

Abstract
As a novel low-side-effect cancer therapy, photo-immunotherapy (PIT) is based on conjugating monoclonal antibody (mAb) with a near-infrared (NIR) phthalocyanine dye IRDye700DX (IR 700). IR700 is not only fluorescent to be used as an imaging agent, but also phototoxic. When illuminating with NIR light, PIT can induce highly-selective cancer cell death while leaving most of tumor blood vessels unharmed, leading to an effect termed super-enhanced permeability and retention (SUPR), which can significantly improve the effectiveness of anti-cancer drug. Currently, the therapeutic effects of PIT are monitored using 2D macroscopic fluorescence reflectance imager, which lacks the resolution and depth information to reveal the 3D distribution of mAb-IR700. In the study, we applied a multi-modal optical imaging approach including high-resolution optical coherence tomography (OCT) and high-sensitivity fluorescence laminar optical tomography (FLOT), to provide 3D tumor micro-structure and micro-distribution of mAb-IR700 in the tumor simultaneously during PIT in situ and in vivo. The multi-wavelength FLOT can also provide the blood vessels morphology of the tumor. Thus, the 3D FLOT reconstructed images allow us to evaluate the IR700 fluorescence distribution change with respect to the blood vessels and at different tumor locations/depths non-invasively, thereby enabling evaluation of the therapeutic effects in vivo and optimization of treatment regimens accordingly. The mAb-IR700 can access more tumor areas after PIT treatment, which can be explained by increased vascular permeability immediately after NIR-PIT. Two-photon microscopy was also used to record the mAb-IR700 on the tumor surface near the blood vessels to verify the results.
Keywords: Fluorescence laminar optical tomography (FLOT), Photoimmunotherapy (PIT), Enhanced permeability and retention effect, Two-photon microscopy (TPM)
Graphical abstract
Multi-model optical imaging apporach for monitering 3D micro-enveronment during and after PIT
1. Introduction
Enhancing drug delivery to cancers can significantly improve the effectiveness of therapy [1]. Targeted cancer therapies offer the promise of more effective tumor treatment with fewer side-effects than conventional cancer therapies. Molecular therapies based on monoclonal antibody (mAb) are highly-selective for tumors expressing particular antigens. However, patient outcomes have been only modestly improved when naked mAb is used as monotherapy or in combination with other therapies [2-4]. Combinations of conventional photosensitizers and mAbs have been tested but the improvement in selectivity is still limited [5-8]. Near-infrared photoimmunotherapy (NIR-PIT) is a newly developed specific cancer treatment that employs a targeted monoclonal antibody (mAb)-photo-absorber conjugate (mAC) [9]. The antibody binds to the appropriate target antigen of the cell surface and the photo-activatable silica-phthalocyanine dye, IRDye700DX (IR700), induces lethal damage to cell membrane after NIR-light exposure. NIR-PIT has been shown to be effective for various types of tumors with a variety of different antibodies [10-16]. Moreover, a first-in-human phase 1 trial of epidermal growth factor receptor (EGFR) targeted NIR-PIT in patients with inoperable head and neck cancer was initiated in June 2015 and has recently advanced to phase 2 [17].
NIR-PIT induces selectively immediate target specific immunogenic cell death (ICD) and the non-target cells which locate next to the target cells are not damaged by therapy [9, 18]. Immediate cell death leads to immediate increases in vascular permeability of treated tumors which can result in 10- to 24-fold enhancement of macromolecules or nano-drugs delivery [19]. This phenomenon has been termed super enhanced permeability and retention (SUPR) effects because it is substantially greater than conventional “enhanced permeability and retention (EPR)” which is commonly seen in untreated tumors [1, 19-21]. We have investigated the mechanism for increased permeability of NIR-PIT into tumor tissue following treatment [19-21]. SUPR effects result in redistribution of already circulating mACs, at least partly overcoming baseline heterogeneity in drug delivery commonly observed in untreated tumors [1, 22]. Therefore, additional exposures of NIR-light can further improve therapeutic effects by depositing additional mACs in the tumor bed after initial NIR-PIT [23]. Real-time monitoring of theranostic agent distribution and therapeutic effects within the tumor micro-environment non-invasively will be critical for further understanding of PIT/SUPR mechanism and optimizing the effectiveness of treatment [22].
Current approach for monitoring mACs fluorescence are based on macroscopic fluorescence reflectance imaging which lacks the resolution and depth information to reveal mAb-IR700 distribution in situ [19, 22]. Histological analysis can reveal intra-tumor inhomogeneity, but in an invasive and terminal manner [22]. We has investigated the micro-distribution of mACs at different locations during/after PIT in situ and in vivo using a two-channel fluorescence fiber imaging system and a high-resolution two-photon microscope interfaced with micro-prism previously [22]. However, both approaches are still invasive. Fluorescence laminar optical tomography (FLOT) [24-32], or mesoscopic fluorescence molecular tomography (MFMT) [33-37], is a promising three-dimensional (3-D) imaging approach that has a resolution of ∼100 μm with 2-3 mm penetration depth, which has the potential to quantify depth-resolved distribution of mACs in tumor. Optical coherence tomography (OCT) is an established biomedical imaging method for subsurface imaging of tissues microstructure with high resolution (<10 μm) and 1-2 mm penetration depth [26, 38-43]. Herein, we applied a multi-modal optical imaging approach including OCT and FLOT, to provide three-dimensional (3-D) tumor micro-structure and micro-distribution of mACs in the tumor simultaneously during cancer treatment in situ and in vivo. Multi-wavelength FLOT could also provide the blood vessels morphology on the tumor. Thus, 3D FLOT/OCT reconstructed images allow us to evaluate the real-time drug distribution change with respect to the blood vessels and at different tumor locations/depths in living mice. The high resolution two-photon microscopy was also used to record the mAb-IR700 on the tumor surface near the blood vessels to verify the results.
2. Materials and methods
2.1. Reagents
Water soluble, silica-phthalocyanine derivative, IRDye 700DX NHS ester was obtained from LI-COR Biosciences (Lincoln, NE, USA). Panitumumab, a fully humanized IgG2 monoclonal antibody (mAb) directed against EGFR, was purchased from Amgen (Thousand Oaks, GA, USA). All other chemicals were of reagent grade [22].
2.2. Synthesis of IR700-conjugates
Conjugation of dyes with mAb was performed according to previous methods [9, 22]. In brief, panitumumab (1.0 mg, 6.8 nmol) was incubated with IR700 NHS ester (66.8 μg, 34.2 nmol) in 0.1 M Na2HPO4 (pH 8.6) at room temperature for 1 h. The mixture was purified with a Sephadex G25 column (PD-10; GE Healthcare, Piscataway, NJ, USA). The protein concentration was determined with Coomassie Plus protein assay kit (Thermo Fisher Scientific Inc, Rockford, IL, USA) by measuring the absorption at 595 nm with UV-VIS (8453 Value System; Agilent Technologies, Santa Clara, CA, USA). The concentration of IR700 was quantified by measuring the absorption at 689 nm to confirm the number of fluorophore molecules per mAb. The synthesis was controlled so that an average of two IR700 molecules was bound to a single antibody. We abbreviate IR700 conjugated to panitumumab as pan-IR700 [22].
2.3. Cell culture
EGFR-expressing A431 cells (epidermoid carcinoma cells) and A431 cells stably expressing green fluorescent protein (GFP) cells were established by Dr. Kobayashi's laboratory. Cells were grown in RPMI 1640 medium (Life Technologies, Gaithersburg, MD USA) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Life Technologies) in tissue culture flasks in a humidified incubator at 37 °C at an atmosphere of 95% air and 5% carbon dioxide [1, 22].
2.4. Animal and tumor models
All in vivo procedures were conducted in compliance with the Guide for the Care and Use of Laboratory Animal Resources (1996), US National Research Council, approved by the Institutional Animal Care and Use Committees (IACUCs) at the University of Maryland and NIH. Six- to eight-week old female homozygous athymic nude mice were purchased from Charles River (NCI-Frederick, Frederick, MD, USA). Two-million A431/A431-GFP cells were subcutaneously injected in the right and left dorsum of each mouse. Tumor volume was based on caliper measurements and calculated using the following formula: tumor volume = length × width2 × 0.5. Mice were monitored daily for their general health. Tumors reaching approximately 100 mm3 in volume were selected for study [1, 22].
2.5. Treatment regimens
We used pan-IR700 as the agent for A431/A431-GFP tumor bearing mice. Tumor bearing mice were randomized into 4 groups of at least 5 animals per group for each imaging experiment. A431 tumor bearing mice were used for PIT FLOT/OCT imaging experiment (group 1) and control FLOT/OCT imaging experiment (group 2). A431-GFP tumor bearing mice were used for PIT two-photon imaging (group 3) and control two-photon imaging experiment (group 4). 24 hours after i.v. injection of 100 μg of pan-IR700 via the tail vein, 50 J/cm2 NIR light was administered at group 1 and 3 while group 2 and 4 (without NIR light) were used as control.
2.6. Imaging
During the procedure, all mice were anesthetized with inhaling 2% isoflurane mixed in O2 and were then immobilized using a customized stereotaxic frame with body temperature maintained at 37 °C by a heating blanket. A small incision was performed to remove the skin covering the tumor for imaging. In FLOT/OCT experiments, 50 μL oregon green labeled dextran with 70000 MW (Dextran, Oregon Green™ 514, Thermo Fisher Scientific Inc, Rockford, IL, USA) was injected i.v. (25 mg/mL) before NIR light administration to indicate the vascular distribution of the tumors. The mice were euthanized and tumors were extracted after imaging.
2.6.1. OCT system
Supplementary Fig. 1A illustrated the schematic of the OCT imaging system. This OCT imaging system was equipped with a wavelength-swept laser as light source which was centered at 1310 nm with 100 nm bandwidth as previously described [26, 44-47]. The wavelength-swept frequency of the laser was 16 kHz with 17 mW output power [26, 46]. A fiber-based Michelson interferometer was used in the OCT system. About 97% of the laser power was split evenly to the sample and reference arms [26, 45]. The remaining 3% of the laser output power was obtained by a Mach-Zehnder interferometer (MZI) to generate a frequency-clock signal with uniformly spaced optical frequency to trigger the sampling of the OCT signal [27]. The reflected/backscattered signals from both the sample and reference arms encountered at the fiber couple (FC) and formed interference fringes [26]. The interference signals were then received by a balanced detector (BD). Depth-resolved tomography can finally be realized by performing fast Fourier transform of the interference fringes [26] as light reflected from different sample depths resulted in interference fringes with different frequencies.
2.6.2. FLOT system
The schematic of the FLOT system shown in Supplementary Fig. 1B was similar to that reported previously [29, 48, 49]. In brief, two pigtailed laser diodes centered at 658-nm and 520-nm (LP660-SF60 and LP520-SF15, Thorlabs Inc, Newton NJ, USA) were used as the light sources to illuminate the mice tumor at two wavelengths in a time-shared manner. The line-field illumination was achieved by a cylindrical lens shaping the collimated light. The line illumination had a full line-width at the half maximum of ∼26 μm at the focal plane [29, 49]. The backscattered light and emitted fluorescent light from the mice tumor passed through an objective lens, a filter wheel (695-nm and 561-nm long-pass emission filters for fluorescence imaging and no filter applied for reflectance images), and finally collected by a 12-bit CCD camera (EM-CCD, PCO-TECH Inc, Romulus MI, USA) [29, 49]. The mice tumor was scanned by translating the mice laterally in the scanning direction X (perpendicular to the line illumination direction Y) using a motorized stage (ESP301, Newport Corp, Irvine CA, USA). The acquired 3D OCT and FLOT datasets were co-registered as previously described [26]. In this study, IR700 was employed as fluorescence biomarkers for FLOT and as a surrogate of mACs accumulation.
2.6.3. Two-photon microscope (TPM) system
In this study, we employed a lab-customized two-photon system previously reported [22]. Briefly, the TPM system utilized a Ti:sapphire laser (Coherent Mira 900-F) operating at 790 nm wavelength with 120 fs pulse width. A 20× water immersion objective (Olympus Corporation) was applied. A customized dichroic mirror (760 nm, single edge dichroic beam splitter; T760LPXXR-UF3; Chroma Technology Corporation) was used to separate the excitation laser and fluorescence signals [22]. The emitted fluorescence signals were further separated into two channels by another 550 nm dichroic beam splitter. Emission filters (525±39 nm and 716±42 nm for the GFP signal in the green channel and the IR700 signal in the red channel, respectively) were employed before the photomultiplier tubes (H7422P-40 for green channel and H7422P-50 for red channel, Hamamatsu Corporation). In the experiments, the skin covering the tumor was removed and the tumor was exposed under the 20× objective lens. TPM can provide higher resolution but limited field of view. Herein, TPM was applied to support our FLOT results. In the TPM experiments, we employed GFP fluorescence as a surrogate of treatment and IR700 fluorescence as a surrogate of mAC s accumulation as previously reported [1, 22].
2.6.4. Data recording protocol
The NIR LED used for the PIT treatment in the experiments has a spectrum of 680-700 nm (Tech-LED, Marubeni America Co.) [22]. In order to deliver the dose of ∼50 J/cm2 light on the tumor, the NIR LED need to irradiate for ∼20 minutes [50, 51]. Since the LED spectrum has overlap with the emission filter spectral range in both FLOT and TPM systems, to record the IR700 fluorescence signal change during and after PIT, the data acquisition protocol is designed as following: one dataset was acquired before turning on the NIR LED, then each dataset was taken after every 5-minutes' NIR exposure on tumor. The NIR LED was turned off during data acquisition (∼30 s). After a total of 20 minutes' NIR irradiation, the PIT treatment was finished and one dataset was taken every 5 minutes in the next 60 minutes to record the post-treatment redistribution of the mACs in the tumor. For the control group, one dataset was acquired every 5 minutes for 80 minutes without NIR LED exposure. In FLOT/OCT experiments, before the acquisition protocol described above, the tumor was first imaged by OCT system to provide the morphology of the tumor. In this study, the area within 500 μm depth from tumor surface was defined as tumor surface (tumor_s) and the area beyond 500 μm depth from tumor surface was defined as deep tumor (tumor_d) [22].
2.7. Ex-vivo evaluation
To detect the antigen-specific micro-distribution in the tumor, fluorescence microscopy was performed. Extracted tumors were frozen with OCT compound (SAKURA Finetek Japan Co., Tokyo, Japan) and frozen sections (10 μm thick) were prepared. Fluorescence microscopy was performed using the BX61 microscope with the following filters; excitation wavelength 590 to 650 nm, emission wavelength 665 to 740 nm for IR700 fluorescence. Transmitted light differential interference contrast (DIC) images were also acquired. Extracted tumors were also placed in 10% formalin and serial 10 μm slice sections were fixed on glass slide with Hematoxylin and Eosin (H&E) staining. Light microscopy study was also performed using Olympus BX61 to evaluate histological changes.
2.8. Statistical analysis
There are no missing data observed in this study. Descriptive statistics (e.g. mean and standard deviation) were calculated. Student t-test was used and statistical analyses were performed using MATLAB (The MathWorks Inc., Natick, MA) with statistical significance P = 0.05.
3. Results and Discussion
3.1. Calibration of the FLOT system
In our previous paper, quantum-dot-em bedded EH-PEG hydrogel was used to characterize the PSF (point spread function) of our FLOT system [29]. To further characterize the FLOT system, liquid phantoms were fabricated. IRDye 700DX NHS ester (IR700), intralipid solution (Hospira Inc.), and PBS buffer were mixed and stirred well. The intralipid concentration was set to provide a reduced scattering coefficient μs′= ∼0.5/mm at 635 nm, which is very similar to optical properties of the tumors used in this experiment determined from the reflectance data using oblique-incidence spectroscopy [52-54]. Different IR700 concentrations between 0-5 μM were diluted to test the fluorescence signals at different depths of our FLOT system and the depth profiles do not change much for different IR700 concentrations as shown in Supplementary Fig.1C. Since the liquid phantoms are homogeneous, the fluorescence signals are supposed to be the same at different depths, while we can notice that there are some decreases in fluorescence as depth increases due to the imperfect reconstruction nature of our FLOT system. We further plotted the normalized fluorescence intensity of homogenous phantoms and tumors at different depths as shown in Supplementary Fig.1D. To improve the accuracy of our results, we extracted a weight matrix from the normalized fluorescence intensity of homogenous phantoms at different depths to compensate for the depth-dependent signal decrease from the FLOT system. All the datasets in the following studies were compensated by the weight matrix before further processing to eliminate the signal decrease. Normalized fluorescence intensity of tumors after compensation at different depths was also shown in Supplementary Fig.1D. Different IR700 concentrations from 0-10 μM were diluted to test the sensitivity of our FLOT system in Supplementary Fig.1E, showing an excellent linear response with R2=0.9918. To make our FLOT results more quantitative and select a proper threshold, we imaged 6 blank tumors without any drug injected. The limit of quantification (LOQ) is defined as: LOQ = M + 10σ, where M is the average intensity of the blank tumors after compensation (background) and σ is the standard deviation of the blank tumors [55]. Supplementary Fig.1F shows the fluorescence intensity change at different depths in the mice tumors after compensation. From this definition we can estimate the imaging depth of our FLOT system is ∼2 mm in the mouse tumor with current setup. Then we applied the LOQ values as a global threshold for the following quantitative analysis. All the pixels in the 3D FLOT data over the global threshold (LOQ) are defined as the ROIs.
3.2. In vivo 3D imaging the distribution of the mACs using FLOT/OCT system
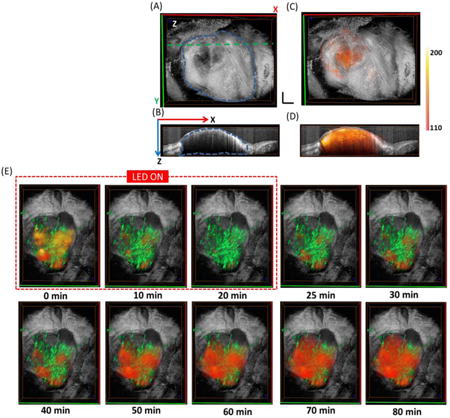
Fig.1A shows the 3D tumor structure obtained from OCT, with the tumor outlined by the blue dashed margin. High-resolution OCT can reveal the microstructure surrounding the tumor (e.g. the fiber tissue covering the tumor can be clearly seen). Fig.1B is the cross-sectional OCT image from the green dashed line in Fig.1A. The tumor area is easy to be distinguished from the surrounding tissues. The muscle tissue on the left of the tumor tissue featured with bright and dark horizontal stripes due to birefringence. The tumor tissue has less penetration depth compared with normal tissue due to increased tissue scattering [26]. Fig.1C shows the fused OCT/FLOT image with red-orange color indicating the distribution of mACs. Fig.1D shows fused cross-sectional OCT/FLOT image with depth-resolved distribution of the mACs scattered inside the tumor. We first evaluated the fluorescence intensity from tumor surface (indicated in blue dashed box in Fig.1D) and deep tumor (indicated in red dashed box in Fig.1D) in the anesthetized mice before PIT treatment. From the results of FLOT, IR700 fluorescence intensity in the tumor surface was significantly higher than that of the deep tumor (Fig.1E), which is consistent with our previous results [22]. Moreover, we plotted the normalized fluorescence intensity of homogenous phantoms and tumors at different depths before compensation as shown in Supplementary Fig.1D. The quicker drop of fluorescence intensity in tumors compared to the homogenous phantoms also implied more mACs in tumor surface. Ex-vivo DIC images confirmed relatively homogeneous cell distribution across the tumor surface and deep tumor, while IR700 fluorescence intensity in tumor surface was also significantly higher than that of the deep tumor (Supplementary Fig. 2A-B).
Fig. 1.
(A) 3D OCT tumor image. The three-dimensional tumor volume is indicated by the blue dashed margin. (B) Cross-sectional OCT images from the green dashed line in Fig. 1 (A). (C) Fused OCT/FLOT image (red-orange indicates the distribution of mACs). (D) Cross-sectional fused OCT/FLOT image. (E) IR700 Fluorescence intensity value of tumor surface (tumor_s) and deep tumor (tumor_d) before PIT as indicated by the dashed blue and red squares in Fig. 1 (D). Tumors surface showed significantly higher fluorescence intensity value than that of the deep tumor (p<0.05, n=5). (F) Representative 3D FLOT images of the micro-distribution of mACs in tumor at different time points during and after PIT Treatment. PIT treatment starts at 0 min and lasts for 20 min as indicated by the red dashed line. Scale bar 1 mm.
The mAb-IR700 distribution during and after PIT treatment was shown in Fig.1F. At 0 min, NIR LED irradiation began and the IR700 fluorescence intensity dropped quickly at both tumor surface and deep tumor regions in the first 20 min. After 20 min, the NIR LED was turned off and the PIT treatment finished. The IR700 fluorescence intensity began to recover and interestingly the fluorescence distribution achieved a bigger volume than the initial value (0 min), meaning the drug accessed more tumor areas after NIR-PIT treatment.
To quantify the 3D images acquired by FLOT, the IR700 fluorescence intensity was averaged and normalized to the initial level (0 min). The normalized IR700 fluorescence intensity showed a clearly drop in the first 20 min during PIT treatment and a recovery in the following 60 min, while there was only gradual decrease in controls without any fluorescence recovery which may result from dye photo-bleaching (Fig. 2A). To investigate the 3D micro-distribution of mACs, we plotted out the histogram of the pixels in the tumor. One pixel can represent a voxel in the tumor and the pixel intensity can reflect the density of the mACs in this voxel. The histograms of the pixel intensity before and 60 min after PIT in the tumor were shown in Fig. 2B. 60 min after PIT treatment, the whole histogram shifted to the middle range while the control group did not have such trend (Fig. 2C). Before PIT treatment, due to the heterogeneous vascularity, increased interstitial pressure and structural barriers imposed by the extracellular matrix, the mACs had limited delivery inside the tumor which resulted most of the voxels in the tumor had low fluorescence values [56-58]. While on the other hand, benefit from the intrinsically leaky nature of tumor vessels compared with healthy vessels in normal organs, which is known as the enhanced permeability and retention (EPR) effect, there was modest delivery for the mACs and the delivered mACs retained mainly near the blood vessels [22], tumor surface (Fig. 1D) and in some areas subject to vascular heterogeneity [58, 59]. This may help to explain the appeared another peak on the right end of the histograms in Fig 2B. We further analyzed the standard deviations (SDs) of the histograms. SD value of tumor of 60 min after PIT showed a significant larger SD than that of before PIT (Fig. 2D), which indicated the fluorescence values in the histogram transformed towards less variance and may indicate that mACs has a more homogeneous distribution 60 min after PIT. PIT treatment leaded to quick decrease of mACs in the tumor [1]. On one hand, P IT facilitated unbound mAb–IR700 to redistribute into the remnant target tumor. While the areas initially filled with clustered fluorescence signals did not recover to the initial intensity due to the limited recovery time and circulated mACs. This explained the shift to homogeneous distribution of mACs after PIT treatment. We also calculated the number of pixels over the value of LOQ in the tumor before and 60 min after PIT treatment. There were significantly more pixels with values over the threshold in the tumor 60 min after PIT treatment than that of before PIT (Fig. 2E). PIT selectively destructed the perivascular layers of cancer cells leading to expansion of the tumor vessel diameter and allowing more fully distribution of circulated mACs throughout the treated tumor than before PIT, which can explain the increased pixels over the threshold. On the other hands, we can notice that the volume intensity after PIT treatment did not recover to the initial value (Fig. 2A), which may be due to the limited quantity of mAb-IR700 conjugate in the circulation system and limited recovery time. Entropy measures the randomness of the intensities in the image [60]. Entropies of pixel values before and 60 min after PIT treatment were also presented. The pixels in the tumor 60 min after PIT have significant smaller entropy than that of tumor before PIT (Fig. 2F). The pixels in the tumor before PIT had more contrast with different values due to the heterogeneous distribution of the mACs and significant smaller entropy 60 min after PIT resulting from the more homogeneous distribution of the mACs. One distinct advantage of the FLOT/OCT system is that we can quantitatively monitor the trend of the 3D mACs micro-distribution in vivo and in situ, thus providing a multi-parameters analysis about the change of the tumor microenvironment during and after PIT treatment.
Fig. 2.
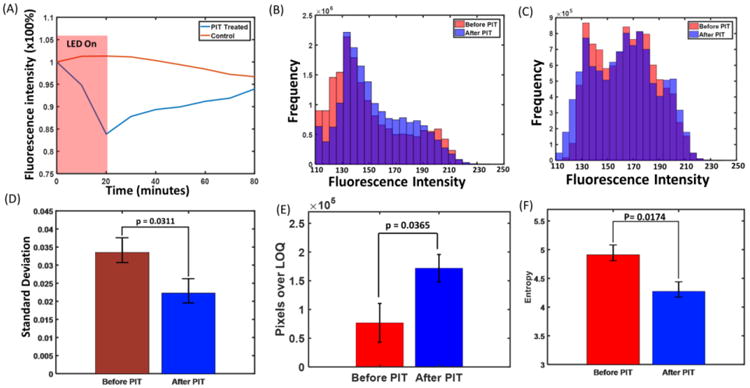
(A) Representative normalized average IR700 fluorescence intensity of the tumor during and after PIT treatment. (B) Histograms of the pixel intensity before and 60 min after PIT in the tumor. (C) Histograms of the pixel intensity at 0 min and 80 min in the control tumor (No PIT treatment). (D) Standard deviations (SDs) of the histograms of tumor before and 60 min after PIT treatment. Tumor before PIT shows a significant larger SD than that of tumor 60 min after PIT. (E) Pixels with values over the LOQ in the tumor before and 60 min after PIT treatment. Tumor 60 min after PIT has a significant larger pixel number with value over the background than that of tumor before PIT. (F) Entropy of pixels values in the tumor before and 60 min after PIT treatment. The pixels in the tumor 60 min after PIT have significant smaller entropy than that of tumor before PIT (n=5).
3.3. In vivo imaging the distribution of the mACs at different intra-tumor depths using FLOT/OCT system
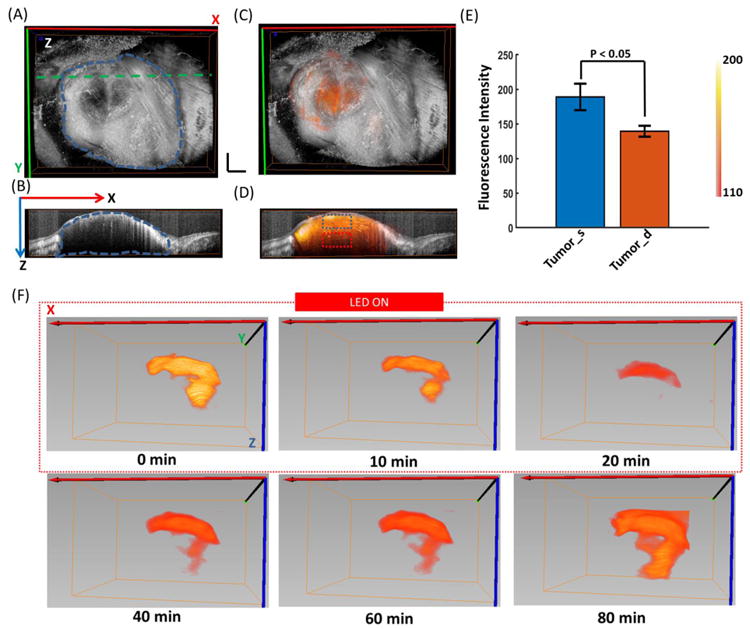
Since the FLOT/OCT system can provide depth-resolved information of the tumor structure and drug distribution non-invasively, representative cross-sections from the 3D FLOT/OCT tumor images at different time points during and after PIT treatment are shown in Fig. 3A. At 0 min, the scattered mACs in the tumor at different depths can be clearly resolved. The mACs distributed in a heterogeneous manner and some areas filled with clustered fluorescence signals especially in the tumor surface. When the NIR LED irradiation began, the IR700 fluorescence intensity dropped quickly at both tumor surface and deep tumor regions. After 20 min PIT treatment, the IR700 fluorescence intensity recovered as described before. The IR700 fluorescence intensity in the areas with clustered fluorescence signals seems undergoing a quicker recovery. After 60 minutes, the IR700 fluorescence intensity showed a more homogeneous distribution in the tumor. To indicate the heterogeneity of the recovery in IR700 fluorescence intensity, region of interest (ROI) (50 × 30 × 15 pixels) from tumor surface (dashed blue box as shown in the Fig. 3A at 80 min) and deep tumor (dashed red box as shown in the Fig. 3A at 80 min) were averaged to show the changes in fluorescence during and after PIT. The IR700 fluorescence intensity was further normalized to the corresponding initial level (0 min). The normalized IR700 fluorescence intensity clearly dropped more in the tumor surface at 20 min (Fig. 3B). The recovery of IR700 fluorescence intensity (at 80 min) in deep tumor was significantly higher than that of the tumor surface (Fig. 3C). There was not any clear change of the mACs distribution in control tumor (without PIT treatment) (Supplementary Fig. 3A).
Fig. 3.
(A) Representative cross-sections from the 3D FLOT/OCT images of the mACs' micro-distribution in tumor at different time points during and after PIT Treatment. PIT treatment starts at 0 min and lasts for 20 min as indicated by the red dashed line. (B) Representative normalized average IR700 fluorescence intensity of tumor surface and deep tumor (as indicated by the dashed blue and red squares in Fig. 3A at 80 min) during and after PIT treatment. (C) IR700 fluorescence recovery value of the tumor surface and deep tumor. Deep tumors showed a significantly higher fluorescence recovery intensity value than that of the tumor surface (n=5). Scale bar 1 mm. (D) Histological specimens of A431 tumors from the PIT-treated group. There is more cell necrosis in tumor surface compared to deep tumor. The image on the right hand is the zoomed in image from the yellow square.
With H&E staining, there is more cell necrosis in tumor surface compared to deep tumor (Fig. 3E). More mAb-IR700 was found in the deep tumor after PIT treatment (Supplementary Fig. 2D). In the control tumor, no obvious cell necrosis was observed in either tumor surface or the deep tumor (Supplementary Fig. 3B).
With the proposed FLOT/OCT system, we evaluated the 3D micro-distribution of mACs at different depths with a very minimally invasive manner (since we need to remove the skin over the tumor). And currently we are working on improving the penetration depth of the system thus we could have the skin intact in the near future [49]. As observed before, tumors surface showed significantly higher IR700 fluorescence intensity value than that of the deep tumor before PIT due to the penetration limitation of the mACs (Fig. 1E), while the recovery of IR700 fluorescence intensity (at 80 min) in deep tumor was significantly higher than that of the tumor surface (Fig. 3C). After PIT treatment, the perivascular layers of cancer cells were selectively destructed and the mACs had easier access to the deep tumor's extravascular space compared to before [22]. Although less mACs were needed to recover or outnumber its initial value for deep tumor, the higher recovery of mACs in deep tumor did provide some cues about the SUPR effect [22].
3.4. In vivo imaging the distribution of the mACs and tumor vasculature using FLOT/OCT system
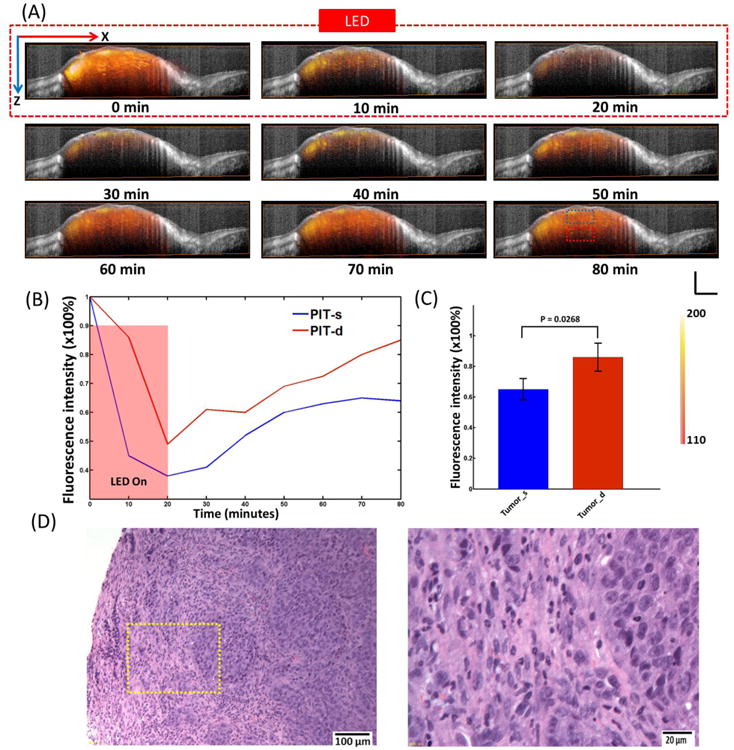
In this study, the tumor vasculature was also obtained using the multi-wavelength FLOT system to further investigate the mACs distribution during and after PIT. The left image of Fig. 4A shows the 3D tumor structure obtained from OCT, with the shape of tumor clearly displayed. The middle image of Fig. 4A shows the 3D vasculature on the same tumor acquired from multi-wavelength FLOT system. The tortuous blood vessels (green) with different sizes are clearly presented. The right image of Fig. 4A fuses the tumor structure with tumor vasculature. The mACs distribution (orange-red) during and after PIT treatment is superimposed with 3D tumor structure and 3D vasculature in Fig. 4B. Before PIT treatment at 0 min, as observed before, the distribution of mACs was heterogeneous with some areas filling with clustered fluorescence signals. The relative distribution of mACs with respect to the blood vessels in different tumor areas could be visualized clearly. After NIR irradiation began, the expected drop of IR700 fluorescence intensity occurred quickly in the entire tumor. From 25 min to 40 min, the IR700 fluorescence intensity in the areas with clustered fluorescence signals began to recover. From 50 min, the mACs started to fill most of the tumor areas in a more homogeneous way. Interestingly the fluorescence distribution achieved a bigger area coverage than that of the initial point (0 min), meaning the drug accessed more tumor areas after NIR-PIT treatment. MACs in the control tumor did not show obvious change (Supplementary Fig. 4B). Different small regions (5 × 5 × 5 pixels) from various locations of the tumor were picked to show the changes in fluorescence during and after PIT (Fig. 4C). We intended to label the areas having clustered fluorescence signals initially with small numbers (e.g. 1-5), the areas having weak fluorescence signals initially with big numbers (e.g. 6-10). The areas with small numbers had a quick but less recovery while the recovery for areas with large numbers mainly happened after 40 min and the recovery reached to a higher value than that of the initial status (0 min). There are no significant changes of IR700 fluorescence in different areas in the control mice but some small fluctuations (Supplementary Fig. 4C and D).
Fig. 4.
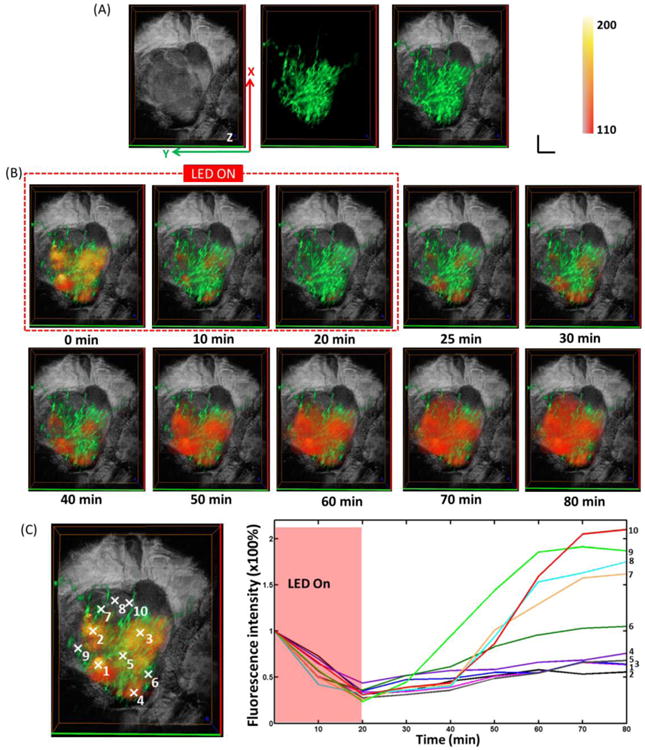
(A) Left: 3D OCT tumor image. Middle: FLOT images of blood vessel (green) on the tumor surface labelled by Oregon Green. Right: Fused OCT/FLOT image. (B) Representative 3D FLOT images of the micro-distribution of mACs in tumor at different time points during and after PIT Treatment (red-orange) superimposed with 3D tumor microstructure and corresponding 3D blood vessel on the tumor surface. PIT treatment starts at 0 min and lasts for 20 min as indicated by the red dashed line. Scale bar 1 mm. (C) Changes in fluorescence of the 3D tumor area during and after PIT treatment. Fluorescence signal was calculated from the positions (centered by the white marks in the left images: with 5 × 5 × 5 pixels size) numbered accordingly.
3.5. In vivo imaging the distribution of the mACs using two-photon microscopy
To verify the results obtained from FLOT/OCT system, we further applied TPM system to investigate the mACs distribution during and after PIT. GFP was also employed as a surrogate of cell death and to make the blood vessels more visible. The nearly intact tumor was placed under the two-photon objective lens for imaging after removing the skin over it. The imaging ROIs were selected to include blood vessels/capillaries. As shown in Fig. 5A, before NIR irradiation, most of the mACs clustered around the blood vessels. Immediately after turning on the NIR LED, the IR700 fluorescence decreased quickly. The GFP signals also decreased while the blood vessels can still be clearly distinguished. After NIR LED irradiation finished, the IR700 fluorescence showed quick recovery surrounding the blood vessels and extended to a bigger region from the blood vessel. In the control mice receiving no NIR irradiation, there were no significant changes of IR700 distribution and GFP fluorescence (Supplementary Fig. 5A). Similarly, changes in IR700 fluorescence from selected areas showed higher recovery in the areas initially (0 min) with less mACs (Fig. 5B). The areas with larger distances from the blood vessels (e.g. areas 6 and 7) also achieved higher recovery of IR700 fluorescence after PIT treatment. We can conclude that PIT can promote the delivery of mACs especially into the areas where were initially difficult for mACs to reach (Fig. 4C and Fig. 5B), which resulted from enhancement of permeability and retention. There were no significant changes of IR700 fluorescence in different selected areas in the control mice without NIR irradiation (Supplementary Fig. 5B and C).
Fig. 5.
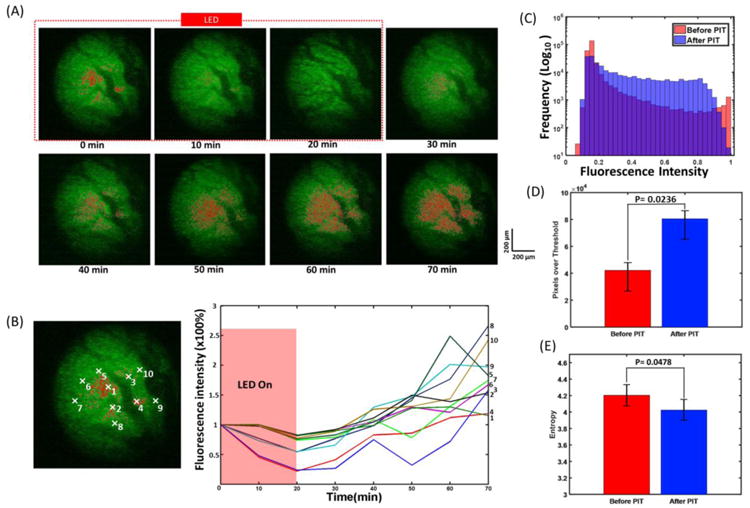
(A) Tumor surface during and after PIT treatment imaged by TPM system. PIT treatment starts at 0 min and lasts for 20 min as indicated by the red dashed line. (B) Changes in fluorescence of the tumor during and after PIT treatment. Fluorescence signal was calculated from the positions (centered by the white marks in the left images: with 10 × 10 pixels size) numbered accordingly. (C) Histograms of the pixel intensity before and 60 min after PIT in the tumor. (D) Pixels with values over the background in the tumor before and 60 min after PIT treatment. Tumor 60 min after PIT has a significant larger pixel number with value over the background than that of tumor before PIT. (E) Entropy of pixels values in the tumor before and 60 min after PIT treatment. The pixels in the tumor 60 min after PIT have significant smaller entropy than that of tumor before PIT (n=5).
The histograms of the pixels before and 60 min after PIT in the tumor were plotted in Fig. 5C. Before PIT treatment, there were two peaks at left and right edges of the histogram. And 60 min after PIT treatment, the entire histogram shifted to the middle range while the control group did not have such trend (Supplementary Fig. 5D). The pixels with values over background in the tumor were also calculated before and 60 min after PIT treatment. There were significantly more pixels with values over background in the tumor 60 min after PIT treatment than that of before PIT (Fig. 5D). We further analyzed the entropy of pixels in the tumor before and 60 min after PIT treatment. The pixels in the tumor 60 min after PIT have significant smaller entropy than that of tumor before PIT (Fig. 5E). Moreover, in this study, we also investigated the micro-distribution of mACs in the tumor-bearing mice implanted with MDAMB468 cells except A431 cells and they provided very consistent results.
4. Conclusions
We investigated the 3D micro-distribution of mACs during/after PIT in situ and in vivo using FLOT/OCT imaging and high-resolution two-photon microscopy. The distribution of mACs was improved after PIT treatment with more mACs reaching more areas in the tumor. Being able to quantitatively monitor 3D theranostic agent distribution and therapeutic effects including cellular necrosis within the tumor in vivo and in real time, these novel imaging methods can serve as new platforms for further understanding the PIT/SUPR mechanism as well as other tumor-targeted drug delivery systems, thus optimizing the effectiveness of treatment. Due to the low penetration depth, this new multi-modal imaging technology is applicable predominantly for applications in which tissues of interest are superficial, such as the exposed mouse brain, skin, and preclinical research at current stage. While, the two imaging modalities could be integrated into a functional endoscopy system using microelectromechanical system (MEMS), Gradient-index (GRIN) cylindrical lens and imaging fiber bundle in the future for more studies on different disease models and potentially clinical applications [61].
Supplementary Material
Acknowledgments
This research was supported by NIH grant (R03 CA176027) and UMD-NCI Partnership for Integrative Cancer Research. We thank Sunghoon Lee from University of Maryland for data analysis and discussion.
Abbreviations
- PIT
Photo-immunotherapy
- mAb
monoclonal antibody
- NIR
near-infrared
- NIR-PIT
Near-infrared photoimmunotherapy
- IR 700
IRDye700DX
- EPR
enhanced permeability and retention
- SUPR
super-enhanced permeability and retention
- OCT
optical coherence tomography
- FLOT
fluorescence laminar optical tomography
- TPM
two-photon microscopy
- mAb
monoclonal antibody
- mAC
photo-absorber conjugate
- EGFR
epidermal growth factor receptor
- ICD
immunogenic cell death
- 3-D
three-dimensional
- pan-IR700
panitumumab-IRDye700DX
- GFP
green fluorescent protein
- IACUCs
Institutional Animal Care and Use Committees
- MZI
Mach-Zehnder interferometer
- FC
fiber couple
- BD
balanced detector
- tumor_s
tumor surface
- tumor_d
deep tumor
- H&E
Hematoxylin and Eosin
- DIC
differential interference contrast
- ROI
region of interest
- GRIN
Gradient-index
- MEMS
microelectromechanical system
Footnotes
Disclosure of Potential Conflicts of Interest: There are no potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nagaya T, Nakamura Y, Sato K, Harada T, Choyke PL, Kobayashi H. Improved micro-distribution of antibody-photon absorber conjugates after initial near infrared photoimmunotherapy (NIR-PIT) Journal of Controlled Release. 2016;232:1–8. doi: 10.1016/j.jconrel.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiner LM, Surana R, Wang SZ. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parish CR. Cancer immunotherapy: The past, the present and the future. Immunol Cell Biol. 2003;81:106–113. doi: 10.1046/j.0818-9641.2003.01151.x. [DOI] [PubMed] [Google Scholar]
- 4.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nature biotechnology. 2005;23:1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 5.Mew D, Wat CK, Towers GH, Levy JG. Photoimmunotherapy: treatment of animal tumors with tumor-specific monoclonal antibody-hematoporphyrin conjugates. Journal of immunology. 1983;130:1473–1477. [PubMed] [Google Scholar]
- 6.Rosenkranz AA, Jans DA, Sobolev AS. Targeted intracellular delivery of photosensitizers to enhance photodynamic efficiency. Immunol Cell Biol. 2000;78:452–464. doi: 10.1046/j.1440-1711.2000.00925.x. [DOI] [PubMed] [Google Scholar]
- 7.Carcenac M, Dorvillius M, Garambois V, Glaussel F, Larroque C, Langlois R, Hynes NE, van Lier JE, Pelegrin A. Internalisation enhances photo-induced cytotoxicity of monoclonal antibody-phthalocyanine conjugates. Brit J Cancer. 2001;85:1787–1793. doi: 10.1054/bjoc.2001.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vrouenraets MB, Visser GWM, Stewart FA, Stigter M, Oppelaar H, Postmus PE, Snow GB, van Dongen GAMS. Development of meta-tetrahydroxyphenylchlorin-monoclonal antibody conjugates for photoimmunotherapy. Cancer Res. 1999;59:1505–1513. [PubMed] [Google Scholar]
- 9.Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nature medicine. 2011;17:1685–1691. doi: 10.1038/nm.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanaoka H, Nagaya T, Sato K, Nakamura Y, Watanabe R, Harada T, Gao W, Feng M, Phung Y, Kim I, Paik CH, Choyke PL, Ho M, Kobayashi H. Glypican-3 targeted human heavy chain antibody as a drug carrier for hepatocellular carcinoma therapy. Molecular pharmaceutics. 2015;12:2151–2157. doi: 10.1021/acs.molpharmaceut.5b00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagaya T, Nakamura Y, Okuyama S, Ogata F, Maruoka Y, Choyke PL, Kobayashi H. Near-Infrared Photoimmunotherapy Targeting Prostate Cancer with Prostate-Specific Membrane Antigen (PSMA) Antibody. Molecular cancer research : MCR. 2017;15:1153–1162. doi: 10.1158/1541-7786.MCR-17-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagaya T, Nakamura Y, Sato K, Harada T, Choyke PL, Hodge JW, Schlom J, Kobayashi H. Near infrared photoimmunotherapy with avelumab, an anti-programmed death-ligand 1 (PD-L1) antibody. Oncotarget. 2017;8:8807–8817. doi: 10.18632/oncotarget.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagaya T, Nakamura Y, Sato K, Harada T, Choyke PL, Kobayashi H. Near infrared photoimmunotherapy of B-cell lymphoma. Molecular oncology. 2016;10:1404–1414. doi: 10.1016/j.molonc.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagaya T, Nakamura Y, Sato K, Zhang YF, Ni M, Choyke PL, Ho M, Kobayashi H. Near infrared photoimmunotherapy with an anti-mesothelin antibody. Oncotarget. 2016;7:23361–23369. doi: 10.18632/oncotarget.8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagaya T, Sato K, Harada T, Nakamura Y, Choyke PL, Kobayashi H. Near Infrared Photoimmunotherapy Targeting EGFR Positive Triple Negative Breast Cancer: Optimizing the Conjugate-Light Regimen. PloS one. 2015;10:e0136829. doi: 10.1371/journal.pone.0136829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato K, Sato N, Xu B, Nakamura Y, Nagaya T, Choyke PL, Hasegawa Y, Kobayashi H. Spatially selective depletion of tumor-associated regulatory T cells with near-infrared photoimmunotherapy. Science translational medicine. 2016;8:352ra110. doi: 10.1126/scitranslmed.aaf6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.https://clinicaltrials.gov/ct2/show/NCT02422979.
- 18.Ogawa M, Tomita Y, Nakamura Y, Lee MJ, Lee S, Tomita S, Nagaya T, Sato K, Yamauchi T, Iwai H, Kumar A, Haystead T, Shroff H, Choyke PL, Trepel JB, Kobayashi H. Immunogenic cancer cell death selectively induced by near infrared photoimmunotherapy initiates host tumor immunity. Oncotarget. 2017;8:10425–10436. doi: 10.18632/oncotarget.14425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sano K, Nakajima T, Choyke PL, Kobayashi H. Markedly enhanced permeability and retention effects induced by photo-immunotherapy of tumors. ACS nano. 2013;7:717–724. doi: 10.1021/nn305011p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81–89. doi: 10.7150/thno.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sano K, Nakajima T, Choyke PL, Kobayashi H. The effect of photoimmunotherapy followed by liposomal daunorubicin in a mixed tumor model: a demonstration of the super-enhanced permeability and retention effect after photoimmunotherapy. Molecular cancer therapeutics. 2014;13:426–432. doi: 10.1158/1535-7163.MCT-13-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang Q, Nagaya T, Liu Y, Lin J, Sato K, Kobayashi H, Chen Y. Real-time monitoring of microdistribution of antibody-photon absorber conjugates during photoimmunotherapy in vivo. Journal of Controlled Release. 2017;260:154–163. doi: 10.1016/j.jconrel.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitsunaga M, Nakajima T, Sano K, Choyke PL, Kobayashi H. Near-infrared theranostic photoimmunotherapy (PIT): repeated exposure of light enhances the effect of immunoconjugate. Bioconjugate chemistry. 2012;23:604–609. doi: 10.1021/bc200648m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan BH, Burgess SA, Iranmahboob A, Bouchard MB, Lehrer N, Bordier C, Hillman EMC. A system for high-resolution depth-resolved optical imaging of fluorescence andabsorption contrast. Rev Sci Instrum. 2009;80 doi: 10.1063/1.3117204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hillman EMC, Bernus O, Pease E, Bouchard MB, Pertsov A. Depth-resolved optical imaging of transmural electrical propagation in perfused heart. Optics express. 2007;15:17827–17841. doi: 10.1364/oe.15.017827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang Q, Wang J, Frank A, Lin J, Li Z, Chen Cw, Jin L, Wu T, Greenwald BD, Mashimo H, Chen Y. Depth-resolved imaging of colon tumor using optical coherence tomography and fluorescence laminar optical tomography. Biomedical optics express. 2016;7:5218–5232. doi: 10.1364/BOE.7.005218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan S, Li Q, Jiang J, Cable A, Chen Y. Three-dimensional coregistered optical coherence tomography and line-scanning fluorescence laminar optical tomography. Optics letters. 2009;34:1615–1617. doi: 10.1364/ol.34.001615. [DOI] [PubMed] [Google Scholar]
- 28.Hillman EMC, Boas DA, Dale AM, Dunn AK. Laminar optical tomography: demonstration of millimeter-scale depth-resolved imaging in turbid media. Optics letters. 2004;29:1650–1652. doi: 10.1364/ol.29.001650. [DOI] [PubMed] [Google Scholar]
- 29.Tang Q, Tsytsarev V, Frank A, Wu Y, Chen CW, Erzurumlu RS, Chen Y. In Vivo Mesoscopic Voltage-Sensitive Dye Imaging of Brain Activation. Scientific reports. 2016;6:25269. doi: 10.1038/srep25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Yuan S, Wierwille J, Naphas R, Li Q, Blackwell TR, Winnard PT, Raman V, Glunde K. Integrated Optical Coherence Tomography (OCT) and Fluorescence Laminar Optical Tomography (FLOT) IEEE J Selected Topics in Quantum Electronics. 2010;16:755–766. [Google Scholar]
- 31.Tang Q, Lin J, Tsytsarev V, Erzurumlu RS, Liu Y, Chen Y. Review of mesoscopic optical tomography for depth-resolved imaging of hemodynamic changes and neural activities. Neurophotonics. 2016;4:011009–011009. doi: 10.1117/1.NPh.4.1.011009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen CW, Chen Y. Optimization of Design Parameters for Fluorescence Laminar Optical Tomography. Journal of Innovative Optical Health Sciences. 2011;4:309–323. [Google Scholar]
- 33.Ozturk MS, Chen CW, Ji R, Zhao LL, Nguyen BNB, Fisher JP, Chen Y, Intes X. Mesoscopic Fluorescence Molecular Tomography for Evaluating Engineered Tissues. Ann Biomed Eng. 2016;44:667–679. doi: 10.1007/s10439-015-1511-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozturk MS, Rohrbach D, Sunar U, Intes X. Mesoscopic Fluorescence Tomography of a Photosensitizer (HPPH) 3D Biodistribution in Skin Cancer. Academic radiology. 2014;21:271–280. doi: 10.1016/j.acra.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 35.Zhao LL, Lee VK, Yoo SS, Dai GH, Intes X. The integration of 3-D cell printing and mesoscopic fluorescence molecular tomography of vascular constructs within thick hydrogel scaffolds. Biomaterials. 2012;33:5325–5332. doi: 10.1016/j.biomaterials.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozturk MS, Lee VK, Zhao LL, Dai GH, Intes X. Mesoscopic fluorescence molecular tomography of reporter genes in bioprinted thick tissue. Journal of biomedical optics. 2013;18 doi: 10.1117/1.JBO.18.10.100501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bjorn S, Ntziachristos V, Schulz R. Mesoscopic Epifluorescence Tomography: Reconstruction of superficial and deep fluorescence in highly-scattering media. Optics express. 2010;18:8422–8429. doi: 10.1364/OE.18.008422. [DOI] [PubMed] [Google Scholar]
- 38.Huang D, Swanson EA, Lin CP, Schuman JS, Stinson WG, Chang W, Hee MR, Flotte T, Gregory K, Puliafito CA, et al. Optical coherence tomography. Science. 1991;254:1178–1181. doi: 10.1126/science.1957169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kut C, Chaichana KL, Xi J, Raza SM, Ye X, McVeigh ER, Rodriguez FJ, Quiñones-Hinojosa A, Li X. Detection of human brain cancer infiltration ex vivo and in vivo using quantitative optical coherence tomography. Science translational medicine. 2015;7:292ra100–292ra100. doi: 10.1126/scitranslmed.3010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Y, Wang S, Guo Q, Kessel S, Rubinoff I, Chan LL, Li P, Liu Y, Qiu J, Zhou C. Optical Coherence Tomography Detects Necrotic Regions and Volumetrically Quantifies Multicellular Tumor Spheroids. Cancer Res. 2017;77:6011–6020. doi: 10.1158/0008-5472.CAN-17-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujimoto JG, Brezinski ME, Tearney GJ, Boppart SA, Bouma B, Hee MR, Southern JF, Swanson EA. Optical Biopsy And Imaging Using Optical Coherence Tomography. Nat Med. 1995;1:970–972. doi: 10.1038/nm0995-970. [DOI] [PubMed] [Google Scholar]
- 42.Tearney GJ, Brezinski ME, Bouma BE, Boppart SA, Pitris C, Southern JF, Fujimoto JG. In vivo endoscopic optical biopsy with optical coherence tomography. Science. 1997;276:2037–2039. doi: 10.1126/science.276.5321.2037. [DOI] [PubMed] [Google Scholar]
- 43.Bouma BE, Yun SH, Vakoc BJ, Suter MJ, Tearney GJ. Fourier-domain optical coherence tomography: recent advances toward clinical utility. Curr Opin Biotech. 2009;20:111–118. doi: 10.1016/j.copbio.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding ZY, Liang CP, Tang QG, Chen Y. Quantitative single-mode fiber based PS-OCT with single input polarization state using Mueller matrix. Biomedical optics express. 2015;6:1828–1843. doi: 10.1364/BOE.6.001828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang Q, Liang CP, Wu K, Sandler A, Chen Y. Real-time epidural anesthesia guidance using optical coherence tomography needle probe. Quantitative Imaging in Medicine and Surgery. 2014;5:118–124. doi: 10.3978/j.issn.2223-4292.2014.11.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding Z, Tang Q, Liang CP, Wu K, Sandlerc A, Li H, Chen Y. Imaging Spinal Structures With Polarization-Sensitive Optical Coherence Tomography. IEEE Photonics Journal. 2016;8:1–8. [Google Scholar]
- 47.Ding Z, Jin L, Wang HW, Tang Q, Guo H, Chen Y. Multi-modality Optical Imaging of Rat Kidney Dysfunction: In Vivo Response to Various Ischemia Times. In: Luo Q, Li LZ, Harrison DK, Shi H, Bruley DF, editors. Oxygen Transport to Tissue XXXVIII. Springer International Publishing; Cham: 2016. pp. 345–350. [DOI] [PubMed] [Google Scholar]
- 48.Chen CW, Yeatts AB, Coates EE, Fisher JP, Chen Y. Biomedical Optics and 3-D Imaging. Optical Society of America; Miami, Florida: 2012. Experimental Demonstration of Angled Fluorescent Laminar Optical Tomography for Tissue Engineering; p. BTu4A.5. [Google Scholar]
- 49.Tang Q, Liu Y, Tsytsarev V, Lin J, Wang B, Kanniyappan U, Li Z, Chen Y. High-dynamic-range fluorescence laminar optical tomography (HDR-FLOT) Biomedical optics express. 2017;8:2124–2137. doi: 10.1364/BOE.8.002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakajima T, Sano K, Mitsunaga M, Choyke PL, Kobayashi H. Real-time monitoring of in vivo acute necrotic cancer cell death induced by near infrared photoimmunotherapy using fluorescence lifetime imaging. Cancer Res. 2012;72:4622–4628. doi: 10.1158/0008-5472.CAN-12-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitsunaga M, Nakajima T, Sano K, Choyke PL, Kobayashi H. Near-infrared Theranostic Photoimmunotherapy (PIT): Repeated Exposure of Light Enhances the Effect of Immunoconjugate. Bioconjugate Chem. 2012;23:604–609. doi: 10.1021/bc200648m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johns M, Giller CA, German DC, Liu HL. Determination of reduced scattering coefficient of biological tissue from a needle-like probe. Optics express. 2005;13:4828–4842. doi: 10.1364/opex.13.004828. [DOI] [PubMed] [Google Scholar]
- 53.Sandell JL, Zhu TC. A review of in-vivo optical properties of human tissues and its impact on PDT. Journal of biophotonics. 2011;4:773–787. doi: 10.1002/jbio.201100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang LH, Jacques SL. Use Of a Laser-Beam with an Oblique Angle Of Incidence To Measure the Reduced Scattering Coefficient Of a Turbid Medium. Applied optics. 1995;34:2362–2366. doi: 10.1364/AO.34.002362. [DOI] [PubMed] [Google Scholar]
- 55.Long GL, Winefordner JD. Limit of Detection A Closer Look at the IUPAC Definition. Analytical chemistry. 1983;55:712A–724A. [Google Scholar]
- 56.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 57.Hambley TW. Is Anticancer Drug Development Heading in the Right Direction? Cancer Res. 2009;69:1259. doi: 10.1158/0008-5472.CAN-08-3786. [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi H, Choyke PL. Super enhanced permeability and retention (SUPR) effects in tumors following near infrared photoimmunotherapy. Nanoscale. 2016;8:12504–12509. doi: 10.1039/c5nr05552k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maeda H. Tumor-Selective Delivery of Macromolecular Drugs via the EPR Effect: Background and Future Prospects. Bioconjugate Chem. 2010;21:797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 60.Anantrasirichai N, Achim A, Morgan JE, Erchova I, Nicholson L. SVM-based texture classification in Optical Coherence Tomography. 2013 IEEE 10th International Symposium on Biomedical Imaging. 2013:1332–1335. [Google Scholar]
- 61.Xie T, Mukai D, Guo S, Brenner M, Chen Z. Fiber-optic-bundle-based optical coherence tomography. Optics letters. 2005;30:1803–1805. doi: 10.1364/ol.30.001803. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.