Hz) resulting in a near-DC/AC-ECoG typical of routine recordings in neurocritical care (Dreier et al., 2006; Fabricius et al., 2006; Dohmen et al., 2008; Hartings et al., 2011) (Fig. 3A and B). At the caudal recording site, linear regression found that the near-DC/AC-amplitude ratio between first and second SD significantly correlated with the cortical infarct size (Table 1). To determine a cut-off value a ROC analysis was performed using Groups 1A and 2A. Both rostral and caudal AUCs were 0.847. As best cut-off value between the two groups, we chose a value of 83% (Table 1).
In the scalp EEG recordings of Groups 1C and 2B, DC shifts failed to reach statistical significance (Fig. 1B). Thus, the scalp DC-EEG in contrast to the brain surface DC-ECoG was not sufficient to identify the ischaemic event or isolated SDs.
Depression of activity as an electrophysiological biomarker of cerebral ischaemia and infarction
Non-spreading depression of activity occurred 15 (9, 24) s after the CBF drop, and, thus, significantly earlier than the first ischaemia-triggered SD (cf. above) (Mann-Whitney U-test, P < 0.001) (Fig. 2G, H, J and K). Linear regression found that the mean AC-ECoG power over the first 15 min, the first 30 min and the full 90-min period significantly correlated with the cortical infarct volume (Table 1). The best cut-off value separating Group 1A (90 min filament occlusion) from control Group 2A was a reduction of the mean AC-ECoG power to 54% from baseline (100%) over at least 15 min (Table 1).
In the scalp EEG recordings, non-spreading depression occurred 18 (14, 38) s after the drop in CBF (Fig. 2L). Linear regression found that also the mean scalp AC-EEG power correlated with the infarct volume (Table 1). The best cut-off value separating Group 1C (90 min filament occlusion) from control Group 2B was a reduction of the mean scalp AC-EEG power to 65% from baseline (100%) over at least 15 min (Table 1).
The first spreading depolarization in the ischaemic core only initiates cell death when it outlasts a threshold duration
All eight animals with 15 min of filament occlusion showed a full recovery of the negative DC shift (Fig. 1B). The negative DC shift lasted for 19.4 (19.1, 20.0) min rostrally and 19.4 (18.4, 20.2) min caudally in Group 1B. At 75 min after the onset of reperfusion, ECoG power had recovered to 70 (59, 80) % rostrally and 80 (61, 89) % caudally in this group. Only at the rostral recording site, this was significantly different from the value at the same time point in control Group 2A (rostral: 106 (97, 118) %; caudal: 98 (77, 127) (t-test, P = 0.01). No infarct was found in either cortex or striatum in the 15 min ischaemia Group 1B (Fig. 2C and D). In three of eight animals, a few isolated pyknotic neurons were seen in cortex, hippocampus and/or striatum ipsilateral to the occlusion.
Correlation between neurological and histological outcomes
The neurological score was significantly worse in the 90 min filament occlusion Groups 1A [8.0 (2.0, 9.3) of 12 points, n = 28] and 1C [7.5 (5.3, 10.0), n = 12] compared with either the 15 min occlusion Group 1B [0.5 (0.0, 1.0), n = 8] or the two control Groups 2A and 2B [0.0 (0.0, 0.0)] (Kruskal-Wallis one-way ANOVA on Ranks with Dunn post hoc tests, P < 0.05). Linear regression found that the neurological score in the pooled data of Groups 1A and 1C significantly correlated with the infarct volumes (Table 1). Various electrophysiological and CBF parameters correlated not only with the infarct volumes but also with the neurological outcome (Table 1).
Analyses of electrographic seizure activity in Groups 1A and 1C, the post-ischaemic hyperperfusion in Groups 1A–C and more detailed descriptions of the control Groups 2A and 2B are given in the Supplementary material.
Human clusters of spreading depolarizations containing a negative ultraslow potential in patients with aSAH
Patient characteristics are given in Table 2.
Table 2.
Summary of demographic, treatment and SD-related data of the patients
| No. | Age (years), sex | WFNS grade | Fisher grade | Location of aneurysm | Intervention | Recording time (h) | Total number of SDs | PTDDD (min) | Peak total number of SDs of a recording day | Peak total number of spreading depressions of a recording day | Peak total number of isoelectric SDs of a recording day | eGOS at 6 months |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 61, m | 5 | 3 | ACoP | Aneurysm clipping | 183.7 | 36 | 560.1 | 40.4 | 8.7 | 31.7 | 1 |
| 2 | 50, f | 3 | 3 | ACoA | Aneurysm clipping | 228.7 | 65 | 647.3 | 34.7 | 25.1 | 9.6 | 1 |
| 3 | 38, m | 5 | 3 | ACoA | Aneurysm clipping | 153.3 | 21 | 430.8 | 13.2 | 7.1 | 6.1 | 5 |
| 4 | 68, f | 4 | 3 | ACoP | Aneurysm coiling | 258.7 | 150 | 1103.8 | 47.1 | 13.2 | 36.1 | 1 |
| 5 | 33, f | 3 | 3 | MCA | Aneurysm clipping | 313.6 | 30 | 179.7 | 12.6 | 12.6 | 1.7 | 7 |
| 6 | 44, f | 4 | 3 | ACoP | Aneurysm clipping | 239.9 | 174 | 1407.0 | 53.5 | 30.2 | 53.5 | 1 |
| 7 | 64, f | 4 | 3 | BCA | Aneurysm clipping | 233.6 | 59 | 301.1 | 13.0 | 8.0 | 5.0 | 2 |
| 8 | 61, f | 5 | 3 | MCA | Aneurysm clipping | 222.4 | 42 | 302.7 | 12.0 | 9.0 | 6.0 | 1 |
| 9 | 33, f | 1 | 3 | ACoA | Aneurysm coiling | 73.6 | 22 | 603.7 | 15.6 | 6.7 | 8.9 | 1 |
| 10 | 51, f | 4 | 3 | MCA | Aneurysm clipping | 230.4 | 24 | 779.9 | 30.4 | 10.1 | 20.3 | 4 |
| 11 | 57, m | 5 | 3 | ACoA | Aneurysm coiling | 175.2 | 92 | 1440.0 | 51.0 | 3.2 | 47.8 | 1 |
ACoA = anterior communicating artery; ACoP = posterior communicating artery; BCA = basilar cerebral artery; eGOS = extended Glasgow Outcome Scale; f = female; m = male; PTDDD = peak total SD-induced depression duration of a recording day (cf. Materials and methods); WFNS = World Federation of Neurosurgical Societies.
In contrast to the findings in rats after MCAO, the cluster containing the NUP typically started with several SDs before the NUP (Fig. 4A and 4B). The median number of SDs before the NUP was 6 (3, 7, n = 11). The median interval between these SDs was 61.7 (45.5, 92.9) min.
Figure 4.

SD-initiated NUPs associated with persistent spreading depression of activity correlate with a new infarct on serial MRI scans. (A) A 61-year-old male (Table 1, Patient 1) developed a Fisher grade 3 haemorrhage with blood in the left Sylvian fissure, basal cisterns and left lateral ventricle due to rupture of an aneurysm of the left posterior communicating artery. Other findings included a mild diffuse brain oedema and beginning hydrocephalus. Accordingly, an external ventricular drain was placed in the right lateral ventricle. On Day 0, the aneurysm was clipped after craniotomy and an electrode strip was placed subdurally over the left frontal lobe. A CT control scan on the same day showed no differences from the initial CT except for a subdural hygroma on both sides of the tentorium and a large (20 × 35 mm) blood clot in the left Sylvian fissure. The NUP occurred here at electrodes 5 and 6 (DC/AC-ECoG traces) on Day 3 (onset: 83 h 34 min) after the initial haemorrhage. The first SD (red asterisk) of the cluster started at electrode 6 ∼1 h before the NUP. From electrode 6, it propagated to electrode 1. It induced spreading depression of the spontaneous activity (cf. AC-ECoG power). Subsequent SD-induced spreading depressions already evolved into persistent depression of the spontaneous activity at electrodes 4–6 before the NUP started. There was no recovery of the spontaneous activity after the NUP at electrode 5. Note that ptiO2 started to decrease (light green asterisk) before the cluster of SDs in this case. Cerebral perfusion pressure = mean arterial pressure − intracranial pressure. (B) shows the same DC/AC-ECoG data from A on an expanded time scale to visualize the transition to the NUP at a higher temporal resolution than in A. The temporal delay between the onsets at different electrodes of the NUP-initiating SD (blue arrows in A and B) evidences that this SD spread in the brain similar to the other SDs. Notably, the NUP-initiating SD originated in this case closer to electrode 4 than electrodes 5 and 6 and, thus, in the periphery rather than the centre of the tissue at risk. (C) Representative MRI slices of the left cerebral hemisphere. The ADC map 59 h after the initial haemorrhage reveals inter alia two small hypointense lesions in the left frontal lobe and diffusion restriction in the splenium of the corpus callosum. Furthermore, the FLAIR image reveals the large clot in the left Sylvian fissure, surrounded by perihaematomal oedema. On Day 7 (178 h after the initial haemorrhage), new large band-like cortical infarctions had evolved throughout the left hemisphere (hypointense in the ADC map and hyperintense in FLAIR images). The new infarcts involved the tissue underlying electrodes 5 and 6 in contrast to the tissue underlying the other electrodes of the subdural recording strip. The lowest row shows the location of the subdural electrode strip over the left frontal lobe and the lesions projected from the MRI scans onto the CT scan. The red-labelled area indicates the blood clot in the Sylvian fissure, the blue regions the early infarcts in the MRI on Day 2 and the green regions the delayed infarcts on Day 7. On Day 7, the patient also underwent digital subtraction angiography. Vasospasm was found in distal rather than proximal arteries. By contrast, transcranial Doppler sonography did not indicate vasospasm (Vora et al., 1999). Hyperdynamic therapy (hypertension, hypervolaemia) was initiated. On Day 8, an asystole necessitated cardio-pulmonary resuscitation. On Day 15, another MRI scan showed further infarct progression. The patient died 149 days after the initial haemorrhage. All images were registered (6 degrees of freedom) to the first postoperative T1 spin echo scan by using the FMRIB’s Linear Image Registration Tool (FLIRT) of the FSL (Jenkinson and Smith, 2001; Jenkinson et al., 2002).
The longest duration of the negative DC shift among the different electrodes significantly increased between first and last SD before the NUP [3.8 (3.6, 7.2) versus 7.4 (6.0, 16.3) min, paired t-test, P = 0.037, n = 10]. The largest amplitude of the negative DC shift among the different electrodes rather increased between first and last SD before the NUP, but this did not reach statistical significance [5.5 (4.9, 7.9) versus 7.6 (5.6, 9.6) mV, n = 10]. The largest positivity following the negative DC shift did not change significantly before the NUP [2.8 (1.7, 4.0) versus 2.1 (0.4, 2.4) mV, n = 10].
Description of the negative ultraslow potential in human recordings with platinum/iridium electrodes
The NUP-initiating SD developed in two (2, 4) electrodes 5.0 (2.5, 13.9) h after the onset of the cluster (n = 11). The shape of the NUP was rather homogeneous in the human recordings but peculiar compared to the rodent recordings because it reached its maximum negativity 86 (80, 108) min after the onset. This produced a tent-like appearance (Figs 3C, 4A and 6). The median number of secondary SDs riding on the NUP was 4 (3, 6). Similar to the rodent findings after MCAO, the amplitude of the near-DC/AC shifts decreased from the first to the second SD riding on the NUP [1.0 (0.7, 1.7) versus 0.6 (0.2, 1.0) mV, Wilcoxon Signed Rank Test, P = 0.016, n = 8] (Fig. 3C). Notably, the median amplitude ratio between these first two SDs riding on the NUP was 53 (37, 82) % (n = 8) in the human recordings, similar to the values of 48 (26, 72) % (caudal recording site, n = 21) and 52 (31, 74) % (rostral recording site, n = 20) in the rodent recordings. Both in animals and patients, the rundown in amplitudes of SDs riding on the NUP thus provides one of the criteria to differentiate the NUP from large negative DC deflections of other nature (Fig. 3).
Figure 6.

SD inducing NUP and spreading ischaemia. (Ai) The NUP of Patient 6, a 44-year-old previously healthy female, was recorded on Day 9 (onset: 218 h 4 min) after the initial haemorrhage. The second SD at electrode 4 not only resulted in a NUP but also induced the characteristic drop in CBF typical of spreading ischaemia in rat experiments when the NO availability is low and the extracellular baseline potassium concentration ([K+]o) is elevated (Dreier et al., 1998; Dreier, 2011) (cf. grey inset). This rodent model mimicking the conditions present following aSAH is also typically characterized by a marked hyperaemia succeeding the spreading ischaemia in contrast to the rat model of proximal arterial occlusion in which spreading ischaemia typically occurs with no or only a minor succeeding hyperaemia (cf. Figure 3 in Feuerstein et al., 2014). A marked, long-lasting hyperaemia succeeding the spreading ischaemia of 50-min duration is seen at optode 3–4 in this patient corresponding with the model mimicking aSAH. The duration of the NUP correlates well with the duration of the spreading ischaemia, as in previous animal experiments (Dreier et al., 2002; Sukhotinsky et al., 2008), because the decreases in perfusion and energy supply limit Na,K-ATPase activity and prolong depolarization (Dreier et al., 1998; Dreier, 2011; Major et al., 2017). Animal experiments suggested that, if sufficiently prolonged, spreading ischaemia causes cell death (Dreier et al., 2000). Accordingly, the patient developed a new infarct at electrodes 4–6 between Days 8 and 12 (Supplementary Fig. 2). After the spreading ischaemia shown in the figure, subsequent ones also involved (opto-) electrodes 4 to 6. The patient died due to the progressive brain infarctions on Day 13. (Aii) CBF and DC/AC-ECoG data from Ai on an expanded time scale to visualize the propagation of the SD and the SD-induced spreading ischaemia (blue arrow). Note that the durations of the negative DC shifts correlate well with the durations of the SD-induced hypoperfusions at the two different recording sites. (B) The NUP of Patient 2, a 50-year-old previously healthy female, was recorded on Day 1 (onset: 39 h 37 min) after the initial haemorrhage. It developed out of a long cluster of recurrent SDs. Note that the SD-induced drops in ptiO2 became progressively more pronounced during the cluster until the SD-initiating NUP finally induced a long-lasting hypoxia/ischaemia. The MRI on Day 2 showed early band-like infarcts of the cortex and underlying white matter in the left middle cerebral artery territory where the electrode strip was located (involving electrodes 4–6) and band-like infarcts in the medial frontal area bilaterally (Supplementary Fig. 3). The second MRI scan on Day 7 revealed new infarcts in the left middle cerebral artery and posterior cerebral artery territories, as well as growth of the previously established infarcts in the right middle cerebral artery and both anterior cerebral artery territories. On Day 9, the patient presented with dilated, fixed pupils. A CT scan showed progressive brain oedema. The patient died on Day 11. (Bii) Panel shows the same ptiO2 and DC/AC-ECoG data from Bi on an expanded time scale to visualize the correlation between the durations of the negative DC shift and the SD-induced hypoxia/ischaemia, similar to the illustration in Aii.
The longest duration of the NUP among different electrodes was 3.7 (3.3, 5.3) h (n = 11). Thus, the NUP-initiating SD gave rise to a negative DC shift that lasted 34.9 (12.7, 56.9) times longer than the negative DC shift of the preceding SD. The largest amplitude of the NUP among different electrodes was 45.0 (39.0, 69.4) mV (n = 11) and was thus 6.6 (4.2, 10.8) times larger than the largest negative DC shift of the preceding SD (Fig. 4A).
In different electrodes, the largest delay between the onsets of the NUP-initiating SD [25.0 (14.2, 43.3) min] was not significantly different from the largest delay between the onsets of the first clustered SD preceding the NUP [24.0 (16.0, 30.3) min]. This suggests that the NUP-initiating SD propagated in a similar fashion. Largely overlapping SD velocities between adequately nourished and energetically compromised tissue correspond well with previous in vitro and in vivo studies in rodents (Aitken et al., 1998; Jarvis et al., 2001; Farkas et al., 2010; Bere et al., 2014).
Relation of the negative ultraslow potential to the depression of spontaneous activity
In 10 cases the cluster of SDs containing the NUP started in electrically-active tissue. All first SDs in electrically-active tissue caused spreading depression of spontaneous activity (Figs 4A and 6A). This could be variably followed by recovery of the activity. Also during the cluster, all depressions of spontaneous activity were by nature SD-induced spreading depressions. No depression of spontaneous activity was observed in a given electrode when no SD appeared in this electrode. In contrast to the findings in rats after MCAO, non-spreading depression of spontaneous activity was not observed in any of the cases.
Even before the NUP-initiating SD, SD-induced depression of spontaneous activity often persisted between subsequent SDs (Fig. 4A). In electrodes displaying the NUP, such persistent depression of activity started with a median of 16 (6, 49) min before the NUP. Yet whenever tissue remained electrically active until the NUP-initiating SD, this SD not only involved the NUP but also persistent depression of spontaneous activity. SDs could also induce persistent depression of spontaneous activity in neighbouring electrodes that did not display the NUP (13/25 electrodes). The belt of tissue showing persistent SD-induced depression was in other words larger than the region of tissue showing the NUP, which corresponds well with previous animal findings (Oliveira-Ferreira et al., 2010). Electrodes displaying the NUP showed significantly worse recovery of neuronal function than electrodes not displaying the NUP (Fig. 5B). Accordingly, linear regression found a highly significant correlation between the day of occurrence of the NUP and the day of occurrence of the peak total SD-induced depression duration of a recording day (Dreier et al., 2017) (adjusted R2 = 0.871, P < 0.001, n = 11) (Fig. 5A).
Figure 5.

The NUP is associated with persistent depression of spontaneous activity, worse recovery of neuronal function, decline in ptiO2 and new neuroimaging-evidenced brain infarcts at the recording site. (A) The day of occurrence of the cluster of SDs containing the NUP correlated with the day of occurrence of the peak total SD-induced depression duration of a recording day (PTDDD). (B) Electrodes displaying the NUP showed a significantly lower AC-ECoG power of spontaneous activity during and after the NUP than electrodes not displaying the NUP indicating that the NUP was associated with worse recovery of neuronal function (Wilcoxon Signed Rank Tests). (C) Mean arterial pressure (MAP) and cerebral perfusion pressure (CPP) showed a statistical trend towards lower values during the cluster of SDs (one-way repeated measures ANOVA). Intracranial pressure did not change significantly. ptiO2 was significantly lower during the NUP (one-way repeated measures ANOVA with Fisher LSD post hoc tests, *P < 0.05, **P < 0.01, ***P < 0.001). (D) Electrodes displaying the NUP were significantly more likely to overlie a developing ischaemic lesion (90.0%, 27/3) than those not displaying a NUP (0.0%, 0/20) (GEEs applied to a logistic regression model).
Neuroimaging
In all 11 patients, serial neuroimaging studies evidenced early and/or delayed ischaemic lesions. In 10 patients, a CT scan was available that revealed the exact position of the six subdural electrodes. DC/AC-ECoG recordings of sufficient quality were available in 50 of 60 electrodes in the 10 patients. Using GEEs applied to a logistic regression model, as explained in the ‘Materials and methods’ section, we found that the presence of infarcts was significantly associated with NUPs (P = 0.004 corrected for the cluster effect of patients). In 20 regions without NUP, no infarction was observed in contrast to 27 of 30 regions with NUP that showed infarction. Thus the positive predictive value of NUPs with respect to infarction was 90%, and the negative predictive value was even 100% (Fig. 5D). Based on serial neuroimaging studies, it was possible to determine that the lesions under the electrodes occurred within a time period of 72 (56, 134) h. The NUP occurred in this time period in 9 of 10 patients (90.0%). In one case, a CT scan already revealed a small hypodensity below electrodes 3 and 4 before these two electrodes displayed the NUP and persistent depression of spontaneous activity. However, the subsequent MRI scan suggested a progression of this lesion in addition to new lesions at other locations in the same hemisphere.
Other neuromonitoring and spreading ischaemia
Mean arterial pressure and intracranial pressure were recorded in all patients, ptiO2 in eight patients and CBF in four (Fig. 5C). Notably, ptiO2 was significantly lower during the NUP (Fig. 5C). In three cases, a gradual decline of ptiO2 started before the cluster containing the NUP (Fig. 4A). The pattern of sharp SD-induced ptiO2 and CBF decreases during the cluster was typical of spreading ischaemia as previously described (Fig. 6) (Dreier et al., 2009). During the NUP, 2 (1, 4) spreading ischaemias were identified in seven of nine cases. The longest spreading ischaemia (hypoperfusion/hypoxic phase) during NUP was significantly longer than the longest spreading ischaemia during the cluster before the NUP [40.0 (28.0, 76.5) min versus 9.3 (6.0, 12.0) min, paired t-test, P = 0.026, n = 7]. CBF fell from 57 (53, 65) % to 26 (16, 42) % (n = 4) and ptiO2 from 12.5 (9.2, 15.2) to 3.3 (2.4, 7.4) mmHg (n = 5) during NUP-associated spreading ischaemia. During spreading ischaemia before the NUP, CBF fell from 95 (89, 97) % to 53 (44, 70) % (n = 4) and ptiO2 from 20.4 (16.7, 24.0) to 12.5 (10.5, 14.0) mmHg (n = 4). During NUP-associated spreading ischaemia, CBF and ptiO2 were significantly lower (paired t-test, P = 0.040 and t-test, P = 0.038, respectively). In six patients, the Licox probe recorded the brain temperature and found a significantly higher level during the NUP than before the cluster of SDs [38.2 (37.7, 38.4) °C versus 37.2 (36.1, 37.6) °C, paired t-test, P = 0.036, n = 5]. All NUPs apart from one case occurred between 8:30 pm and 8:30 am (91%), with a clear preponderance in the early morning hours.
Electrographic seizure activity during the NUP was seen in 1/11 patients (onset: 76 min after NUP onset, amplitude: up to 220 µV, 3/s rhythmic δ-activity in four neighbouring electrodes of which two displayed the NUP, duration: 46 min with one propagating interruption of 8 min due to an SD at the two electrodes not displaying the NUP; duration of the NUP: 3 h 48 min; Patient 11, Table 2).
The patient outcome using the extended Glasgow Outcome Scale (eGOS) at 6 months was poor in patients displaying a NUP (Table 2) and significantly worse than that of two previously published samples (Dreier et al., 2012; Winkler et al., 2017) comprising a total of 58 patients that originated from the same aSAH population and contained seven patients of the present study. The eGOS of patients with NUP was 1 (1, 3) whereas the eGOS of the historical samples less the patients with NUP was 4 (2, 6) (Mann-Whitney Rank Sum Test, P = 0.020). In contrast, age [51 (41, 61) versus 50 (47, 52) years], sex [eight females of 11 patients (72.7%) versus 35 of 51 (67.2%)] and World Federation of Neurosurgical Societies (WFNS) grade on admission [4 (3.5, 5) versus 4 (2, 5)] were not different. One of the historical samples comprised the first 25 patients consecutively recruited into the COSBID study in Berlin (Dreier et al., 2012). The other one contained all patients prospectively enrolled in Berlin between September 2009 and August 2013 in which the neurosurgeon had decided to implant an intraparenchymal oxygen sensor.
Discussion
The present paper reveals that severe focal cerebral ischaemia of the brain cortex is associated with an SD-initiated NUP not only in animals but also in humans. Notably, serial neuroimaging scans evidenced in patients that electrodes displaying the NUP were significantly more likely to overlie a newly developing ischaemic lesion than neighbouring electrodes not displaying the NUP.
In human recordings with platinum/iridium electrodes at the brain surface, the SD-initiated NUP has a peculiar tent-like shape that is easily recognized. It was about seven times larger and 35 times longer than the negative DC shift of SDs in less metabolically compromised tissue and 75 times larger than the DC shift of typical electrocorticographic seizures (Revankar et al., 2017). During SD in hypoxic rats, we have observed this peculiar shape in a similar fashion using brain surface platinum/iridium plate electrodes in contrast to intracortical Ag/AgCl-based glass microelectrodes (data not shown). It may at least partially result from platinum’s strong catalytic capacity for many chemical reactions in biological media: the SD process is associated with the largest possible changes of not only the intraneuronal but also the extracellular microenvironment in living neural tissue (Dreier and Reiffurth, 2015). It is hence conceivable that factors produced or released during SD and affect platinum recordings increasingly reach the electrodes at the brain surface and distort the DC signal when the SD process is abnormally prolonged. For example, molecular oxygen and pH change during SD and affect platinum recordings. A complementary hypothesis posits that the tent-like appearance of the NUP is in part biological rather than solely of artificial origin because the DC potential generated at the blood–brain barrier is strongly influenced by pH and ptiO2 changes (Lehmenkühler et al., 1999; Voipio et al., 2003; Kang et al., 2013). In this hypothesis, the flattened shape of the NUP in the rodent recordings following MCAO in contrast to the human recordings could, possibly, be related to species differences in DC potential generation at the blood–brain barrier between rats, rabbits, goats and dogs on the one hand and cats and non-human primates on the other (Besson et al., 1970; Woody et al., 1970). This deserves further study.
The rundown in amplitudes between subsequent SDs riding on the NUP can be used as an important criterion to differentiate the NUP from large negative DC shifts of other nature such as DC potential deflections generated at the blood–brain barrier by systemic changes in partial pressures of CO2 and O2 (Woody et al., 1970; Lehmenkühler et al., 1999; Voipio et al., 2003; Kang et al., 2013). For a simple quantitative analysis of amplitude ratios between subsequent SDs during the NUP in humans, near-DC-ECoG showed the advantage over DC-ECoG in that the subjacent continuously rising NUP was filtered out of the signal. The accompanying persistent depression of AC-ECoG power was yet another crucial criterion to differentiate the NUP from large negative DC shifts of other causes.
The animal experiments revealed that the NUP, quantified as DC integral over time, the amplitude ratio between subsequent SDs during the NUP, the persistent depression of AC-ECoG power and the number of SDs correlated in a similar fashion to the drop in CBF with the cortical infarct volume and the neurological outcome after MCAO. This further supports the notion that the electrophysiological measures not only qualitatively but also quantitatively indicate disruptions in brain energy metabolism and evolution of brain injury (Mies et al., 1993; Hossmann, 1996; Dijkhuizen et al., 1999; Somjen, 2001; Strong and Dardis, 2005; Dreier, 2011; Hartings et al., 2017b). A long-term goal is the advancement of non-invasive technologies (Dreier et al., 2017). If combined with other technologies, such as near-infrared spectroscopy or diffuse correlation spectroscopy, that measure CBF or its surrogates (Kohl et al., 1998; Selb et al., 2014), continuous scalp EEG may hold promise for non-invasive monitoring (Claassen et al., 2005; Drenckhahn et al., 2012; Hartings et al., 2017a). In this respect, our finding was encouraging that the persistent depression in scalp AC-EEG power rather than brain surface AC-ECoG power correlated more strongly with cortical infarct volume and neurological outcome after MCAO in rats. Contributions of volume-conducted signals from more widespread cortical generators to the scalp AC-EEG power may explain this (Drenckhahn et al., 2012). On the other hand, it was disappointing that DC shifts failed to reach statistical significance in the scalp EEG recordings. This suggests that DC-EEG shifts of SD were observable in human scalp recordings only because of prior craniotomy in these patients (Drenckhahn et al., 2012; Hartings et al., 2014).
The long-term goal of an automated neuromonitoring system
SDs are characterized by abrupt, near-complete breakdown of the neuronal transmembrane ion gradients with marked loss of Gibbs free energy (Kraig and Nicholson, 1978; Windmuller et al., 2005), including a toxic intraneuronal calcium and sodium surge (Dietz et al., 2008), neuron swelling with dendritic beading and signal decrease on ADC maps (cytotoxic oedema) (Takano et al., 1996, 2007; Risher et al., 2010; Dreier and Reiffurth, 2017), depolarization of mitochondria (Bahar et al., 2000; Zhou et al., 2010), marked glutamate release (excitotoxicity) (Fabricius et al., 1993; Zhou et al., 2013; Hinzman et al., 2015) and depolarization of astrocytes secondary to the extracellular potassium surge (Muller and Somjen, 2000; Chuquet et al., 2007). Notably, SD propagation rate is similar between severely ischaemic and normal tissue (Aitken et al., 1998; Jarvis et al., 2001; Farkas et al., 2008; Bogdanov et al., 2016), but SD duration is significantly longer in ischaemic tissue (Hartings et al., 2017b).
SD does not mark the onset of cell death, but rather starts the clock on the countdown to cell death. Only if SD outlasts a threshold duration, the so-called ‘commitment point’, neurons will die (Somjen, 2004). This implies that neurons can survive SD even in the ischaemic core, if the tissue is reperfused and repolarizes before the commitment point (Memezawa et al., 1992; Ayad et al., 1994; Nozari et al., 2010). The phenomenon of NUP is to a certain degree similar to an abnormally prolonged SD, but is hypothesized to include a further persistent negative DC shift component that results from cell death, after the commitment point is reached in an increasing fraction of neurons, rather than merely a reversible depolarized state (Dreier et al., 2013). Unfortunately, there is currently no marker in either animals or humans that allows the exact identification of the commitment point during the SD-initiated negative DC shift, but its occurrence may be estimated. Thus, we found in the MCAO model in rats that an SD-initiated negative DC shift of 15-min duration was not sufficient to lead to a brain infarct. Irreversible injury rather required a negative DC shift between 15- and 90-min duration. This time window corresponds well with previous observations in the MCAO model (Heiss and Rosner, 1983; Memezawa et al., 1992; Shen et al., 2005; Pignataro et al., 2007) and with brief ischaemia in mice by injection of air micro-emboli into the carotid circulation (Nozari et al., 2010). Further, it corresponds well with our rodent model mimicking the conditions present following aSAH in which nitric oxide (NO) availability is low, baseline [K+]o is elevated, baseline CBF is normal or mildly to moderately reduced and SD triggers spreading ischaemia (Dreier et al., 1998, 2000). Only when the circulation completely ceases such as after cardiac arrest, the commitment point seems to be shifted closer to the onset of SD and may be reached within less than 10 min (Ayad et al., 1994; Somjen, 2004).
Neurocritical care doctors would like to start an intervention before SD has led to irreversible damage while avoiding any potential side effects. To enable this, the MCAO model would suggest that an automated system should emit an alarm when variables such as persistent depression of AC-ECoG power or the NUP remain within the pathological range for a time period of 15 min. Infarct evolution in aSAH patients, however, showed several differences to the MCAO model. For instance, there was no sharp initial decrease in CBF followed first by non-spreading depression of activity and then by SD, but the first alteration in aSAH patients was often a cluster of recurrent SDs with progressively increasing durations of negative DC shifts, spreading depressions and spreading ischaemias whose phenomenology were similar to the above mentioned rodent model mimicking the conditions present following aSAH (Dreier et al., 1998; Dreier, 2011). The pattern of clustered SDs and an SD-initiated NUP following the cluster’s onset with some delay was previously observed in a significant proportion in rats when the vasoconstrictor endothelin-1 was brain topically applied at a concentration of 1 µM (Oliveira-Ferreira et al., 2010). This model investigates the effects of mild but prolonged focal cerebral ischaemia, in contrast to the abrupt, severe ischaemia from arterial occlusion. Though, in a small fraction of recording sites (11%), the same pattern was also noted previously after MCAO when intracortical glass microelectrodes were used (Higuchi et al., 2002), its predominance in our human recordings supports the notion that infarcts in aSAH patients generally evolve in a slower fashion than infarcts after embolic or thrombotic arterial occlusion.
It was of particular interest in the patients that the evolution of injury involved complex cross-talks between different zones via SDs. For example, we observed SDs invading adjacent zones with profoundly disturbed neurovascular coupling from less disrupted ones, thereby converting the adjacent zones into ischaemic regions. Overall, there seemed to be a spiralling trend towards increasing ischaemia and risk of injury. We suggest that an automated system should take these peculiarities into account through a multi-level alarm function progressively activated by the here described hierarchy of events such as a decreasing interval between SDs, progressively longer depression periods and progressively longer DC shifts that finally emerge into the NUP. In addition, SD-triggered increasing durations of ptiO2 and CBF decreases could be valuable to increase the diagnostic accuracy. Also, variables such as brain temperature could be considered, which was significantly higher during the NUP than before the cluster of SDs, corresponding with previous clinical observations of low-grade fever during delayed cerebral ischaemia (Adams and Love, 1992) and positive correlations between body/brain temperature and SD occurrence in patients with traumatic brain injury and spontaneous intracranial haemorrhage (Hartings et al., 2009; Schiefecker et al., 2018).
The ultimate goal would be to develop interventions capable of preventing the NUP and the infarct. With respect to such interventions, it would be interesting to know inter alia whether or not the pathogenesis of early cerebral ischaemia and delayed cerebral ischaemia is in fact fundamentally different. Our data cannot exclude this but they did not provide evidence for this either. In terms of electrophysiology, ptiO2 and CBF, the patterns of early and delayed cerebral ischaemia were similar. To a certain degree, early and delayed cerebral ischaemia could be part of the same incrementally developing disease process, which might be the main determinant of poor outcome after aSAH once the aneurysm is treated (Broderick et al., 1994; Vergouwen et al., 2011). This notion is supported by the poor outcome of the here-described patient population and deserves further study.
Potential treatment approaches targeting SDs have been discussed elsewhere (Dreier, 2011; Pietrobon and Moskowitz, 2014; Dreier and Reiffurth, 2015; Lauritzen and Strong, 2017). In this debate, it is of importance that there are large variations in mechanistic aspects and pharmacological sensitivity of SDs along the SD continuum, as dictated by local tissue conditions, and these have implications for the efficacy of therapeutic targeting. For example, it is well known that N-methyl-d-aspartate receptor (NMDAR) antagonists effectively abolish SDs in well supplied tissue (Marrannes et al., 1988), but are ineffective to block SD or the SD-initiated NUP in severely ischaemic or hypoxic tissue (Hernandez-Caceres et al., 1987; Lauritzen and Hansen, 1992; Muller and Somjen, 1998; Madry et al., 2010). However, in sedation requiring patients such as in the present study, it is currently unknown whether moderately hypoperfused tissue at risk could still benefit from NMDAR inhibiting sedative drugs such as ketamine (Sakowitz et al., 2009; Hertle et al., 2012). This is a question that takes on particular significance at the moment when the neurocritical care specialist records a newly developing SD cluster in a sedated patient. This and other related questions deserve further extensive clinical and experimental investigations.
Funding
This work was supported by grants of the Bundesministerium für Bildung und Forschung (BMBF) Center for Stroke Research Berlin 01 EO 0801 to J.P.D., Deutsche Forschungsgemeinschaft (DFG) DFG DR 323/5-1 to J.P.D. and J.W., DFG WO 1704/1-1 to J.W.
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
Abbreviations
- aSAH
aneurysmal subarachnoid haemorrhage
- CBF
cerebral blood flow
- ECoG
electrocorticography
- MCAO
middle cerebral artery occlusion
- NUP
negative ultraslow potential
- ptiO2
tissue partial pressure of oxygen
- SD
spreading depolarization
References
- Adams HP, Love BB. Medical management of aneurismal subarachnoid hemorrhage. In: Barnett HJM, Mohr JP, Stein BM, Yatsu FM, editors. Stroke pathophysiology, diagnosis, and management. New York: Churchill Livingstone; 1992. p. 1029–54. [Google Scholar]
- Aitken PG, Tombaugh GC, Turner DA, Somjen GG. Similar propagation of SD and hypoxic SD-like depolarization in rat hippocampus recorded optically and electrically. J Neurophysiol 1998; 80: 1514–21. [DOI] [PubMed] [Google Scholar]
- Ayad M, Verity MA, Rubinstein EH. Lidocaine delays cortical ischemic depolarization: relationship to electrophysiologic recovery and neuropathology. J Neurosurg Anesthesiol 1994; 6: 98–110. [DOI] [PubMed] [Google Scholar]
- Bahar S, Fayuk D, Somjen GG, Aitken PG, Turner DA. Mitochondrial and intrinsic optical signals imaged during hypoxia and spreading depression in rat hippocampal slices. J Neurophysiol 2000; 84: 311–24. [DOI] [PubMed] [Google Scholar]
- Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke 1996; 27: 1616–22; discussion 1623. [DOI] [PubMed] [Google Scholar]
- Bere Z, Obrenovitch TP, Bari F, Farkas E. Ischemia-induced depolarizations and associated hemodynamic responses in incomplete global forebrain ischemia in rats. Neuroscience 2014; 260: 217–26. [DOI] [PubMed] [Google Scholar]
- Besson JM, Woody CD, Aleonard P, Thompson HK, Albe-Fessard D, Marshall WH. Correlations of brain d-c shifts with changes in cerebral blood flow. Am J Physiol 1970; 218: 284–91. [DOI] [PubMed] [Google Scholar]
- Bogdanov VB, Middleton NA, Theriot JJ, Parker PD, Abdullah OM, Ju YS, et al. Susceptibility of primary sensory cortex to spreading depolarizations. J Neurosci 2016; 36: 4733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosche B, Graf R, Ernestus RI, Dohmen C, Reithmeier T, Brinker G, et al. Recurrent spreading depolarizations after SAH decrease oxygen availability in human cerebral cortex. Ann Neurol 2010; 67: 607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994; 25: 1342–7. [DOI] [PubMed] [Google Scholar]
- Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci 2007; 27: 4036–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citerio G, Oddo M, Taccone FS. Recommendations for the use of multimodal monitoring in the neurointensive care unit. Curr Opin Crit Care 2015; 21: 113–19. [DOI] [PubMed] [Google Scholar]
- Claassen J, Mayer SA, Hirsch LJ. Continuous EEG monitoring in patients with subarachnoid hemorrhage. J Clin Neurophysiol 2005; 22: 92–8. [DOI] [PubMed] [Google Scholar]
- Dietz RM, Weiss JH, Shuttleworth CW. Zn2+ influx is critical for some forms of spreading depression in brain slices. J Neurosci 2008; 28: 8014–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen RM, Beekwilder JP, van der Worp HB, Berkelbach van der Sprenkel JW, Tulleken KA, Nicolay K. Correlation between tissue depolarizations and damage in focal ischemic rat brain. Brain Res 1999; 840: 194–205. [DOI] [PubMed] [Google Scholar]
- Dohmen C, Sakowitz OW, Fabricius M, Bosche B, Reithmeier T, Ernestus RI, et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol 2008; 63: 720–8. [DOI] [PubMed] [Google Scholar]
- Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 2011; 17: 439–47. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Ebert N, Priller J, Megow D, Lindauer U, Klee R, et al. Products of hemolysis in the subarachnoid space inducing spreading ischemia in the cortex and focal necrosis in rats: a model for delayed ischemic neurological deficits after subarachnoid hemorrhage? J Neurosurg 2000; 93: 658–66. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Fabricius M, Ayata C, Sakowitz OW, William Shuttleworth C, Dohmen C, et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J Cereb Blood Flow Metab 2017; 37: 1595–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Isele T, Reiffurth C, Offenhauser N, Kirov SA, Dahlem MA, et al. Is spreading depolarization characterized by an abrupt, massive release of gibbs free energy from the human brain cortex? Neuroscientist 2013; 19: 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Korner K, Ebert N, Gorner A, Rubin I, Back T, et al. Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab 1998; 18: 978–90. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009; 132: 1866–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Major S, Pannek HW, Woitzik J, Scheel M, Wiesenthal D, et al. Spreading convulsions, spreading depolarization and epileptogenesis in human cerebral cortex. Brain 2012; 135: 259–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Reiffurth C. The stroke-migraine depolarization continuum. Neuron 2015; 86: 902–22. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Reiffurth C. Exploitation of the spreading depolarization-induced cytotoxic edema for high-resolution, 3D mapping of its heterogeneous propagation paths. Proc Natl Acad Sci USA 2017; 114: 2112–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Windmuller O, Petzold G, Lindauer U, Einhaupl KM, Dirnagl U. Ischemia caused by inverse coupling between neuronal activation and cerebral blood flow in rats. In: Tomita M, Kanno I, Hamel E, editors. Brain activation and CBF control. Amsterdam: Elsevier; 2002. p. 487–92. [Google Scholar]
- Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 2006; 129: 3224–37. [DOI] [PubMed] [Google Scholar]
- Drenckhahn C, Windler C, Major S, Kang EJ, Scheel M, Vajkoczy P, et al. Complications in aneurysmal subarachnoid hemorrhage patients with and without subdural electrode strip for electrocorticography. J Clin Neurophysiol 2016; 33: 250–9. [DOI] [PubMed] [Google Scholar]
- Drenckhahn C, Winkler MKL, Major S, Scheel M, Kang EJ, Pinczolits A, et al. Correlates of spreading depolarizations in human scalp electroencephalography. Brain 2012; 135: 853–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain 2006; 129: 778–90. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Jensen LH, Lauritzen M. Microdialysis of interstitial amino acids during spreading depression and anoxic depolarization in rat neocortex. Brain Res 1993; 612: 61–9. [DOI] [PubMed] [Google Scholar]
- Farkas E, Bari F, Obrenovitch TP. Multi-modal imaging of anoxic depolarization and hemodynamic changes induced by cardiac arrest in the rat cerebral cortex. Neuroimage 2010; 51: 734–42. [DOI] [PubMed] [Google Scholar]
- Farkas E, Pratt R, Sengpiel F, Obrenovitch TP. Direct, live imaging of cortical spreading depression and anoxic depolarisation using a fluorescent, voltage-sensitive dye. J Cereb Blood Flow Metab 2008; 28: 251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein D, Takagaki M, Gramer M, Manning A, Endepols H, Vollmar S, et al. Detecting tissue deterioration after brain injury: regional blood flow level versus capacity to raise blood flow. J Cereb Blood Flow Metab 2014; 34: 1117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6: 1–9. [DOI] [PubMed] [Google Scholar]
- Hartings JA, Bullock MR, Okonkwo DO, Murray LS, Murray GD, Fabricius M, et al. Spreading depolarisations and outcome after traumatic brain injury: a prospective observational study. Lancet Neurol 2011; 10: 1058–64. [DOI] [PubMed] [Google Scholar]
- Hartings JA, Li C, Hinzman JM, Shuttleworth CW, Ernst GL, Dreier JP, et al. Direct current electrocorticography for clinical neuromonitoring of spreading depolarizations. J Cereb Blood Flow Metab 2017a; 37: 1857–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA, Shuttleworth CW, Kirov SA, Ayata C, Hinzman JM, Foreman B, et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leao's legacy. J Cereb Blood Flow Metab 2017b; 37: 1571–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA, Strong AJ, Fabricius M, Manning A, Bhatia R, Dreier JP, et al. Spreading depolarizations and late secondary insults after traumatic brain injury. J Neurotrauma 2009; 26: 1857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA, Wilson JA, Hinzman JM, Pollandt S, Dreier JP, DiNapoli V, et al. Spreading depression in continuous electroencephalography of brain trauma. Ann Neurol 2014; 76: 681–94. [DOI] [PubMed] [Google Scholar]
- Hartings JA, York J, Carroll C, Hinzman JM, Krueger B, Winkler MKL, et al. Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction. Brain 2017c; 140: 2673–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss WD, Rosner G. Functional recovery of cortical neurons as related to degree and duration of ischemia. Ann Neurol 1983; 14: 294–301. [DOI] [PubMed] [Google Scholar]
- Hernandez-Caceres J, Macias-Gonzalez R, Brozek G, Bures J. Systemic ketamine blocks cortical spreading depression but does not delay the onset of terminal anoxic depolarization in rats. Brain Res 1987; 437: 360–4. [DOI] [PubMed] [Google Scholar]
- Hertle DN, Dreier JP, Woitzik J, Hartings JA, Bullock R, Okonkwo DO, et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain 2012; 135: 2390–8. [DOI] [PubMed] [Google Scholar]
- Higuchi T, Takeda Y, Hashimoto M, Nagano O, Hirakawa M. Dynamic changes in cortical NADH fluorescence and direct current potential in rat focal ischemia: relationship between propagation of recurrent depolarization and growth of the ischemic core. J Cereb Blood Flow Metab 2002; 22: 71–9. [DOI] [PubMed] [Google Scholar]
- Hinzman JM, Andaluz N, Shutter LA, Okonkwo DO, Pahl C, Strong AJ, et al. Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain 2014; 137: 2960–72. [DOI] [PubMed] [Google Scholar]
- Hinzman JM, DiNapoli VA, Mahoney EJ, Gerhardt GA, Hartings JA. Spreading depolarizations mediate excitotoxicity in the development of acute cortical lesions. Exp Neurol 2015; 267: 243–53. [DOI] [PubMed] [Google Scholar]
- Hossmann KA. Periinfarct depolarizations. Cerebrovasc Brain Metab Rev 1996; 8: 195–208. [PubMed] [Google Scholar]
- Jarvis CR, Anderson TR, Andrew RD. Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cereb Cortex 2001; 11: 249–59. [DOI] [PubMed] [Google Scholar]
- Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage 2002; 17: 825–41. [DOI] [PubMed] [Google Scholar]
- Jenkinson M, Smith S. A global optimisation method for robust affine registration of brain images. Med Image Anal 2001; 5: 143–56. [DOI] [PubMed] [Google Scholar]
- Kang EJ, Major S, Jorks D, Reiffurth C, Offenhauser N, Friedman A, et al. Blood-brain barrier opening to large molecules does not imply blood-brain barrier opening to small ions. Neurobiol Dis 2013; 52: 204–18. [DOI] [PubMed] [Google Scholar]
- Kohl M, Lindauer U, Dirnagl U, Villringer A. Separation of changes in light scattering and chromophore concentrations during cortical spreading depression in rats. Opt Lett 1998; 23: 555–7. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Nicholson C. Extracellular ionic variations during spreading depression. Neuroscience 1978; 3: 1045–59. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab 1992; 12: 223–9. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Strong AJ. ‘Spreading depression of Leao’ and its emerging relevance to acute brain injury in humans. J Cereb Blood Flow Metab 2017; 37: 1553–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão AAP. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol 1947; 10: 409–14. [DOI] [PubMed] [Google Scholar]
- Lehmenkühler A, Richter F, Poppelmann T. Hypoxia- and hypercapnia-induced DC potential shifts in rat at the scalp and the skull are opposite in polarity to those at the cerebral cortex. Neurosci Lett 1999; 270: 67–70. [DOI] [PubMed] [Google Scholar]
- Madry C, Haglerod C, Attwell D. The role of pannexin hemichannels in the anoxic depolarization of hippocampal pyramidal cells. Brain 2010; 133: 3755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major S, Petzold GC, Reiffurth C, Windmuller O, Foddis M, Lindauer U, et al. A role of the sodium pump in spreading ischemia in rats. J Cereb Blood Flow Metab 2017; 37: 1687–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrannes R, Willems R, De Prins E, Wauquier A. Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res 1988; 457: 226–40. [DOI] [PubMed] [Google Scholar]
- Memezawa H, Smith ML, Siesjo BK. Penumbral tissues salvaged by reperfusion following middle cerebral artery occlusion in rats. Stroke 1992; 23: 552–9. [DOI] [PubMed] [Google Scholar]
- Mies G, Iijima T, Hossmann KA. Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. Neuroreport 1993; 4: 709–11. [DOI] [PubMed] [Google Scholar]
- Milakara D, Grozea C, Dahlem M, Major S, Winkler MKL, Lückl J, et al. Simulation of spreading depolarization trajectories in cerebral cortex: correlation of velocity and susceptibility in patients with aneurysmal subarachnoid hemorrhage. NeuroImage Clin 2017; 16: 524–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Somjen GG. Inhibition of major cationic inward currents prevents spreading depression-like hypoxic depolarization in rat hippocampal tissue slices. Brain Res 1998; 812: 1–13. [DOI] [PubMed] [Google Scholar]
- Muller M, Somjen GG. Na(+) and K(+) concentrations, extra- and intracellular voltages, and the effect of TTX in hypoxic rat hippocampal slices. J Neurophysiol 2000; 83: 735–45. [DOI] [PubMed] [Google Scholar]
- Nozari A, Dilekoz E, Sukhotinsky I, Stein T, Eikermann-Haerter K, Liu C, et al. Microemboli may link spreading depression, migraine aura, and patent foramen ovale. Ann Neurol 2010; 67: 221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira-Ferreira AI, Milakara D, Alam M, Jorks D, Major S, Hartings JA, et al. Experimental and preliminary clinical evidence of an ischemic zone with prolonged negative DC shifts surrounded by a normally perfused tissue belt with persistent electrocorticographic depression. J Cereb Blood Flow Metab 2010; 30: 1504–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 2014; 15: 379–93. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Simon RP, Boison D. Transgenic overexpression of adenosine kinase aggravates cell death in ischemia. J Cereb Blood Flow Metab 2007; 27: 1–5. [DOI] [PubMed] [Google Scholar]
- Revankar GS, Winkler MKL, Major S, Schoknecht K, Heinemann U, Woitzik J, et al. Spreading depolarizations and seizures in clinical subdural electrocorticographic recordings. In: Varelas PN, Claassen J, editors. Seizures in critical care a guide to diagnosis and therapeutics. New York: Springer; 2017. p. 77–90. [Google Scholar]
- Risher WC, Ard D, Yuan J, Kirov SA. Recurrent spontaneous spreading depolarizations facilitate acute dendritic injury in the ischemic penumbra. J Neurosci 2010; 30: 9859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakowitz OW, Kiening KL, Krajewski KL, Sarrafzadeh AS, Fabricius M, Strong AJ, et al. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke 2009; 40: e519–22. [DOI] [PubMed] [Google Scholar]
- Sarrafzadeh AS, Sakowitz OW, Kiening KL, Benndorf G, Lanksch WR, Unterberg AW. Bedside microdialysis: a tool to monitor cerebral metabolism in subarachnoid hemorrhage patients? Crit Care Med 2002; 30: 1062–70. [DOI] [PubMed] [Google Scholar]
- Schiefecker AJ, Kofler M, Gaasch M, Beer R, Unterberger I, Pfausler B, et al. Brain temperature but not core temperature increases during spreading depolarizations in patients with spontaneous intracerebral hemorrhage. J Cereb Blood Flow Metab 2018; 38: 549–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selb J, Boas DA, Chan ST, Evans KC, Buckley EM, Carp SA. Sensitivity of near-infrared spectroscopy and diffuse correlation spectroscopy to brain hemodynamics: simulations and experimental findings during hypercapnia. Neurophotonics 2014; 1: pii: 015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Ren H, Cheng H, Fisher M, Duong TQ. Functional, perfusion and diffusion MRI of acute focal ischemic brain injury. J Cereb Blood Flow Metab 2005; 25: 1265–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 2001; 81: 1065–96. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Irreversible hypoxic (ischemic) neuron injury. In: Somjen GG, editor. Ions in the brain. New York: Oxford University Press; 2004. p. 338–72. [Google Scholar]
- Strong AJ, Dardis R. Depolarisation phenomena in traumatic and ischaemic brain injury. Adv Tech Stand Neurosurg 2005; 30: 3–49. [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Dilekoz E, Moskowitz MA, Ayata C. Hypoxia and hypotension transform the blood flow response to cortical spreading depression from hyperemia into hypoperfusion in the rat. J Cereb Blood Flow Metab 2008; 28: 1369–76. [DOI] [PubMed] [Google Scholar]
- Takano K, Latour LL, Formato JE, Carano RA, Helmer KG, Hasegawa Y, et al. The role of spreading depression in focal ischemia evaluated by diffusion mapping. Ann Neurol 1996; 39: 308–18. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, et al. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 2007; 10: 754–62. [DOI] [PubMed] [Google Scholar]
- Vergouwen MD, Etminan N, Ilodigwe D, Macdonald RL. Lower incidence of cerebral infarction correlates with improved functional outcome after aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab 2011; 31: 1545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergouwen MD, Vermeulen M, van Gijn J, Rinkel GJ, Wijdicks EF, Muizelaar JP, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41: 2391–5. [DOI] [PubMed] [Google Scholar]
- Voipio J, Tallgren P, Heinonen E, Vanhatalo S, Kaila K. Millivolt-scale DC shifts in the human scalp EEG: evidence for a nonneuronal generator. J Neurophysiol 2003; 89: 2208–14. [DOI] [PubMed] [Google Scholar]
- Vora YY, Suarez-Almazor M, Steinke DE, Martin ML, Findlay JM. Role of transcranial Doppler monitoring in the diagnosis of cerebral vasospasm after subarachnoid hemorrhage. Neurosurgery 1999; 44: 1237–47; discussion 1247–8. [PubMed] [Google Scholar]
- Waziri A, Claassen J, Stuart RM, Arif H, Schmidt JM, Mayer SA, et al. Intracortical electroencephalography in acute brain injury. Ann Neurol 2009; 66: 366–77. [DOI] [PubMed] [Google Scholar]
- Windmuller O, Lindauer U, Foddis M, Einhaupl KM, Dirnagl U, Heinemann U, et al. Ion changes in spreading ischaemia induce rat middle cerebral artery constriction in the absence of NO. Brain 2005; 128: 2042–51. [DOI] [PubMed] [Google Scholar]
- Winkler MK, Dengler N, Hecht N, Hartings JA, Kang EJ, Major S, et al. Oxygen availability and spreading depolarizations provide complementary prognostic information in neuromonitoring of aneurysmal subarachnoid hemorrhage patients. J Cereb Blood Flow Metab 2017; 37: 1841–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woody CD, Marshall WH, Besson JM, Thompson HK, Aleonard P, Albe-Fessard D. Brain potential shift with respiratory acidosis in the cat and monkey. Am J Physiol 1970; 218: 275–83. [DOI] [PubMed] [Google Scholar]
- Zeger SL, Liang KY. Longitudinal data analysis for discrete and continuous outcomes. Biometrics 1986; 42: 121–30. [PubMed] [Google Scholar]
- Zhou N, Gordon GR, Feighan D, MacVicar BA. Transient swelling, acidification, and mitochondrial depolarization occurs in neurons but not astrocytes during spreading depression. Cereb Cortex 2010; 20: 2614–24. [DOI] [PubMed] [Google Scholar]
- Zhou N, Rungta RL, Malik A, Han H, Wu DC, Macvicar BA. Regenerative glutamate release by presynaptic NMDA receptors contributes to spreading depression. J Cereb Blood Flow Metab 2013; 33: 1582–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.