

Abstract
Motivation
Molecular subtypes of cancers and autoimmune disease, defined by transcriptomic profiling, have provided insight into disease pathogenesis, molecular heterogeneity and therapeutic responses. However, technical biases inherent to different gene expression profiling platforms present a unique problem when analyzing data generated from different studies. Currently, there is a lack of effective methods designed to eliminate platform-based bias. We present a method to normalize and classify RNA-seq data using machine learning classifiers trained on DNA microarray data and molecular subtypes in two datasets: breast invasive carcinoma (BRCA) and colorectal cancer (CRC).
Results
Multiple analyses show that feature specific quantile normalization (FSQN) successfully removes platform-based bias from RNA-seq data, regardless of feature scaling or machine learning algorithm. We achieve up to 98% accuracy for BRCA data and 97% accuracy for CRC data in assigning molecular subtypes to RNA-seq data normalized using FSQN and a support vector machine trained exclusively on DNA microarray data. We find that maximum accuracy was achieved when normalizing RNA-seq datasets that contain at least 25 samples. FSQN allows comparison of RNA-seq data to existing DNA microarray datasets. Using these techniques, we can successfully leverage information from existing gene expression data in new analyses despite different platforms used for gene expression profiling.
Availability and implementation
FSQN has been submitted as an R package to CRAN. All code used for this study is available on Github (https://github.com/jenniferfranks/FSQN).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Molecular subtypes of human disease have been defined in various cancers and autoimmune diseases. These subtypes are based on conserved changes in gene expression across groups of individuals and reveal patterns of disease heterogeneity and insights into disease pathogenesis that may not be apparent from clinical information alone. Molecular subtypes have previously been identified using de novo analyses and unsupervised clustering methods on large study cohorts in cancer and autoimmune diseases (Milano et al., 2008; Perou et al., 2000), but assigning these subtypes in clinical trials or for diagnostic testing requires supervised classification methods that can assign an individual, single sample to a subtype.
For many diseases, the volume of DNA microarray data available in NCBI GEO is far greater than the volume of RNA-seq data, thus it is important that we can leverage well-validated DNA microarray datasets to analyze data generated on newer platforms with updated technology, such as RNA-seq. Although DNA microarray and RNA-seq data have been shown to have a high correlation (Guo et al., 2013; Zhao et al., 2014), overall processing and standard quantification methods specific to each platform result in different data distributions. This violates the assumption of identical distributions that is required for many statistical and machine learning approaches. Consequently, there is a need for methods that allow accurate molecular subtype assignments using RNA-seq data, which leverages DNA microarray experiments for training of the classification models.
Several methods have been proposed to translate RNA-seq data into DNA microarray-comparable values. Probe Region Expression estimation Based on Sequencing (PREBS) (Uziela and Honkela, 2015) was designed to map RNA-seq reads to probes used for microarrays. This method requires raw reads and can be computationally time-consuming and memory intensive. Moreover, it is often necessary to use data reported as gene-level expression when considering publicly available large-scale analyses. In this study, we focus on utilizing log2 transformed RPKM (Reads Per Kilobase of transcript per Million mapped reads) values, a commonly reported measure of gene expression which are normalized based on gene length and total counts (Robinson and Oshlack, 2010).
Feature scaling is an important and often overlooked component of cross-platform gene expression analyses. It is well-established that features which have been rescaled to a determined interval train a better-performing model (Jain and Dubes, 1988; Milligan and Cooper, 1988). However, feature scaling is considered most appropriate in cases when the features are not measured on the same scale, and thus the comparative difference is irrelevant (e.g. comparing an individual’s income and height). The relative differences in values between genes are certainly not meaningless and are essential for biological interpretation. Additionally, many studies that apply classification algorithms to gene expression achieve good results by using z-score transformations, which does not rescale each genes value to a defined interval (Sorlie et al., 2001; Tibshirani et al., 2002, 2003).
In order to render RPKM values comparable to DNA microarray data, it is necessary to consider both center and spread of the data. With these criteria in mind, we introduce a novel method termed Feature Specific Quantile Normalization (FSQN) for normalizing RNA-seq data for optimal comparability when analyzing data generated from different gene expression profiling platforms. We benchmark our approach against other methods reported for this purpose including Quantile Normalization (QN) (Bolstad et al., 2003), Training Distribution Matching (TDM) (Thompson et al., 2016), Non-paranormal transformation (NPN) (Liu et al., 2009) and untransformed data (LOG2). Also, we explore the effects of feature scaling on classification accuracy by rescaling each feature following each normalization method. The methods we have chosen for our analyses are those that either are explicitly designed to use DNA microarray data as the target distribution (e.g. QN, TDM) or assume that the reference data follow a normal distribution (e.g. NPN). It should be noted that NPN should be considered an inappropriate normalization method in cases where training data do not follow a normal distribution but despite this caveat, has been shown to give good results. The goal of the study was to define a robust method to render two datasets, originating from different platforms, comparable for the purposes of machine learning classification.
2 Materials and methods
2.1 TCGA dataset curation and pre-processing
DNA microarray data were from TCGA level 3 breast cancer (BRCA) 3.1.8.0 data that were downloaded from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/) and included 590 samples with molecular subtype reported in the 2012 paper (Cancer Genome Atlas, 2012). RNA-seq data were re-calculated from RSEM estimated read counts to log2RPKM. For scaled feature analysis, data were scaled by gene to an interval [0, 1] using the rescale function as part of the plotrix package (Lemon, 2006) in R version 3.3.2. For the CRC dataset, DNA microarray data, RNA-seq data and molecular subtype labels were downloaded from the data repository as described (Guinney et al., 2015). CRC datasets included an overlap of 186 samples for TCGA microarray and RNA-seq datasets and an overlap of zero samples for KFSYSCC microarray and TCGA RNA-seq datasets.
2.2 Model training and classification
The KernSmooth (Wand, 2015), glmnet (Friedman et al., 2010), random forest (Liaw and Wiener, 2002) and caret (Kuhn, 2008) packages implemented in R were used to train the supervised classifiers. The SVM classifier was trained with a linear kernel. GLMnet and random forest were run with default parameters. Repeated cross-validation (10x, 3-fold) was used to train the model and simultaneously assess robustness based on classification accuracy metrics. Overall accuracy and Cohen’s kappa coefficient were measured across all repeated cross-validated folds. Sensitivity and sensitivity for each subset were calculated and recorded for each repeated fold. Average value was calculated for accuracy, and error bars represent SEM.
2.3 Normalization procedures
For quantile normalization (QN), we utilized the normalize.quantiles.use.target function from the preprocessCore package (Bolstad, 2016) in R with the entire microarray dataset matrix as the target distribution. For training distribution matching (TDM), we downloaded the package and followed implementation notes from Github (https://github.com/greenelab/TDM). For non-paranormal transformation (NPN), we used the huge package (Zhao et al., 2012) implemented in R. For all scaled analyses, feature scaling was performed post-normalization for all methods.
2.4 Feature specific quantile normalization (FSQN)
For each corresponding feature (gene), we quantile normalized log2RPKM counts from RNA-seq data using DNA microarray data as the target distribution. When N = number of samples in the target distribution, d is the 1 x N unit diagonal:
| (1) |
and qi is a vector of expression values from one gene:
| (2) |
then the resulting vector for the corresponding gene in the new dataset is
| (3) |
This projection is equivalent to substituting the average of the quantile for each value in the new dataset.
2.5 Bootstrapping procedure
For each sample size (n = 5–54), samples were randomly drawn with replacement from the corresponding RNA-seq dataset. RNA-seq data were normalized using each method described above and classified with each machine learning model trained from DNA microarray data. Average values of classification accuracy were calculated from 100 trials of each sample size and plotted along with error bars representing SEM.
2.6 Statistical tests
We used the Kolmogorov–Smirnov (K–S) test statistic for two samples to determine if two datasets were drawn from the same distribution. We utilized the Guided Principal Components Analysis (gPCA) package (Reese et al., 2013) implemented in R to plot principal components and determine statistically significant batch effects between platforms. P-values < 0.05 were considered significant.
3 Results
3.1 Data curation and processing
We identified samples from The Cancer Genome Atlas breast invasive carcinoma (TCGA-BRCA) study (Cancer Genome Atlas, 2012), which included both DNA microarray and RNA-seq gene expression data in addition to PAM50 subtype (Parker et al., 2009) information determined from the DNA microarray data. We chose this dataset because it is well characterized, breast cancer molecular subtypes have been validated in multiple cohorts (Carey et al., 2006; Perou et al., 2000; Sorlie et al., 2001) and PAM50 subtype labels determined from DNA microarray are a true gold standard to test the accuracy of subtype assignments for corresponding RNA-seq samples. These criteria resulted in 539 samples (Table 1; Supplementary Tables S1, S2). DNA microarray data were collected as processed gene-level data from Agilent custom 244 K whole genome DNA microarrays and median centered by gene as previously described (Cancer Genome Atlas, 2012). RNA-seq data were obtained as RSEM (RNA-seq by Expectation Maximization; Li and Dewey, 2011) data generated from Illumina HiSeq, then recalculated as RPKM values and log2 transformed. Similarly, we obtained data from The Cancer Genome Atlas colorectal cancer samples, which included DNA microarray, RNA-seq gene expression data and molecular subtypes (Guinney et al., 2015). CRC datasets included an overlap of 186 samples across TCGA microarray and RNA-seq datasets and an overlap of zero samples for KFSYSCC microarray and TCGA RNA-seq datasets (Table 1; Supplementary Table S3).
Table 1.
Summary of training and testing gene expression datasets
| Dataset | Training data | Testing data | Sample overlap |
|---|---|---|---|
| BRCA | TCGA | TCGA | 539 |
| Agilent 244K | Illumina HiSeq | ||
| CRC | TCGA | TCGA | 186 |
| Agilent | Illumina HiSeq | ||
| CRC | KFSYSCC | TCGA | 0 |
| Affymetrix HG133plus2 | Illumina HiSeq |
All data were derived from breast tissue samples (TCGA) analyzed by The Cancer Genome Atlas (2012) or colorectal cancer samples (TCGA, KFSYSCC) analyzed in Guinney et al. (2015).
3.2 Model training and initial evaluation
Classifiers were trained using three different machine learning methods on both scaled and unscaled gene expression data from DNA microarrays. Classification was based on previously described molecular subtypes: Luminal A, Luminal B, Basal-like, HER2-enriched and Normal-like for the BRCA dataset. For the CRC dataset, classification was based on the molecular subtype labels: CMS1, CMS2, CMS3 and CMS4. Performance of the classifiers was evaluated using sensitivity and specificity of assigning each class label through repeated 3-fold cross-validation (Fig. 1). Overall, SVM, GLMnet and random forest achieved high classification accuracy for DNA microarray data in repeated cross-validation analyses using either unscaled (Fig. 1A–C) or scaled data (Fig. 1D–E). All three classifiers demonstrated strength in accurate classification of Basal-like and Luminal A subtypes. However, performance suffered slightly for HER2-enriched, Luminal-B and Normal-like subtypes. Overall, GLMnet and SVM performed best with very comparable average sensitivity and specificity for all subtypes. GLMnet trained on scaled DNA microarray values demonstrated a very slight increase in performance over the other methods. The numbers of genes included in the final models as well as the models trained on the CRC dataset are described in Supplementary Material (Supplementary Fig. S1, Supplementary Table S4).
Fig. 1.
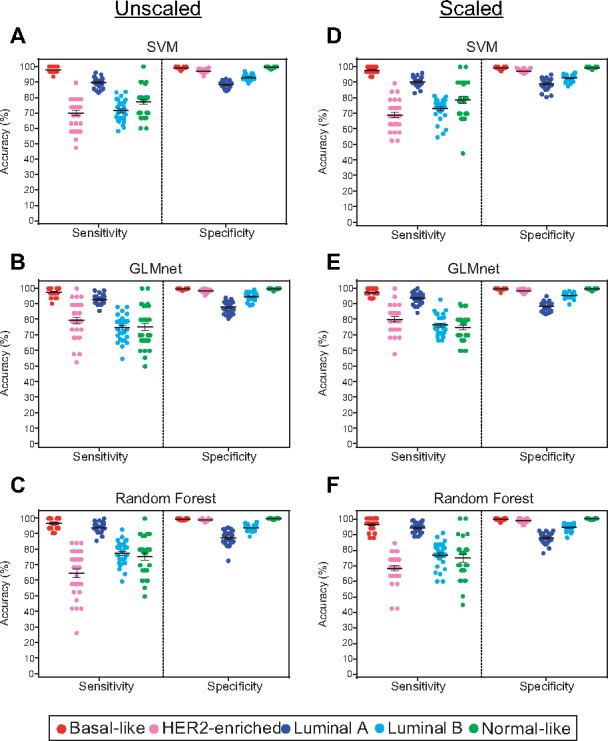
Training dataset performance. Training dataset performance for TCGA-BRCA (sensitivity and specificity for each subtype) across repeated cross-fold validation for support vector machine, GLMnet and random forest using unscaled (A–C) and scaled (D–F) microarray data. Bold lines indicate mean value and error bars represent SEM
3.3 Normalizing RNA-seq data
Processed DNA microarray data have a different distribution compared to LOG2 values from RNA-seq data (Fig. 2A;Supplementary Fig. S2A). However, it is important to note that the shapes observed in Figure 2 are global depictions of the expression distributions. If, instead, we consider distributions from a local level (Fig. 2B;Supplementary Fig. S2B), we see that they can differ both in center and spread for each gene. Notably, some genes may even take on a distribution markedly different from the overall global distribution. These distribution differences persist to varying degrees in the RNA-seq data as shown for multiple example genes in Figure 2.
Fig. 2.

Comparison of BRCA DNA microarray and RNA-seq data distributions. DNA microarray and RNA-seq distributions displayed from a global perspective (A) and local perspective of individual genes (B). Center points indicate mean and lines indicate SD
3.4 Performance of normalization techniques on unscaled data
We normalized data using FSQN, QN, TDM and NPN using both unscaled and scaled data to investigate how each normalization technique impacts the global and local distributions of each feature. Classification models were trained on DNA microarray data alone, and normalized RNA-seq data was used to assign molecular subtype labels and assess overall accuracy and robustness of normalization methods.
In the unscaled data, there were statistical differences between DNA microarray data and LOG2 data distributions (p = 2.2E–16, K–S). For global distributions, all methods make visual improvements in rendering the RNA-seq data to resemble that of the DNA microarray data (Fig. 3A;Supplementary Fig. S3A). There was no difference in distribution for DNA microarray compared to RNA-seq data normalized with FSQN (p = 1, K–S) indicating that normalization resulted in a nearly indistinguishable match between distributions. However, statistically significant differences remained in the distributions between DNA microarray and QN (p = 7.1E–11, K–S), TDM (p = 2.2E–16, K–S) and NPN (p = 2.2E–16, K–S) normalized datasets. Additionally, we see that each normalization method produces widely ranging results for a single gene (Fig. 3B;Supplementary Figs S3B and S4). Based on these results, we find that FSQN is the only method that preserves feature-level distribution information.
Fig. 3.
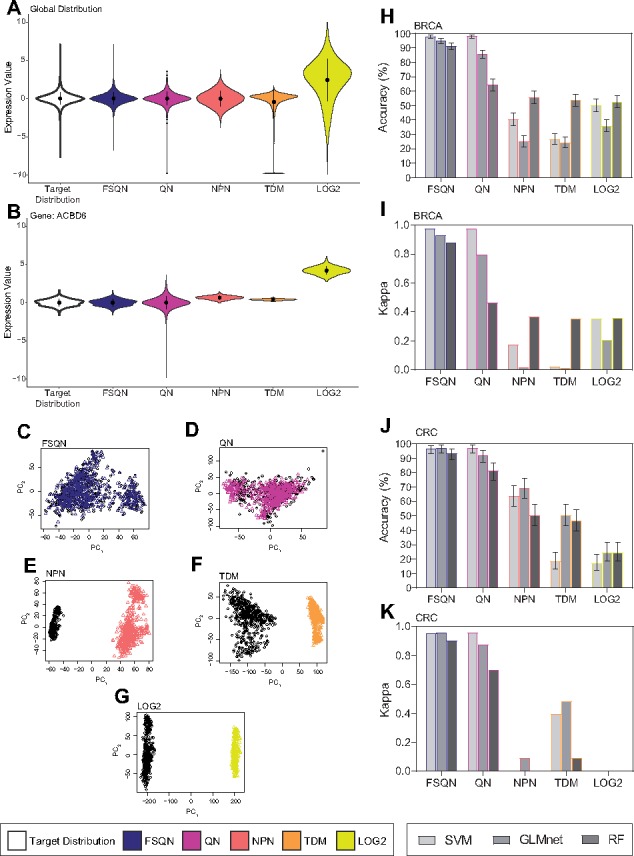
Normalization results with unscaled data. Different normalization techniques using unscaled BRCA data from both a global perspective of the data (A) and a local perspective from a randomly chosen gene (B). Principal components plot (PC1 v. PC2) for FSQN (C), MC (D), QN (E), NPN (F), TDM (G) and LOG2 (H) normalized BRCA RNA-seq and microarray data. Performance of classifications using normalized unscaled data classified by SVM, GLMnet and RF are reported as overall accuracy and kappa for the BRCA dataset (I, J) and the CRC dataset (K, L)
Next, we examined platform-related batch effects on DNA microarray and pre-normalized RNA-seq data using principal components analysis (PCA). In comparing untransformed RNA-seq data to DNA microarray, PC1 clearly separates data based on the gene expression profiling system used, and there is a statistically significant batch effect (P < 0.001, gPCA; Fig. 3G). We evaluated each normalization technique in ability to remove platform-specific bias (Fig. 3D–F;Supplementary Figs S3D–F). For QN (P < 0.001, gPCA), TDM (P < 0.001, gPCA) and NPN (P < 0.001, gPCA), normalization is insufficient for eliminating platform-based bias. We find that FSQN successfully integrates the data without significant platform bias (P = 1, gPCA).
We assigned subtypes to all samples in each normalized dataset using the classifiers trained on unscaled DNA microarray data (Fig. 3H–K, Supplementary Table S4). In both BRCA and CRC datasets, the highest classification accuracies are attained with FSQN and QN normalization methods. However, across datasets and machine learning algorithms, FSQN is consistently the most accurate. Although SVM is able to classify FSQN and QN data equally well, there are substantial differences in the GLMnet and RF models, indicating that it may be more important to use feature specific estimators, like FSQN, in contexts where classifiers use small numbers of genes for classification. NPN, TDM and LOG2 perform with very low accuracy, often barely surpassing the no-information rate for each dataset (0.4286 for BRCA, 0.4677 for CRC).
3.5 Performance of normalization techniques on scaled data
The distributions for DNA microarray data and LOG2 RNA-seq data were scaled [0, 1] and plotted (Fig. 4A;Supplementary Fig. S5A). When comparing data distributions at the global level, statistical differences exist between DNA microarray data and LOG2 RNA-seq data (p = 2.2E–16, K–S). These differences persisted despite QN (p = 7.82E–11, K–S), TDM (p = 2.2E–16, K–S) and NPN (p = 2.2E–16, K–S) normalization. Again, there were no statistically significant differences detected between DNA microarray and FSQN normalized RNA-seq data (p = 1, K–S). These trends are conserved at the feature level as distribution differences are evident for several examples genes that are normalized using each method (Fig. 4B;Supplementary Figs S5B and S6).
Fig. 4.
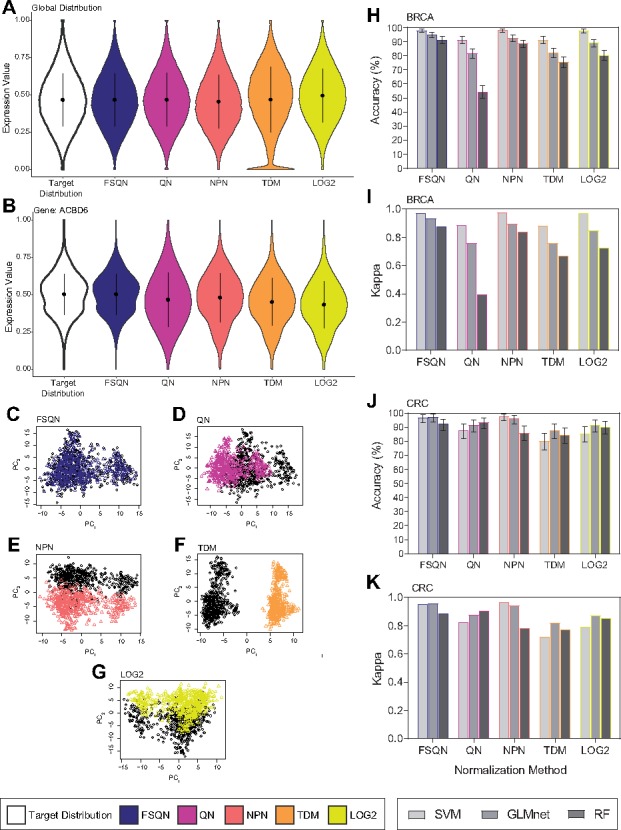
Normalization results with scaled data. Different normalization methods displayed from both a global (A) and a local perspective (B) of BRCA data. Principal components plot for FSQN (C), NPN (D), TDM (E) and LOG2 (G) normalized data and microarray data from BRCA datasets. Performance of classifications using normalized unscaled data classified by SVM, GLMnet and RF are reported as overall accuracy and kappa for the BRCA dataset (H, I) and the CRC dataset (J, K)
We then evaluated each normalization technique with regard to its ability to remove platform-specific bias on scaled data (Fig. 4C–G;Supplementary Figs S5C–G). For analyzing the existence of platform-related batch effects on DNA microarray and pre-normalized RNA-seq data, we used gPCA based on the expression profiling system used. LOG2 (P < 0.001, gPCA), QN (P < 0.001, gPCA), NPN (P < 0.001, gPCA) and TDM (P < 0.001, gPCA) normalization methods are insufficient for eliminating platform-based bias and this is evidenced by distinct clusters of data corresponding to dataset. FSQN (Fig. 4C) is the only method which shows no significant platform bias post-normalization (P = 1, gPCA).
Finally, we assigned subtypes to all samples in each normalized and scaled dataset using the classifiers trained using scaled DNA microarray data (models described in Fig. 1D–E). The classification results for scaled data classified using SVM, GLMnet and RF for the BRCA and CRC datasets are summarized in Fig. 4I–K and Supplementary Table S5. With scaled data, we see that all normalization methods result in high classification accuracy. For both BRCA and CRC datasets, high accuracy in classification (Fig. 4H and J) and kappa (Fig. 4I and K) are reached in nearly every model. The SVM classifies all datasets with high accuracy regardless of normalization method. However, differences in overall classification accuracy and kappa are evident in the GLMnet and RF models. For the GLMnet model, FSQN and NPN are the most accurate, followed by QN. For the RF model, FSQN and NPN are the most accurate in the BRCA dataset, but in the CRC dataset, FSQN and QN are the most accurate. Overall, FSQN is the most reliable normalization method for accurate classification, regardless of dataset or machine learning classifiers.
3.6 Considerations of sample size and matching
We performed a bootstrapping procedure to determine the minimum sample size to reach maximum classification accuracy and to evaluate the power of classification on possible composition change of subtypes due to random selection and small sample size (Fig. 5). For each sample size, we randomly selected samples from the RNA-seq dataset, used each normalization method to normalize the values using the full DNA microarray dataset as the target distribution, and classified the samples to assess accuracy. In this way, we simulate comparing a small novel dataset, with varying distribution of molecular subtypes, to a larger legacy dataset used for training the original model while still maintaining gold standard classification labels to assess accuracy. We find our bootstrap analyses largely mimic the results shown in our earlier analysis: the best performances are reached with FSQN, QN and NPN. In unscaled analyses (Fig. 5A), FSQN and QN result in the most accurate classifications. In SVM, there is a slight increase in accuracy for FSQN at lower sample sizes. For GLMnet and RF, FSQN is clearly superior to the other normalization methods. In scaled analyses (Fig. 5B), FSQN, NPN and LOG2 perform the best overall. For SVM, TDM quickly gains power in classification accuracy with increasing samples sizes, while FSQN and LOG2 remain consistently accurate. For GLMnet and RF, FSQN, LOG2 and NPN perform very comparably. FSQN displays a slight edge in performance at larger sample size in RF. Importantly, FSQN is consistently in the top performing range regardless of dataset, feature scaling or machine learning method.
Fig. 5.
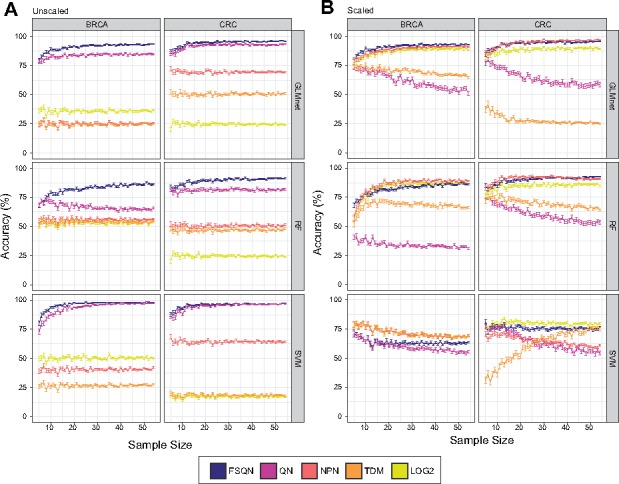
Bootstrapping results. Classification accuracy is shown over varying sample size for unscaled (A) and scaled (B) data for each dataset and machine learning classifier. Points represent average classification accuracy and error bars show SEM
To apply the methods outlined in our analysis, it is unlikely that there will be matched samples with gene expression data from both platforms. Thus, we further explore the utility of our method in assigning colorectal subtype labels with machine learning classifiers trained on a set of DNA microarray samples (KFSYSCC) and tested on a set of RNA-seq samples from a different study (TCGA). Importantly, there were no matched samples between the two datasets (Guinney et al., 2015) (Table 1). In this analysis, we find that FSQN results in the overall highest and most consistent subtype classification accuracy and kappa when compared to QN, NPN, TDM and LOG2 regardless of feature scaling and machine learning algorithm (Supplementary Fig. S7). Additional information for this analysis is included in Supplementary Material.
In conclusion, we find that FSQN often reaches maximum classification accuracy and low standard error with sample sizes of n = 25 and that matched samples are not necessary to achieve accurate classification rates. Taken together, these results support FSQN as the most robust normalization method in our analysis.
4 Discussion
This study introduces FSQN as a robust method of rendering RNA-seq data comparable to DNA microarray data. We trained three classifiers, which represent a spectrum of supervised machine learning classifiers, and have the ability to assign samples to molecular subtypes using gene expression data. We then show that FSQN is a simple and effective method of normalizing RNA-seq data to a target DNA microarray distribution. We achieve extremely high accuracy in classifying RNA-seq data that has been normalized using FSQN with all classifiers regardless of feature scaling. SVM is the most reliable classifier we tested and demonstrates strength in using large compendia of data for classifying genome-wide expression.
In our analysis, feature scaling improves the results of classification accuracy for every normalization method except for FSQN. This is because FSQN capitalizes on the rigorous computational processing of the original DNA microarray dataset by preserving distribution information about the center and spread of each individual gene. Furthermore, the results of this series of experiments highlight an important nuance of our initial hypothesis; considering data in terms of center and spread on a feature-specific level is crucial to proper normalization, and in general, feature-specific estimators may perform better. Ultimately, the simplicity of our normalization method eliminates the need for explicit feature scaling, which preserves biological interpretation and results in a highly robust and reliable method.
We tested sample sizes which are appropriate for applying FSQN as well as a scenario with no matched samples. Our study shows that datasets with small sample numbers and datasets with no matched samples both benefit from FSQN normalization, especially when comparing to a large target distribution that contains the full spectrum of interest; in this case, all subtypes of breast invasive carcinoma or colorectal cancer.
Although we have demonstrated strength in assigning subtypes to normalized RNA-seq data using DNA microarray reference data, several considerations should be taken into account. The distribution of the training dataset (in this case, DNA microarray data) should be carefully considered when selecting data that will be used to develop a supervised classifier and used as a target distribution for normalization techniques. In cases where there are clinical covariates associated with molecular subtypes, it is logical to limit the target distribution to appropriate samples matched according to those covariates, as described (Zhao et al., 2015). For example, a test distribution consisting solely of estrogen receptor positive (ER+) breast cancer patients should use a target distribution that only includes ER+ patients and excludes ER- patients and healthy controls. These considerations will result in a more accurate target distribution for each feature and likely will produce correct subtype classifications overall.
Molecular subtypes have proven to be an effective method of grouping patients, especially in the context of complex diseases which often exhibit heterogeneous phenotypes. In diseases such as breast invasive carcinoma, intrinsic molecular subtypes have improved many diagnostic and prognostic measures. Intrinsic molecular subtypes have been implicated in many other diseases including systemic sclerosis (Milano et al., 2008) and psoriasis (Ainali et al., 2012). Especially for rare diseases, like systemic sclerosis, it is very important to utilize existing data compendia and validated subtypes for new analyses while embracing technology advancements. With our newly developed approach, we can successfully leverage information from validated gene expression datasets despite the different platforms used for gene expression profiling.
5 Conclusions
We have developed a cross-platform normalization method using FSQN to improve comparability of DNA microarray and RNA-seq datasets. We have shown that using datasets composed entirely of legacy DNA microarray data can be effectively used for training machine learning classifiers. Using FSQN for RNA-seq data ensures that the testing data follow the same distribution as the training data, even when inspected on a feature-specific level. This improved comparability provides data distributions that do not violate assumptions in statistical and machine learning methods and, overall, results in extremely accurate subtype assignments.
These methods are most relevant when investigators wish to leverage large compendia of validated data to analyze new studies. Moreover, our method is widely applicable to any situation aiming to render a novel dataset comparable to an existing and validated target distribution that represents the underlying spectrum of interest.
Supplementary Material
Acknowledgement
The authors would like to thank Jaclyn N. Taroni for helpful discussions in conceptualizing the study.
Funding
This work has been supported by the Burroughs-Wellcome Big Data in the Life Sciences Training Program, the Dr. Ralph and Marian Falk Medical Research Trust Catalyst and Transformational Awards, the National Institutes of Health [P30 AR061271, P50 AR060780]; and the Scleroderma Research Foundation.
Conflict of Interest: none declared.
References
- Ainali C. et al. (2012) Transcriptome classification reveals molecular subtypes in psoriasis. BMC Genomics, 13, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad B.M. (2016) preprocessCore: a collection of pre-processing functions. R package version 1.40.0.
- Bolstad B.M. et al. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics, 19, 185–193. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas (2012) Comprehensive molecular portraits of human breast tumours. Nature, 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey L.A. et al. (2006) Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA, 295, 2492–2502. [DOI] [PubMed] [Google Scholar]
- Friedman J. et al. (2010) Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw., 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- Guinney J. et al. (2015) The consensus molecular subtypes of colorectal cancer. Nat. Med., 21, 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y. et al. (2013) Large scale comparison of gene expression levels by microarrays and RNAseq using TCGA data. PLoS One, 8, e71462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A.K., Dubes R.C. (1988) Algorithms for Clustering Data. Prentice Hall, Englewood Cliffs, New Jersy. [Google Scholar]
- Kuhn M. (2008) Building predictive models in R using the caret package. J. Stat. Softw., 28, 1–26.27774042 [Google Scholar]
- Lemon J. (2006) Plotrix: a package in teh red light district of R. R-News, 6, 8–12. [Google Scholar]
- Li B., Dewey C.N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A., Wiener M. (2002) Classification and regerssion by random forest. R. News, 2, 18–22. [Google Scholar]
- Liu H. et al. (2009) The non-paranormal: semiparametric estimation of high dimensional undirected graphs. J. Mach. Learn Res., 10, 2295–2328. [Google Scholar]
- Milano A. et al. (2008) Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One, 3, e2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G.W., Cooper M.C. (1988) A study of standardization of variables in cluster-analysis. J. Classif., 5, 181–204. [Google Scholar]
- Parker J.S. et al. (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol., 27, 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou C.M. et al. (2000) Molecular portraits of human breast tumours. Nature, 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Reese S.E. et al. (2013) A new statistic for identifying batch effects in high-throughput genomic data that uses guided principal component analysis. Bioinformatics, 29, 2877–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., Oshlack A. (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol., 11, R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T. et al. (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA, 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.A. et al. (2016) Cross-platform normalization of microarray and RNA-seq data for machine learning applications. Peer J., 4, e1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R. et al. (2002) Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA, 99, 6567–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R. et al. (2003) Class prediction by nearest shrunken centroids, with applications to DNA microarrays. Stat. Sci., 18, 104–117. [Google Scholar]
- Uziela K., Honkela A.. et al. (2015) Probe region expression estimation for RNA-Seq data for improved microarray comparability. PLoS One, 10, e0126545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand M. (2015) KernSmooth: Functions for Kernel Smoothing Supporting Wand & Jones (1995). R package version 2.23-15.
- Zhao S. et al. (2014) Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One, 9, e78644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T. et al. (2012) The huge package for high-dimensional undirected graph estimation in R. J. Mach. Learn Res., 13, 1059–1062. [PMC free article] [PubMed] [Google Scholar]
- Zhao X. et al. (2015) Molecular subtyping for clinically defined breast cancer subgroups. Breast Cancer Res., 17, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.