

Abstract
2′-Deoxy-5-methylisocytidine is widely used in assays to personalize the care of patients infected with HIV, hepatitis, and other infectious agents. However, oligonucleotides that incorporate 2′-deoxy-5-methylisocytidine are expensive, because of its intrinsic chemical instability. We report here a C-glycoside analog that is more stable and, in oligonucleotides, pairs with 2′-deoxyisoguanosine, contributing to duplex stability about as much as a standard 2′-deoxycytidine and 2′-deoxyguanosine pair.
Keywords: isocytidine, pseudocytidine, synthetic biology, expanded genetic alphabets, clinical diagnostics
Introduction
One success story arising from efforts to create artificially expanded genetic information systems (Aegis) come from the use of 2′-deoxy-5-methylisocytidine (isoC, 1, Figure 1) and 2′-deoxyisoguanosine (isoG, 2, Figure 1) as fifth and sixth “letters” in the DNA genetic alphabet. 1,2 The isoC presents a hydrogen bond “acceptor-acceptor-donor” hydrogen bonding pattern on a small pyrimidine; isoG presents a hydrogen bond “donor-donor-acceptor” hydrogen bonding pattern on a large purine nucleobase. Therefore, isoC and isoG can form a Watson-Crick pair having the same geometry as the T:A and C:G base pairs, but are joined by a different hydrogen bonding scheme. This gives DNA molecules containing isoC and isoG the property known as “orthogonality”; they pair with each other without pairing with DNA built only from natural dA, T, dC and dG. At the same time, the close structural similarity of the nonstandard and standard pairs means that polymerases and other enzymes used as standard tools in molecular biology can also be used with this synthetic genetic system.
Figure 1.

The chemical structures of 2′-deoxyisocytidine (1), 2′-deoxyisoguanosine (2) and 2′-deoxy-1-methylpseudocytidine (3).
Orthogonality enabled by a fifth and sixth letters of a genetic alphabet can, in principle, support low-noise microarrays, multiplexed PCR, and low cost re-sequencing of the human genome, among other applications. In commerce, the orthogonality provided by the isoC:isoG pair underlies the high sensitivity and broad dynamic range of the branched DNA Versant diagnostics systems that received FDA approval in 2003 and 2004.3 Developed by scientists at Chiron and Bayer and now sold by Siemens Medical Solutions, these permit the very sensitive detection of the nucleic acids from infectious agents in complex biological media. These tools personalize the care of some 400,000 patients infected with the HIV, hepatitis B, and hepatitis C viruses.4
The isoC:isoG pair has also been used as a fifth and sixth letter in a PCR-amplifiable artificial genetic system.5,6 This represents one of the first artificial chemical systems capable of Darwinian evolution, meeting some of the criteria for artificial life.
As one of its disadvantages, however, 2′-deoxy-5-methylisocytidine undergoes facile cleavage of the glycosidic bond under acidic conditions. This propensity for acid-catalyzed depyrimidinylation can be used as a tool to detect isoC in oligonucleotides5 and is managed during phosphoramidite synthesis by careful handling of formamidine protected phosphoramidite precursors. Nevertheless, these requirements increase the cost of oligonucleotides that contain 2′-deoxy-5-methylisocytidine, especially in human diagnostics where regulatory standards are rigorous. An analog of isoC that still pairs with 2′-deoxyisoguanosine but does not suffer depyrimidinylation is desirable.
One analog that might have these properties is 2′-deoxy-1-methylpseudocytidine (ψC, 3, Figure 1), the C-glycoside analog of 2′-deoxy-5-methylisocytidine. Further, a methyl group on the pyrimidine nitrogen should prevent tautomerization and epimerization, undesired reactions seen with other AEGIS components.7 However, given the known propensity of C-glycosides to change the conformation of the 2′-deoxyribose ring, we were concerned that incorporating a significant number of C-glycosides would significantly alter the conformation of a DNA duplex that is made by this strand.
Very recently, we reported experiments that showed that the conformation of a duplex is not altered even if twelve C-glycosides are incorporated sequentially.8 Further, we showed that that certain polymerases could incorporate five consecutive C-glycosides in template-directed synthesis from triphosphates. This provided the motivation to examine the analogous C-glycoside of iso-cytidine.
We report here the synthesis of this species, a new compound, in protected form, as well as the synthesis of a test set of oligonucleotides. We report that when incorporated into an oligonucleotide at the appropriate position, 2′-deoxy-1-methylpseudocytidine forms nucleobase pairs with isoG that contribute to duplex stability about as well as the natural C:G nucleobase pair. Further, discrimination against mismatches is at the same level as seen with standard nucleobases. Therefore, this compound is a cost-effective replacement for a 2′-deoxyisocytidine that will support the synthesis and use of Aegis-containing oligonucleotides at lower cost.
Results and Discussion
Pseudothymidine (7), prepared from pseudouridine,9 is potentially a precursor for protected 2′-deoxy-1-methylpseudocytidine (9) (Figure 2). As pseudouridine is expensive and the N(1)-methylation of pseudouridine requires 5 days at reflux, an alternative route was sought. Coupling of glycal (4) and 5-iodo-1-methyluracil (5) via a palladium-catalyzed Heck reaction, followed by desilylation and diastereoselective reduction, gave pseudothymidine (7) in 51% yield (Figure 2), a result consistent with results obtained similar palladium-mediated glycal-aglycone couplings.10
Figure 2.

Synthesis of protected 2′-deoxypseudocytidine (9). Conditions: (a) Pd(OAc)2, Ph3As, Bu3N, DMF, 60 °C, 18h; (b) TBAF, HOAc; (c) NaBH(OAc)3, HOAc, CH3CN, 0 °C, 2h (51% for 3 steps); (d) TBDMSCl, Imidazole, DMF (93%); (e) 2,4,6-triisopropylbenzenesulfonyl chloride, DMAP, TEA, CH3CN, rt, 24h; (f) NH4OH, rt, 4h (75% for 2 steps).
Pseudothymidine having both sugar oxygens protected (8) was converted to the correspondingly protected 2′-deoxy-1-methylpseudocytidine (9) by treatment first with 2,4,6-triisopropylbenzenesulfonyl chloride, DMAP and triethylamine in CH3CN, and then with ammonium hydroxide (75% yield). The structure of 9 was established by 1H NMR and high resolution mass spectrometry.
With the 2′-deoxy-1-methylpseudocytidine 9 in hand, the phosphoramidite derivative 13 suitable for oligonucleotide synthesis was prepared as shown in Figure 3. Compound 9 was treated with benzoyl chloride in pyridine/CH2Cl2 to give dibenzoylated compound 10. Treatment of 10 with excess triethylamine trihydrofluoride in THF at 45 °C for 2 days gave 11 in 85% yield. The phosphoramidite derivative 13 for DNA synthesis was synthesized via dimethoxytritylation of 11 and subsequent phosphitylation of 12 using a standard procedure.
Figure 3.

Synthesis of 2′-deoxypseudocytidine phosphoramidite (13), Conditions: (a) BzCl, Pyridine (74%); (b) Et3N•3HF, THF, 45 °C, 2 days (86%); (c) DMTrCl, Pyridine (66%); (d) 2-cyanoethyl diisopropylchlorophosphoramidite, DIPEA, CH2Cl2 (78%).
Oligonucleotides were constructed to permit melting studies with both perfectly matched duplexes and duplexes containing mismatches. The standard sequence was identical to that used by Geyer et al.11 and by their predecessor, Horn et al.12 This allowed direct comparison of the results obtained here with their earlier results. Melting temperatures are illustrated in Table 1.
Table 1.
Melting temperatures (°C) for nucleobase pairs. The sequence of the DNA duplex used in the thermal denaturation experiments are
| 5′—C A C N1 A C T T T C T C C T-3′ | |||||
|---|---|---|---|---|---|
| 3′-TG T G N2 T G A A AG A G G—5′ | |||||
| Matched nucleobase pairs | Mismatched nucleobase pairs | ||||
| N1-N2 | Measured | Geyer (2003)11 | N1-N2 | Measured | Geyer (2003)11 |
| iG-iC | 62.5 | 63.3 | iG-C | 52.5 | 52.6 |
| iG-ψC | 60.2 | - | iG-A | 42.6 | 43.4 |
| G-C | 59.6 | 59.5 | iG-T | 51.9 | 52.3 |
| iC-iG | 61.3 | 61.5 | iG-G | 53.2 | 52.7 |
| ψC-iG | 59.0 | - | |||
| C-G | 60.1 | 58.5 | |||
In a variety of contexts, the isoCisoG base pair contributes more to duplex stability than any of the other standard nucleobase pairs. The reason for this is unclear, but the isoC: isoG pair appears to be the strongest of any joined by three hydrogen bonds.
At least in the context in the two test oligonucleotides, the pair between 2′-deoxy-1-methylpseudocytidine and 2′-deoxyisoguanosine contributes to duplex stability like the standard dC:dG. This result is consistent with other results obtained by Geyer et al.,11 who found that an oligonucleotide duplex having a C-glycoside had a melting temperature 1-2 degrees lower than an analogous duplex with an N-glycoside.
To demonstrate the improved stability of oligonucleotides containing 2′-deoxy-1-methylpseudocytidine above that displayed by analogous oligonucleotides containing 2′-deoxyisocytidine, four oligonucleotides were prepared (Table 2) and purified by PAGE.
Table 2. Oligonucleotides used in thermal and acidic stability study.
| Oligomer No. | Sequence | Comment |
|---|---|---|
| 1 | 5′—GGAGAAAGT-iC-GTGT—3′ | iC at position 10 |
| 2 | 5′—CAC-iC-ACTTTCTCCT—3′ | iC at position 4 |
| 4 | 5′-GGAGAAAGT-ΨC-GTGT-3′ | ΨC at position 10 |
| 5 | 5′-CAC-ΨC-ACTTTCTCCT-3′ | ΨC at position 4 |
Treatment of the oligonucleotides with gamma-labeled ATP generated 5′-kinased oligonucleotides. These oligonucleotides were treated in parallel experiments with (a) heat at 95 °C for various times (Figure 4) and (b) 0.05 M acetic acid followed by 0.1 M ammonium hydroxide (Figure 5).
Figure 4.
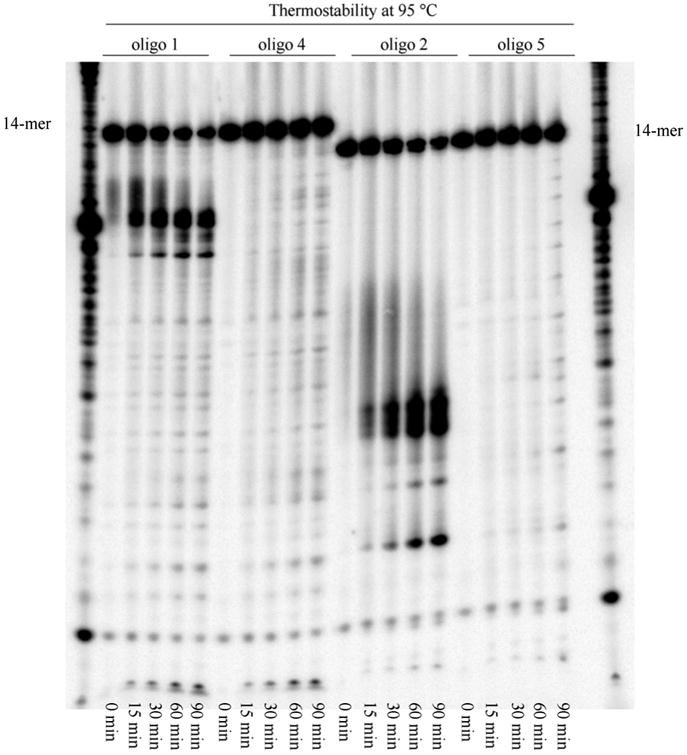
Thermostability of oligonucleotides containing 2′-deoxyisocytidine and 2′-deoxy-1-methylpseudocytidine. Samples (0.3 pmol) were resolved on a 20% PAGE.
Figure 5.
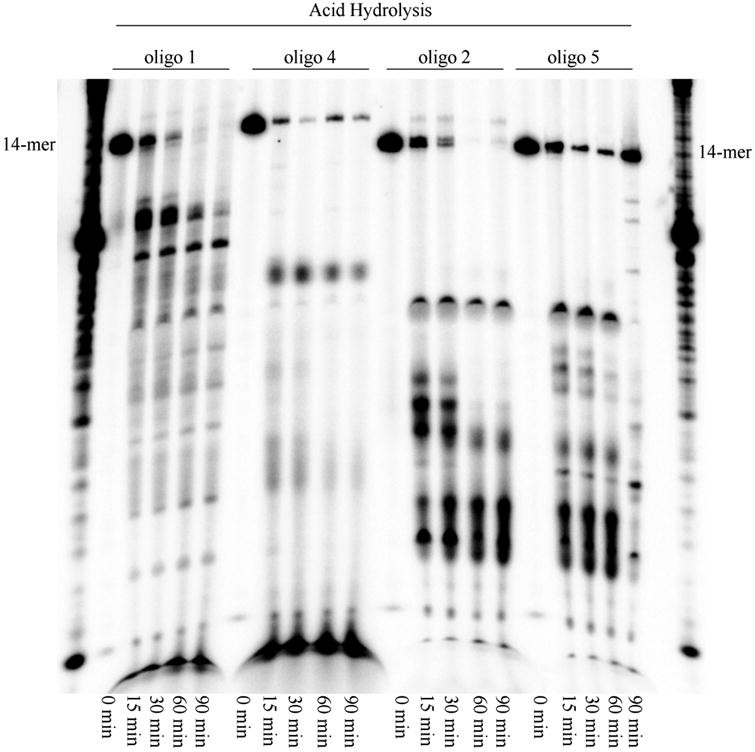
Acid-catalyzed hydrolysis of oligonucleotides containing 2′-deoxyisocytidine and 2′-deoxy-1-methylpseudocytidine. Starting 14-mers moved in electrophoresis at slightly different rates.
Treatment of oligonucleotides containing 2′-deoxyisocytidine (oligo 1 and 2 in Figure 4) with heat at 95 °C gave fragmentation of the oligonucleotides and the intensity of fragments were increased with prolonged treatment time. However, treatment of oligonucleotides containing 2′-deoxy-1-methylpseudocytidine instead of isocytidine (oligo 4 and 5 in Figure 4) with heat at 95 °C gave no cleavage up to 90 min treatment. This result showed 2′-deoxy-1-methylpseudocytidine is more stable than 2′-deoxyisocytidine under thermal condition at neutral pH.
The acid-stability of oligonucleotides containing 2′-deoxyisocytidine and 2′-deoxy-1-methylpseudocytidine was investigated by the incubation of the oligonucleotides in 0.05 M acetic acid at 90 °C for 0 to 90 min. Treatment of oligonucleotides containing 2′-deoxyisocytidine (oligo 1 and 2 in Figure 5) in 0.05 M acetic acid gave fragmentation of the oligonucleotides and complete disappearance of the starting 14-mer after 60 min. The acid treatment of oligonucleotides containing 2′-deoxy-1-methylpseudocytidine instead of isocytidine (oligo 4 and 5 in Figure 5) in 0.05 M acetic acid gave fragmentation of the oligonucleotides but even after 90 min, small fraction of the starting 14mer of oligo 4 and 5 was not cleaved. This result showed 2′-deoxy-1-methylpseudocytidine is more resistant to acidic depyrimidinylation.
Conclusion
2′-Deoxy-1-methylpseudocytidine (1-deaza-5-aza-5-methylisocytidine) is more robust chemically than the 2′-deoxy-5-methylisocytidine that is widely used in assays to personalize the care of patients infected with HIV, hepatitis, and other infectious agents, assay for genetic disease, and support advanced architectures for human genome sequencing and analysis. It joins with the 6-aza-2′-deoxyisocytidine, which is also reported to display acid stability.13 Further, it supports duplex pairing well with complementary nucleotides as part of an expanded genetic information system. Oligonucleotides that incorporate it are considerably less expensive to prepare in high quality than oligonucleotides that incorporate 2′-deoxy-5-methylisocytidine. Therefore, we expect 2′-deoxy-1-methylpseudocytidine to become an important new addition to synthetic genetic systems, especially in practical application.
Experimental
Pseudothymidine (7)
A mixture of Pd(OAc)2 (36 mg, 0.16 mmol) and AsPh3 (98 mg, 0.32 mmol) in anhydrous DMF (10 mL) was stirred at rt for 30 min. The solution was then added to a mixture of glycal 4 (280 mg, 0.80 mmol), 5-iodo-1-methyluracil (5) (200mg, 0.80 mmol), and tributylamine (0.28 mL, 0.12 mmol) in DMF (10 mL). The resulting mixture was stirred at 60 °C for 18h. After being cooled, the mixture was treated with HOAc (0.2 mL) and TBAF (1M in THF, 2 mL) and stirred at rt for 1h. Volatiles were removed by rotary evaporation under reduced pressure. The product was purified by flash chromatography (silica, gradient CH2Cl2 : MeOH = 15:1 to 10:1). The appropriate fraction (by tlc) was collected, evaporated and dissolved in acetic acid/acetonitrile (7 mL/7 mL). To this solution were added NaBH(OAc)3 (370 mg, 1.75 mmol) at 0 °C. The mixture was stirred for 2 h, volatiles were removed by rotary evaporation under reduced pressure, and the residue was resolved by flash chromatography (silica, gradient CH2Cl2 : MeOH = 7:1 to 4:1) to give 7 as a white solid (100 mg, 51 %). TLC (CH2Cl2/MeOH, 5:1): Rf 0.25.
3′,5′-Di-O-tert-butyldimethylsilyl-pseudothymidine (8)
To a mixture pseudothymidine (7) (100 mg, 0.41 mmol) and imidazole (113 mg, 1.65 mmol) in anhydrous DMF (4 mL) was added TBDMSCl (154 mg, 1.03 mmol). The mixture was stirred at rt overnight, volatiles were removed by rotary evaporation under reduced pressure, and the product was isolated by flash chromatography (silica, gradient ethyl acetate:hexane = 1:2 to 1:1) to give 8 as a colorless viscous liquid (180 mg, 93%). TLC (EtOAc/Hexane, 1:1): Rf 0.6. 1H NMR (300 MHz, CDCl3) δ 8.43 (br s, 1H), 7.32 (d, 1H, J = 1.2 Hz), 5.02 (m, 1H), 4.32 (m, 1H), 3.87 (m, 1H), 3.60-3.72 (m, 2H), 3.34 (s, 3H), 2.31 (ddd, 1H, J = 12.8, 6.0, 2.4 Hz), 1.70-1.82 (m, 1H), 0.90 (s, 9H), 0.89 (s, 9H), 0.07 (s, 6H), 0.06 (s, 6H).
2′-Deoxy-3′,5′-di-O-tert-butyldimethylsilyl-1-methylpseudocytidine (9)
To a mixture of 3′,5′-di-O-tert-butyldimethylsilyl-pseudothymidine (8) (200 mg, 0.42 mmol), 2,4,6-triisopropylbenzenesulfonyl chloride (257 mg, 0.85 mmol) and DMAP (51 mg, 0.42 mmol) in CH3CN (15 mL) was added triethylamine (0.24 mL, 1.70 mmol) at rt and stirred for 24h. The mixture was treated with saturated NH4OH (7 mL) and stirred at rt for 4h. Volatiles were removed by rotary evaporation under reduced pressure and the residue was dissolved in CH2Cl2 (40 mL) and washed with brine. The organic layer was collected, dried and evaporated. The product was purified from the residue by flash chromatography (silica, EtOAc:hexane = 1:1 to CH2Cl2 : MeOH = 20:1) to give 9 as a light yellow solid (150 mg, 75%). TLC (CH2Cl2/MeOH, 20:1): Rf 0.15. 1H NMR (300 MHz, CDCl3) δ 7.20 (s, 1H), 4.82 (dd, 1H, J = 11.1, 5.4 Hz), 4.39 (d, 1H, J = 6.6 Hz), 3.7-4.0 (m, 3H), 3.41 (s, 3H), 2.2-2.3 (m, 1H), 1.75-1.85 (m, 1H), 0.90 (s, 18H), 0.08 (m, 12H). 13C NMR (75 MHz, CDCl3) δ 163.99, 144.87, 103.93, 88.71, 77.84, 74.04, 63.51, 41.27, 37.65, 26.09, 26.00, 25.23, 18.74, 18.23, -4.37, -4.49, -5.28, -5.31. HRMS calculated for C22H43N3O4Si2 + H+: 470.2865, found: 470.2907.
2′-Deoxy-3′,5′-di-O-tert-butyldimethylsilyl-1-methyl-N,N′-dibenzoyl-pseudocytidine (10)
A mixture of 2′-deoxy-3′,5′-di-O-tert-butyldimethylsilyl-1-methylpseudocytidine (9) (75 mg, 0.16 mmol), benzoyl chloride (74 μL, 0.64 mmol) and anhydrous pyridine (0.5 mL) in CH2Cl2 (5 mL) was stirred at rt overnight. Volatiles were removed by rotary evaporation under reduced pressure. The product was purified by flash chromatography (silica, ethyl acetate : hexane = 1:5 to 1:3) to give 10 as a white solid (80 mg, 74%). TLC (EtOAc/Hexane, 1:3): Rf 0.3. 1H NMR (300 MHz, CDCl3) δ 8.28 (m, 2H), 8.12 (m, 2H), 7.36-7.64 (m, 7H), 5.35 (dd, 1H, J = 10.2, 5.4 Hz), 4.38 (m, 1H), 3.98 (m, 1H), 3.60-3.80 (m, 2H), 3.43 (s, 3H), 2.59 (m, 1H), 1.70 (m, 1H), 0.97 (s, 9H), 0.91 (s, 9H), 0.14 (s, 3H), 0.12 (s, 3H), 0.10 (s, 6H). 13C NMR (75 MHz, CDCl3) 179.44, 158.85, 148.94, 141.75, 137.34, 133.90, 132.63, 130.41, 130.10, 128.69, 128.33, 116.91, 87.82, 74.51, 74.16, 63.87, 43.22, 37.07, 26.14, 26.02, 18.56, 18.24, -4.40, -4.47, -5.11, -5.13.
2′-Deoxy-1-methyl-N-benzoyl-pseudocytidine (11)
A mixture of 2′-deoxy-3′,5′-di-O-tert-butyldimethylsilyl-1-methyl-N,N′-dibenzoyl-pseudocytidine (10) (310 mg, 0.46 mmol) and triethylamine trihydrofluoride (0.6 mL) in THF (15 mL) was stirred at 45 °C for two days. Volatiles were removed by rotary evaporation under reduced pressure, and the product was purified by flash chromatography (silica, CH2Cl2:MeOH = 15:1 to 12:1) to give 11 as a white solid (137 mg, 86%). TLC (CH2Cl2/MeOH, 12:1): Rf 0.3. 1H NMR (300 MHz, CDCl3/CD3OD) δ 8.16 (d, 2H, J = 7.8 Hz), 7.70 (s, 1H), 7.3-7.5 (m, 3H), 5.24 (m, 1H), 4.21 (m, 1H), 3.84 (m, 1H), 3.5-3.7 (m, 2H), 3.37 (s, 3H), 2.4-2.6 (m, 1H), 1.8-1.9 (m, 1H).
2′-Deoxy-5′-dimethoxytrityl-1-methyl-N-benzoyl-pseudocytidine (12)
A mixture of 2′-deoxy-1-methyl-N-benzoyl-pseudocytidine (11) (129 mg, 0.37 mmol) and 4,4′-dimethoxytritylchloride (128 mg, 0.38 mmol) in anhydrous pyridine (6 mL) was stirred at rt overnight. Volatiles were removed by rotary evaporation under reduced pressure. The product was purified by flash chromatography (silica, EtOAc:hexane = 1:2 to 1:1 then CH2Cl2:MeOH = 20:1) to give 12 as a white solid (160 mg, 66%). TLC (CH2Cl2/MeOH, 20:1): Rf 0.2. 1H NMR (300 MHz, CDCl3) δ 8.2-8.3 (m, 2H), 7.61 (d, 1H, J = 1.5 Hz), 7.2-7.6 (m, 12H), 6.8-6.9 (m, 4H), 5.39 (t, 1H, J = 7.5 Hz), 4.45 (m, 1H), 4.06 (m, 1H), 3.80 (s, 6H), 3.35 (m, 1H), 3.17 (s, 3H), 2.6-2.7 (m, 1H), 2.0-2.2 (m, 1H).
2′-Deoxy-5′-dimethoxytrityl-1-methyl-N-benzoyl-pseudocytidine 3′-(2-cyanoethyl diisopropylphosphoramidite) (13)
To a mixture of 2′-deoxy-5′-dimethoxytrityl-1-methyl-N-benzoyl-pseudocytidine (12) (120 mg, 0.185 mmol) and DIPEA (97 μL, 0.55 mmol) in anhydrous CH2Cl2 (5 mL) was added 2-cyanoethyl diisopropylchlorophosphoramidite (62 μL, 0.278 mmol) at 0 °C. The mixture was allowed to warm to rt and stirred for 2h. The mixture was poured into a saturated sodium bicarbonate solution (10 mL) and extracted with CH2Cl2. The organic layer was washed with brine, dried over anhydrous sodium sulfate and evaporated. The product was purified from the residue by flash chromatography (neutral silica, EtOAc:hexane = 1:3 to 1:1 with 0.5% Et3N) to give diastereomeric mixture of 13 as a white foam (123 mg, 78%). TLC (EtOAc/Hexane, 1:1): Rf 0.5, 0.53. 1H NMR (300 MHz, CDCl3) δ 8.2-8.3 (m, 2H), 7.2-7.8 (m, 14H), 6.8-6.9 (m, 4H), 5.39 (m, 1H), 4.57 (m, 1H), 4.19 (m, 1H), 3.79 (m, 6H), 3.5-3.7 (m, 3H), 3.2-3.5 (m, 2H), 3.15 (m, 3H), 2.7-2.9 (m, 1H), 2.4-2.6 (m, 2H), 2.0 (m, 1H), 1.6 (m, 1H), 1.0-1.3 (m, 12H). 31P NMR (121 MHz, CDCl3) δ 150.26 (s), 149.36 (s).
Oligonucleotide synthesis
Oligonucleotide synthesis was performed by the DNA synthesizer ABI 394, at the 1.0 μmol scale. The synthesis was done using the manufacturer's recommended procedure except extended (600 sec) coupling time for isoG, isoC and ψC. All natural (A, G, C, and T) and unnatural (isoG and isoC) nucleobase phosphoramidites were purchased from Glen Research. At the completion of the synthesis, all protective groups in oligonucleotides were removed by heating in 30% ammonium hydroxide at 55 °C overnight. The mixture was then filtered through 0.2 μm filter and the remaining ammonia was removed by rotary evaporation under reduced pressure at rt. The sample was then diluted with water and lyophilized. The residue after lyophilization was dissolved in water and filtered through 0.2 μm filter. The oligonucleotide was purified from the filtrate by ion exchange HPLC (Dionex DNAPac PA-100 9 X 250 mm Semi-Prep column, eluent A = 25 mM NaOH, eluent B = 25 mM NaOH, 1.0 M NaCl, gradient from 10 to 80% B in 40 min, flow rate 3 mL/min). The appropriate fractions were collected, neutralized by aqueous acetic acid, desalted by Sep-PaK cartridges (Waters) and lyophilized. The purified oligonucleotides were dissolved in water at a concentration of 50 μM.
HPLC profile of synthetic oligonucleotides
HPLC condition; Dionex DNAPac PA-100 4 X 250 mm analytical column, eluent A = 25 mM NaOH, eluent B = 25 mM NaOH, 1.0 M NaCl, gradient from 10 to 80% B in 50 min, flow rate 0.5 mL/min
| 5′—C A C N1 A C T T T C T C C T-3′ | |||
| 3′-TG T G N2 T G A A AG A G G—5′ | |||
| N1 | Retention time | N2 | Retention time |
| iG | 36.6 | iG | 43.5 |
| iC | 34.4 | iC | 41.8 |
| ψC | 34.3 | ψC | 41.8 |
Thermal DNA duplex denaturation
Thermal DNA duplex denaturation experiments were done in 0.45 M NaCl, 0.045 M sodium citrate buffer (pH 7.9). Each oligonucleotide was present at 1.6 μM. Absorbance was monitored at 260 nm over a range of 25 °C -85 °C with a change in temperature of 1 °C /min-1 for five heating and cooling cycles on a Varian Cary 300 (Palo Alto, CA). Initial heating and cooling cycles were discarded and the Tm was determined by averaging the temperature of the maximum derivatives of each cooling and heating cycle.
Acid stability
The 5′-radiolabelled oligonucleotides (10 μL, 0.2 pmol /μL) were mixed with an equal volume of 0.1 M acetic acid. Mixtures were incubated for 0, 15, 30, 60 or 90 min at 95°C in a thermocycler with a heated lid. The tubes containing the mixtures were then heated with the tubes open to let volatile materials evaporate (2 min). Two volumes (20 μL) of ammonium hydroxide (0.1 M) were then added, and heating continued at 95 °C for 5 min. The tubes were then opened, and the NH3 was evaporated by heating at 95 °C for 2 min. Cleavage products were resuspended in formamide (3×) and resolved by PAGE (20%, 0.3 pmol samples).
Thermal stability
To determine stability to heating at near neutral pH, oligonucleotides (10 μL, 0.2 pmol /μL) in Tris buffer (10 mM, pH 8.5) were incubated for 0, 15, 30, 60 or 90 min at 95°C in a thermocycler with a heated lid. The tubes containing the mixtures were then heated with the tubes open to let volatiles evaporate (2 min). Cleavage products were resuspended in formamide (3×) and resolved by PAGE.
Acknowledgments
This work was supported by the National Institutes of Health 1R01GM081527-01 and a grant from SAB.
References and Notes
- 1.Sismour AM, Benner SA. Nat Rev Genet. 2005;6:533. doi: 10.1038/nrg1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elbeik T, Markowitz N, Nassos P, Kumar U, Beringer S, Haller B, Ng V. J Clin Microbiol. 2004;42:3120. doi: 10.1128/JCM.42.7.3120-3127.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elbeik T, Surtihadi J, Destree M, Gorlin J, Holodniy M, Jortani SA, Kuramoto K, Ng V, Valdes R, Valsamakis A. J Clin Microbiol. 2004;42:563. doi: 10.1128/JCM.42.2.563-569.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benner SA. Accounts Chem Res. 2004;37:784. doi: 10.1021/ar040004z. [DOI] [PubMed] [Google Scholar]
- 5.Sismour AM, Benner SA. Nucl Acids Res. 2005;33:5640. doi: 10.1093/nar/gki873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson SC, Sherrill CB, Marshall DJ, Moser MJ, Prudent JR. Nucl Acids Res. 2004;32:1937. doi: 10.1093/nar/gkh522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutter D, Benner SA. J Org Chem. 2003;68:9839. doi: 10.1021/jo034900k. [DOI] [PubMed] [Google Scholar]
- 8.Havemann SA, Hoshika S, Hutter D, Benner SA. Nucleos Nucleot Nucl Acids. 2008;27:261. doi: 10.1080/15257770701853679. [DOI] [PubMed] [Google Scholar]
- 9.Bhattacharya BK, Devivar RV, Revankar GR. Nucleosides & Nucleotides. 1995;14:1269. [Google Scholar]
- 10.Zhang HC, Daves DD., Jr J Org Chem. 1992;57:4690. [Google Scholar]
- 11.Geyer CR, Battersby TR, Benner SA. Structure. 2003;11:1485. doi: 10.1016/j.str.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 12.Horn T, Chang CA, Collins ML. Tetrahedron Lett. 1995;36:2033. [Google Scholar]
- 13.Seela F, He Y. J Org Chem. 2003;68:367. doi: 10.1021/jo020507n. [DOI] [PubMed] [Google Scholar]