

ABSTRACT
Sindbis virus (SINV) infection of neurons in the brain and spinal cord in mice provides a model system for investigating recovery from encephalomyelitis and antibody-mediated clearance of virus from the central nervous system (CNS). To determine the roles of IgM and IgG in recovery, we compared the responses of immunoglobulin-deficient activation-induced adenosine deaminase-deficient (AID−/−), secretory IgM-deficient (sIgM−/−), and AID−/− sIgM−/− double-knockout (DKO) mice with those of wild-type (WT) C57BL/6 mice for disease, clearance of infectious virus and viral RNA from brain and spinal cord, antibody responses, and B cell infiltration into the CNS. Because AID is essential for immunoglobulin class switch recombination and somatic hypermutation, AID−/− mice produce only germ line IgM, while sIgM−/− mice secrete IgG but no IgM and DKO mice produce no secreted immunoglobulin. After intracerebral infection with the TE strain of SINV, most mice recovered. Development of neurologic disease occurred slightly later in sIgM−/− mice, but disease severity, weight loss, and survival were similar between the groups. AID−/− mice produced high levels of SINV-specific IgM, while sIgM−/− mice produced no IgM and high levels of IgG2a compared to WT mice. All mice cleared infectious virus from the spinal cord, but DKO mice failed to clear infectious virus from brain and had higher levels of viral RNA in the CNS late after infection. The numbers of infected cells and the amount of cell death in brain were comparable. We conclude that antibody is required and that either germ line IgM or IgG is sufficient for clearance of virus from the CNS.
IMPORTANCE Mosquito-borne alphaviruses that infect neurons can cause fatal encephalomyelitis. Recovery requires a mechanism for the immune system to clear virus from infected neurons without harming the infected cells. Antiviral antibody has previously been shown to be a noncytolytic means for alphavirus clearance. Antibody-secreting cells enter the nervous system after infection and produce antiviral IgM before IgG. Clinical studies of human viral encephalomyelitis suggest that prompt production of IgM is associated with recovery, but it was not known whether IgM is effective for clearance. Our studies used mice deficient in production of IgM, IgG, or both to characterize the antibody necessary for alphavirus clearance. All mice developed similar signs of neurologic disease and recovered from infection. Antibody was necessary for virus clearance from the brain, and either early germ line IgM or IgG was sufficient. These studies support the clinical observation that prompt production of antiviral antibody is a determinant of outcome.
KEYWORDS: Sindbis virus, antibody, mice, neurons, viral encephalomyelitis, virus clearance
INTRODUCTION
Sindbis virus (SINV) is an enveloped, positive-sense RNA virus in the Togaviridae family that causes summertime outbreaks of fever, rash, and arthritis in northern Europe and South Africa (1). In mice, SINV infects neurons in the brain and spinal cord and provides a model system for the study of recovery from alphavirus encephalomyelitis (2). The types of neurons that are most susceptible to infection are cortical and hippocampal neurons in the brain and motor neurons in the brain stem and spinal cord (3, 4). Virus replication is maximal between 3 and 5 days after infection, and most neurons infected with relatively avirulent strains of SINV (e.g., strain TE) survive. During recovery, virus clearance is initiated and three phases of noncytolytic virus clearance are recognized: (i) rapid clearance of infectious virus (days 5 to 8) and (ii) gradual clearance of viral RNA (weeks 2 to 8), followed by (iii) sustained low levels of persistent viral RNA (after 8 weeks) (5–7). Previous studies have shown that the innate type I interferon (IFN) response is essential for initial control of virus replication and neuronal survival (8, 9) and that the primary effectors of adaptive immune response-mediated virus clearance from brain and spinal cord neurons are antiviral antibody and gamma IFN (IFN-γ) (2, 10–12). Although these mechanisms usually act in concert, IFN-γ alone can clear the virus from motor neurons in the spinal cord, while antibody alone is also effective for clearance from the brain (10, 12).
Both polyclonal antibody in immune serum and monoclonal antibody specific for E2, the virion surface glycoprotein responsible for cellular attachment, can clear infectious virus and viral RNA from the nervous system of persistently infected mice with severe combined immunodeficiency (SCID mice) and inhibit SINV replication in persistently infected primary rat dorsal root ganglion neurons in culture (11, 13). Antibody treatment of infected cells restores host cell protein synthesis, membrane potential, and the antiviral activity of IFN-α late in infection to infected cells (14, 15). In vitro studies have also shown that the IgG3 and IgG1 isotypes of SINV-specific anti-E2 monoclonal antibodies are equally effective at inducing virus clearance from infected neurons. Furthermore, bivalent antibody, but not the Fc portion of antibody, is required for clearance, as demonstrated by the fact that F(ab′)2 fragments, but not Fab fragments, of IgG have antiviral activity (13). Therefore, it is likely that anti-E2 antibody acts by cross-linking glycoproteins on the surface of infected cells.
In vivo clearance studies using adoptive transfer of antibody to infected SCID mice, during which antibody needs to enter the central nervous system (CNS) from plasma by crossing the blood-brain barrier to reach infected neurons, have shown that IgG is effective (11, 16). However, several pieces of information suggest that IgM may be most important for in vivo clearance of SINV in immunologically normal mice. For instance, only IgM-secreting SINV-specific B cells have infiltrated the CNS when clearance of infectious virus is occurring during the first 7 to 8 days after infection (7). IgG is not detected in the CNS until 10 to 14 days and then steadily increases during the gradual clearance of viral RNA 2 to 8 weeks after infection (6, 7). In addition, athymic nu/nu mice and thymectomized mice that produce IgM but little to no IgG can clear infectious virus from the brain as quickly as wild-type (WT) mice (17–19).
To better understand antibody-mediated virus clearance, mice with deficits in immunoglobulin synthesis have been studied. For instance, μMT mice have a targeted defect in the μ heavy chain, leading to a failure of both B cell maturation and antibody production (20). Studies of μMT mice have shown that antibody is required for virus clearance from brain cortical and hippocampal neurons, but not for clearance from spinal cord motor neurons, where clearance can also be accomplished by T cell production of IFN-γ that acts through a JAK/STAT signaling pathway to inhibit virus replication (10, 12, 21–23). Because B cells have immune functions in addition to antibody production during the response to virus infection (24), we sought to further evaluate the immunoglobulin requirements for in vivo clearance of SINV, particularly the relative importance of secreted IgM and IgG, by using mice with targeted defects in antibody synthesis.
The responses of three types of antibody-deficient mice, activation-induced cytidine deaminase (AID)-deficient (AID−/−) mice, secretory IgM-deficient (sIgM−/−) mice, and double-knockout (DKO) mice deficient in both AID and sIgM (AID−/− sIgM−/− mice) were compared to those of WT C57BL/6 mice. AID is a key enzyme for both class switch recombination and somatic hypermutation, so AID−/− mice can produce and secrete only germ line IgM (25). sIgM−/− mice lack alternative splicing for secretory IgM but produce membranous IgM and undergo class switch recombination to secrete IgG, IgA, and IgE, but not IgM (26). AID−/− sIgM−/− DKO mice have mature B cells that can differentiate into plasmablasts, but not plasma cells, and cannot secrete any immunoglobulin (27). These mice were infected intracerebrally with SINV and compared for the development of neurologic signs of disease, the production of antiviral antibody in the CNS, and the clearance of infectious virus and viral RNA from brain and spinal cord. These studies show that either IgM or IgG can clear SINV from the CNS.
RESULTS
Development of neurologic disease.
AID−/−, sIgM−/−, and AID−/− sIgM−/− DKO and WT mice were infected intracerebrally with 1,000 PFU of the TE strain of SINV and evaluated daily for evidence of neurologic disease (Fig. 1). Signs of disease appeared slightly later in sIgM−/− mice at a median of 6 days after infection, whereas they appeared at 5 days after infection for WT, AID−/−, and DKO mice (Fig. 1A), and once they were initiated, signs of disease developed more rapidly in AID−/− mice than WT mice (Fig. 1B). For all groups, disease was mostly mild, with peak average clinical scores being 1.5 (characterized primarily by mild gait abnormalities). The recovery of DKO mice was the slowest (Fig. 1B). Weight loss did not differ between groups; it began 3 to 5 days after infection and reached a maximum at 7 to 8 days, with recovery to the original weight occurring by 2 weeks after infection (Fig. 1C). Five to 10% of the animals died between 8 and 14 days after infection (Fig. 1D). Therefore, clinical evidence of neurologic disease was of similar severity in the different groups and showed only slight differences in onset between groups.
FIG 1.

Morbidity and mortality of SINV-infected C57BL/6 (B6) mice. WT (n = 22), AID−/− (n = 31), sIgM−/− (n = 33), and DKO (n = 29) mice were inoculated intracerebrally with 1,000 PFU SINV TE and followed daily for the onset of clinical signs (A) (**, P < 0.01, Kaplan-Meier log-rank test), clinical scores (B) (*, P < 0.05; ***, P < 0.001 for AID−/− versus sIgM−/− mice, 2-way analysis of variance with Tukey's multiple-comparison test), percent body weight change (C), and survival (D).
Clearance of infectious virus from brain and spinal cord.
To determine the effect of the deficiency of secreted IgM, IgG, or both on the clearance of infectious virus from the brain and spinal cord, tissue homogenates were assayed for SINV by plaque assay (Fig. 2A and B). Virus replication and clearance from brain in sIgM−/− and WT mice were similar. AID−/− mice had lower peak levels of virus and a delay in clearance compared to WT and sIgM−/− mice, while DKO mice had higher brain virus titers at day 3 and delayed clearance (days 5 to 14), followed by persistent virus replication (day 21 onward) (Fig. 2A). Most DKO mice, but not AID−/−, sIgM−/−, or WT mice, had detectable infectious virus in brains by plaque assay at days 10, 14, 45, and 60. All groups of mice cleared infectious virus from the spinal cord, although clearance was slightly delayed for AID−/− mice and substantially delayed for DKO mice compared to WT mice (Fig. 2B).
FIG 2.

Virus clearance from brain and spinal cord. WT, AID−/−, sIgM−/−, and DKO mice were inoculated intracerebrally with 1,000 PFU SINV TE, and brain (A) and spinal cord (B) homogenates from 3 mice from each group at each time point were assayed for infectious virus by plaque assay. RNA was extracted from brain (C) and spinal cord (D) tissues from 6 to 8 mice from each group at each time point and assayed for viral RNA by RT-qPCR. P values were determined by 2-way analysis of variance with Bonferroni's posttest. *, P < 0.05; **, P < 0.01, ***, P < 0.001.
Clearance of viral RNA from brain and spinal cord.
The effect of a deficiency of secreted IgM, IgG, or both on the clearance of viral RNA was assessed by reverse transcription-quantitative PCR (RT-qPCR) for the SINV E2 gene. All mouse groups had similar peak levels and initial clearance of viral RNA from both brains (Fig. 2C) and spinal cords (Fig. 2D). However, for DKO mice, the decrease in viral RNA slowed after day 30 in brains and day 45 in spinal cords, resulting in higher sustained levels of viral RNA in the CNS at late time points. The state of this RNA is not known.
Numbers and distribution of infected and dying cells during virus clearance from brain.
To determine the effect of a deficiency of secreted IgM, IgG, or both on the distribution of SINV-infected cells and induction of cell death, brain sections from 5, 7, and 10 days after infection were stained for SINV proteins by immunofluorescence (white), for SINV RNA by fluorescence in situ hybridization (FISH; red), and for evidence of cell death by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL; green) in the presence of DAPI (4′,6-diamidino-2-phenylindole; blue) to label nuclei (Fig. 3A). The distribution patterns of virus-infected cells were similar between the groups, although individual mice differed, with more abundant clusters of infected cells, as indicated by immunofluorescence (IF) and FISH, being found in the hippocampus and cortex at day 5 than at day 7 or 10 (Fig. 3B). TUNEL-positive cells were in regions of virus-infected cells. The numbers of cells positive for viral proteins (Fig. 4A) and viral RNA (Fig. 4B) declined from day 5 to day 10. The numbers of TUNEL-positive cells were the highest on day 7 and remained high at day 10 for DKO mice (Fig. 4C). In general, AID−/− and sIgM−/− mice had fewer viral RNA-positive cells than WT mice, and sIgM−/− mice had the fewest TUNEL-positive cells at all time points.
FIG 3.

Distribution of viral protein, RNA, and cell death in brain. WT, AID−/−, sIgM−/−, and AID−/− sIgM−/− (DKO) mice were inoculated intracerebrally with 1,000 PFU SINV TE. Formalin-fixed paraffin-embedded brain slices were stained by use of a combination of RNA fluorescence in situ hybridization (FISH) for SINV E1 and E2 genes (red) and fluorescent antibody for SINV proteins (white) and by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL-TSA; green) for dead cells in the presence of DAPI. (A) Brain section from a WT mouse 5 days after infection showing individual channels and merged channels; (B) merged images of brains from WT, AID−/−, sIgM−/−, and DKO mice collected 5, 7, and 10 days (D) after infection.
FIG 4.

Quantification of cells positive for viral protein, RNA, and TUNEL staining in brain. WT, AID−/−, sIgM−/−, and AID−/− sIgM−/− (DKO) mice were inoculated intracerebrally with 1,000 PFU SINV TE. Formalin-fixed paraffin-embedded brain slices from 5, 7, and 10 days after infection were stained by use of a combination of RNA fluorescence in situ hybridization (FISH) for SINV E1 and E2 genes and antibody for SINV proteins (IF) and by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL-TSA) for dead cells (Fig. 3). The numbers of cells positive for SINV protein (A), viral RNA (B), and TUNEL-TSA (C) per slice of right brain hemisphere are shown. Data are from one of two experiments and are for 3 mice per group per time point. *, P < 0.05; **, P < 0.01.
Ifng mRNA expression in brain and spinal cord.
IFN-γ functions in conjunction with antibody for virus clearance from neurons, particularly spinal cord motor neurons (10, 12). To determine the effect of a deficiency of secreted IgM, IgG, or both on the production of IFN-γ in the CNS after SINV infection, Ifng mRNA was quantified in brain and spinal cord homogenates by RT-qPCR (Fig. 5A and B). In both tissues, Ifng mRNA expression peaked 5 to 7 days after infection. In brain, this peak was the highest in AID−/− mice and the lowest in DKO mice (Fig. 5A), while in spinal cord, DKO mice had higher levels at day 7 than all other groups (Fig. 5B). The levels of Ifng mRNA in brains and spinal cords at later time points were similar between groups.
FIG 5.

Ifng and Aid mRNA expression after infection. WT, AID−/−, sIgM−/−, and AID−/− sIgM−/− (DKO) mice were inoculated intracerebrally with 1,000 PFU SINV TE. RNA was extracted from brain and spinal cord homogenates, and RT-qPCR was performed to determine the change (fold increase) in Ifng mRNA expression in brains (A) and spinal cords (B) from that before infection (day 0). Because AID−/− and DKO mice do not express the AID gene, only WT and sIgM−/− mice were tested for Aid mRNA expression in brains (C) and spinal cords (D). *, P < 0.05; ***, P < 0.001.
Aid mRNA expression in brain and spinal cord.
To determine the effect of infection on expression of AID in the CNS, Aid mRNA was quantified in brains and spinal cords of WT and sIgM−/− mice by RT-qPCR. Infection led to a greater relative increase in the amount of Aid mRNA in the brain (Fig. 5C) than the spinal cord (Fig. 5D). Aid mRNA expression reached a plateau 7 days after infection, a time of maximal CNS inflammation (7), and this level was maintained for 3 to 4 weeks, with more expression by WT mice than sIgM−/− mice at day 21 in spinal cord and day 30 in brain.
Levels of SINV-specific IgM and IgG.
To determine the effect of immunoglobulin deficiencies on production of antibodies to SINV in serum, brain, and spinal cord after infection, SINV-specific IgM (Fig. 6A to C) and IgG (Fig. 6D to F) were quantified by enzyme immunoassay (EIA) in the relevant mice. AID−/− mice had high baseline levels of IgM in serum that rapidly increased after infection (Fig. 6A). The baseline levels of IgM in the CNS tissues of AID−/− mice did not differ from those in WT mice; however, after infection, AID−/− mice produced high levels of SINV-specific IgM in brains (Fig. 6B) and spinal cords (Fig. 6C) that initially paralleled the levels of IgM produced by WT mice but kept increasing to day 14 rather than plateauing at day 10 and then decreasing, as observed in WT mice. Serum levels of SINV-specific IgG increased earlier in sIgM−/− mice, were comparable through day 30, and then lower than those in WT mice (Fig. 6D), while in brains (Fig. 6E) and spinal cords (Fig. 6F), the levels were similar. Antibody increases after infection in serum (Fig. 6A and D), but not in the CNS (Fig. 6B, C, E, and F), were more rapid in AID−/− and sIgM−/− mice than WT mice.
FIG 6.

Production of SINV-specific IgM and IgG. WT, AID−/−, sIgM−/−, and AID−/− sIgM−/− (DKO) mice were inoculated intracerebrally with 1,000 PFU SINV TE. Serum and brain and spinal cord homogenates were tested by EIA for SINV-specific IgM (A to C) and IgG (D to F). The graphs show the optical density (OD) of mouse serum (1:100 dilution) (A and D), 20% brain homogenates (1:8 dilution) (B and E), and 10% spinal cord homogenates (1:4 dilution) (C, F). P values represent the significance of differences between WT and AID−/− mice for IgM (A to C) and WT and sIgM−/− mice for IgG (D to F) and were determined by 2-way analysis of variance. Asterisks show significant differences at particular time points (**, P < 0.01, ***, P < 0.001).
Subclasses of SINV-specific IgG.
To determine the effect of sIgM deficiency on the isotypes of IgG antibodies produced after infection, SINV-specific IgG1, IgG2a, IgG2b, and IgG3 were quantified in serum, brains, and spinal cords by EIA (Fig. 7). Although the total levels of SINV-specific IgG were similar (Fig. 6D to F), sIgM−/− mice had higher levels of SINV-specific IgG2a and lower levels of IgG1 and IgG2b than WT mice in sera, brains, and spinal cords (Fig. 7). Very little SINV-specific IgG3 was produced by either sIgM−/− or WT mice.
FIG 7.

Production of SINV-specific IgG isotypes. WT and sIgM−/− mice were inoculated intracerebrally with 1,000 PFU SINV TE. The levels of each IgG subclass in serum (1:100 dilution), 20% brain homogenates (1:8 dilution), and 10% spinal cord homogenates (1:4 dilution) reactive with SINV by EIA are shown. (A) IgG1; (B) IgG2a; (C) IgG2b; (D) IgG3. P values represent the significance of the differences between mouse groups and were determined by 2-way analysis of variance. Asterisks show significant differences at particular time points (*, P < 0.05; **, P < 0.01, ***, P < 0.001).
Cells with cytoplasmic immunoglobulin in brain.
To assess the effects of immunoglobulin deficiency on infiltration of immunoglobulin-producing cells into the CNS, brain sections were stained for IgM or IgG. Cells with cytoplasmic IgM and IgG infiltrated areas of infected brain and nearby thickened pia mater (Fig. 8A). Over time, the numbers of these cells differed between mouse groups (Fig. 8B and C). WT mice had more IgM-positive cells than IgG-positive cells at 10 days after infection, and the numbers of IgM-positive cells decreased rapidly. AID−/− mice retained large numbers of IgM-positive cells for at least 30 days (Fig. 8B). Although sIgM−/− mice had almost no IgM-positive cells, they had the same pattern and numbers of IgG-positive cells as WT mice (Fig. 8C). DKO mice had only IgM-positive cells, which were still prevalent 45 days after infection (Fig. 8B).
FIG 8.
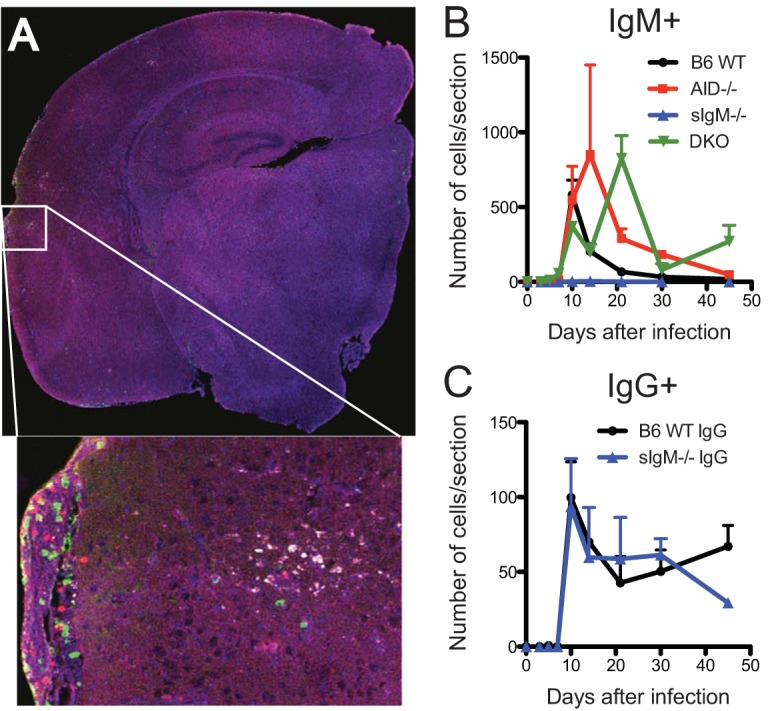
Localization and quantification of IgM- and IgG-secreting cells in brain. (A) A 4-by-4 tile scan image (stitching of a 7-by-7 tile scan with 50% overlap) of the right hemisphere of a WT mouse at 10 days after infection shows the distribution of cells with viral proteins (white), IgG (red), and IgM (green). (Inset) A higher-power image shows IgG- and IgM-positive cells infiltrating the pia mater and brain parenchyma near infected cells. (B, C) Numbers of IgM-positive cells (B) and IgG-positive cells (C) per slice of right brain hemisphere (3 mice per group per time point) at 10 days after infection.
DISCUSSION
In these studies, the role of antiviral IgM in the recovery of mice from viral encephalomyelitis after intracerebral infection with the relatively avirulent TE strain of SINV was assessed. AID−/− mice that produced only germ line IgM, sIgM−/− mice that produced IgG but no secretory IgM, and AID−/− sIgM−/− DKO mice that secreted neither IgM nor IgG resolved the signs of neurologic disease after infection. Development of disease was slightly later in sIgM−/− mice and recovery was slower in DKO mice, but in general, disease severity, weight loss, and survival were similar between the groups. Antibody, either germ line IgM or IgG, was required for the sustained clearance of infectious virus and viral RNA from the brain. As previously observed with antibody-deficient μMT mice (10, 12), antibody was not required for clearance of infectious virus from the spinal cord, but DKO mice had persistently higher levels of viral RNA at late times after infection, indicating that antibody contributes to long-term suppression of viral RNA. Both IgM- and IgG-secreting cells entered the CNS, localized near sites of virus infection, and sustained continued production of antiviral antibody. Therefore, antiviral antibody is essential for effective control of virus replication in the CNS, and this control can be accomplished by germ line IgM as well as IgG.
The TE strain of SINV causes relatively mild disease in both immunocompetent and immunocompromised mice (10), and most signs of neurologic disease are mediated by the inflammatory response to infection rather than virus replication per se (28–30). Both T and B cells infiltrate the CNS in response to virus infection (7, 31). Delayed recovery in DKO mice was associated with sustained virus replication in the brain, with DKO mice having higher levels of viral RNA in both brain and spinal cord than WT mice, accompanied by a prolonged B cell presence in brain and higher levels of IFN-γ in the spinal cord than WT mice. IFN-γ has previously been shown to compensate for a lack of antibody for clearance of infectious virus from the spinal cord (12, 23), and the levels of Ifng mRNA were the highest in DKO mice (Fig. 5), likely due to expression by T cells infiltrating the CNS. IFN-γ suppresses virus production in neurons, but the specific mechanism(s) and the reason for the greater efficacy for infected motor neurons are not known (21, 22).
Although IFN-γ contributes to virus clearance both in a direct manner and by inducing the local production of chemokines that attract B cells (23), antibody remains the most important means of virus clearance from the CNS in immunologically normal mice. Either IgM or IgG antibody alone was sufficient for clearance of infectious virus from brains. IgM is the first antibody to be produced after infection, and the specific role of germ line IgM was assessed using AID−/− mice. AID is induced in activated B cells through a Toll-like receptor, CD40, or cytokine signaling and is necessary for class switch recombination and somatic hypermutation (25, 32). The amount of Aid mRNA was increased in the CNS of WT mice and, to a lesser extent, sIgM−/− mice during the inflammatory response to SINV infection and then sustained for several weeks (Fig. 5). In the absence of AID, IgM retains germ line variable region sequences characteristic of natural antibody (25). Germ line IgM has a low affinity but a broad specificity and is produced constitutively by B-1 cells (natural IgM) and by conventional B-2 cells after antigen stimulation (antigen-induced IgM) (33). Because of its size, IgM cannot cross the blood-brain barrier and needs to be produced by antibody-secreting cells in the CNS or, potentially, transported early after infection by the high-affinity IgM receptor FcμR on lymphocytes (34, 35). AID−/− mice produced high levels of SINV-reactive IgM in serum that increased quickly after infection, with a subsequent increase in CNS IgM levels occurring at the same time that IgM-secreting B cells entered the CNS (Fig. 8) and CNS IgM levels increased in WT mice (Fig. 6). AID−/− mice retain IgM-positive cells longer than WT mice and have prolonged elevation of IgM in both plasma and the CNS, consistent with AID deficiency as a cause of hyper-IgM syndrome in humans (36). Infectious virus was cleared only slightly more slowly in AID−/− mice than WT mice, and this may have been enhanced by higher levels of IFN-γ in brain (Fig. 2 and 5). Viral RNA clearance was not identifiably different, demonstrating the effectiveness of germ line IgM for viral clearance. Previous studies have shown that virus-induced IgM protects against infection with vesicular stomatitis virus and that the presence of antiviral IgM is predictive of survival in humans with arboviral encephalomyelitis (37, 38). IgM produced by AID−/− mice is sufficient to protect against lethal influenza virus (39) and Friend virus (40) infections and also against mouse hepatitis virus-induced chronic demyelination (41). Therefore, germ line IgM plays an important role in recovery from most virus infections that have been examined.
Natural antibody is important for early protection from infection and also facilitates trapping of antigen by follicular dendritic cells important for development and maturation of IgG to T cell-dependent antigens (26, 42). To determine whether IgM was required for recovery from SINV infection, sIgM−/− mice, which were unable secrete IgM but able to produce IgG, were analyzed. In general, sIgM−/− mice produced SINV-specific serum IgG more rapidly (Fig. 6), developed clinical evidence of disease later (Fig. 1), and had less CNS cell death (Fig. 4) than WT or AID−/− mice. In humans, intravenous immunoglobulin enhances expression of the high-affinity Fc-γ receptor 1 on monocytes (43) that might bind SINV-specific IgG in plasma before infiltrating brain parenchyma to contribute to virus clearance. The levels of total SINV-specific serum IgG were lower in sIgM−/− mice than in WT mice late during recovery, with substantial differences in isotype distribution being detected (Fig. 7). Although the amounts of total IgG were similar in brain and spinal cord, sIgM−/− mice had higher levels of IgG2a and lower levels of IgG1 and IgG2b than WT mice, similar to observations after antigen immunization (26). The level of AID gene mRNA expression in both brains and spinal cords of sIgM−/− mice was lower than that in WT mice, while most immunoglobulin-containing cells in sIgM−/− mouse brains were IgG positive. Thus, class switching in sIgM−/− mice may have occurred before the cells infiltrated the CNS. Unlike IgM, it is possible for IgG antibody in plasma to enter the CNS and contribute to early virus clearance, but it might be undetectable by EIA due to the viral antigen present in the homogenates tested. Previous studies have shown that sIgM−/− mice have various degrees of increased susceptibility to vesicular stomatitis virus, influenza virus, and West Nile virus infections compared to WT mice, with delayed IgG antibody responses and an altered isotype distribution being common features (44–48). However, the isotypes affected were distinct.
The viral clearance observed in DKO mice (no secreted antibody) was similar to that observed in μMT mice (no B cells) in previous studies, with clearance from the spinal cord but not the brain (10, 12). Interestingly, DKO mice cannot secrete IgM but still had IgM-containing cells in the brain. Even without antibody, a few DKO mice had no detectable infectious virus in the brain. In contrast, in all mice, infectious virus was cleared from spinal cords. The amounts of infectious virus and viral RNA in the spinal cord were less than those in the brain, so a more comprehensive immune response may be needed to clear the higher burden of SINV in brains and to prevent viral reactivation. In fact, even SCID mice, which lack adaptive immune responses, can decrease the levels of infectious virus in the spinal cord before reactivation (10). Table 1 provides a summary of the current data on the role of the adaptive immune response in SINV clearance from the brain and spinal cord of infected mice.
TABLE 1.
Summary of mouse models used in SINV clearance studiesa
| Mouse [reference(s)] | Presence of the following: |
Status of virus in: |
|||||
|---|---|---|---|---|---|---|---|
| T cells | IFN-γ | B cells | IgM | IgG | Brain | SC | |
| SCID (10–12) | − | + | − | − | − | P | P |
| Athymic nude (17, 19) | − | + | + | + | − | C | NA |
| μMT (10, 12) | + | + | − | − | − | P | C |
| IFN-γ−/− (10, 23) | + | − | + | + | + | C | C |
| μMT IFN-γ−/− (10) | + | − | − | − | − | P | P |
| AID−/− | + | + | + | + | − | C | C |
| sIgM−/− | + | + | + | − | + | C | C |
| AID−/− sIgM−/− | + | + | + | − | − | P | C |
| Wild type | + | + | + | + | + | C | C |
SC, spinal cord; P, persistent; C, cleared; NA, not available.
In conclusion, immunoglobulin, regardless of its class, helps clear infectious virus from the brain but is not required for clearance from the spinal cord. The largest effect of antibody on the clearance of viral RNA is seen late in recovery, suggesting an important role in prevention of viral reactivation. Thus, both IFN-γ and antibody contribute to clearance and can to some degree compensate for each other, suggesting some overlap and redundancy of their functions.
MATERIALS AND METHODS
Cells and virus.
BHK-21 cells (ATCC) were cultured at 37°C in 5% CO2 in Dulbecco's modified Eagle medium (DMEM; Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin-streptomycin (PS), and 2 mM glutamine and were used to grow and assay the TE strain of SINV (49) by plaque formation. The virus stock had a titer of 2.63 × 109 PFU/ml and was stored in aliquots at −80°C.
Mice.
All mice were of the C57BL/6 background. WT mice were obtained from The Jackson Laboratory and bred in-house. AID+/− mice (25) were a gift from Matthew Scharff (Albert Einstein College of Medicine), and sIgM+/− mice (42) were a gift from Nicole Baumgarth (University of California, Davis). Heterozygous mice were mated to produce homozygous AID−/− or sIgM−/− mice. Male AID−/− mice were mated with female sIgM−/− mice to produce double heterozygous (AID+/− sIgM+/−) mice, which were subsequently mated with their siblings to produce DKO (AID−/− sIgM−/−) mice. To confirm mouse phenotypes, blood was collected by either facial vein puncture or tail snip under isoflurane anesthesia, and serum was analyzed for IgM, IgG, and IgA by enzyme immunoassay (EIA). Because AID+/− mothers could vertically transfer IgG to their pups, a serum IgA EIA was used instead of an IgG EIA for typing before 8 weeks. AID−/− mice had serum IgM without IgG or IgA, while sIgM−/− mice had serum IgG and IgA without IgM. DKO mice lacked all immunoglobulins in serum. All experiments were performed according to protocols approved by the Johns Hopkins University Animal Care and Use Committee.
Infection, disease evaluation, and tissue collection.
Twenty microliters of phosphate-buffered saline (PBS) containing 1,000 PFU SINV TE was injected into the left cerebral hemisphere of male and female 4- to 6-week-old mice under light isoflurane anesthesia. The mice were weighed and scored for evidence of disease daily for 21 days. The following clinical scoring system was used: 0, normal; 1, slightly hunched and abnormal gait and tail position; 2, very hunched and walking on hind limbs; 3, sitting on hind limbs, discharge from eyes, and ruffled fur on nose; 4, death. For scoring, observers were blind as to the identity of each mouse group. Data from two independent experiments were pooled.
Brain and spinal cord tissues were collected before infection and at 3, 5, 7, 10, 14, 21, 30, 45, 60, and 90 days after infection. Heart blood was collected from mice under deep isoflurane anesthesia, and serum was separated and frozen at −20°C. After intracardiac perfusion with 15 to 20 ml ice-cold PBS, the left hemisphere of the brain and the spinal cord were collected, frozen in liquid nitrogen, and stored in Lysing Matrix D tubes (MP Biomedicals) at −80°C for subsequent homogenization and RNA extraction. The brain right hemisphere was sliced into five sections 2 mm thick and fixed in 4% paraformaldehyde (PFA) in PBS (pH 7.2) at 4°C overnight. Collected tissues were paraffin embedded, sectioned (5 μm), and mounted on positively charged slides by the Johns Hopkins Pathology Reference Laboratory. These formalin-fixed paraffin-embedded (FFPE) tissue sections were used for immunofluorescence (IF), RNA fluorescence in situ hybridization (FISH), and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL).
Tissue homogenates, RNA isolation, and RT-qPCR.
Ice-cold PBS was added to the tissues in Lysing Matrix D tubes and homogenized with a FastPrep-24 homogenizer (MP Biomedicals) at a 6-m/s speed for 40 s to produce either a 20% (wt/vol) brain homogenate or a 10% (wt/vol) spinal cord homogenate. Homogenates were centrifuged at 13,200 rpm at 4°C for 15 min. Supernatants were collected and stored at −80°C for virus and antibody assays. One milliliter of QIAzol lysis reagent was added to the pellet, which was rehomogenized with the FastPrep-24 homogenizer (MP Biomedicals) at a 6-m/s speed for 40 s. RNA was extracted with an RNeasy lipid minikit (Qiagen) according to the manufacturer's protocol, quantified with a NanoDrop spectrophotometer, and stored at −80°C.
For analysis of RNA, 2 μg was reverse transcribed in a 20-μl reaction mixture using a High Capacity cDNA reverse transcription kit (Thermo Scientific) with random primers and an Applied Biosystems 2720 thermocycler (25°C for 10 min, 37°C for 120 min, and 85°C for 5 s). Quantitative PCR was performed using 2.5 μl cDNA and an Applied Biosystems 7500 real-time PCR system (50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min) with TaqMan Universal master mix (Thermo Scientific) in a 25-μl reaction mixture. Primers and probes were as follows: for SINV E2, primers SV8732F (5′-TGG GAC GAA GCG GAC GAT AA-3′; Integrated DNA Technologies [IDT] 43567022) and SV8805R (5′-CTG CTC CGC TTT GGT CGT AT-3′; IDT 43567023) and 6-carboxytetramethylrhodamine (TAMRA) probe SVE2-8760 (catalog number 5283081; Applied Biosystems); for GAPDH (glyceraldehyde-3-phosphate dehydrogenase; TaqMan rodent GAPDH control reagent), VIC probe (catalog number 4308313; Life Technologies); for IFN-γ (exon location 1-2), TAMRA probe (catalog number IDT 141107662); and for AID (TaqMan gene expression assays), Mm01184115_m1 Aicda (Applied Biosystems). Standards for SINV E2 (TE plasmid) and GAPDH (TaqMan rodent GAPDH control reagent) were serially diluted 10-fold from 3 × 107 to 3 copies. The SINV E2 RNA copy number was calculated as the absolute number per 106 Gapdh RNA copies. The change in expression of Ifng and Aid mRNAs was calculated as the fold increase (2−ΔΔCT) using the day 0 samples and Gapdh as controls.
EIAs.
SINV antigen for enzyme immunoassay (EIA) was prepared by adding a 1/3 volume of 40% polyethylene glycol (PEG) 8000 in 2 M NaCl to the TE-containing supernatant from BHK-21 cells, with precipitation taking place at 4°C overnight on a rocker. The PEG precipitate was pelleted at 10,000 rpm and resuspended in PBS (titer = 9 × 109 PFU/ml). The wells of a MaxiSorp (Nunc) 96-well plate were coated with 106 PFU PEG-concentrated TE in 50 μl coating buffer (50 mM NaHCO3, pH 9.6) at 4°C overnight. The plate was washed with PBS–0.05% Tween 20 (PBST)–2% FBS and blocked with PBST–10% FBS at 37°C for 2 h. Samples (50 μl/well in triplicate) were diluted in blocking buffer at 1:100 for serum, 1:8 for brain homogenate, and 1:4 for spinal cord homogenate and incubated at room temperature for 2 h. After 4 washes, horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-mouse IgG, IgG1, IgG2a, IgG2b, IgG3, IgM, or IgA; Southern Biotech) diluted 1:1,000 were added and the mixture was incubated at room temperature for 1 h. Color was developed with tetramethylbenzidine (TMB) substrate (OptEIA TMB substrate reagent set; BD Biosciences) at room temperature for up to 20 min, and the reaction was stopped with H2SO4. The optical density (OD) was read at 450 nm on a Multiskan MCC spectrophotometer (Thermo Scientific).
To screen for the presence of immunoglobulin in mouse serum, EIA plates were coated with a 1:400 dilution of serum in coating buffer and detected with HRP-conjugated anti-mouse IgG, IgM, or IgA, as described above.
Immunofluorescence (IF).
For localization of viral antigen and immunoglobulin-secreting cells, FFPE sections were baked at 58°C and then immersed in xylene substitute and rehydrated through an ethanol gradient of 100%, 95%, and 70%, followed by running distilled H2O (dH2O). For antigen retrieval, the rehydrated sections were boiled in 0.01 M sodium citrate (pH 6.0) for 9 min. Sections were cooled at room temperature for 20 min with intermittent shaking and then immersed in PBS. For staining, sections were blocked with 0.04% Triton X-100 in PBS with 10% normal goat serum (NGS) at room temperature for 20 min. Staining for viral antigen was with rabbit anti-SINV (50) diluted 1:200 in blocking buffer at 4°C overnight. Sections were washed and incubated at room temperature for 30 min with anti rabbit IgG-CF633 (Biotium) diluted 1:200 in blocking buffer with 25 ng/ml DAPI to detect SINV-infected cells, anti-mouse IgG-Dylight594 (gamma chain specific; catalog number ab98738; Abcam) to detect IgG-producing cells, and anti-mouse IgM-AF488 (mu chain specific; catalog number A21042; Invitrogen) to detect IgM-producing cells. Sections were mounted in ProLong Gold antifade reagent.
For combined TUNEL, RNA FISH, and IF, diethyl pyrocarbonate-treated H2O (Quality Biological) replaced dH2O for all solutions. Sections were rehydrated as described above but left in 70% ethanol at 4°C for at least 1 h. For antigen retrieval, sections were immersed in PBS for 10 min and then incubated with prewarmed proteinase K (10 μg/ml in PBS). TUNEL staining used a TACS 2 terminal deoxynucleotidyl transferase (TdT) core kit (catalog number 4810-30-CK; Trevigen) and was performed according to the manufacturer's protocol. Briefly, sections were immersed in TdT labeling buffer for 5 min, incubated with labeling reaction buffer (1 μl deoxynucleoside triphosphate mix, 1 μl TdT enzyme, 1 μl Mn2+, 50 μl TdT labeling buffer), immersed in TdT stop buffer, washed, and incubated with streptomycin-HRP for 10 min at 37°C. For detection and signal amplification with a tyramide signal amplification (TSA) kit, sections were incubated with tyramide-Alexa Fluor 488 (1:100) diluted in amplification buffer containing 0.0015% H2O2 according to the manufacturer's protocol (TUNEL-TSA).
Following TUNEL-TSA, sections were washed twice with 10% deionized formamide in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) for combined IF and RNA FISH. Stocks of 25 μM custom Stellaris RNA FISH probes with Quasar 570 dye (LGC Biosearch Technologies) specific for the SINV E1 and E2 genes in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) were diluted 1:1,000 in hybridization buffer (10% dextran and 10% deionized formamide in 2× SSC), and rabbit anti-SINV (1:200) (50) was added. Sections were incubated at 37°C overnight with FISH probes and antibody and then washed for 30 min at 37°C and incubated with 100 μl of 1:200 anti-rabbit IgG-CF633 (Biotium) in hybridization buffer at 37°C for 30 min. A second wash was done at 37°C for 30 min with 1 ml of 25 ng/ml DAPI (Sigma) in the washing buffer. Sections were mounted in ProLong Gold antifade reagent.
Imaging and cell quantification.
Brain sections that contained the hippocampus were imaged in a 4-by-4 tile scan (objective lens, 10×; zoom out, 0.6; Zeiss model LSM780FCS laser scanning confocal microscope on an Axio Observer Z1 inverted microscope). The lasers used were 405 nm (DAPI), 488 nm (Alexa Fluor 488), 561 nm (Dylight594 or Quasar 570), and 633 nm (CF633). The digital files of the captured images were coded, and the cells positive for SINV RNA, TUNEL, endogenous IgG, and IgM were counted manually, while cells positive for SINV protein were counted by set threshold in ImageJ software.
ACKNOWLEDGMENTS
This study was funded by research grants R01 NS038932 (to D.E.G.), T32 OD011089 (to V.K.B.), T32 AI007417 (to E.M.T. and J.X.Y.), and F31 NS101775 (to E.M.T.) from the National Institutes of Health and the Ananda Mahidol Foundation Scholarship in Medicine (to V.N.).
We are grateful to the Microscope Facility of the Johns Hopkins School of Medicine for the use of a confocal microscope and to Patricia Gearhart and Kirsten Kulcsar for helpful discussions of this work.
REFERENCES
- 1.Adouchief S, Smura T, Sane J, Vapalahti O, Kurkela S. 2016. Sindbis virus as a human pathogen—epidemiology, clinical picture and pathogenesis. Rev Med Virol 26:221–241. doi: 10.1002/rmv.1876. [DOI] [PubMed] [Google Scholar]
- 2.Griffin DE, Metcalf T. 2011. Clearance of virus infection from the CNS. Curr Opin Virol 1:216–221. doi: 10.1016/j.coviro.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson AC, Moench TR, Trapp BD, Griffin DE. 1988. Basis of neurovirulence in Sindbis virus encephalomyelitis of mice. Lab Invest 58:503–509. [PubMed] [Google Scholar]
- 4.Havert MB, Schofield B, Griffin DE, Irani DN. 2000. Activation of divergent neuronal cell death pathways in different target cell populations during neuroadapted Sindbis virus infection of mice. J Virol 74:5352–5356. doi: 10.1128/JVI.74.11.5352-5356.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine B, Griffin DE. 1992. Persistence of viral RNA in mouse brains after recovery from acute alphavirus encephalitis. J Virol 66:6429–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tyor WR, Wesselingh S, Levine B, Griffin DE. 1992. Long term intraparenchymal Ig secretion after acute viral encephalitis in mice. J Immunol 149:4016–4020. [PubMed] [Google Scholar]
- 7.Metcalf TU, Griffin DE. 2011. Alphavirus-induced encephalomyelitis: antibody-secreting cells and viral clearance from the nervous system. J Virol 85:11490–11501. doi: 10.1128/JVI.05379-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. 2000. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol 74:3366–3378. doi: 10.1128/JVI.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrnes AP, Durbin JE, Griffin DE. 2000. Control of Sindbis virus infection by antibody in interferon-deficient mice. J Virol 74:3905–3908. doi: 10.1128/JVI.74.8.3905-3908.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burdeinick-Kerr R, Wind J, Griffin DE. 2007. Synergistic roles of antibody and interferon in noncytolytic clearance of Sindbis virus from different regions of the central nervous system. J Virol 81:5628–5636. doi: 10.1128/JVI.01152-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine B, Hardwick JM, Trapp BD, Crawford TO, Bollinger RC, Griffin DE. 1991. Antibody-mediated clearance of alphavirus infection from neurons. Science 254:856–860. doi: 10.1126/science.1658936. [DOI] [PubMed] [Google Scholar]
- 12.Binder GK, Griffin DE. 2001. Interferon-gamma-mediated site-specific clearance of alphavirus from CNS neurons. Science 293:303–306. doi: 10.1126/science.1059742. [DOI] [PubMed] [Google Scholar]
- 13.Ubol S, Levine B, Lee SH, Greenspan NS, Griffin DE. 1995. Roles of immunoglobulin valency and the heavy-chain constant domain in antibody-mediated downregulation of Sindbis virus replication in persistently infected neurons. J Virol 69:1990–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Despres P, Griffin JW, Griffin DE. 1995. Antiviral activity of alpha interferon in Sindbis virus-infected cells is restored by anti-E2 monoclonal antibody treatment. J Virol 69:7345–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Despres P, Griffin JW, Griffin DE. 1995. Effects of anti-E2 monoclonal antibody on Sindbis virus replication in AT3 cells expressing bcl-2. J Virol 69:7006–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stanley J, Cooper SJ, Griffin DE. 1986. Monoclonal antibody cure and prophylaxis of lethal Sindbis virus encephalitis in mice. J Virol 58:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirsch RL, Griffin DE. 1979. The pathogenesis of Sindbis virus infection in athymic nude mice. J Immunol 123:1215–1218. [PubMed] [Google Scholar]
- 18.Park MM, Griffin DE, Johnson RT. 1981. Studies of immune responses during recovery from Sindbis virus encephalitis in selectively reconstituted, thymectomized, lethally irradiated mice. Infect Immun 34:306–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyor WR, Moench TR, Griffin DE. 1989. Characterization of the local and systemic B cell response of normal and athymic nude mice with Sindbis virus encephalitis. J Neuroimmunol 24:207–215. doi: 10.1016/0165-5728(89)90118-5. [DOI] [PubMed] [Google Scholar]
- 20.Kitamura D, Roes J, Kuhn R, Rajewsky K. 1991. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 21.Burdeinick-Kerr R, Griffin DE. 2005. Gamma interferon-dependent, noncytolytic clearance of Sindbis virus infection from neurons in vitro. J Virol 79:5374–5385. doi: 10.1128/JVI.79.9.5374-5385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burdeinick-Kerr R, Govindarajan D, Griffin DE. 2009. Noncytolytic clearance of Sindbis virus infection from neurons by gamma interferon is dependent on Jak/STAT signaling. J Virol 83:3429–3435. doi: 10.1128/JVI.02381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baxter VK, Griffin DE. 2016. Interferon gamma modulation of disease manifestation and the local antibody response to alphavirus encephalomyelitis. J Gen Virol 97:2908–2925. doi: 10.1099/jgv.0.000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Homann D, Tishon A, Berger DP, Weigle WO, von Herrath MG, Oldstone MB. 1998. Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from muMT/muMT mice. J Virol 72:9208–9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102:553–563. doi: 10.1016/S0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 26.Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, Chen J. 1998. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J Immunol 160:4776–4787. [PubMed] [Google Scholar]
- 27.Kumazaki K, Tirosh B, Maehr R, Boes M, Honjo T, Ploegh HL. 2007. AID−/−mus−/− mice are agammaglobulinemic and fail to maintain B220-CD138+ plasma cells. J Immunol 178:2192–2203. doi: 10.4049/jimmunol.178.4.2192. [DOI] [PubMed] [Google Scholar]
- 28.Baxter VK, Glowinski R, Braxton AM, Potter MC, Slusher BS, Griffin DE. 2017. Glutamine antagonist-mediated immune suppression decreases pathology but delays virus clearance in mice during nonfatal alphavirus encephalomyelitis. Virology 508:134–149. doi: 10.1016/j.virol.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potter MC, Baxter VK, Mathey RW, Alt J, Rojas C, Griffin DE, Slusher BS. 2015. Neurological sequelae induced by alphavirus infection of the CNS are attenuated by treatment with the glutamine antagonist 6-diazo-5-oxo-l-norleucine. J Neurovirol 21:159–173. doi: 10.1007/s13365-015-0314-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manivannan S, Baxter VK, Schultz KL, Slusher BS, Griffin DE. 2016. Protective effects of glutamine antagonist DON in mice with alphaviral encephalomyelitis. J Virol 90:9251–9262. doi: 10.1128/JVI.01045-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metcalf TU, Baxter VK, Nilaratanakul V, Griffin DE. 2013. Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J Virol 87:2420–2429. doi: 10.1128/JVI.01769-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem 274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 33.Baumgarth N, Herman OC, Jager GC, Brown L, Herzenberg LA, Herzenberg LA. 1999. Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proc Natl Acad Sci U S A 96:2250–2255. doi: 10.1073/pnas.96.5.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang DW, Gartland GL, Bertoli LF, Mori H, Takatsu H, Kitamura T, Ohno H, Wang JY. 2009. Identity of the elusive IgM Fc receptor (FcmuR) in humans. J Exp Med 206:2779–2793. doi: 10.1084/jem.20091107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kubagawa H, Kubagawa Y, Jones D, Nasti TH, Walter MR, Honjo K. 2014. The old but new IgM Fc receptor (FcmuR). Curr Top Microbiol Immunol 382:3–28. doi: 10.1007/978-3-319-07911-0_1. [DOI] [PubMed] [Google Scholar]
- 36.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell 102:565–575. doi: 10.1016/S0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 37.Gunther G, Haglund M, Lindquist L, Skoldenberg B, Forsgren M. 1997. Intrathecal IgM, IgA and IgG antibody response in tick-borne encephalitis. Long-term follow-up related to clinical course and outcome Clin Diagn Virol 8:17–29. [DOI] [PubMed] [Google Scholar]
- 38.Burke DS, Lorsomrudee W, Leake CJ, Hoke CH, Nisalak A, Chongswasdi V, Laorakpongse T. 1985. Fatal outcome in Japanese encephalitis. Am J Trop Med Hyg 34:1203–1210. doi: 10.4269/ajtmh.1985.34.1203. [DOI] [PubMed] [Google Scholar]
- 39.Harada Y, Muramatsu M, Shibata T, Honjo T, Kuroda K. 2003. Unmutated immunoglobulin M can protect mice from death by influenza virus infection. J Exp Med 197:1779–1785. doi: 10.1084/jem.20021457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kato M, Tsuji-Kawahara S, Kawasaki Y, Kinoshita S, Chikaishi T, Takamura S, Fujisawa M, Kawada A, Miyazawa M. 2015. Class switch recombination and somatic hypermutation of virus-neutralizing antibodies are not essential for control of Friend retrovirus infection. J Virol 89:1468–1473. doi: 10.1128/JVI.02293-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gil-Cruz C, Perez-Shibayama C, Firner S, Waisman A, Bechmann I, Thiel V, Cervantes-Barragan L, Ludewig B. 2012. T helper cell- and CD40-dependent germline IgM prevents chronic virus-induced demyelinating disease. Proc Natl Acad Sci U S A 109:1233–1238. doi: 10.1073/pnas.1115154109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ehrenstein MR, O'Keefe TL, Davies SL, Neuberger MS. 1998. Targeted gene disruption reveals a role for natural secretory IgM in the maturation of the primary immune response. Proc Natl Acad Sci U S A 95:10089–10093. doi: 10.1073/pnas.95.17.10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ling ZD, Yeoh E, Webb BT, Farrell K, Doucette J, Matheson DS. 1993. Intravenous immunoglobulin induces interferon-gamma and interleukin-6 in vivo. J Clin Immunol 13:302–309. doi: 10.1007/BF00920238. [DOI] [PubMed] [Google Scholar]
- 44.Kopf M, Brombacher F, Bachmann MF. 2002. Role of IgM antibodies versus B cells in influenza virus-specific immunity. Eur J Immunol 32:2229–2236. doi:. [DOI] [PubMed] [Google Scholar]
- 45.Lutz C, Ledermann B, Kosco-Vilbois MH, Ochsenbein AF, Zinkernagel RM, Kohler G, Brombacher F. 1998. IgD can largely substitute for loss of IgM function in B cells. Nature 393:797–801. doi: 10.1038/31716. [DOI] [PubMed] [Google Scholar]
- 46.Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. 2000. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med 192:271–280. doi: 10.1084/jem.192.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. 2003. A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med 198:1853–1862. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baumgarth N, Chen J, Herman OC, Jager GC, Herzenberg LA. 2000. The role of B-1 and B-2 cells in immune protection from influenza virus infection. Curr Top Microbiol Immunol 252:163–169. [DOI] [PubMed] [Google Scholar]
- 49.Lustig S, Jackson AC, Hahn CS, Griffin DE, Strauss EG, Strauss JH. 1988. Molecular basis of Sindbis virus neurovirulence in mice. J Virol 62:2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jackson AC, Moench TR, Griffin DE, Johnson RT. 1987. The pathogenesis of spinal cord involvement in the encephalomyelitis of mice caused by neuroadapted Sindbis virus infection. Lab Invest 56:418–423. [PubMed] [Google Scholar]