

Abstract
The aim of this study is to characterize the genotoxicity of depleted uranium (DU) in Chinese Hamster Ovary cells (CHO) with mutations in various DNA repair pathways. CHO cells were exposed to 0 – 300 μM of soluble DU as uranyl acetate (UA) for 0 – 48 hr. Intracellular UA concentrations were measured via inductively coupled mass spectrometry (ICP-MS) and visualized by transmission electron microscopy (TEM). Cytotoxicity was assessed in vitro by clonogenic survival assay. DNA damage response was assessed via Fast Micromethod® to determine UA-induced DNA single strand breaks. Results indicate that UA is entering the CHO cells, with the highest concentration localizing in the nucleus. Clonogenic assays show that UA is cytotoxic in each cell line with the greatest cytotoxicity in the base excision repair deficient EM9 cells and the nuclear excision repair deficient UV5 cells compared to the non-homologous end joining deficient V3.3 cells and the parental AA8 cells after 48 hr. This indicates that UA is producing single strand breaks and forming UA-DNA adducts rather than double strand breaks in CHO cells. Fast Micromethod® results indicate an increased amount of single strand breaks in the EM9 cells after 48 hr UA exposure compared to the V3.3 and AA8 cells. These results indicate that DU induces DNA damage via strand breaks and uranium-DNA adducts in treated cells. These results suggest that: (1) DU is genotoxic in CHO cells, and (2) DU is inducing single strand breaks rather than double strand breaks in vitro.
Keywords: depleted uranium, uranyl acetate, DNA damage, cytotoxicity
INTRODUCTION
Uranium is an alpha-emitting, radioactive, heavy metal that occurs naturally in nearly all rocks and soils. Depleted uranium (DU) is the byproduct of the uranium enrichment process, resulting in a relatively weak radioactive heavy metal, which is used in tank armor and ammunition rounds due to the metal’s high density (Bleise 2003). The use of DU in military action is a cause for concern, as there is an increased risk for exposure to DU from entry into wounds (embedded shrapnel), inhalation of dust from ammunition rounds, and ingestion of DU from food and water sources. Uranium also poses as an environmental pollutant that contaminates local water reserves used by populations that reside near the numerous abandoned mines from the mining industry. Despite uranium being a weak radioactive heavy metal, the International Agency for Research on Cancer (IARC) currently has no classification on the carcinogenicity ratings for depleted uranium and it is currently under further review (ATSDR 2013).
There is an increased concern for the possible association between an increased risk of cancer and adverse health effects due to the occupational and environmental exposures to depleted uranium. Although epidemiological studies have not yet reported significant increases in cancer mortality in exposed populations, there are a limited number of reports that indicate an increase in cancer incidences, birth defects, and chromosomal instability (Fathi 2013, Zhivin 2014, Prabhavathi 2003). Many in vitro and in vivo studies have established that DU induces a chemical genotoxic response dependent upon several factors including cell type, speciation, and solubility (Carriere 2004, Prat 2005, LaCerte 2010, Holmes 2014, Asic 2017). Like most heavy metals, uranium has been shown to produce oxidative stress, DNA strand breaks, chromosome instability, cell transformation, apoptosis, and cell death at and below the recommended limits for genotoxicity testing of (>500 μM or 119 ppm U) to determine the mechanism of action (Parry 2010, Garmash 2014, Hao 2014, Guéguen 2015, LaCerte 2010). However, the current maximum contaminant level of uranium in drinking water is 30 ppb and the range for reported contaminated groundwater from naturally occurring uranium and uranium mill tailings can reach 210 – 250 μM or 50 – 60 ppm U (EPA 2017, Abdelouas 2000, Cardenas 2008). Therefore, it is important to determine cellular responses to uranium-induced toxicity at more environmentally relevant concentrations ranging from 50 – 300 μM or 12 – 72 ppm U. Several studies have shown that uranium is not genotoxic at this lower concentration range and activates cellular stress responses rather than cell-mediated death responses (Wilson 2015, Guéguen 2015, Garmash 2014).
The mechanism of DU induced toxicity remains unclear. Several studies have proposed that depleted uranium may indirectly cause oxidative DNA damage via a Fenton-type redox mechanism or directly generate U-DNA adduct formation via a DNA hydrolysis mechanism (Stearns 2005, Yazzie 2003). While DU has been shown to cause DNA damage, there has not been a systematic identification of types of DNA lesions caused by uranium at an environmentally relevant concentration range and at a longer exposure duration. The purpose of the current study was to characterize uranium-induced DNA damage. It was hypothesized that DU in the form of uranyl acetate (UA) will localize in the nucleus and generate significant cytotoxicity. This novel systematic identification approach utilizes three DNA repair-deficient CHO cell lines that allows for the characterization of the type of DNA damage caused by UA, as each cell type is sensitive to specific types of DNA lesions. A parental cell line was used as a control (CHO AA8), and compared to CHO EM9 (base excision repair (BER) deficient) cell line, CHO UV5 (nucleotide excision repair (NER) deficient) cell line, and lastly CHO V3.3 (non-homologous end joining (NHEJ) deficient) cell line. By the process of elimination to characterize the type of DNA damage in repair sensitive cell lines, this work further examines if DU-induced DNA damage is altered in complemented CHO cells that re-express the human cloned genes of the mutant repair deficient cell lines. Results indicate that UA is capable of inducing single strand breaks and UA-DNA adducts at lower concentrations and are consistent with previous studies.
MATERIALS AND METHODS
Reagents and chemicals
Depleted uranium as uranyl acetate dihydrate [6159-44-0] (UA) was obtained from International Bio-Analytical Industries, Inc. (Boca Raton, FL).
Preparation of DU compounds
Uranyl acetate (UA) was used as a soluble DU compound. Solutions of UA were prepared by weighing out the desired amount of UA and dissolving it in double distilled water. Dilutions were made for appropriate treatment concentrations and then filter sterilized through a 10 ml syringe with a 0.2 μm filter.
General cell culture conditions
Chinese Hamster Ovary (CHO) AA8, EM9, V3.3, and UV5 cell lines were purchased from the American Type Culture Collection (Manassas, VA). The Chinese Hamster Ovary (CHO) H9T3, and 5T4-12 cell lines were a kind gift from the Dr. Larry H. Thompson Laboratory (Lawrence Livermore National Laboratory, Livermore CA). Table 1 and Table 2 show the type of mutant CHO cells utilized in this study. Cells were thawed from cryopreservation, cultured in α-MEM (Hyclone, Logan, UT) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), antibiotic/antimycotic (100 U/ml penicillin, 100 mg/ml streptomycin) (Sigma) and 1 mM glutamine (Gibco-BRL, Rockville, MD). Cells were maintained at 37 °C in a 5% CO2/air humidified incubator calibrated with a Fryrite analyzer (Bacharach Co., Pittsburgh, PA).
Table 1.
Mutant Chinese hamster ovary cell lines. The repair deficient cell lines that prevent specific DNA damage repair mechanisms. As a result, the cell lines are sensitive to specific types of DNA lesions.
| Cell lines: | Gene Deficiency: | Protein Function: | Sensitivity: | References: |
|---|---|---|---|---|
| AA8 (parental) | – | – | – | – |
| EM9 | XRCC1 | Acts as scaffolding protein, ligation, gap filling (BER) | Single strand breaks | (Thompson 1990) |
| V3.3 | XRCC7 | Non-homologous end joining repair (NHEJ); reduced expression of the DNA-PKCS | Double strand breaks | (Rothkamm 2003) |
| UV5 | ERCC2 (XPD) | NER | Bulky adducts | (Cullinane 1997) |
Table 2.
Complemented Chinese hamster ovary cell lines. The repair proficient cell lines that re-express the human cloned genes of the mutant repair deficient cell lines. These cells have been shown to have full (H9T3) and partial (5T4-12) recovery of its respective DNA repair mechanisms.
| Cell lines: | Human Gene Cloned: | Protein Function: | Sensitivity: | References: |
|---|---|---|---|---|
| H9T3 | XRCC1 into the EM9 cell line | Acts as scaffolding protein, ligation, gap filling (BER) | – | (Thompson 1990) |
| 5T4-12 | ERCC2 into the UV5 cell line | NER | – | (Weber 1988, Cullinane 1997) |
Intracellular DU measurements
Inductively coupled plasma mass spectrometry (ICP-MS) was used to determine U concentration in the total cell (“Total Cell Fraction”) and intracellular U concentrations in the cytosol (“Cytosol Fraction”) and nucleus (“Nuclear Fraction”) as previously described (Guillemin 2005). Briefly, 8 × 106 cells were seeded and allowed to adhere for 24 hr. Cells were then treated with UA for 0, 24 or 48 hr. After treatment, cells were washed three times with 1X phosphate buffered saline (PBS) and harvested by trypsinization with Accutase® (Innovative Cell Technologies, San Diego, CA). Cells were pelleted by centrifugation at 3500 rpm in a Beckman T-23R centrifuge for 5 min and resuspended in 0.5 ml of CBL buffer (10 mM HEPES, 10 mM NaCl, 1 mM KH2PO4, 5 mM NaHCO3, 5 mM EDTA, 1 mM CaCl2, 0.5 mM MgCl2) and incubated for 5 min on ice (4 °C). The cell suspension was homogenized by applying 50 strokes with a motorized homogenizer at 250 rpm. Thereafter, 50 μl of 2.5 M sucrose was added to the cell suspension to restore isotonic conditions and 100 μl of the cell suspension was set aside to determine total DU cell content (“Total Cellular Fraction”). The remaining cell suspension was centrifuged at 6,300 × g for 10 min at 4 °C. The supernatant was removed and set aside for further processing (supernatant 1). The remaining pellet was resuspended in 1 ml of TSE buffer (10 mM Tris-HCl, 300 mM sucrose, 1 mM EDTA, 0.1% IGEPAL-CA 630 v/v, pH 7.5). The cell suspension was homogenized by applying 30 strokes with a motorized homogenizer at 250 rpm cells and centrifuged at 4,000 × g for 10 min at 4 °C. The supernatant was removed and the resulting pellet was washed with TSE buffer. The pellet was then resuspended in 100 μl TSE buffer (“Nuclear Fraction”). The supernatant 1 fraction was then centrifuged at 14,000 × g for 150 min and the supernatant was collected (“Cytosol Fraction”). Aliquots of the cell homogenate, nucleus, and cytoplasm fractions were kept for protein determination by using the microassay procedure, Coomassie Protein Assay Reagent Kit per instructions by manufacturer (Pierce Biotechnology, Rockford, IL). The colormetric intensity was immediately measured at an absorbance of 595 nm on a plate reader (Molecular Devices, Sunnyvale, CA). Protein content was expressed as micrograms per milliliter (μg/ml) using bovine serum albumin (BSA) as a standard. The remainder of each fraction was mixed with nitric acid (1:1 v/v) and digested at room temperature for 48 hr for further ICP-MS (ICP-MS Agilent 7500, Agilent Technologies) analysis. Results of the intracellular U contents were normalized by cellular proteins and expressed as micrograms of uranium per milligram protein (μg U/mg protein) for n = 3 independent experiments.
CHO AA8 processing
Chinese hamster ovary (CHO) AA8 cells were seeded onto 13 mm Thermanox cover slips at a density of 2.5 × 105 cells per 60 mm dish. Cells were grown for 24 hr and then treated for 24 hr with 220 μM of UA, or left untreated as controls. Medium was then removed, and cells were washed with 1X PBS once and primary fixation done with ~4 ml of 2% glutaraldehyde (EM grade, distillation purified; Electron Microscopy Sciences, Hatfield, PA) in 1X PBS for 1 hr. The glutaraldehyde was then removed and the cover slips were washed with 1X PBS. Cover slips were then transferred into 20 ml glass scintillation vials and post-fixed with 1% osmium tetraoxide (OsO4; Electron Microscopy Sciences, Hatfield, PA) in 1X PBS for 30 min and then rinsed with 1x PBS. Cover slips were then dehydrated in an ethanol series (50, 70, 95, 3x 100%). Infiltration began with neat propylene oxide (PO) and then mixtures of 1:1 and 1:3 PO to resin (Embed 812) and rotated for at least 4 hr, and then rotated in pure resin for at least 4 hr. Cover slips were then placed into embedding molds (Chang monolayer) and embedded with fresh resin and sufficiently polymerized in a 60 °C oven overnight. Embedded cover slips were then cut and adhered with resin to a resin block.
Sectioning for transmission electron microscopy
Sample blocks were trimmed to 1.5 mm2 sections with a safety-edge razor blade. Thick sections (0.92 μm) were trimmed from each block and wet-mounted to a glass slide, and were examined by compound light microscopy to identify the positions of cells within the sample block. Sample blocks were then thinly sectioned (0.72 nm) with a Diatome 45° diamond knife on an Ultra 2 microtome.
Approximately 2–4 thin sections of each block were placed onto a 200 square mesh copper grid (Electron Microscopy Sciences) with Formvar film coating using a looped copper grid with a droplet of water to capture the sections. Three grids were made for each sample. Each grid was stained with saturated uranyl acetate in 50% EtOH for 20 min and then lead citrate for 5 min. The stained samples were then analyzed with a JEOL 1200 II TEM at 60 kV.
Cytotoxicity measurements
Cytotoxicity, as decreased cell survival, was determined by measuring colony forming ability in the CHO AA8, EM9, V3.3, UV5, H9T3, and 5T4-12 cell lines. Cytotoxicity measurements were carried out after incubation with UA. Cells were seeded at 8 × 105 cells per 100 mm plate, allowed to adhere for over 24 hr, and treated with sterile-filtered aqueous solutions of UA (0 – 300 μM) for 24 or 48 hr. Upon completion of exposure, cells were trypsinized to remove the cells from the plates, quantified on a hemocytometer and reseeded at 200 cells per 60 mm dish in quadruplicate. After 7 days, all dishes were stained with crystal violet and the colonies were counted. Cell survival was calculated as percent colonies in treated dishes relative to untreated controls. Data are presented as percentages of controls, for n = 8 – 11 independent experiments. Plating efficiency for each CHO cell line was: 91%, 83%, 71%, 63%, 95%, and 79% for the CHO AA8, EM9, V3.3, UV5, H9T3, and 5T4-12 cell lines respectively.
Detection of DNA damage in CHO cells
The Fast Micromethod® was used to detect DNA damage (strand breaks, alkali-labile sites and incomplete excision repair) as previously described (Batel 1999, Schröder 2006, Ullmann 2008). The Fast Micromethod® determines DNA integrity in cell suspensions in single microplates. PicoGreen® (Molecular Probes, Eugene, OR) is a fluorescence dye that binds specifically with double stranded DNA (dsDNA), in the presence of single strand DNA (ssDNA) and proteins. Following DNA denaturation in a high alkaline buffer (pH 12.40), DNA unwinding occurs and PicoGreen® is released. Due to the specificity of PicoGreen® to dsDNA, the amount of DNA double strands in a given sample is directly proportional to the intensity of DNA-bound PicoGreen® fluorescence. The more damaged DNA present, the more DNA strand breaks there are to serve as sites of DNA unwinding in the high alkaline solution. The unwinding of DNA releases PicoGreen® over a certain period of time during denaturation in an alkaline buffer (i.e. the intensity of the measured fluorescence). This reflects the extent of DNA damage. The specific dsDNA binding allows for the direct fluorometric measurements of dsDNA denaturation without sample handling and stepwise DNA separations compared to the single cell gel electrophoresis assay (Comet assay) (Bihari 2002). All solutions were prepared according to Schröder et al. (2006). Briefly, cells were seeded and allowed to adhere for 24 hr. Cells were then treated with UA for 24 or 48 hr. After treatment, cells were washed with 1X PBS and harvested by trypsinization. Cells were resuspended in PBS, counted, and a cell concentration of 150,000 cells/ml was added to a 96-well plate. Lysis solution mixed with PicoGreen® was added to each well for 40 min without mixing and protected from light to prevent further DNA damage. After the cells were lysed, freshly prepared NaOH-EDTA solution (pH 12.40 ± 0.2) was added. The fluorescence intensity was immediately measured at an excitation wavelength of 480 nm and an emission wavelength of 520 nm for 20 min in 30 sec intervals on a fluorescence plate reader (Molecular Devices Gemini-X fluorescence spectrophotometer, Molecular Devices, Sunnyvale, CA). After measuring the fluorescence, 50 μl aliquots of each cell suspension was transferred to a 96-well plate to determine protein content by using the microassay procedure, Coomassie Protein Assay Reagent Kit, per instructions by manufacturer (Pierce Biotechnology, Rockford, IL). Protein content was expressed as micrograms per milliliter (μg/ml) using bovine serum albumin (BSA) as a standard. The colormetric intensity was immediately measured at an absorbance of 595 nm on a plate reader (Molecular Devices, Sunnyvale, CA). Protein content was calculated as a function of a BSA standard curve. Blank values of cell-free wells were subtracted from each sample value (corrected fluorescence). Thereafter, the normalized fluorescence was calculated by taking into account the protein content in each well as follows:
Strand Scission Factor (SSF) is the parameter for DNA damage, and was calculated as the log10 of the ratio of the fluorescence of the sample to the untreated samples after 20 min of unwinding. These values were then multiplied by −1 to obtain positive values as shown for n = 4 independent experiments:
Detection of Abasic Sites
Abasic site analysis was carried out utilizing the OxiSelect Oxidative DNA Damage Quantitation Kit (AP Sites) (Cell BioLabs, San Diego, CA) according to the manufacturer’s instructions. An apurinic/apyrimidinic site (AP or abasic sites) is one of the most frequently encountered DNA lesions in cells that can be spontaneously generated or induced by exposure to oxidative stress or to genotoxic agents such as ionizing radiation (Loeb 1986). Briefly, cells were seeded and allowed to adhere for 24 hr. Cells were then treated with 0 – 300 μM UA for 24 or 48 hr. After treatment, cells were washed with 1X PBS and harvested by trypsinization. Genomic DNA was isolated with the DNeasy® total DNA isolation kit according to manufacturers’ instructions (Qiagen, Valencia, CA) and tagged with an Aldehyde Reactive Probe (ARP) to react specifically with an aldehyde group on the open ring form of AP sites. The AP sites were then tagged with biotin and detected with a streptavidin-enzyme conjugate. The colormetric intensity was immediately measured at an absorbance of 450 nm on a plate reader (Molecular Devices, Sunnyvale, CA). AP sites were then determined by comparing the absorbance with a standard curve generated by the provided DNA standard containing predetermined AP sites for n = 3 independent experiments.
Statistical analysis
Data were expressed as mean ± SEM and analyzed by either one-way or two-way ANOVA, followed by post-hoc Tukey or Bonferroni posttests for multiple comparisons between treatment and untreated groups; p < 0.05 was considered to be significant.
RESULTS
UA is internalized in the nucleus of CHO AA8 cells
It is unclear where soluble uranium is distributed once it enters the cell at low concentrations. Therefore, UA cell content was measured in confluent CHO AA8 cells that were exposed to 50 – 300 μM UA for 0 – 48 hr by ICP-MS to determine where UA is internalized and accumulated after treatment (Figure 1). After 24 hr, the total cellular content of U was shown to accumulate in a concentration dependent manner with 332 and 429 μg U/mg protein measured at the 200 μM and 300 μM (p < 0.05 and p < 0.01, respectively) concentrations (Figure 1A). The 48 hr time point also indicated that U accumulates in the cell with 334 and 208 μg U/mg protein measured at the 200 μM and 300 μM UA doses, respectively. It was observed that UA did not accumulate in the total cell at the 0 hr time point as the concentrations of UA were increased.
Figure 1.
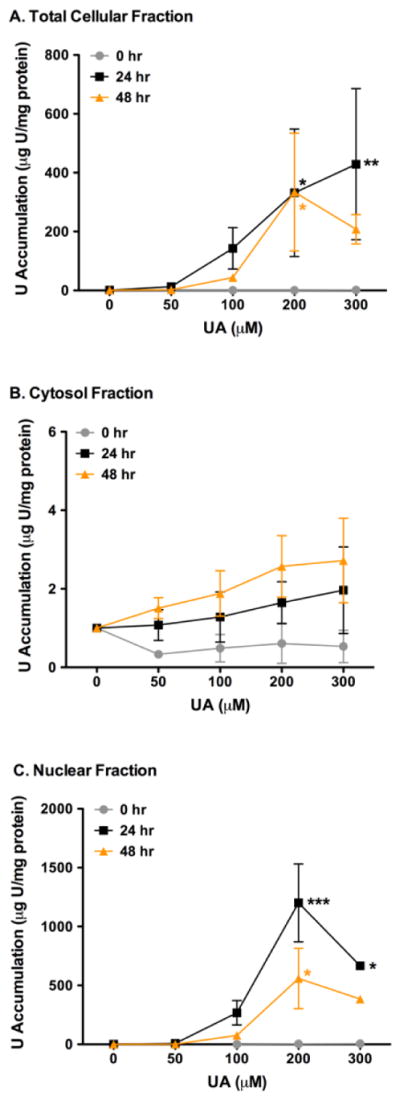
Time course of total intracellular U content (μg U/mg protein) in CHO AA8 cells after 0 (●), 24 (■), and 48 (▲) hr. A. U accumulation is seen after 24 hr in the whole total cellular fractions. B. No changes were seen in U in the cytosol after 0 – 48 hr exposure compared to the accumulation of U in the total cellular fractions and nuclear fractions. C. Increasing amounts of U is accumulated in the nuclear fraction after 24 hr. Cells were treated and UA concentrations were determined by ICP-MS and normalized for the total protein content as described in the text. P values were considered to be statistically significant as indicated with * (p < 0.05), ** (p < 0.01) and *** (p < 0.001) compared to untreated cells.
The amount of U measured in the cytosol fractions increased as time and concentrations increased with UA treatment, however the accumulation of U was low compared to the total cellular and nuclear fractions. Uranium accumulation was measured at 1.6 and 2.0 μg U/mg protein and 2.6 and 2.7 μg U/mg protein at 200 μM and 300 μM UA after 24 hr and 48 hr, respectively (Figure 1B).
Interestingly, UA was observed to be accumulating in the nuclear fraction as the concentrations and time exposure increased in cells treated up to 200 μM after 24 and 48 hr and decrease with cells treated with 300 μM UA (Figure 1C). After 24 hr, U was measured to accumulate 1201 and 667 μg U/mg protein at the 200 μM (p < 0.001) and 300 μM (p < 0.05) concentrations. The 48 hr time indicated uranium accumulation of 559 and 384 μg U/mg protein at the 200 μM (p < 0.05) and 300 μM UA doses in the nucleus, respectively. As observed in the total cellular fraction, UA did not accumulate in the nuclear fraction at the 0 hr time point as UA concentrations were increased.
Electron transmission microscopy was utilized to investigate the accumulation of UA in confluent CHO AA8 cells that were exposed to 220 μM UA for 24 hr (Figure 2). Untreated AA8 cells did not have uranium deposits in the cells, despite the staining process with uranyl acetate (Figure 2A). After 24 hr of UA treatment, uranium deposits in the cell consisted of particulates, presumed to be uranyl phosphate, either isolated or grouped in clusters (Salanga and Stearns, unpublished). The electron dense materials were present in the nucleus (Figure 2B). Collectively, the data indicates that uranium can readily enter cells and localize in the nucleus after 24 and 48 hr and begin to precipitate at higher concentrations (300 μM UA) for clearance or overt toxicity.
Figure 2.

Representative TEM micrograph of CHO AA8 cells. A. An untreated CHO AA8 cell. (15,000x magnification, bar = 500 nanomicron) N= nucleus, C=chromatin, MV= microvilli, R= ribosomes. B. Representative TEM micrograph of a CHO AA8 cell exposed to 220 μM UA for 24 hr. (6000x magnification, bar= 1 micron) N= nucleus, M= mitochondria, UP= uranyl phosphate, V= vacuoles.
UA is cytotoxic in DNA repair-deficient CHO cells
Clonogenic assays were utilized to measure cytotoxicity in both repair-proficient and repair-deficient CHO cells lines listed in Tables 1 and 2. The overall hypothesis is that if a test agent causes DNA damage, then cell lines deficient in the repair of the specific lesion(s) should be more sensitive to the test agent and decreased cell survival would be observed relative to the wild-type or repair-proficient cell line. More specifically, the types of UA-induced DNA damage in vitro should be more apparent in the DNA repair-deficient CHO lines (EM9, V3.3, and UV5) compared to the parental CHO AA8 line, as shown in Figure 3. The single strand break sensitive (SSB) cell line, EM9, exhibited the most sensitivity to UA treatment compared to untreated EM9 cells after 24 hr (Figure 3A). At the higher 100, 200, and 300 μM UA concentrations, the EM9 cells had significant increases in cell death of 24% (p < 0.001), 44%, and 52% (p < 0.001), respectively. In the DNA-adduct sensitive cell line, UV5, treatment with 100 – 300 μM UA doses caused significant cytotoxicity compared to untreated UV5 cells (p < 0.001). The higher UA doses (200 and 300 μM; p < 0.01) produced significant cytotoxicity in the double strand break (DSB) sensitive cell line, V3.3, compared to untreated V3.3 cells. The parental AA8 cell line did not have significant change in cytotoxicity in UA-treated cells compared to untreated AA8 cells after 24 hr.
Figure 3.

Cytotoxicity of UA in CHO AA8 (●), EM9 (■), V3.3 (▲), and UV5 (▼) cells after 24 hr (A) and 48 hr (B). Cells were treated and assayed for the 7-day colony formation as described in the text. Compared to no uranium exposures, UA induced significant cytotoxicity after 24 hr in the repair deficient EM9, UV5 and V3.3 cell lines. At the higher doses, UA did not produce significant cytotoxicity in the parental AA8 cell line compared to the untreated cells. Data represents mean ± SEM for n = 8 – 11 independent experiments. P values were considered to be statistically significant as indicated with * (p < 0.05), ** (p < 0.01) and *** (p < 0.001) compared to untreated cells in the same cell line after 24 hr; a (p < 0.05) and b (p < 0.001) are the p values of UA treated cells compared to cells treated with equivalent UA doses in the parental AA8 cell line after 24 or 48 hr.
After 48 hr exposure, the cytotoxicity profiles for the UV5 and the EM9 cell lines significantly increased compared to the V3.3 and the AA8 cell lines (Figure 3B). The DNA-adduct sensitive cell line, UV5, had the most significant changes in cytotoxicity after 48 hr of UA exposure. Cells treated with 100 – 300 μM UA resulted in significant (p < 0.001) cell death compared to untreated UV5 cells and to the parental AA8 cells treated with 100 – 300 μM UA after 48 hr. The increased cytotoxicity of UA-treated UV5 cells at 48 hr was also significant compared to the 24 hr UA exposures. The SSB sensitive EM9 cells treated with 200 – 300 μM UA had significant increases in cell death (p < 0.001) compared to untreated EM9 cells and to the parental AA8 cells treated with 200 – 300 μM UA after 48 hr. Collectively, these results inferred that UA produced DNA adducts and single strand breaks after 48 hr of exposure.
Both the parental AA8 cells and DSB sensitive V3.3 cells showed similar effects after UA treatment. Treatment of 300 μM UA produced a significant (p < 0.001) increase in cell death compared to untreated cells after 48 hr. Despite the significant increase in cytotoxicity at 300 μM for both cell lines, there was no significant change in cytotoxicity after 48 hr time exposures in the AA8 and V3.3 cell lines at the lower concentrations of UA. These results inferred that UA does not induce DNA double strand breaks at concentrations lower than 200 μM UA after 48 hrs.
UA cytotoxicity is decreased in complemented DNA repair-deficient CHO cells
Based on the cytotoxic responses of the repair-deficient EM9 and UV5 cells, the complemented mutant CHO H9T3 and 5T4-12 cell lines were used to investigate if the observed UA effects were directly related to the repair-deficiency. The complemented H9T3 cell line is the CHO EM9 cell line cloned with the human XRCC1 gene and has been shown to have full XRCC1 functional recovery (Thompson 1990). The clonogenic assay was used to investigate the cytotoxic response of H9T3 cells treated with increasing concentrations of UA compared to UA treated EM9 cells for 24 and 48 hr (Figure 4). After 24 hr exposure, H9T3 cells treated with 50 – 300 μM UA did not have a significant cytotoxic response compared to untreated H9T3 cells (Figure 4A). A significant decrease in cytotoxicity was observed in comparison between the H9T3 cells and EM9 cells treated with 300 μM UA after 24 hr (p < 0.001). The 48 hr UA exposure did not produce significant cytotoxicity in the H9T3 cells compared to untreated H9T3 cells (Figure 4B). The UA cytotoxicity in the H9T3 was significantly reduced compared to the EM9 cells (p < 0.001) in the 200 and 300 μM UA doses. Based on the current observations, it was inferred that the significant increase in UA-induced cytotoxicity in the EM9 cells were directly related to the BER deficiency.
Figure 4.

Cytotoxicity of UA in CHO AA8 (●), EM9 (■), and H9T3 (▲) cells after 24 hr (A) or 48 hr (B). Cells were treated and assayed for 7-day colony formation as described in the text. UA-treated corrected mutant H9T3 cells, showed a significant increase in survival compared to the repair-deficient EM9 cells after 24 hr. Data represents mean ± SEM for n = 8 – 11 independent experiments. P values were considered to be statistically significant as indicated with * (p < 0.05), ** (p < 0.01), and *** (p < 0.001) compared to untreated cells for the respective cell line; a (p < 0.05) and b (p < 0.001) is the p values of UA-treated EM9 cells compared to cells treated with equivalent UA doses in the parental AA8 cell line after 24 and 48 hr; c (p < 0.01) and d (p < 0.001) are the p values of UA-treated EM9 cells compared to cells treated with equivalent UA doses in the repair proficient H9T3 cell line after 24 and 48 hr.
The complemented 5T4-12 cell line is the CHO UV5 cell line cloned with the human ERCC2 gene and has been shown to have partial ERCC2 functional recovery (Weber 1988). 5T4-12 cells treated with UA did not have a significant cytotoxic response compared to untreated 5T4-12 cells with the 200 and 300 μM doses (Figure 5). Although a decrease in cytotoxicity was observed in comparison between the 5T4-12 cells and UV5 cells treated with 300 μM UA after 24 hr (25% vs. 38%), it was not significant (Figure 5A). After 48 hr of exposure, 200 and 300 μM UA generated a significant cytotoxic response in the 5T4-12 cells compared to untreated 5T4-12 cells (Figure 5B). The 5T4-12 cells had an intermediate response compared to the parental cell line (AA8) and the ERCC2 gene deficient UV5 cell line. The transfection of the human ERRC2 gene back into the UV5 cells (5T4-12) resulted in decreased cytotoxicity. The reduction in UA cytotoxicity in the 5T4-12 cells was significant compared to the UV5 cells treated with 100 μM UA (p < 0.001). These results suggested that the significant increase in UA-induced cytotoxicity in the UV5 cells was directly related to the NER deficiency.
Figure 5.

Cytotoxicity of UA in CHO AA8 (●), UV5 (■), and 5T4-12 (▲) cells after 24 hr (A) and 48 hr (B). Cells were treated and assayed for 7-day colony formation as described in the text. UA-treated corrected mutant cells, 5T4-12, showed a significant increase in survival compared to the repair-deficient UV5 cells after 24 hr. Data represents mean ± SEM for n = 8 – 11 independent experiments. P values were considered to be statistically significant as indicated with ** (p < 0.01) and *** (p < 0.001) compared to untreated cells for the respective cell line after 24 or 48 hr; a (p < 0.05) and b (p < 0.001) are the p values of UA treated UV5 and 5T4-12 cells compared to cells treated with equivalent UA doses in the parental AA8 cell line after 48 hr; d (p < 0.001) is the p value of UA treated UV5 cells compared to cells treated with equivalent UA doses in the repair proficient 5T4-12 cell line after 48 hr.
UA induces single strand breaks in CHO cells in vitro
Increased cytotoxicity in the BER-deficient EM9 cell line indicates that UA may cause DNA strand breaks in vitro. The presence of UA induced strand breaks was confirmed by the Fast Micromethod® Assay. DNA strand breaks were measured after 24 and 48 hr exposures with increasing doses of UA (Figure 6). Interestingly, the results of 24 hr UA exposures in CHO AA8 and EM9 with the Fast Micromethod® Assay were similar to the results previously published (Stearns 2005). Stearns et al. utilized the comet assay to determine the effects of UA exposures in CHO AA8 and EM9 cell lines. It was observed that 50 – 300 μM of UA produced similar increases in tail moment relative to untreated controls after 24 hr exposures (Stearns 2005). Following 24 hr exposure to UA, there were no dose dependent differences in the SSF between the various cell lines tested. Single strand breaks were produced in the parental cell line, AA8, and the DSB-sensitive cell line, V3.3, at the highest UA dose (300 μM) (Figure 6A). The SSB-sensitive cell line, EM9, and the XRCC1-complemented cell line, H9T3, produced similar amounts of SSBs after exposure to 200 and 300 μM UA after 24 hr. The adduct sensitive cell line, UV5, was not utilized in this study as this cell line has functional BER mechanisms which yield similar results to the parental AA8 cell line.
Figure 6.

Strand breaks detected by the Fast Micromethod® Assay in terms of the strand scission factor (SSF) in CHO AA8 (●), EM9 (■), V3.3 (▲), and H9T3 (▼) cells treated with UA after 24 hr (A) and 48 hr (B) exposures. UA induced significant single strand breaks in the repair deficient cell line, EM9, after 48 hr compared to untreated cells. Cells were treated and assayed for strand breaks as described in the text. Data represents mean ± SEM for n = 4 independent experiments. P values were considered to be statistically significant as indicated with * (p < 0.01) and ** (p < 0.001) compared to untreated cells for the respective cell line.
After 48 hr, there were significant increases in strand breaks in the SSB-sensitive EM9 cell line at 100 – 300 μM of UA (p < 0.01 and p < 0.001) (Figure 6B). There were no differences in strand breaks with the parental, AA8, and DSB repair-deficient, V3.3, cell lines as the concentration of UA increased. Interestingly, the XRCC1-complemented H9T3 cell line did not have a significant amount of SSBs compared to the mutant EM9 cell line. It was concluded that UA induces the formation of SSBs.
UA induces abasic sites in repair-deficient cells
UA induced SSBs in the CHO cell lines. An apurinic/apyrimidinic site (AP or abasic sites) is one of the most frequently encountered DNA damage lesions in cells that can be spontaneously generated or induced by exposure to oxidative stress or to genotoxic agents such as ionizing radiation (Loeb 1986). AP sites are also created by DNA glycosylases during BER in response to DNA damage from a wide variety of damaging agents and proposed to come from U-DNA adduct formation (Dianov 2003, Wilson 2014). AP sites may affect cell viability and genomic integrity by interfering with normal DNA replication, thereby making AP sites cytotoxic and mutagenic (Loeb 1986, Dianov 2003). The formation of AP (or abasic) sites was measured as shown in Figure 7. The parental cell line, AA8, did not produce significant AP sites at any of the doses of UA treatment compared to untreated cells after 24 and 48 hr (Figure 7A). This may indicate that UA does not induce AP site formation or these types of lesions are readily repaired by BER mechanisms.
Figure 7.
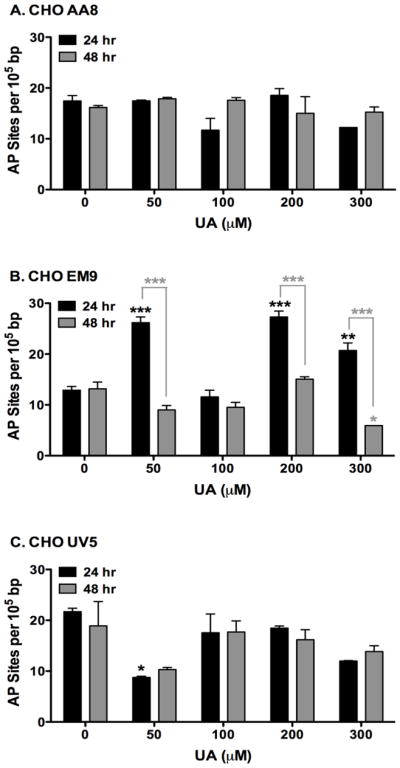
Therefore, repair-deficient cell lines were utilized to determine the formation of AP sites. In the SSB-sensitive cell line, EM9, cells treated with 50, 200, and 300 μM UA significantly induced the formation of AP sites compared to untreated cells after 24 hr (Figure 7B). It is unclear as to the lack of response in the 100 μM UA. The measured amount of AP sites was significantly reduced after 48 hr UA exposure compared to 24 hr UA exposures (p < 0.0001). UA did not significantly induce AP site production in the adduct sensitive cell line, UV5, after 24 hr and 48 hr exposures compared to untreated cells (Figure 7C). These results indicate that UA is able to induce AP sites, however, due to functional BER mechanisms, these lesions are readily repaired.
DISCUSSION
The purpose of the study was to characterize the types of DNA damage produced by DU in the form of soluble uranyl acetate at environmentally relevant concentration ranges. UA was shown to readily enter CHO cells and localize in the nucleus in a time dependent manner as previously observed (DiSpirito 1983, Kalinich 2001, Rouas 2010, Guéguen 2015). Although the mechanism is unclear, it is postulated that uranium enters the cell via endocytosis as observed with nickel (Mirto 1999, Kasprzak 2003). Prat et al. (2005) has shown that nonselective vesicle transporters were up-regulated in human renal HEK293 cells treated with 250 μM UA (Prat 2005). The amount of the uranium accumulation in the total cellular fraction of the CHO AA8 cells was low compared to the nuclear fraction of treated cells. This phenomenon may be explained due to the common method of differential centrifugation method used in this study. This approach is effective in separating the majority of subcellular components, without requiring pre-concentration procedures or the addition of reagents. However, the differential centrifugation approach can produce clumping or breakage of organelles, leading to leakage of their soluble constituents into other fractions (Rosabal 2014). In this case, the leakage may have added to the accumulation in the nuclear fraction, rather than in the cytosol fraction. Additionally, cells that did not undergo the differential centrifugation formed uranyl phosphate precipitates in the nucleus rather than in the cytosol indicates that UA accumulates in the nucleus as previously shown (Guéguen 2015).
The mechanism of DNA interaction is unclear. The clonogenic assay showed that UA had a significant cytotoxic effect in the BER-deficient EM9 and the NER-deficient UV5 cell lines, compared to the parental AA8 cells as previously observed. The x-ray repair cross-complementing gene I protein (XRCC1) deficiency in the EM9 cell line prevents formation of a complex with ligase IIIα, poly(ADP-ribose) polymerase 1 (PARP-1), PARP-2, and several other DNA repair proteins to prevent the repair of single strand breaks (Thompson 1990). ERCC2, is a protein involved in the incision step of NER (Cullinane 1997). The ERCC2-deficiency in the UV5 cell line prevents the removal of bulky chemical adducts and UV-induced photoproducts from DNA. The observation that UA was significantly cytotoxic in EM9 and UV5 cell lines suggests that UA induces SSBs and DNA adducts in CHO cells. To further validate this observation, the mutant EM9 and UV5 cells were compared to the complemented H9T3 and 5T4-14 cell lines. The H9T3 cells have the human XRCC1 gene clone which fully corrects the repair deficiency in the mutant EM9 cell line (Thompson 1990). The 5T4-12 cells have the human ECRR2 gene clone that partially corrects the repair deficiency in the mutant UV5 cell line (Weber 1988). The decreased cytotoxicity in the corrected CHO mutant cell lines, H9T3 and 5T4-12, further suggests that UA produces bulky adducts and SSBs rather than DSBs in CHO cells at lower concentrations. The most sensitive DNA damage was produced after exposure to 100 μM of UA in the UV5 cell line which infers that UA is producing UA-DNA adducts after 48 hr of exposure as previously observed (Stearns 2005). These adducts could result in DNA hydrolysis leading to SSBs as observed with the lanthanides (Franklin 2001, Wilson 2014).
The XRCC7-deficient V3.3 cell line did not show any additional cytotoxicity above what was observed in the parental AA8 cell line. A deficiency in XRCC7 in the V3.3 cell line leads to a reduced expression of the DNA-PKCS and results in defective DSB repair via NHEJ repair (Rothkamm 2003). The lack of a significant cytotoxic response in the NHEJ-deficient V3.3 cell line after 24 and 48 hr UA exposures was expected below 200 μM. Similar cytotoxicity results were observed in XRCC3-deficient and RAD51D-deficient CHO cells treated with particulate DU (Holmes 2014). Interestingly, XRCC3 and RAD51D are involved in homologous recombination (HR) DNA repair. It was inferred that HR repair has a minor role in cell survival after particulate DU exposure. The lack of additional cytotoxicity in the repair deficient cell lines were attributed to the formation of chromatid lesions and chromosome fragmentation. Another previous study has shown that uranyl nitrate (UN) induced single- and double-strand breaks in NRK-52E cells at sub-lethal (300 μM UN) and lethal (400 μM+ UN) concentrations, respectively (Thiébault 2007). NRK-52E cells treated with UN had a significant increase in γ-H2AX staining (an indication of DSB damage) as UN concentrations increased (400 – 500 μM). γH2AX has an important role in the processing and repair of DSBs. We have also performed γ-H2AX immunostaining experiments in human bronchial epithelial cells (16HBE14o−) treated with 0 – 0.7 μM of UA for 0 – 48 hr. At low UA concentrations, there was no observable difference in γ-H2AX staining between UA-treated and untreated cells after 48 hr (data not shown).
Previous results have shown a significant difference between the AA8 and EM9 cell lines in terms of DNA strand break production after 24 hr UA (0 – 300 μM) exposures (Yazzie 2003). Similar results were seen in this current study. It was previously suggested that strand breaks and the presence of other forms of damage (ie. DNA adducts and DNA crosslinks) may have interfered in the migration of DNA in the comet assay, thereby decreasing tail migration, as has been observed for platinum and other cross-linking agents (Kawanishi 2002). After a 48 hr time exposure, 100 – 300 μM UA doses caused a significant increase in SSBs in the BER-deficient EM9 line compared to the NHEJ-deficient V3.3, XRCC1-complemented H9T3, and parental AA8 cell lines. It is proposed that if UA is forming UA-DNA adducts in a time dependent manner in CHO cells, then it may suggest two possible outcomes: (1) the accumulation of UA-DNA adducts may directly induce SSBs after 48 hr exposure via DNA hydrolysis, or (2) the amount of indirectly-induced SSBs are significant and cannot be repaired, leading to cell death.
To further explore this idea, AP sites were measured in the parental, AA8, and repair-deficient, EM9 and UV5, cell lines. Uranyl acetate was shown to produce a significant amount of AP sites in the BER-deficient EM9 cell line after 24 hr. AP sites are readily repaired in the early stages of BER (Vidal 2001), therefore a deficient BER mechanism may account for this observation. Additionally, Vidal et. al. (2001) demonstrated that AP sites are repaired in early stages of BER, which may explain the decreased amount of AP sites measured in the EM9 cell line after 48 hr UA exposure. It has been proposed that the formation of uranyl-DNA adducts may act as a parent lesion that maybe further convert to stand breaks, AP sites, or possibly DNA cross-links which supports the current and previously published results (Wilson 2014). The parental, AA8, and NER deficient, UV5, cells did not have a significant increase in AP sites production, which suggests that (1) that these lesions are readily repaired by the fully functional BER mechanisms, and (2) that oxidative stress may not be a major pathway of DNA damage induction, as AP sites are a prevalent lesion of oxidative DNA damage.
As observed in other heavy metals, uranium is considered carcinogenic, mutagenic, and genotoxic by more than one mechanism. Based on current results and previously published studies, we postulate that UA maybe inducing DNA damage by primarily forming UA-DNA adducts, which may directly cause SSBs rather than indirectly inducing oxidative stress strand breaks at low UA concentrations. Previous studies have also shown that the uranyl ion (UO22+) is known to bind strongly to oligonucleotides (Wu 1996) and DNA to form a DU-phosphodiester moiety in the minor grooves of DNA, more specifically, the N3 position of adenine (Nielsen 1992, Wilson 2014). Wu et al. (1996) has also shown that one uranyl ion is able to displace either two protons or two sodium ions (Na2+) to bind to two phosphates of DNA phosphate backbone. The resulting U-DNA phosphate complex observed did not promote significant structural changes in the small oligonucleotides studied (Wu 1996). Interestingly, the proposed uranium binding position of the N3 position of adenine in the minor groove has been shown to a binding site for d-block divalent metals Cu(II), Zn(II), and Cd(II) (Nielsen 1992). Although the specific mechanisms are unclear, current studies have shown that uranium induced cytotoxicity is significantly decreased with pre-treatments of zinc (Hao 2014, Cooper 2016). Further studies should not only focus on the formation of U-DNA adducts, but to determine if the adducts are responsible for the observed increased SSBs.
Further efforts are ongoing to determine if the observed UA-induced DNA damage is possibly due to the uranium-induced inhibition of DNA repair pathways. Many heavy metals are known to have weak carcinogenic and/or mutagenic potentials, or rather act as co-mutagens to disrupt different DNA repair systems (Hartwig 2002). DU has been previously shown to induce cell transformation in human osteoblast cells, leading to increased levels of k-ras oncogene expression, altered pRB phosphorylation, and increased sister chromatid exchanges (Miller 1998a). Further research needs to address DU exposure on genes and protein interactions at the molecular level to determine the long-term effects on possible neoplasia formation. A basic understanding of the molecular mechanisms involved in damage and repair of these lesions is critical to understanding the events leading to DU-induced carcinogenesis and possible neoplasia.
HIGHLIGHTS.
UA localizes in the nucleus of CHO cells
UA is significantly cytotoxic in CHO repair deficient cells sensitive to single strand breaks and bulky adduct formation
UA cytotoxicity is decreased in complemented DNA repair-deficient CHO cells
UA induces abasic sites in CHO repair deficient cells
Acknowledgments
FUNDING INFORMATION:
This work was supported by the National Institutes of Health [CA096281 (RCL), CA096320 (DS), ES019703 (DS), and F31ES014971 (MY)], Southwest Environmental Health Sciences Centers [P30ES006694 (RCL)], The Alfred P. Sloan Foundation (MY), and the More Graduate Education at Mountain States Alliance (MY).
We thank Mike Kopplin of the University of Arizona Superfund Basic Research Program Hazard Identification Core [P42ES004940] for performing the ICP-MS total metal analysis. We would also like to thank Dr. Larry H. Thompson for the use of the CHO H9T3 and 5T4-12 cell lines. The results of this manuscript are part of a 2011 dissertation in partial fulfillment of requirements for Dr. Monica Yellowhair, Ph.D., from the University of Arizona (http://arizona.openrepository.com/arizona/bitstream/10150/202509/1/azu_etd_11664_sip1_m.pdf).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelouas A, Lutze W, Gong W, Nuttall EH, Strietelmeier BA, Travis BJ. Biological reduction of uranium in groundwater and subsurface soil. Sci Total Environ. 2000;250:21–35. doi: 10.1016/s0048-9697(99)00549-5. [DOI] [PubMed] [Google Scholar]
- Asic A, Kurtovic-Kozaric A, Besic L, Mehinovic L, Hasic A, Kozaric M, Hukic M, Marjanovic D. Chemical toxicity and radioactivity of depleted uranium: The evidence from in vivo and in vitro studies. Environmental Research. 2017;156:665–673. doi: 10.1016/j.envres.2017.04.032. [DOI] [PubMed] [Google Scholar]
- ATSDR. Toxicological Profile for Uranium. U.S. Department of Health and Human Services, Agency for Toxic Substance and Disease Registry; Atlanta, GA: 2013. [PubMed] [Google Scholar]
- Batel R, Jaksic Z, et al. A Microplate Assay for DNA Damage Determination (Fast Micromethod) in Cell Suspensions and Solid Tissues. Analytical Biochemistry. 1999;270:195–200. doi: 10.1006/abio.1999.4098. [DOI] [PubMed] [Google Scholar]
- Bihari N, Batel R, et al. Comparison between the Comet Assay and Fast Micromethod for Measuring DNA damage in HeLa Cells. Croatica Chemica Acta. 2002;75(3):793–804. [Google Scholar]
- Bleise A, Danesi PR, et al. Properties, Use and Health Effects of Depleted Uranium (DU): A General Overview. Journal of Environmental Radioactivity. 2003;64:93–112. doi: 10.1016/s0265-931x(02)00041-3. [DOI] [PubMed] [Google Scholar]
- Cardenas E, Wu WM, Leigh MB, Carley J, Carroll S, Gentry T, Luo J, Watson D, Gu B, Ginder-Vogel M, Kitanidis PK, Jardine PM, Zhou J, Criddle CS, Marsh TL, Tiedje JM. Microbial Communities in Contaminated Sediments, Associated with Bioremediation of Uranium to Submicromolar Levels. Appl Environ Microbiol. 2008;74(12):3718–3729. doi: 10.1128/AEM.02308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere M, Avoscan L, Collins R, Carrot F, Khodja H, Ansoborlo E, Gouget B. Influence of uranium speciation on normal rat kidney (NRK-52E) proximal cell cytotoxicity. Chemical Research Toxicology. 2004;17:446–452. doi: 10.1021/tx034224h. [DOI] [PubMed] [Google Scholar]
- Cooper KL, Dashner EJ, Tsosie R, Cho YM, Lewis J, Hudson LG. Inhibition of poly(ADP-ribose)polymerase-1 and DNA repair by uranium. Toxicol Appl Pharmacol. 2016;291:13–20. doi: 10.1016/j.taap.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coryell VH, Stearns DM. Molecular Analysis of hprt Mutations Generated in Chinese Hamster Ovary EM9 Cells by Uranyl Acetate, by Hydrogen Peroxide, and Spontaneously. Molecular Carcinogenesis. 2006;45:60–72. doi: 10.1002/mc.20155. [DOI] [PubMed] [Google Scholar]
- Cullinane C, Weber CA, Dianov G, Bohr VA. Restoration of preferential and strand specific gene repair in group 2 Chinese hamster ovary mutants (UV5) by the XPD (ERCC2) gene. Carcinogenesis. 1997;18(4):681–686. doi: 10.1093/carcin/18.4.681. [DOI] [PubMed] [Google Scholar]
- Dianov GL, Sleeth KM, Dianova II, Allinson SL. Repair of abasic sites in DNA. Mutation Research. 2003;531(1–2):157–163. doi: 10.1016/j.mrfmmm.2003.09.003. [DOI] [PubMed] [Google Scholar]
- DiSpirito AA, Talnagi JW, Jr, Tuovinen OH. Accumulation and cellular distribution of uranium in Thiobacillus ferroxidans. Archives of Microbiology. 1983;135(4):250–253. [Google Scholar]
- EPA. [Accessed 1 30, 2017];n.d https://www.epa.gov/ground-water-and-drinking-water/table-regulated-drinking-water-contaminants#one.
- Fathi RA, Matti LY, Al-Salih HS, Godbold D. Environmental pollution by depleted uranium in Iraq with special reference to Mosul and possilbe effects on cancer and birth defect rates. Medicine, Conflict and Survival. 2013;29(1):7–25. doi: 10.1080/13623699.2013.765173. [DOI] [PubMed] [Google Scholar]
- Franklin SJ. Lanthanide- mediated DNA hydrolysis. Curr Opin Chem Bio. 2001;5:201–208. doi: 10.1016/s1367-5931(00)00191-5. [DOI] [PubMed] [Google Scholar]
- Garmash SA, V, Smirnova S, Karp OE, Usacheva AM, Berezhnov AV, Ivanov VE, Chernikov AV, Bruskov VI, Gudkov SV. Pro-oxidative, genotoxic and cytotoxic properties of uranyl ions. Journal of Environmental Radioactivity. 2014;127:163–170. doi: 10.1016/j.jenvrad.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Guéguen Y, Suhard D, Poisson C, Manens L, Elie C, Landon G, Bouvier-Capely C, Rouas C, Benderitter M, Tessier C. Low-concentration uranium enters the HepG2 cell nucleus rapidly and induces cell stress response. Toxicology in Vitro. 2015;30(1B):552–560. doi: 10.1016/j.tiv.2015.09.004. [DOI] [PubMed] [Google Scholar]
- Guillemin I, Becker M, Ociepka K, Friauf E, Nothwang HG. A subcellular prefractionation protocol for minute amounts of mammalian cell cultures and tissue. Proteomics. 2005;5(1):35–45. doi: 10.1002/pmic.200400892. [DOI] [PubMed] [Google Scholar]
- Hao Y, Ren J, Liu C, Li H, Liu J, Yang Z, Li R, Su Y. Zinc protects human kidney cells from depleted uranium-induced apoptosis. Basic & Clinical Pharmacology & Toxicology. 2014;114:271–280. doi: 10.1111/bcpt.12167. [DOI] [PubMed] [Google Scholar]
- Hartwig A, Schwerdtle T. Interactions by carcinogenic metal compounds with DNA repair processes: toxicological implications. Toxicology Letters. 2002;127(1–3):47–54. doi: 10.1016/s0378-4274(01)00482-9. [DOI] [PubMed] [Google Scholar]
- Holmes AL, Joyce K, Xie H, Falank C, Hinz JM, Wise JP., Sr The impact of homologous recombination repair deficiency on depleted uranium clastogenicity in Chinese hamster ovary cells: XRCC3 protects cells from chromosome aberrations, but increases chromosome fragmentation. Mutation Research. 2014;762:1–9. doi: 10.1016/j.mrfmmm.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Kalinich JF, McClain DE. Staining of Intracellular Deposits of Uranium in Cultured Murine Macrophages. Biotechnic & Histochemistry. 2001;76(5–6):247–252. doi: 10.1080/bih.76.5-6.247.252. [DOI] [PubMed] [Google Scholar]
- Kasprzak KS, Sunderman FW, Jr, Salnikow K. Nickel Carcinogenesis. Mutation Research. 2003;533(1–2):67–97. doi: 10.1016/j.mrfmmm.2003.08.021. [DOI] [PubMed] [Google Scholar]
- Kawanishi S, Hiraku Y, Murata M, Oikawa S. The Role of Metals in Site-Specific DNA Damage with Reference to Carcinogenesis. Free Radic Biol Med. 2002;32(9):822–832. doi: 10.1016/s0891-5849(02)00779-7. [DOI] [PubMed] [Google Scholar]
- Löbrich M, Rief N, Kühne M, Heckmann M, Fleckenstein J, Rübe C, Uder M. In vivo formation and repair of DNA double-strand breaks after computed tomography examinations. Proc Natl Acad Sci USA. 2005;102(25):8984–8989. doi: 10.1073/pnas.0501895102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCerte C, Xie H, Aboueissa A, Wise JP., Sr Particulate depleted uranium is cytotoxic and clastogenic to human lung epithelial cells. Mutation Research. 2010;697:33–37. doi: 10.1016/j.mrgentox.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Loeb LA, Preston BD, Snow ET, Schaaper RM. Apurinic sites as common intermediates in mutagenesis. Basic Life Sciences. 1986;38:341–347. doi: 10.1007/978-1-4615-9462-8_37. [DOI] [PubMed] [Google Scholar]
- Martin IV, MacNeill SA. ATP-dependent DNA ligases. Genome Biology. 2002;3:3005.1–3005.7. doi: 10.1186/gb-2002-3-4-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AC, Blakely WF, Livengood D, Whittaker T, Xu J, Ejnik JW, Hamilton MM, et al. Transformation of human osteoblast cells to the tumorigenic phenotype by depleted uranium-uranyl chloride. Environmental Health Perspectives. 1998a;106(8):465–471. doi: 10.1289/ehp.98106465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirto H, Hengé-Napoli MH, Gibert R, Ansoborlo E, Fournier M, Cambar J. Intracellular behaviour of uranium(VI) on renal epithelial cell in culture (LLC-PK1): influence of uranium speciation. Toxicol Lett. 1999;104(3):249–256. doi: 10.1016/s0378-4274(98)00371-3. [DOI] [PubMed] [Google Scholar]
- Nielsen PE, Hiort C, Sonnichsen SH, Buchardt O, Dahl O, Norden B. DNA binding and photocleavage by uranyl salts. J Am Chem Soc. 1992;114(13):4967–4975. [Google Scholar]
- Parry JM, Parry E, Phrakonkham P, Coriv R. Analysis of published data for top concentration considerations in mammalian cell gentoxicity testing. Mutagenesis. 2010;25:531–538. doi: 10.1093/mutage/geq046. [DOI] [PubMed] [Google Scholar]
- Prabhavathi PA, Padmavathi P, et al. Chromosomal Aberrations in the Leucocytes of Men Occupationally Exposed to Uranyl Compounds. Bulletin of Environmental Contamination and Toxicology. 2003;70:322–327. doi: 10.1007/s00128-002-0194-3. [DOI] [PubMed] [Google Scholar]
- Prat O, Berenguer F, Malard V, Tavan E, Sage N, Steinmetz G, Quemeneur E. Transcriptomic and proteomic responses of human renal HEK293 cells to uranium toxicity. Proteomics. 2005;5:297–306. doi: 10.1002/pmic.200400896. [DOI] [PubMed] [Google Scholar]
- Rübe CE, Grudzenski S, Kühne M, Dong X, Rief N, Löbrich M, Rübe C. DNA double-strand break repair of blood lymphocytes and normal tissue analysed in a preclinical mouse model: implications for radiosensitivity. Clin Cancer Research. 2008;14(20):6546–6555. doi: 10.1158/1078-0432.CCR-07-5147. [DOI] [PubMed] [Google Scholar]
- Rosabal M, Hare L, Campbell PGC. Assessment of a subcellular metal partitioning protocol for aquatic invertebrates: preservation, homogenization, and subcellular fractionation. Limnology and Oceanography: Methods. 2014;12:507–518. [Google Scholar]
- Rothkamm K, Krüger I, Thompson LH, Löbrich M. Pathways of DNA Double-Strand Break Repair during the Mammalian Cell Cycle. Mol Cell Biol. 2003;23(16):5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouas C, Bensoussan H, Suhard D, Tessier C, Grandcolas L, Rebiere F, Dublineau I, Taouis M, Pallardy M, Lestaevel P, Gueguen Y. Distribution of Soluble Uranium in the Nuclear Cell Compartment at Subtoxic Concentrations. Chemical Research Toxicology. 2010;23(12):1883–1889. doi: 10.1021/tx100168c. [DOI] [PubMed] [Google Scholar]
- Schröder HC, Batel R, Schwertner H, Boreiko O, Müller WEG. Fast Micromethod DNA Single-Strand-Break Assay. Methods in Molecular Biology: DNA Repair Protocols: Mammalian Systems. 2006;314:287–305. doi: 10.1385/1-59259-973-7:287. [DOI] [PubMed] [Google Scholar]
- Stearns DM, Yazzie M, Bradley AS, Coryell VH, Shelley JT, Ashby A, Asplund CS, Lantz RC. Uranyl Acetate Induces hprt mutations and Uranium-DNA adducts in Chinese Hamster Ovary EM9 Cells. Mutagenesis. 2005;20:417–423. doi: 10.1093/mutage/gei056. [DOI] [PubMed] [Google Scholar]
- Thiébault C, Carrière M, Milgram S, Simon A, Avoscan L, Gouget B. Uranium induces apoptosis and is genotoxic to normal rat kidney (NRK-52E) proximal cells. Toxicol Sci. 2007;98(2):479–487. doi: 10.1093/toxsci/kfm130. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Brookman KW, Jones NJ, Allen SA, Carrano AV. Molecular Cloning of the Human XRCC1 Gene, Which Corrects Defective DNA Strand Break Repair and Sister Chromatid Exchange. Mol Cell Biol. 1990;10(12):6160–6171. doi: 10.1128/mcb.10.12.6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmann K, Müller C, Steinberg P. Two essential modifications strongly improve the performance of the Fast Micromethod to identify DNA single- and double-strand breaks. Arch Toxicol. 2008;82(11):861–867. doi: 10.1007/s00204-008-0356-0. [DOI] [PubMed] [Google Scholar]
- Vidal AE, Boiteux S, Hickson ID, Radicella JP. XRCC1 Coordinates the Initial and Late Stages of DNA Abasic Site Repair Through Protein-Protein Interactions. EMBO J. 2001;20(22):6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CA, Salazar EP, Stewart SA, Thompson LH. Molecular Cloning and Biological Characterization of a Human Gene, ERCC2, that Corrects the Nucleotide Excision Repair Defect in CHO UV5 Cells. Mol Cell Biol. 1988;8(3):1137–1146. doi: 10.1128/mcb.8.3.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J, Young A, Civitello ER, Stearns DM. Analysis of Heat-Labile Sites Generated by Reactions of Depleted Uranium and Ascorbate in Plasmid DNA. J Biol Inorg Chem. 2014;19(1):45–57. doi: 10.1007/s00775-013-1057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J, Zuniga MC, Yazzie F, Stearns DM. Synergistic cytotoxicity and DNA strand breaks in cells and plasmid DNA exposed to uranyl acetate and ultraviolet radiation. J Appl Toxicol. 2015;35(4):338–349. doi: 10.1002/jat.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Cheng X, Hofstadler SA, Smith RD. Specific Metal-Oligonucleotide Binding Studied by High Resolution Tandem Mass Spectrometry. J Mass Spectrom. 1996;6:669–675. doi: 10.1002/(SICI)1096-9888(199606)31:6<669::AID-JMS340>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Yazzie M, Gamble SL, Civitello ER, Stearns DM. Uranyl acetate causes DNA single strand breaks in vitro in the presence of ascorbate (Vitamin C) Chemical Research Toxicology. 2003;16:524–530. doi: 10.1021/tx025685q. [DOI] [PubMed] [Google Scholar]
- Zhivin S, Laurier D, Guseva Canu I. Health effects of occupational exposure to uranium: do physicochemical properties matter? International Journal of Radiation Biology. 2014;90(11):1104–1113. doi: 10.3109/09553002.2014.943849. [DOI] [PubMed] [Google Scholar]