

Abstract
Motivation
In cancer, clonal evolution is assessed based on information coming from single nucleotide variants and copy number alterations. Nonetheless, existing methods often fail to accurately combine information from both sources to truthfully reconstruct clonal populations in a given tumor sample or in a set of tumor samples coming from the same patient. Moreover, previously published methods detect clones from a single set of variants. As a result, compromises have to be done between stringent variant filtering [reducing dispersion in variant allele frequency estimates (VAFs)] and using all biologically relevant variants.
Results
We present a framework for defining cancer clones using most reliable variants of high depth of coverage and assigning functional mutations to the detected clones. The key element of our framework is QuantumClone, a method for variant clustering into clones based on VAFs, genotypes of corresponding regions and information about tumor purity. We validated QuantumClone and our framework on simulated data. We then applied our framework to whole genome sequencing data for 19 neuroblastoma trios each including constitutional, diagnosis and relapse samples. We confirmed an enrichment of damaging variants within such pathways as MAPK (mitogen-activated protein kinases), neuritogenesis, epithelial-mesenchymal transition, cell survival and DNA repair. Most pathways had more damaging variants in the expanding clones compared to shrinking ones, which can be explained by the increased total number of variants between these two populations. Functional mutational rate varied for ancestral clones and clones shrinking or expanding upon treatment, suggesting changes in clone selection mechanisms at different time points of tumor evolution.
Availability and implementation
Source code and binaries of the QuantumClone R package are freely available for download at https://CRAN.R-project.org/package=QuantumClone.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The principal cause of cancer is believed to be the accumulation of somatic variants and structural variations (SVs) in the genome. Recently, many efforts have focused on the identification of driver mutations; nonetheless, passenger variants, although they are not directly linked to the disease, may provide additional evidence from which to infer the phylogeny of a tumor and so help uncover the basis for its proliferative activity (Marusyk et al., 2014). Indeed high confidence passenger set of variants shared by a clonal population should be observed at the same cellular prevalence at any given point in time, allowing statistical models to cluster variants together and define a clone.
To understand the role that driver mutations play in clonal expansion and cancer progression, it is essential to accurately reconstruct the clonal structure and assign functional variants to it. We define a clone as a cell population that harbors a unique pattern of mutations and SVs. Such clones are related to each other and share a common ancestor. A hierarchical phylogenetic tree, which represents the ancestry of clones, can be constructed to reflect the order of appearance of new sets of mutations defining each clone. Each such set of mutations is expected to contain at least one driver mutation or SV giving a selective advantage to the clone compared to its ancestry. A clone can thus have a different behavior from its ancestral clone when facing the same stimuli. With accumulation of driver mutations, clones are likely to gain hallmarks of cancer such as evading growth suppressors and activating invasion and metastasis (Hanahan and Weinberg, 2011).
High-throughput sequencing of bulk tumor tissues has allowed uncovering genetic differences at the clonal level in primary and relapse/metastatic tumors. Modern computational methods provide ways to reconstruct the structure of the phylogenetic tree from variant allele frequencies (VAFs) in sequenced reads, where VAF is a proportion of reads supporting each given variant among all reads spanning the position of interest (Fischer et al., 2014; Jiao et al., 2014; Kepler, 2013; Malikic et al., 2015; Miller et al., 2014; Qiao et al., 2014; Schwarz et al., 2014). However, existing methods for clonal reconstruction often neglect information about the genotype of each position, which refers to the paternal or maternal inheritance of a locus and the number of copies of each allele. Accounting for the genotype information is especially crucial in the case of hyper-diploid cancers and cancers with highly rearranged genomes, as the cellular prevalence—measured as the proportion of cancer cells carrying a variant—is linked to VAF through such parameters as copy number of the locus and the number of chromosome bearing the mutation. Computationally, we can detect different clones based on the clustering of VAF values (Miller et al., 2014; Roth et al., 2014; Qiao et al., 2014). However, identifying the correct hierarchical tree is a complex task, and this problem often does not have a unique solution. Therefore, in this paper, clones and variant clusters are considered as synonyms.
Here, we show that by combining the genotype and VAF information it is possible to correctly cluster variants and assign them to specific clones, thus reconstructing the clonal architecture of an individual cancer. This may be done with our novel method, QuantumClone, designed to reconstruct clones based on both VAF and genotype information; so we call it ‘genotype-aware’. We demonstrate that our algorithm accurately clusters variants on simulated data, even when cancer is hyper-diploid or contaminated by normal cells. We also propose a general framework based on QuantumClone to detect driver mutations of clonal evolution. This general approach is applied to 19 neuroblastoma cases; each case includes whole genome sequencing (WGS) data from a sample at diagnosis and relapse. We show that mutations possibly affecting the expression level or the structure of the protein (here called damaging or deleterious) in neuroblastoma accumulate at relapse in specific pathways such as cell motility [e.g. cell-matrix adhesion and regulation of epithelial-mesenchymal transition (EMT)] and cell survival (e.g. PI3K/AKT/mTOR, MAPK or noncanonical Wnt pathways).
2 Materials and methods
2.1 Clonal reconstruction
In this section, we describe QuantumClone, a method we have developed for the clonal reconstruction of a tumor. QuantumClone performs clustering of cellular prevalence values of variants defined by:
| (1) |
where is the number of copies of the corresponding locus in cancer cells, is the number of copies of the corresponding locus in normal cells ( for autosomes), NC is the (a priori unknown) number of chromosomal copies bearing the variant and P is the tumor purity. Each VAF value thus corresponds to several possible values of cellular prevalence; each solution is associated with a value of NC. In order to address the problem of non-uniqueness of a solution, we use an expectation-maximization (EM) algorithm based on the probability to observe a specific number of reads confirming a mutation given the number of reads overlapping the position, the contamination and the cellularity of a clone. In more detail, we attribute to each possibility a probability to observe a reads supporting the variant given that the latter belongs to a clone of cellular prevalence θ, based on a binomial distribution:
| (2) |
where d is the depth of coverage of the variant. In the following equation, we note m for variant, k is the cluster, s is the sample and pm is the possibility for variant m in an hyperdiploid loci. We can then write the log likelihood function to maximize:
| (3) |
where are weights of the possibility computed for a corresponding genotype xAyB (major allele A is present x times and the minor allele B is present y times):
By adding weights that, for each variant, sum up to one, we include in our model the fact that variants in low copy number regions bear more information than those in hyper diploid regions. Each variant is then attributed to its most likely possibility, which is the possibility with highest probability to belong to a clone.
The number of clones is determined by minimization of the Bayesian information criterion. Priors can be provided by the user, randomly generated, determined by the k-medoids clustering on mutations in A and AB sites when the latter contain enough mutations, or using a hierarchical clustering based on the probability of two variants to belong to the same distribution (default).
2.2 Datasets
2.2.1 Simulated datasets
In silico validation data were generated using the QuantumCat method from package QuantumClone. For the validation of the QuantumClone method, we generated a phylogenetic tree for each simulated tumor, which was used to compute observed alternative allele read count given the cell fraction of the clone, the ploidy, and the depth of coverage at this position.The following parameters varied within realistic ranges: depth of sequencing (100× to 1000×), fraction of contamination by normal cells (from 0% to 70%), number of variants used for the clonal reconstruction (from 50 to 200), number of tumor samples used for each patient (from 1 to 5) and number of distinct clones per cancer type (from 2 to 10) (Fig. 1).
Fig. 1.

Comparison of QuantumClone to existing methods. (A) NMI is used to assess the quality of variant clustering on simulated data, with a single parameter varying in each test. This measure evaluates correct assignment of two variants to the same cluster. (B)L2 average error is used to assess the error for each clustered variants between its simulated position and its reconstructed position. (C) Computational time necessary to complete the clustering with each algorithm. Default parameters: two tumor samples without contamination sequenced at 100×; 6 clones; 100 mutations used for clustering
For the pipeline validation, we simulated variants coming from six clones observed in two samples per patient, with a purity of 70% for the first sample and 60% for the second. We create 150 variants that pass stringent filters, and an additional 150 variants passing tolerant filters but not stringent filters. All variants passing stringent filters were simulated in diploid regions, with a depth of coverage higher than 50×, whereas mutations passing permissive filters were located either in AB regions with a coverage between 30× and 50× (approximately 1/4 of permissive variants), or in AAB regions with coverage (approximately 1/2 of permissive variants), or in AABB regions with coverage . We then attributed the ‘driver’ characteristic to 100 variants, by sampling without replacement with probability 10/11 to be selected from the variants passing permissive filters.
2.2.2 Neuroblastoma WGS data
We used WGS data for 19 neuroblastoma trios each including constitutional, diagnosis and relapse samples. Data for 15 patients were taken and reanalyzed from the previous study (Eleveld et al., 2015). Additional four were profiled with illumina paired-end sequencing. In total, we had DNA from 11 cases sequenced using Illumina HiSeq2500 to an average depth of coverage of 80× by Beijing Genomics Institute or the Centre National de Génotypage (CNG) and 8 cases sequenced by Complete Genomics with an average read depth of coverage of 50× (unpublished data, Supplementary Table S1).
2.3 Variant calling and filtering in neuroblastoma WGS data
Mutations were called using Varscan2 (Koboldt et al., 2013). Two sets of variants were created for each patient using tolerant and stringent filtering options. The ‘high confidence’ set of variants obtained using stringent filters was further used for clonal reconstruction, while the set of variants obtained with tolerant filters was used for inference of recurrently altered pathways. The total list of somatic variants used for clonal reconstruction in 19 neuroblastoma patients is provided at http://xfer.curie.fr/get/VZs7XCMTvGx/VCF.tar.gz
2.4 Pipeline comparison
The ‘classical’ pipeline used all 300 simulated variants as input for the clonal reconstruction, using direct clustering by QuantumClone. The ‘selective’ pipeline used the 150 variants passing stringent filters and all variants qualified as drivers from the permissive filters as input for direct clustering. The ‘two-step’ pipeline first used the 150 stringent variants as input for direct clustering and then attributed the variants qualified as drivers a posteriori to the clusters, using the characteristics of the clones found by the initial QuantumClone clustering of high confidence variants. All three pipelines searched for two to ten clones, running with two different initializations, on four threads.
Evaluation of the L2 error and normalized mutual information (NMI) was made using only variants from the stringent and driver groups. The displayed computational time takes into account data processing, clustering and when necessary a posteriori attribution to the clonal structure.
3 Results
We extensively validated QuantumClone on simulated data, where we compared it with recently published methods (Deshwar et al., 2015; Miller et al., 2014; Roth et al., 2014). We complemented QuantumClone with a robust framework for the functional assessment of mutations based on signaling pathway analysis combined with the assignment of functional variants to the reconstructed clones. We then applied the framework to neuroblastoma WGS data.
3.1 Assessment of clonal reconstruction accuracy of QuantumClone on in silico data
3.1.1 Accuracy on diploid cancers
Using in silico data, we compared the performance of QuantumClone, sciClone (Miller et al., 2014), pyClone (Roth et al., 2014) and phyloWGS (Deshwar et al., 2015) in inferring the clonal structure of a set of tumors derived from the same patient. sciClone is based on variational Bayesian Mixture Models, while pyClone relies on a hierarchical Bayes statistical model. Similarly to QuantumClone, pyClone leverages copy number information to better infer clonal architecture. phyloWGS adds to the reconstruction a phylogenetic tree constraint and allows for the use of copy number information.
For each set of parameters, we performed and analyzed 50 independent simulation experiments (Section 2). The accuracy of clonal reconstruction was assessed by evaluating the NMI (Manning et al., 2008) and the average error in distance between the estimated cellularity of a clone and its theoretic value. Perfect variant clustering would result in a L2 (or Euclidean distance) mean error of 0, and a NMI value of 1, which would correspond to an identification of the exact number of clones and correct assignment of all the variants of a clone to the same cluster.
Our analysis showed that QuantumClone is equivalent to or better than the best published algorithm in clustering quality (Fig. 1A) for diploid genomes. In terms of NMI QuantumClone showed similar performances compared to pyClone. However, QuantumClone generally outcompeted sciClone (P-value ) and phyloWGS (P-value ) for NMI. On average, QuantumClone decreased the L2 mean error by 39% compared to sciClone, 22% compared to pyClone and 25% compared to phyloWGS, significantly improving predictions compared to both methods (P-value ). At high values of sequencing depth, all methods accurately estimated prevalence of variants (Fig. 1B, L2 mean error < 0.059 at 1000× for all methods). However, a sequencing depth of 100×, which is the depth of sequencing currently used for the majority of WES and WGS experiments, QuantumClone consistently gave better predictions than pyClone (P-value ), phyloWGS (P-value ) and sciClone (P-value ). In addition, compared to the other methods, QuantumClone took the best advantage of data when multiple samples were provided for the analysis (P-value , P-value and for phyloWGS, pyClone and sciClone, respectively, for simulated tumors with five samples).
Also, the average computational time was significantly decreased using QuantumClone compared to sciClone (median 35-fold improvement), phyloWGS (median 95-fold improvement) or pyClone (median 45-fold improvement, Fig. 1C).
3.1.2 Accuracy in hyper-diploid cancers or cancers with highly rearranged genomes
In order to validate QuantumClone on rearranged or hyper-diploid genomes, we simulated variants in loci of genotype AB, AAB, AABB and in a nearly diploid genome, where all possible genotypes can be observed (Fig. 2). In addition to QuantumClone, we tested the performance of pyClone (Section 2). We excluded sciClone from this experiment as it cannot use variants from non-diploid regions, and phyloWGS as in addition to somatic variant read counts it required to generate a complex input dataset: read coverage on single nucleotide polymorphisms along the genome.
Fig. 2.
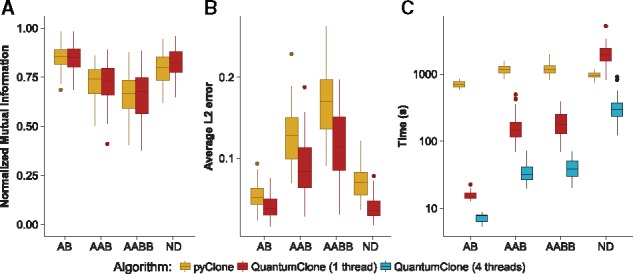
Quality of clonal reconstruction for mutations located in regions of altered copy number. (A) NMI shows equivalent performances of pyClone and QuantumClone in diploid, triploid and tetraploid tumors, or nearly diploid (ND) tumors, whereas the average L2 error (B) shows significantly better performance of QuantumClone. (C) Parallel computing implemented in QuantumClone allows it to significantly decrease computational time and makes QuantumClone remarkably faster than pyClone
In all types of regions, QuantumClone and pyClone performed equally in terms of NMI (Fig. 2A), but QuantumClone outperformed pyClone in terms of mean L2 error with an improvement of 31% (Fig. 2B, P-value ). In addition, QuantumClone without parallelization was faster than pyClone in three out of four settings (from 6.3-fold slower to 61.5 faster; 15.6 times faster on average), while the distributed algorithm outcompeted pyClone in all settings (average computational time decreased by a 43-fold compared to pyClone, Fig. 2C).
In addition, in the majority of cases QuantumClone correctly assumed the exact number of copies of a variant in polyploid regions (average accuracy = 68.9%, P-value , Supplementary Fig. S1).
3.2 Creating a robust framework for clonal assignment of functional mutations
We proposed a novel strategy of reconstruction of the clonal architecture in cancer. Our method combines the identification of clones, using high confidence variants, with the attribution of functional variants (potential drivers) to identified clones (Fig. 3). The approach is based on the different usage of ‘functional’ variants which can potentially affect cell phenotype and ‘high fidelity’ variants that are used to define clones. High fidelity variants can be either drivers or passengers; however, they should have high depth of coverage ( in our implementation), have no strand bias and should not coincide with annotated single-nucleotide polymorphisms (SNPs). As we showed in the simulation studies (Fig. 1), 50 high fidelity variants are sufficient for an accurate clonal reconstruction (Section 2).
Fig. 3.
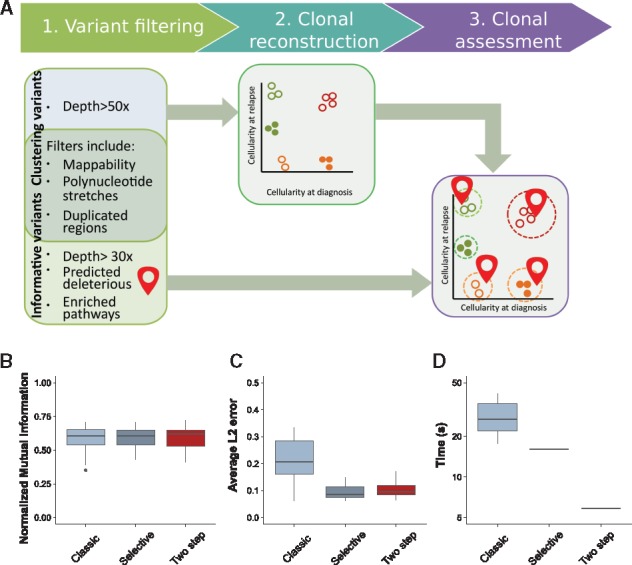
Assessment of the pipeline. (A) Overview of the general clonal reconstruction workflow: steps 1–3. (1) Variants are filtered to remove false positive calls; stringent filters are used to produce mutations that are further employed for clonal reconstruction (step 2), tolerant filters are used to detect functional mutations. (2) Variants that pass stringent filters and have genotype information assigned to the corresponding genomic loci are used as input to QuantumClone to reconstruct clonal populations. (3) Finally, possibly damaging mutations belonging to frequently altered pathways are mapped to the reconstructed clones. Quality of reconstruction. The pipeline aforementioned (two step), or a clustering using all variants called (classic) or a pipeline using only variants of biological interest and variants of high quality (selective) are assessed in terms of NMI (B), average L2 error (C) or computational time (D). The pipelines are evaluated on 20 simulations (Section 2)
High fidelity variants, because they have a lower dispersion of observed VAF compared with other variants, are applied to define clones, i.e. high fidelity variants serve as input to QuantumClone or to an alternative method. Functional mutations are defined here as variants that can possibly alter protein function as predicted by commonly used annotation tools (Adzhubei et al., 2013; Khurana et al., 2013; Ng and Henikoff, 2003) and that can affect either genes reported in the Cancer Census List (Futreal et al., 2004) or genes from gene modules/signaling pathways that are enriched in deleterious variants (Section 2). At the last step of our framework, functional variants are mapped to the clonal structure inferred from high fidelity variants based on the likelihood values.
Here, we demonstrated that having the proposed two-step approach allows for a better reconstruction of the tumor, as well as an important decrease in computational time (Fig. 3D). To test our pipeline, we compared it to two common pipelines: the first one, termed ‘classic’, uses all variants as input for the clustering. The second one, called ‘selective’, only uses variants passing the stringent filters and informative variants as input for the clustering. The third pipeline, termed ‘two-step’, uses a posteriori attribution of the putative drivers to the clones found using only variants passing stringent filters. While all three pipelines had similar outcomes when we compared the quality of reconstruction using NMI (Fig. 3B), the selective and two step pipelines fared significantly better than the classical pipeline (p-value , Fig. 3C). In addition, the two-step analysis resulted in an average 4.9-fold decrease in computational time compared to the classical pipeline and an average 2.7-fold decrease compared to the selective pipeline (Fig. 3D). Furthermore, separating both steps eases iterative improvement of the clonal reconstruction. Once achieved, this reconstruction can be reused to answer questions about the evolution of different pathways separately, while previous pipelines required re-running the whole reconstruction with the new set of data.
3.3 Application of the QuantumClone-based framework: characterization of neuroblastoma clonal evolution from diagnosis to relapse
We applied our framework to investigate the clonal composition of neuroblastoma primary and relapse tumors and to study their clonal evolution. In order to remove false positive variant calls, we used a set of stringent filters (Fig. 3, Section 2) as the initial number of variants in the Varscan2 output was highly dependent on the sequencing technology and platform (Supplementary Fig. S2). Of note, variants called as germline in any of the tumor samples have been removed to focus on the somatic variants only. Indeed, if kept, germline variants will be assigned to the ancestral clone and will not provide additional value in clonal reconstruction.
3.3.1 Clonal reconstruction
We applied QuantumClone on high fidelity variants we defined using stringent filters (Fig. 3A, Section 2). Across our cohort, we did not observe a significant association between the predicted number of clones and the number of mutations per patient (Spearman’s , P-value = 0.84). In addition, the number of clones at relapse was similar to that at diagnosis, even despite the fact that the relapse samples had about twice as many mutations as the diagnosis samples (number of mutation clusters varied from one to four with a median of three for both time points). In 79% of reconstructed clonal structures (15 out of 19 patients, we identified mutations coming from the ancestral clone (Fig. 4A), i.e. the clone that gave rise to all cells in both diagnosis and relapse samples.
Fig. 4.
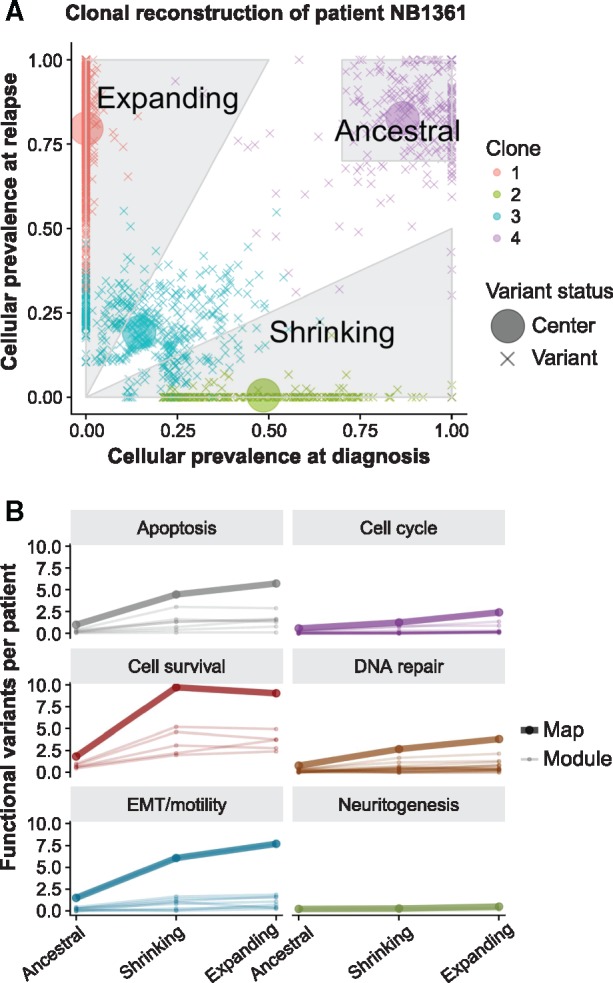
Annotation of clones in neuroblastoma and pathway enrichment analysis. (A) Illustration with data from patient NB1361 of the rules for assignment of variants to (i) the ancestral clone (cellular prevalence of the mutation cluster exceeds 70% both at diagnosis and relapse), (ii) clones expanding after the treatment (cellular prevalence of the mutation cluster increases at least two-fold at relapse) and (iii) shrinking clones (cellular prevalence of such mutation clusters decreases at least two-fold). Here, evaluated cellular prevalence values higher than 1 were set to 1. (B) Evolution of the total number of functional variants for enriched maps and modules, across all 19 patients. The majority of modules show an increase in the number of functional variants between the two time points
3.3.2 Annotation of functional mutations in each sample based on the global pathway enrichment analysis
In our framework, we assumed that functional mutations (i.e. putative drivers) in a given cancer type should target-specific signaling pathways or pathway modules (Fig. 3, Step 2). We attributed annotated deleterious variants obtained with tolerant filters (Fig. 3, Section 2) to the ACSN maps and detected recurrently altered gene modules using the ACSNmineR package (Deveau et al., 2016). Overall, six general gene maps (apoptosis, cell cycle, DNA repair, EMT/cell motility, cell survival and neuritogenesis) and their 53 gene modules were found to be enriched in mutations (Supplementary Table S2). The enrichment of pathways in ACSN was corroborated by enrichment of similar pathways from two other methods (Huang et al., 2009 a, b; Mi et al., 2010; Thomas et al., 2003) (Supplementary Tables S3 and S4). In further analysis, deleterious mutations were annotated as functional when corresponding genes were included in the enriched pathways, or when such genes belonged to the Cancer Census list. The resulting number of functional mutations per patient varied from 2 to 147, with a median of 51.
At this step, the cell survival map registered the highest enrichment in putative drivers, and among its modules, the highest enrichment in putative driver mutations was observed for the non-canonical Wnt pathway (q-value ). We also detected significant enrichment in functional mutations of the Wnt canonical and the MAPK pathways (q-value and , respectively), and of the PI3K/AKT/mTOR and Hedgehog gene modules (q-value and , respectively). Genes coding for the EMT regulators were also significantly affected by the deleterious mutations in our cohort of relapsed neuroblastoma patients (q-value ).
3.3.3 Assignment of functional mutations to the identified clonal structure
Using the results of the mapping of functional mutations on the clonal structure detected for each patient by QuantumClone (Fig. 3A, Step 3), we annotated mutations as (i) those belonging to expanding clones—corresponding to a two-fold cellular prevalence increase between diagnosis and relapse, (ii) those belonging to shrinking clones—cellular prevalence halved between diagnosis and relapse and (iii) those belonging to ancestral clones—cellular prevalence higher than 70% in both samples (Fig. 4A). Overall, 34.4%, 30% and 8.5% of all functional mutations fell in these three categories.
3.3.4 Analysis of pathways enriched in functional mutations in shrinking and expanding clones
Assignment of mutations to clones shrinking or expanding after the treatment resulted in the identification of 331 and 380 possible driver mutations in these clone types, respectively. Expanding clones had more deleterious mutations targeting genes from all six general maps (apoptosis, cell cycle, DNA repair, EMT/cell motility, cell survival and neuritogenesis) than the shrinking clones (Fig. 4B). Similarly, in these expanding clones, most of the corresponding gene modules (e.g. MAPK, Wnt canonical or PI3K/AKT/mTOR) were also more frequently targeted. An extreme example of this behavior can be given with the neuritogenesis substrates module, the RB pathway or the E2F1 pathway in which genes are only found mutated in the expanding clones. The increase in functional variants can partly be explained by the observed doubling of variants at relapse compared to diagnosis. We define μ the functional mutation rate in a module as the number of functional variants per high fidelity variants of the patient by number of genes in a module. The functional mutation rate across modules was significantly different between the three classes of clones according to the z-score computed as suggested by Paternoster et al. (1998) and described in Section 2 (Fig. 5A, P-value between ancestral and shrinking, P-value between ancestral and expanding and P-value between expanding and shrinking). This functional mutation rate has been previously linked to the fitness of a clone (McFarland et al., 2013), and it is interesting to notice that the functional mutation rate is lower in the ancestral clone ( functional variations per 1000 variants per 1000 genes in module, standard error ) and expanding clones () than in the shrinking clones (). The difference in functional mutation rate suggests different selection mechanisms.
Fig. 5.
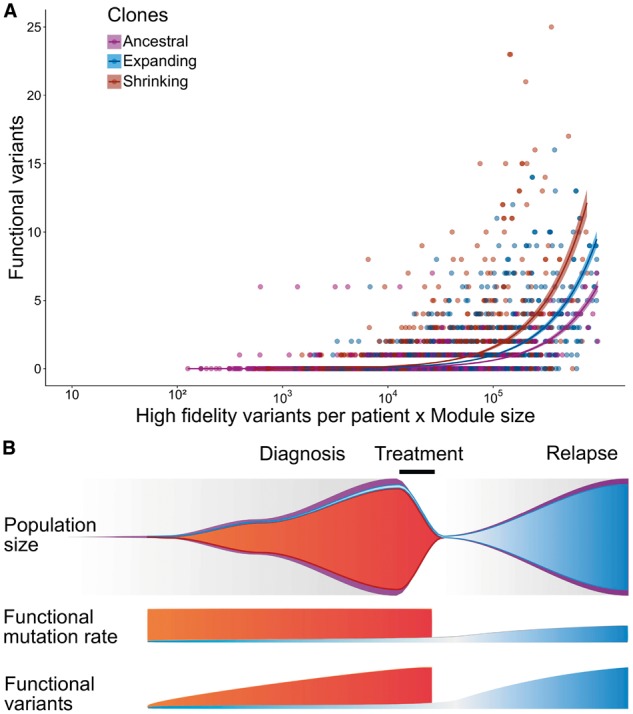
Ancestral, shrinking and expanding clones exhibit different mutation patterns in neuroblastoma relapse tumors. (A) Functional mutation rate is higher in shrinking and expanding clones compared to the ancestral ones. We define the functional mutation rate as a ratio of the number of functional mutations to the number of high fidelity variants. For a given gene module the number of functional mutations in each patient is supposed to linearly depend on the product of the module size and the total number of detected variants. Therefore, we used the product of the module size and number of high fidelity variants as a covariate in a linear regression model evaluating functional mutation rate for neuroblastoma tumors. The rate was defined as the slope of the linear regression. (B) Given the differences in functional mutation rates observed in neuroblastoma relapse tumors we propose the following model for clonal selection in this type of cancer: (1) Clones with high functional mutation rate (red) disappear after the chemotherapy; lower mutational burden provides an advantage in escape from treatment; (2) lower values for functional mutation rate in clones expanding at relapse (blue) compared to the shrinking clones (red) is due to a lower frequency of functional mutations before treatment, followed by a gradual accumulation of functional mutations at relapse. From top to bottom: the number of variants in the clone, number of functional variants in the clone, and population size in the tumor
4 Discussion
Here, we have proposed a pathway-based framework to detect functional mutations in cancer samples and associate the mutations to their corresponding clonal structure. The central part of our framework is represented by the QuantumClone method, which allows reconstruction of clonal populations based on both variant allele frequencies and genotype information. QuantumClone showed stable results on simulated data, significantly outperforming other methods in difficult settings such as highly contaminated samples, heterogeneous tumors and relatively low depth of sequencing coverage. We showed that with the average depth of sequencing of 100×, and with only two biopsies per patient we can reliably reconstruct up to 10 simulated variant clusters corresponding to subclones. To get more fine-grained information about the subclonal structure, we recommend increasing the depth of coverage and, more importantly, the number of biopsies per patient.
The central idea of our analysis framework is to use high fidelity variants to reconstruct the clonal structure of tumor samples; then, map low coverage functional mutations (with high variance in VAFs) onto the inferred clonal structure. Also, we suggest limiting the set of functional mutations to those in genes known to be associated with cancer (e.g. Cancer Census genes) or to those in genes from gene modules/pathways that are frequently disrupted in a given cancer type (Fig. 3). Of note, applying a filter on sequencing depth to determine ‘high fidelity’ variants will not remove low frequency variants corresponding to rare subclones, as the number of reads covering a genomic position and the percentage of reads supporting a variant are statistically independent.
Here, we propose to assign all deleterious variants from gene sets and pathways that are frequently targeted by mutations in a given cancer type to ‘functional’ variants. Of note, many of these genes may never have been associated with oncogenesis previously. Moreover, any variant of a user’s choice can be mapped to the inferred clonal structure using the function ‘Probability.to.belong.to.clone’ of the QuantumClone package.
We applied the proposed analysis framework to decipher clonal structure in neuroblastoma and assign to clones possible driver mutations. In neuroblastoma, until recently no biopsies were performed in case of high risk relapse due to absence of curative therapeutic options. More recently, relapse-specific biopsies have been advocated within precision medicine programs to orient patients to (early) clinical trials based on tumor molecular profiles. Often, the most readily accessible tumor sites are biopsied, which may correspond to either the primary tumor or a metastatic site.
Our analysis of neuroblastoma diagnosis/relapse samples identified genes associated with DNA repair, cell motility, apoptosis and survival to be enriched in functional mutations. For relapsed neuroblastoma samples, we recovered the previously reported enrichment of mutations in the MAPK signaling pathway (Eleveld et al., 2015), while complementing this knowledge with discovery of accumulation of functional mutations at the relapse in such functional gene modules as PI3K/AKT/mTOR, Wnt, Hedgehog signaling and modules consisting of genes responsible for cell-matrix adhesion and EMT.
Previous studies have shown that the number of variants was linked to the number of divisions a cell undergoes (Tomasetti and Vogelstein, 2015). The observed doubling of variants between diagnosis and relapse in neuroblastoma samples suggests that cells have undergone many divisions between diagnosis and relapse and, possibly, DNA repair pathways have been affected.
In addition, the functional mutation rate was significantly lower in the ancestral populations compared to the clones expanding or shrinking at relapse. Chen et al. (2015) have shown that wild-type cells have more adaptive capabilities than mutants, even though a mutant can appear fitter than the wild-type lineage in a specific culture condition. Applied to our results, their finding could suggest that a clone with a low level of functional variants would be more likely to adapt to environment changes during and after treatment. After this selection round and once the tumor environment has returned to physiological state, another set of functional variants would appear, giving selective advantage to the expanding clone.
A direct consequence of this assumption is that the functional mutation rate should be lower at relapse compared to diagnosis, as a period of low functional mutation rate before treatment would be followed by a period of higher functional mutation rate during disease progression (Fig. 5B). This consequence is in line with the 29% functional mutation rate decrease observed between expanding and shrinking clones.
The proposed framework can be applied in the future to any type of cancer. The pre-requirements are sufficient number of candidate mutations (at least 50 mutations per sample) and a minimal read depth of coverage of 50×. These requirements are usually met by WGS or whole exome sequencing datasets. Our simulation results show that increasing the number of mutations used for clonal reconstruction above 50 does not improve significantly the clonal reconstruction accuracy provided that mutations specific for every clone are present in the input. We highlight however that our simulated data were generated for six subclones only. Inferring a more complex clonal structure may require a higher number of input variants.
Our framework will be extremely useful in settings when tumor samples have been sequenced with a limited read depth—from 100× to 200×—and when the contamination level by normal cells is non-negligible, i.e. 10–70%. Our method also runs much faster than previously published methods. A limited number of very high confidence variants can be used to reconstruct the clonal structure; then, in a matter of seconds, all other variants of interest can be mapped to this structure (Fig. 3D).
Study of the clonal evolution and its processes can be highly relevant for drug design. We have described a framework and an algorithm that performs better on in silico data than previously published methods, which should allow for a better analysis of existing datasets. In addition, we showed that the same processes are at play throughout the disease course in our neuroblastoma cohort, targeting similar pathways in diagnosis and relapse.
Acknowledgements
The authors would like to thank Elodie Girard for developing the variant calling pipeline and Pierre Gestraud for his help with statistical analysis of the data.
Funding
This work was supported by Annenberg Foundation and the Nelia and Amadeo Barletta Foundation, SiRIC/INCa [INCa-DGOS-4654 to G.S.], the CEST of Institut Curie, the Associations Enfants et Santé, Association Hubert Gouin Enfance et Cancer, Les Bagouz à Manon, Les amis de Claire, the ATIP-Avenir Program, the ARC Foundation [RAC16002KSA - R15093KS to V.B.], Worldwide Cancer Research [WCR16-1294 R16100KK to V.B.], the ‘Who Am I?’ laboratory of excellence ANR-11-LABX-0071 funded by the French Gouvernement [ANR-11-IDEX-0005-02], the ABS4NGS project of the French Program ‘Investissement d’Avenir’, US National Institutes of Health [RC1MD004418 to J.M. and the TARGET consortium, and CA98543 and CA180899 to J.M. and the Children’s Oncology Group], Federal funds from the National Cancer Institute, National Institutes of Health [HHSN261200800001E to J.M.].
Sequencing of French samples was carried out in a collaboration of Institut Curie with CEA/Jacob/CNRGH financed by France Génomique infrastructure, as part of the program “Investissements d’Avenir” from the ANR [ANR-10-INBS-09].
Conflict of Interest: none declared.
Supplementary Material
References
- Adzhubei I. et al. (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protocol. Hum. Genet., 0 7, Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. et al. (2015) Targeting the adaptability of heterogeneous aneuploids. Cell, 160, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshwar A.G. et al. (2015) PhyloWGS: reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome Biol., 16, 35.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau P. et al. (2016) Calculating biological module enrichment or depletion and visualizing data on large-scale molecular maps with ACSNMineR and RNaviCell packages. R J., 8, 293–306. [Google Scholar]
- Eleveld T.F. et al. (2015) Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet., 47, 864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. et al. (2014) High-definition reconstruction of clonal composition in cancer. Cell Rep., 7, 1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal P.A. et al. (2004) A census of human cancer genes. Nat. Rev. Cancer, 4, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. (2011) Hallmarks of cancer: the next generation. Cell, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Huang D.W. et al. (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl. Acids Res., 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W. et al. (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jiao W. et al. (2014) Inferring clonal evolution of tumors from single nucleotide somatic mutations. BMC Bioinformatics, 15, 35.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepler T.B. (2013). Reconstructing a B-cell clonal lineage. I. Statistical inference of unobserved ancestors. F1000Res., 2, 103. [DOI] [PMC free article] [PubMed]
- Khurana E. et al. (2013) Integrative annotation of variants from 1092 humans: application to cancer genomics. Science (New York, N.Y.), 342, 1235587.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C. et al. (2013) Using VarScan 2 for germline variant calling and somatic mutation detection. Curr Protoc Bioinformatics, 44, 15.4.1–15.4.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikic S. et al. (2015) Clonality inference in multiple tumor samples using phylogeny. Bioinformatics, 31, 1349–1356. [DOI] [PubMed] [Google Scholar]
- Manning C.D. et al. (2008). Introduction to Information Retrieval. Cambridge University Press, New York. [Google Scholar]
- Marusyk A. et al. (2014) Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature, 514, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland C.D. et al. (2013) Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci., 110, 2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H. et al. (2010) PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucl. Acids Res., 38, D204–D210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C.A. et al. (2014) SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput. Biol., 10, e1003665.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P.C., Henikoff S. (2003) SIFT: predicting amino acid changes that affect protein function. Nucl. Acids Res., 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternoster R. et al. (1998) Using the correct statistical test for the equality of regression coefficients. Criminology, 36, 859–866. [Google Scholar]
- Qiao Y. et al. (2014) SubcloneSeeker: a computational framework for reconstructing tumor clone structure for cancer variant interpretation and prioritization. Genome Biol., 15, 443.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A. et al. (2014) PyClone: statistical inference of clonal population structure in cancer. Nat. Methods, 11, 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz R.F. et al. (2014) Phylogenetic quantification of intra-tumour heterogeneity. PLoS Comput. Biol., 10, e1003535.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P.D. et al. (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res., 13, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C., Vogelstein B. (2015) Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science, 347, 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.