

ABSTRACT
The 5′ untranslated region (UTR) of hepatitis C virus (HCV), which is composed of four domains (I, II, III, and IV) and a pseudoknot, is essential for translation and viral replication. Equine nonprimate hepacivirus (EHcV) harbors a 5′ UTR consisting of a large 5′-terminal domain (I); three additional domains (I′, II, and III), which are homologous to domains I, II, and III, respectively, of HCV; and a pseudoknot, in the order listed. In this study, we investigated the roles of the EHcV 5′ UTR in translation and viral replication. The internal ribosome entry site (IRES) activity of the EHcV 5′ UTR was lower than that of the HCV 5′ UTR in several cell lines due to structural differences in domain III. Domains I and III of EHcV were functional in the HCV 5′ UTR in terms of IRES activity and the replication of the subgenomic replicon (SGR), although domain II was not exchangeable between EHcV and HCV for SGR replication. Furthermore, the region spanning domains I and I′ of EHcV (the 5′-proximal EHcV-specific region) improved RNA stability and provided the HCV SGR with microRNA 122 (miR-122)-independent replication capability, while EHcV domain I alone improved SGR replication and RNA stability irrespective of miR-122. These data suggest that the region spanning EHcV domains I and I′ improves RNA stability and viral replication regardless of miR-122 expression. The 5′-proximal EHcV-specific region may represent an inherent mechanism to facilitate viral replication in nonhepatic tissues.
IMPORTANCE EHcV is the closest viral homolog to HCV among other hepaciviruses. HCV exhibits a narrow host range and liver-specific tropism, while epidemiological reports suggest that EHcV infects the liver and respiratory organs in horses, donkeys, and dogs. However, the mechanism explaining the differences in host or organ tropism between HCV and EHcV is unknown. In this study, our data suggest that the 5′ untranslated region (UTR) of EHcV is composed of an internal ribosome entry site (IRES) element that is functionally exchangeable with HCV IRES elements. Furthermore, the 5′-proximal EHcV-specific region enhances viral replication and RNA stability in a miR-122-independent manner. Our data suggest that the region upstream of domain II in the EHcV 5′ UTR contributes to the differences in tissue tropism observed between these hepaciviruses.
KEYWORDS: hepacivirus, hepatitis C virus, horse, internal ribosome entry site
INTRODUCTION
Hepatitis C virus (HCV), which is an enveloped, hepatotropic, positive-sense, single-stranded RNA virus, belongs to the genus Hepacivirus in the family Flaviviridae. HCV is well known as a causative agent of chronic hepatitis, cirrhosis, and hepatocellular carcinoma in humans (1). HCV naturally infects humans and experimentally infects chimpanzees but does not infect other animals (reviewed by Ding et al. [2]). Diverse types of hepaciviruses were recently identified in nonprimate animals, including dogs (3, 4), horses (5–10), donkeys (11), bats (12, 13), rodents (14, 15), and cattle (16, 17), and were temporarily designated nonprimate hepaciviruses (NPHVs). Equine hepacivirus (EHcV) is phylogenetically the closest homolog of HCV among reported NPHVs. Horses are predicted to be a natural host of EHcV. These viral strains are isolated rarely from donkeys and dogs, but not from humans, and are highly homologous with EHcV (3, 4, 11, 18, 19), suggesting that EHcV crosses the species barrier via horizontal transmission. EHcV transiently and asymptomatically infects horses, although several groups reported mild liver damage and inflammatory infiltration in horses naturally or experimentally infected with EHcV (20, 21). EHcV infection is also reported to be involved in canine respiratory disease (3, 4). Thus, EHcV differs from HCV in terms of the course of infection, pathogenicity, and tissue and host tropism.
HCV genomic RNA possesses an open reading frame (ORF) of a polyprotein flanked by 5′ and 3′ untranslated regions (UTRs). The HCV 5′ UTR forms highly conserved secondary and tertiary structures that are termed domains I to IV (22, 23), as shown in Fig. 1A. Domain III consists of the subdomains IIIa to IIIf. Translation initiation of the ORF is directed by the internal ribosome entry site (IRES) element in the 5′ UTR (24), which principally comprises domains II and III. Domain IV harbors the initiation codon of the ORF, and the pseudoknot, consisting of subdomain IIIf and the downstream stem, is essential for positioning of the initiation codon (25). Translation initiation begins with specific interaction of the 40S ribosome with the IRES element involving subdomains IIId and IIIe and the pseudoknot (26–28), followed by recruitment of eukaryotic initiation factor 3 (eIF3) and an eIF2·GTP·tRNA ternary complex (29, 30), resulting in the formation of the 48S initiation complex. Domain II interacts with the ribosomal S5 protein to induce conformational changes of the 40S ribosome (31–34) and promotes eIF5-mediated hydrolysis of GTP to release eIF2 (35), which facilitates the recruitment of the 60S ribosome to the 48S complex. Furthermore, the HCV 5′ UTR contains essential sequences for RNA replication. Domains I and II are required for efficient RNA replication (36, 37), and the GGG motif in subdomain IIId engages in an essential long-range interaction with 5BSL3.2 in the NS5B coding region (38, 39). HCV employs liver-specific microRNA 122 (miR-122) to promote its RNA replication (40). miR-122 binds to two sites upstream of the IRES element, termed S1 and S2 (41). The sequences of S1 and S2 are ACACUCC and CACUCC, respectively, both of which are complementary to the seed sequence of miR-122. The seed sequence and several 3′ overhang nucleotides of miR-122 participate in interactions with the HCV 5′ UTR (42, 43), which significantly facilitates but is not essential for viral replication (44, 45). Several reports suggest that miR-122 function is responsible for the stimulation of IRES activity, recruitment of the P body protein, protection of HCV RNA against cellular exoribonuclease, and displacement of poly(C) binding protein 2 (PCBP2) from HCV RNA (42, 46–53). However, the mechanism by which miR-122 mediates the enhancement of HCV replication is not fully understood. miR-122 represents the majority of microRNA in hepatocytes and is expressed rarely in other tissues (54). Nonhepatic cells exogenously express miR-122, which permits efficient HCV replication (55–57). Expression of miR-122 may contribute to the liver tropism of HCV.
FIG 1.

IRES activities of the EHcV and HCV 5′ UTRs in mammalian cell lines. (A) Schematic diagram of the secondary structures of the HCV and EHcV 5′ UTRs. The stem-loop located between domains I and II of EHcV is designated domain I′ in this study. (B) Schematic diagram of the bicistronic reporter RNA transcripts. (C) The reporter plasmid bearing the indicated transcript was transfected into 293T cells. Total RNA was subjected to Northern blotting using a specific probe against the RL or FL ORF. 28S and 18S rRNAs were stained with ethidium bromide. In vitro-transcribed monocistronic RNA for the RL or FL ORF was applied as the control. NC, mock-transfected cells as a negative control. (D) The bicistronic reporter plasmids were transfected in triplicate into the indicated cell lines. The cells were harvested at the indicated time points and subjected to a dual-luciferase assay after lysate preparation. The ratio of FL to RL was normalized to the mean value of cells expressing RL-FL transcripts, whose expression was defined as 1. RL-HCV-FL-ΔIII is the transcript RL-HCV-FL lacking domain III, as shown in Fig. 3, which was used as a negative control. (E) Relative translation activities of EHcV IRES versus HCV IRES in mammalian cell lines. The FL/RL ratio value of RL-EHcV-FL at 48 or 72 h posttransfection was divided by that of RL-HCV-FL to calculate the ratio of IRES activity. (F) The capped and polyadenylated bicistronic reporter RNAs were in vitro transcribed from the bicistronic reporter plasmids. The bicistronic reporter RNA was transfected in triplicate into 293T cells. The cells were harvested at 6 h posttransfection and subjected to a dual-luciferase assay. The ratio of FL to RL in cells transfected with each RNA was normalized with the mean value of the ratio in cells transfected with RL-FL RNA. (G) The bicistronic reporter RNA was incubated in a rabbit reticulocyte lysate (RRL) at 30°C for 90 min. Then, the RRL mixture was subjected to a dual-luciferase assay. The ratio of FL to RL in each RNA-RRL mixture was normalized with the mean value of the ratio in the RRL incubated with RL-FL RNA. The data are representative of the results of three independent experiments. *, P < 0.05; **, P < 0.01; n.s., not significant. The error bars indicate standard deviations (SD).
Previous reports showed that the EHcV 5′ UTR was predicted to form secondary structures (domains I, I′, II, and III and a pseudoknot), which are similar to those of HCV and exhibit IRES activity (58). S1- and S2-like sequences (CACAUUA and CACUCC, respectively) are located upstream of domain II, while the counterpart to the 3′ overhang nucleotides of miR-122 has not been identified in the 5′ UTR of the EHcV genome (8, 10). In this study, we examined the functions of the EHcV 5′ UTR in terms of IRES activity and RNA replication. Our data suggest that the 5′ UTRs of human and horse hepaciviruses contain a mutual architecture for translation initiation and carry virus-specific elements for RNA replication in the 5′-proximal region comprising domains I and II. Furthermore, domain I of the EHcV 5′ UTR is suggested to be involved in a miR-122-independent mechanism to enhance RNA replication and stability. The virus-specific region in the EHcV 5′ UTR may contribute to the broad tissue or host tropism of EHcV infection.
RESULTS
Efficiency of EHcV IRES activity.
Comparison of the complete nucleotide sequence of the EHcV 5′ UTR (strain JPN3; GenBank accession no. AB863589.1) with that of the HCV 5′ UTR (strain JFH1; GenBank accession no. AB047639.1) using Genetyx software (Nihon Genetyx, Tokyo, Japan) revealed that the EHcV and HCV 5′ UTRs share 58.9% nucleotide homology. Domains II and III of the EHcV 5′ UTR show relatively high homology with those of the HCV 5′ UTR, although the domain I region of the EHcV 5′ UTR showed low similarity to that of the HCV 5′ UTR (4, 10). A bicistronic reporter plasmid carrying the Renilla luciferase (RL) gene, a viral 5′ UTR sequence, and the firefly luciferase (FL) gene (RL-HCV-FL or RL-EHcV-FL) was generated to investigate the IRES activity of the EHcV 5′ UTR (Fig. 1B). We detected the transcript bearing both the RL and FL ORFs in cells transfected with the bicistronic reporter plasmid by Northern blotting (Fig. 1C), suggesting that the cDNAs of the EHcV and HCV 5′ UTRs lack promoter activity. Furthermore, bicistronic reporter plasmids were introduced into several cell lines, including 293T, Huh7.5.1, BRL-3A, A549, and Vero cells. Cells transfected with the bicistronic plasmid bearing the EHcV 5′ UTR showed a higher FL/RL ratio than cells transfected with a bicistronic reporter plasmid lacking a viral UTR, suggesting that the EHcV 5′ UTR exhibited IRES activity in these cell lines (Fig. 1D). However, the IRES activity of the EHcV 5′ UTR was significantly lower than that of the HCV 5′ UTR in these cell lines (Fig. 1D). Although Stewart et al. reported that the EHcV 5′ UTR exhibited higher IRES activity than the HCV 5′ UTR in MDCK cells (58), the IRES activity of the EHcV 5′ UTR was lower than that of the HCV 5′ UTR in MDCK cells and horse fibroblast lines under our experimental conditions (Fig. 1D). The EHcV 5′ UTR tended to exhibit high IRES activity in A549 and Vero cells (Fig. 1E), which suggests that cellular components of A549 and Vero cells are adequate for EHcV translation. Next, we synthesized the capped and polyadenylated bicistronic reporter RNAs from the bicistronic reporter plasmids to investigate the possibility that a posttranscriptional modification of mRNA inhibits the IRES activity of the EHcV 5′ UTR. However, the bicistronic reporter RNA carrying the EHcV 5′ UTR showed IRES activity lower than that of the HCV 5′ UTR in the 293T cells transfected with the bicistronic reporter RNAs (Fig. 1F) and also in the rabbit reticulocyte lysate incubated with the bicistronic reporter RNAs (Fig. 1G). We employed 293T cells for further analyses because the highest levels of IRES activity were observed in 293T cells among the tested cell lines. Because the HCV core-coding region was previously reported to mediate HCV IRES activity (59), the EHcV core-coding region (60 or 120 nucleotides) was inserted in frame upstream of the FL ORF in RL-EHcV-FL (Fig. 2A, B). The EHcV 5′ UTR with the core-coding region showed lower activity than the EHCV 5′ UTR lacking the core-coding region (Fig. 2B). The possibility that N-terminal EHcV core peptides interfered with the enzymatic activity of the FL protein was rejected because insertion of the porcine teschovirus 1 2A peptide sequence between the core protein and FL did not affect the enzymatic activity of FL (Fig. 2A and B) (60). In addition, in trans expression of the EHcV core protein did not affect EHcV IRES activity (Fig. 2C and D). Taken together, our data suggest that the EHcV 5′ UTR includes an IRES, although the IRES activity is lower than that of the HCV 5′ UTR in both host- and non-host-derived cell lines.
FIG 2.

Effects of the core-coding region on EHcV IRES activity. (A) The indicated lengths of the EHcV core-coding region (black boxes) and 2A-cleavage site-coding sequence (hatched boxes) were N-terminally fused to the FL ORF of RL-EHcV-FL in frame (gray boxes). The arrowhead indicates the cleavage site of the P2A sequence. (B) The reporter plasmid carrying a transcript, as shown in panel A, was transfected into 293T cells. The cells were harvested at 48 h posttransfection. (Top) The expression of FL with or without EHcV core peptides was analyzed by Western blotting. (Bottom) IRES activity was estimated in triplicate by a dual-luciferase assay. The ratio of FL to RL was normalized to the mean value of cells expressing RL-EHcV-FL (C0). NC, mock-transfected cells as a negative control. C20 and C40, coding regions of the N-terminal 20 amino acids and 40 amino acids, respectively, of the EHcV core protein. ΔIII, RL-HCV-FL-ΔIII as defined in Fig. 1. The statistical significance versus the cells expressing RL-EHcV-FL (C0) is indicated. **, P < 0.01; n.s., not significant. The error bars indicate standard deviations. (C) The reporter plasmid carrying RL-EHcV-FL was transfected into 293T cells with the plasmid encoding the EHcV core. The total amount of DNA was equilibrated with the plasmid encoding green fluorescent protein (GFP). The transfected cells were harvested at 24 h posttransfection. The EHcV core protein was immunoblotted with a rabbit polyclonal antibody specific for the EHcV core protein (top) (10), while GFP was detected using BZ-9000 fluorescence microscopy (Keyence, Osaka, Japan) (bottom). (D) Each lysate prepared from the transfected cells shown in panel C was subjected to a dual-luciferase assay. The ratio of FL activity to RL activity was normalized with the mean value of cells cotransfected with both RL-EHcV-FL and GFP plasmids. The statistical significance versus the cells cotransfected with both RL-EHcV-FL and GFP plasmids is indicated. The data are representative of the results of three independent experiments.
Requirement for 5′ UTR domains for IRES activity.
The EHcV 5′ UTR is predicted to consist of four domains, designated domains I, I′, II, and III in this study, according to previous reports (8, 10, 24) (Fig. 1A). A structural element corresponding to HCV domain IV is not predicted in the EHcV 5′ UTR (8, 10, 58). To investigate the role of each EHcV domain in IRES activity, we generated a bicistronic reporter plasmid in which each domain of the HCV or EHcV 5′ UTR was deleted. Deletion of HCV domain II or III, but not domain I, abolished IRES activity (Fig. 3A, top), and deletion of EHcV domain II or III abolished IRES activity (Fig. 3A, bottom). Deletion of EHcV domain I, but not I′, reduced IRES activity (Fig. 3A, bottom). All subdomains of EHcV or HCV domain III were also indicated to be necessary for IRES activity (Fig. 3B). The pseudoknot downstream of subdomain IIIf is essential for HCV IRES activity (23). The complementary residue pair within the pseudoknot of EHcV was disrupted by noncomplementary mutation or was replaced by another, different complementary pair as a complementary mutation (Fig. 3C). The noncomplementary mutation impaired EHcV IRES activity, while the complementary mutation partially restored IRES activity (Fig. 3D). We previously identified the 3′-extended (3′ X) region containing an A-rich region and three stem-loops (the 3′ SL region) downstream of the NS5B-coding region (Fig. 3E). Scheel et al. identified a further 3′-extended region containing the poly(U) tract and an additional three stem-loops (the 3′ X region) downstream of the 3′ SL region (10, 61). The monocistronic reporter RNAs shown in Fig. 3E were prepared to investigate the role of each EHcV 3′ UTR domain in IRES activity. The complete EHcV 3′ UTR enhanced IRES activity, which was consistent with the report of Scheel et al. (61), although the 3′ X region or NS5B-coding region was not essential for enhancement (Fig. 3F). Deletion of stem-loop III (SLIII), but not SLI or SLII, of the 3′ SL region impaired IRES activity compared to the original 5′ FL-3′ SL (Fig. 3G). These data suggested that domains I, II, and III and the pseudoknot of the EHcV 5′ UTR are required for complete IRES activity and that SLIII of the 3′ SL region is responsible for the enhancement of IRES activity.
FIG 3.
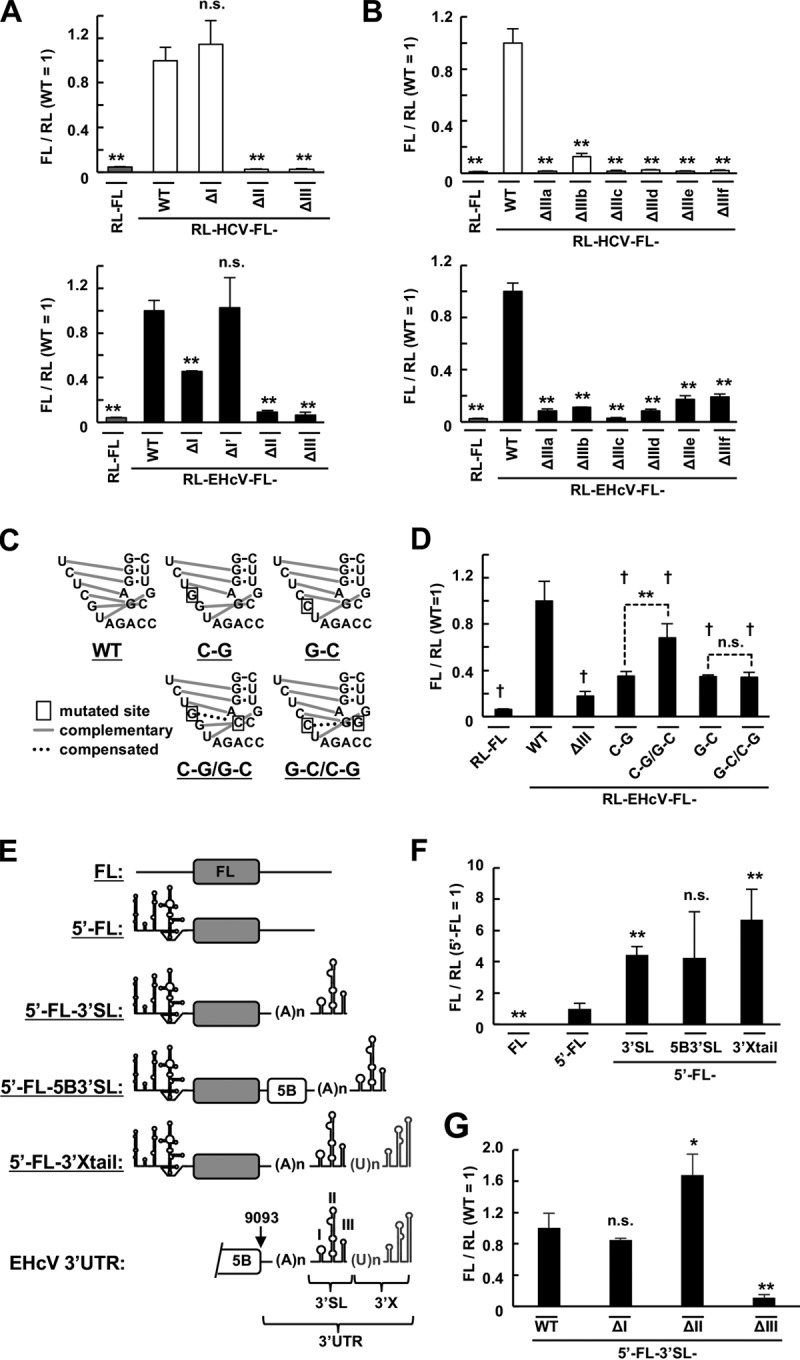
Roles of the secondary structures of HCV and EHcV 5′ UTRs in IRES activity. (A) Plasmids carrying RL-HCV-FL with a deleted domain I (ΔI), II (ΔII), or III (ΔIII) or plasmids carrying RL-EHcV-FL with a deleted domain I (ΔI), I′ (ΔI′), II (ΔII), or III (ΔIII) were transfected in triplicate into 293T cells. The resulting cells were subjected to a dual-luciferase assay. The statistical significance versus the cells transfected with RL-HCV-FL (top) and RL-EHcV-FL (bottom), respectively, is indicated. (B) Plasmids carrying RL-HCV-FL or RL-EHcV-FL with a deleted subdomain IIIa (ΔIIIa), IIIb (ΔIIIb), IIIc (ΔIIIc), IIId (ΔIIId), IIIe (ΔIIIe), or IIIf (ΔIIIf) were transfected in triplicate into the cells. Each lysate prepared from the resulting cells was subjected to a dual-luciferase assay. The statistical significance versus the cells transfected with RL-HCV-FL (top) and RL-EHcV-FL (bottom), respectively, is indicated. (C) Schematic diagram of RL-EHcV-FL carrying the WT pseudoknot and the mutants. Disrupted base pairing in the pseudoknot of the EHcV 5′ UTR was introduced by replacement of C with G (C-G) or G with C (G-C) at the indicated positions. The base pairing of the C-G and G-C mutations was restored by the compensatory C-G/G-C and G-C/C-G mutations, respectively, at the indicated positions. Inherent and restored base pairings are indicated by gray lines and dotted lines, respectively. The boxed nucleotides represent mutated residues. (D) The reporter plasmids carrying the transcripts illustrated in panel C were transfected in triplicate into 293T cells. The cells were harvested at 24 h posttransfection and subjected to a dual-luciferase assay. The statistical significance versus the mean value using RL-EHcV-FL (WT) is indicated (†, P < 0.01). The asterisks indicate statistical significance between two groups (**, P < 0.01). (E) Schematic diagram of the reporter RNAs carrying the EHcV 5′ UTR and 3′ UTR or its mutant. The transcript bearing the EHcV 5′ UTR and FL ORF, in that order, was designated 5′ FL. The transcript 5′ FL contains no polyadenylation site. The transcript 5′ FL-3′ SL is composed of a 5′ FL and SLI, SLII, and SLIII (the 3′ SL region), in that order, while the transcript 5′ FL-3′ X tail is composed of 5′ FL-3′ SL and an additional 3′ X region (GenBank accession no. KP325401). The transcript 5′ FL-5B3′ SL contains the region after nucleotide 9093 but not the 3′ X region. (F) The in vitro-transcribed RNA shown in panel E was cotransfected in triplicate into 293T cells with the in vitro-transcribed capped and polyadenylated RNA including the RL ORF. The cells were harvested at 6 h posttransfection and subjected to a dual-luciferase assay. The ratio of FL to RL was normalized with the mean of that of the 5′ FL RNA. The statistical significance versus the cells expressing 5′ FL is indicated. (G) The 5′ FL-3′ SL transcript or a mutant lacking SLI (ΔI), SLII (ΔII), or SLIII (ΔIII) of the 3′ SL region was cotransfected into 293T cells with the RL mRNA. The cells were harvested at 6 h posttransfection and subjected to a dual-luciferase assay. The relative ratios of FL to RL were normalized with that of the 5′ FL-3′ SL RNA (WT). The statistical significance versus the cells expressing 5′ FL-3′ SL-WT is indicated. The data are representative of the results of three independent experiments. *, P < 0.05; **, P < 0.01; n.s., not significant. The error bars indicate standard deviations.
Cross-species function of the domain structure of hepaciviral 5′ UTRs in IRES activity.
We next investigated the functional compatibility of each domain structure in terms of IRES activity. Each domain of the 5′ UTR in RL-HCV-FL was replaced with the corresponding domain of the EHcV 5′ UTR, while each domain of the 5′ UTR in RL-EHcV-FL was replaced with a corresponding domain of the HCV 5′ UTR, as shown in Fig. 4A. The IRES activity of the HCV 5′ UTR mutant ESLI, which is composed of EHcV domain I and other HCV domains, was higher than that of the wild-type (WT) HCV 5′ UTR, while that of ESLII was lower than that of the WT HCV 5′ UTR (Fig. 4B). However, the IRES activity of ESLIII was similar to that of the WT EHcV 5′ UTR (Fig. 4B). The IRES activities of HSLI and HSLIII were similar to that of the WT HCV 5′ UTR but higher than that of the WT EHcV 5′ UTR (Fig. 4C). The IRES activity of HSLII was similar to that of the WT EHcV 5′ UTR (Fig. 4C). These data suggest that domains II and III are functionally compatible between HCV and EHcV in terms of translation initiation and that domain III contributes to species-specific IRES activity.
FIG 4.
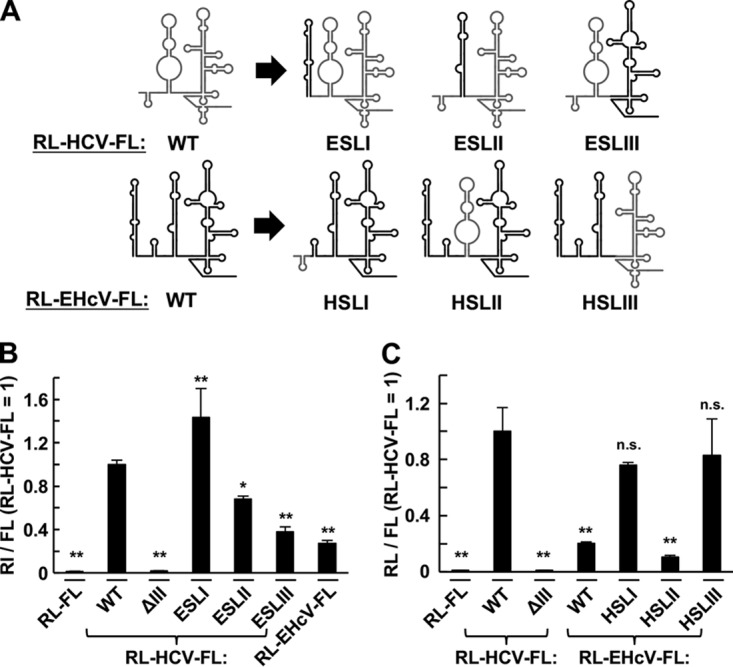
Functional compatibility of each domain structure in the EHcV and HCV 5′ UTRs in terms of its IRES activity. (A) Schematic diagram of the bicistronic reporter transcripts including the chimeric 5′ UTRs. Domain I, II, or III of RL-HCV-FL was replaced with EHcV domain I (ESLI), II (ESLII), or III (ESLIII), respectively. Domains I, II, and III of RL-EHcV-FL were replaced with HCV domains I (HSLI), II (HSLII), and III (HSLIII), respectively. (B and C) The bicistronic reporter plasmids bearing the transcripts shown in panel A were transfected in triplicate into 293T cells. The resulting cells were harvested at 24 h posttransfection and subjected to a dual-luciferase assay. The ratio of FL to RL was normalized with the mean of that of RL-HCV-FL carrying WT HCV IRES. The statistical significance versus the cells transfected with RL-HCV-FL is indicated. *, P < 0.05; **, P < 0.01; n.s., not significant. The data are representative of the results of three independent experiments. The error bars indicate standard deviations.
Functional compatibility of the EHcV 5′ UTR in RNA replication of HCV.
The HCV-based replicon RNA primarily used in this study includes the HCV 5′ UTR, a neomycin resistance gene, the encephalomyocarditis virus (EMCV) IRES, the genetic region encoding HCV NS3 to NS5B, and the HCV 3′ UTR, in that order (HCVSGR). HCVSGR is derived from the genotype 2a strain JFH1, as reported by Kato et al. (62). Two reports showed that the HCVSGR mutant including the EHcV 5′ UTR instead of the intrinsic HCV 5′ UTR (HCVSGR-EHcV in Fig. 5A) was incapable of replicating in Huh7 cells (58, 63). Therefore, we generated an HCVSGR mutant that included each EHcV domain instead of the corresponding domain, as shown in Fig. 5A, to investigate the role of the EHcV 5′ UTR in viral replication. These replicon RNAs were transfected into Huh7.5.1 cells for a colony formation assay. HCVSGR-EHcV and HCVSGR-ESLII were incapable of replicating in the cells, while the colony formation activity of HCVSGR-ESLIII was slightly higher than that of the WT HCVSGR (Fig. 5B and C). HCVSGR-ESLI exhibited 10-fold-higher colony formation activity than WT HCVSGR (Fig. 5B and C). Several colonies were arbitrarily isolated to evaluate the amounts of intracellular SGR RNAs by quantitative reverse transcription (qRT)-PCR analysis using a primer pair targeting the HCV NS3 region (Table 1), and NS5A protein levels were evaluated by Western blotting. Intracellular SGR RNA and NS5A protein levels were comparable among the WT and mutant SGRs (Fig. 5D and E). We carried out RT-PCR analyses using a forward primer specific for HCV or EHcV domain I and a reverse primer specific for HCV or EHcV domain III in combination (Table 1) to confirm whether the chimera structures in the 5′ UTRs of the mutant SGRs were maintained. The RNA of HCVSGR-ESLI was successfully amplified by RT-PCR analysis using forward and reverse primers for EHcV domain I and HCV domain III, respectively, while the RNA of HCVSGR-ESLIII was amplified with forward and reverse primers for HCV domain I and EHcV domain III, respectively (Fig. 5F). Each clone was passaged 10 times to determine the sequence of the 5′ UTR, resulting in no mutations in the 5′ UTRs of HCVSGR-ESLI and HCVSGR-ESLIII (data not shown). These data suggest that domain III, but not domain II, shares functional similarity between HCV and EHcV and that domain I of EHcV enhances the colony formation efficiency of HCVSGR.
FIG 5.

Replication of the subgenomic replicon harboring the HCV-EHcV chimeric 5′ UTR. (A) Schematic diagram of HCVSGR and mutants. Domains I, II, and III-IV of the intrinsic HCVSGR 5′ UTR were replaced with domains I (ESLI), II (ESLII), and III (ESLIII), respectively, of the EHcV 5′ UTR. The domain structures of HCV and EHcV are indicated with gray and black lines, respectively. (B) The WT or mutant HCVSGR RNAs were subjected to a colony formation assay. The HCVSGR RNA carrying inactivated polymerase mutations (GND) was also subjected to the assay as a negative control. (C) Colony counts for the colony formation assay shown in panel B were estimated using ImageJ software to calculate CFU. ND, not detected. **, P < 0.01. (D) The amount of replicon RNA in each clone was estimated by qRT-PCR and normalized with the amount of GAPDH mRNA. The average value of each group is indicated as the mean (thick horizontal line) ± SD. (E) The cell lysates extracted from 4 clones in each group were subjected to Western blotting. Clone numbers are indicated at the top. NC indicates the lysate of nontransfected cells. (F) Total RNA extracted from the 4 clones in each group was subjected to RT-PCR analysis using the indicated primer pairs (Table 1). Clone numbers are indicated at the top. In vitro-transcribed RNAs including the HCV 5′ UTR (lane H) or EHcV 5′ UTR (lane E) were used as positive controls. The total RNA extracted from the nontransfected cells (NC) was used as a negative control. The data are representative of the results of three independent experiments. The error bars indicate standard deviations.
TABLE 1.
Primers used for RT-PCR and qRT-PCR
| Name | Target | Orientation | Sequence (5′–3′) | Purpose |
|---|---|---|---|---|
| HSLI-F | HCV domain I | Forward | GACGCGACCTGCCCCTAATAG | RT-PCR |
| HSLIII-R | HCV domain III | Reverse | ACCCAGTCTTCCCGGCAA | RT-PCR |
| ESLI-F | EHcV domain I | Forward | TACGACACCTCCGTGCTATGCA | RT-PCR |
| ESLI′-F | EHcV domain I′ | Forward | GGAGGGATTCTTCCACATTA | RT-PCR |
| ESLIII-R | EHcV domain III | Reverse | TCGGCTCCGAAGGTCACG | RT-PCR |
| NS3-F | HCV NS3 | Forward | CTGCTTATGCCCAGCAAACA | RT-PCR or qRT-PCR |
| NS3-R | HCV NS3 | Reverse | AACGTCGAGTGTCTCAACGG | RT-PCR or qRT-PCR |
| GAP-F | GAPDH | Forward | GAAGGTGAAGGTCGGAGTC | RT-PCR or qRT-PCR |
| GAP-R | GAPDH | Reverse | GAAGATGGTGATGGGATTTC | RT-PCR or qRT-PCR |
Involvement of EHcV domain I in miR-122-independent replication.
The EHcV 5′ UTR contains S1- and S2-like sites in domain I′ and the upstream tract of domain II, respectively (Fig. 6A) (4, 10). The role of miR-122 in EHcV replication is controversial. To investigate whether the region upstream of domain II in the EHcV 5′ UTR contributes to viral replication, we generated additional HCVSGR mutants, called HCVSGR-ESLI-I′ and HCVSGR-ESLI′ (Fig. 6B). HCVSGR-ESLI and -ESLIII carry intact S1 and S2 HCV sites, while HCVSGR-ESLI-I′ and -ESLI′ carry S1- and S2-like EHcV sequences (Fig. 6B). WT and mutant SGR RNAs were electroporated into Huh7.5.1 cells, Huh7-derived cells deficient in miR-122 (Huh-122KO), and Huh-122KO cells exogenously expressing miR-122 (Huh-122KOR). miR-122 was expressed in the Huh7.5.1 and Huh7-122KOR cell lines but not in the Huh-122KO cell line (Fig. 6C). The colony formation activities of WT HCVSGR and HCVSGR-ESLIII were significantly reduced in the absence of miR-122 (Fig. 6D and E). The colony formation activity of HCVSGR-ESLI was significantly lower in Huh-122KO cells than in Huh7.5.1 cells, while the colony formation activity of HCVSGR-ESLI in Huh-122KO cells was similar to that of WT HCVSGR or HCVSGR-ESLIII in Huh7.5.1 cells (Fig. 6D and E). In contrast, the colony formation activity of HCVSGR-ESLI-I′ was not affected by miR-122 expression (Fig. 6D and E). HCVSGR-ESLI′ did not exhibit any colony formation activity in the absence or presence of miR-122 (Fig. 6D and E). A transient replicon assay revealed that replicon RNA levels of WT HCVSGR, HCVSGR-ESLI, and HCVSGR-ESLIII were decreased by miR-122 depletion, while HCVSGR-ESLI-I′ was not affected by miR-122 depletion (Fig. 6F). Intracellular RNA of HCVSGR-ESLI-I′ was detected by RT-PCR analysis with the forward and reverse primers for EHcV domain I and HCV domain III and with the forward and reverse primers for EHcV domain I′ and HCV domain III (Table 1 and Fig. 6G). These data suggest that the S1- and S2-like sites of EHcV are not involved in miR-122-dependent viral replication and that EHcV domain I maintains colony formation efficiency in a miR-122-independent manner.
FIG 6.

Role of miR-122 in viral replication. (A) (Left) Schematic diagram of the interaction between the HCV 5′ UTR (black letters) and two miR-122 molecules (gray letters). The gray lines indicate the S1 and S2 regions. (Right) Schematic diagram of a putative model for the interaction between the EHcV 5′ UTR (black letters) and two miR-122 molecules (gray letters). The gray lines indicate S1- and S2-like regions. The 5′ UTR nucleotides binding to miR-122 are in boldface. (B) Schematic diagram of the WT HCV subgenomic replicon (HCVSGR-WT) and mutants. The region spanning from the 5′ terminus to the S2 site of HCVSGR-WT was replaced with the region spanning from domains I to I′ of the EHcV 5′ UTR (HCVSGR-ESLI-I′) or with domain I′ of the EHcV 5′ UTR (HCVSGR-ESLI′). The domain structures of HCV and EHcV are illustrated with gray and black lines, respectively. The open stars indicate the S1 and S2 sites of HCV, while the filled stars indicate the S1- and S2-like sites of the EHcV 5′ UTR. (C) Total RNAs were extracted from Huh7.5.1, Huh-122KO, Huh-122KOR, and 293T cells. miR-122 levels were quantified by qRT-PCR and standardized to U6 small nuclear RNA levels. ND, not detected. (D) HCVSGR-WT and each mutant were electroporated into the indicated cell lines. The resulting cells were subjected to colony formation assays. (E) Colony counts were estimated using ImageJ software to calculate the CFU. (F) HCVSGR-WT and the mutant RNAs were electroporated into the indicated cell lines. The resulting cells were harvested at 48 h posttransfection. Total RNAs were extracted from the cells and subjected to qRT-PCR. The amount of intracellular HCVSGR RNA was estimated and normalized to the amount of GAPDH mRNA. (G) Total RNA extracted from the cells was subjected to RT-PCR analysis using the indicated primer pairs (Table 1). Clone numbers are indicated at the top. In vitro-transcribed RNAs including the HCV 5′ UTR (lane H) or EHcV 5′ UTR (lane E) were used as positive controls. The total RNA extracted from the nontransfected cells (NC) was used as a negative control. The data are representative of the results of three independent experiments. *, P < 0.05; **, P < 0.01; n.s., not significant. The error bars indicate standard deviations.
Next, we further investigated the role of EHcV domain I in improved colony formation activity. The precise mechanism of miR-122 in enhancing HCV RNA replication remains controversial, although miR-122 binding was reported to improve translation efficiency and RNA stability (48, 50, 51). A minigenome RNA including the HCV 5′ UTR (or its chimera mutant), FL gene, and HCV 3′ UTR, in that order (HCVmg), was prepared as shown in Fig. 7A. HCVmg or individual mutant RNAs were transfected into Huh-122KO or Huh-122KOR cells together with the RL mRNA. Expression of miR-122 enhanced the IRES activities of WT HCVmg and HCVmg-ESLI, but not those of HCVmg-EHcV and -ESLI-I′ (Fig. 7B), suggesting that the HCV 5′ UTR, but not the EHcV 5′ UTR, functionally maintains IRES activity in a miR-122-dependent manner. Because the translational activity of HCVmg-ESLI tended to be higher than that of WT HCVmg, irrespective of miR-122 expression (Fig. 7B), improved IRES activity may contribute to enhancement of the colony formation efficiency of HCVSGR-ESLI. To investigate the stability of WT HCVmg or the chimeric mutant, an RNA decay assay was conducted using Huh-122KO and Huh-122KOR cells (48). WT HCVmg was more stable in the presence of miR-122 than in the absence of miR-122 (Fig. 7C and D). In contrast, the stability of HCVmg-ESLI-I′ or -ESLI was not affected by miR-122 expression (Fig. 7C and D). Additionally, HCVmg-ESLI-I′ or -ESLI exhibited higher RNA stability than WT HCVmg RNA in Huh-122KO cells (Fig. 7C). These data suggest that EHcV domain I stabilizes RNA in a miR-122-independent manner. Taken together, the data show that EHcV domain I may cooperatively facilitate the colony formation of HCVSGR via miR-122-independent enhancement of IRES activity and RNA stability.
FIG 7.

Effects of miR-122 on IRES activity and RNA stability. (A) Schematic diagram of HCVmg RNA carrying the HCV (WT), EHcV, ESLI, or ESLI-I′ 5′ UTR. The domain structures of HCV and EHcV are illustrated with gray and black lines, respectively. The open stars indicate the S1 and S2 sites of HCV, while the filled stars indicate the S1- and S2-like sites of the EHcV 5′ UTR. (B) HCVmg RNA or each mutant was transfected into Huh-122KO or Huh-122KOR cells with the capped and polyadenylated RNA including the RL ORF. The resulting cells were harvested at 6 h posttransfection and subjected to a dual-luciferase assay or total RNA extraction. The amount of intracellular HCVmg RNA was estimated by qRT-PCR. The ratio of FL to RL was normalized to the amount of HCVmg RNA (WT). (C) Each HCVmg RNA was transfected into Huh-122KO or Huh-122KOR cells. The transfected cells were harvested at the indicated time points and then subjected to Northern blotting. The data are representative of the results of three independent experiments. (D) Quantification of HCVmg RNAs from Northern blotting (C) by densitometric analysis. The intensities of the bands of HCVmg RNA and actin mRNA were quantified from three separate experiments with ImageJ software. Each intensity of HCVmg RNA was normalized with that of actin mRNA. The relative amount of remaining HCVmg RNA is indicated as the mean and SD. *, P < 0.05; **, P < 0.01; n.s., not significant.
DISCUSSION
Recent studies have shown similarities and differences between HCV and EHcV. The core proteins of HCV and EHcV share structural and functional similarities in terms of their maturation and intracellular localization. The HCV and EHcV core proteins are similarly processed by signal peptide peptidase and then translocated on the surfaces of lipid droplets near the endoplasmic reticulum (ER) membrane to assemble as a nucleocapsid (10). EHcV p7, certain domains of which are functionally compatible with the individual domains of HCV p7 viroporin, exhibits ion channel activity (64). Additionally, EHcV NS3/4A cleaves mitochondrial antiviral signaling protein (MAVS) in a manner similar to that of HCV NS3/4A (65). These findings imply that EHcV not only is genetically related to HCV but also employs common strategies for viral particle formation and innate immune evasion. In this study, the EHcV 5′ UTR exhibited IRES activity (Fig. 1), which is partially consistent with the data reported by Stewart et al. (58). The HCV and EHcV IRES regions comprise domains II and III and the pseudoknot, which were responsible for IRES activity (Fig. 3A to D). Domains II and III of the EHcV 5′ UTR were functionally substituted for domains II and III-IV of the HCV 5′ UTR, respectively, in the HCV 5′ UTR (Fig. 4), suggesting that these domains share common translation initiation functions in both species. GB virus B (GBV-B) and pestiviruses also carry similar IRES structures (66–68), which suggests that the fundamental architecture required for IRES-mediated initiation is conserved among the IRES elements of flaviviruses. The EHcV IRES exhibited lower activity than the HCV IRES (Fig. 1). The HCV and EHcV 5′ UTRs exhibited high IRES activities in 293T cells and Huh7.5.1 cells (Fig. 1D), and the EHcV 5′ UTR tended to show high IRES activity in A549 and Vero cells compared to the other cell lines (Fig. 1E). Different cellular components may affect the IRES activity of each hepacivirus. In addition, RL-EHcV-FL-HSLIII exhibited IRES activity at levels similar to that of RL-HCV-FL-WT, and vice versa (Fig. 4). HCV domain IV facilitates translation by positioning initiation codons in the ribosome-decoding center (69), while the EHcV 5′ UTR is thought to lack such a stem-loop domain just behind subdomain IIIf. Indeed, the EHcV core-coding region downstream of the pseudoknot impaired the IRES activity of the EHcV 5′ UTR (Fig. 2), which indicates a similar relationship between the HCV core-coding region and HCV IRES (59). Alternatively, the IRES of classical swine fever virus (a pestivirus) was reported to exhibit higher activity than the HCV IRES due to the potent interaction of domain III with eIF3 and/or the 40S ribosome (66). Thus, domain III and/or IV may contribute to differences between HCV and EHcV in terms of IRES activity.
HCV SGR replication was abolished by replacing the whole 5′ UTR with the EHcV 5′ UTR (Fig. 5B). This region, including domains III and IV of the HCV 5′ UTR, was completely replaced with the corresponding region derived from EHcV and underwent RNA replication, while the mutant SGRs that contained EHcV domains I′ and II instead of HCV domains I and II, respectively (HCVSGR-ESLI′ and HCVSGR-ESLII) lost their colony formation activity (Fig. 5B and C and 6D). This result suggests that the 5′-terminal region containing domains I and II of the HCV 5′ UTR plays a specific role in RNA replication. This region contains sequences responsible for interactions with host factors, including miR-122 and PCBP2 (37, 47). HCVSGR-ESLI showed significantly higher colony formation activity than WT HCVSGR (Fig. 5B and C). The depletion of miR-122 impaired the replication of HCVSGR-ESLI, which harbors intact miR-122 binding sequences of HCV (Fig. 6D). When the miR-122 binding sequences in HCVSGR-ESLI were replaced with EHcV-derived S1- and S2-like sequences, the mutant SGR (HCVSGR-ESLI-I′) showed colony formation activity levels similar to those of WT HCVSGR in the presence of miR-122 and was resistant to miR-122 depletion (Fig. 6D and E). It remains unclear whether the S1- and S2-like sequences in the EHcV 5′ UTR play a role in the replication of EHcV. HCVSGR-ESLI-I′ showed replication levels similar to those of HCVSGR-ESLI in the absence of miR-122 (Fig. 6D and E), suggesting that the S1- and S2-like sequences in the EHcV 5′ UTR have no significant effects on replication of the HCVSGR mutant. A recent report by Yu et al. (63) showed that an HCV chimera including the region upstream of domain II of EHcV propagated in a miR-122-independent manner (63), which supports our data. The tissue tropism of HCV is likely determined by miR-122, because exogenous expression of miR-122 enhanced the replication of HCV in nonhepatic cells (55–57). Although the liver tropism of EHcV is observed in horses (20), the involvement of miR-122 in liver tropism remains debatable, because infection of canine respiratory organs by EHcV has been reported. Thus, the prediction of miR-122-independent EHcV replication is plausible to explain the differences in tissue and host tropism observed between HCV and EHcV. Unfortunately, a robust EHcV in vitro replicon or infection system has not yet been established. Therefore, additional research will be required to elucidate miR-122-independent EHcV replication. Furthermore, the precise mechanism by which miR-122 binding enhances HCV replication remains unclear. Thus, the HCV chimera and our replicon system provide a loss-of-function system that will allow us to investigate the role of miR-122 binding in replication.
EHcV domain I, whose nucleotide sequence is not similar to that of HCV domain I (4, 10), augmented IRES activity, although HCV domain I and EHcV domain I′ were entirely dispensable for IRES activity (Fig. 3A). The S1- and S2-like sequences of EHcV are located between domains I′ and II. These data suggest that EHcV domain I′ is homologous to HCV domain I and EHcV domain I is a unique structure within the EHcV 5′ UTR. The 5′ UTR of rodent hepacivirus contains domain III, which is similar to those of the HCV and EHcV 5′ UTRs, while the region upstream of domain II is structurally unique in the 5′ UTR of rodent hepacivirus (14). The region upstream of domain II in the hepacivirus 5′ UTR may be specific for individual hepaciviruses and may act as a determinant of host specificity and/or organ tropism rather than viral gene expression. GBV-B SGR in Hep3B cells, in which miR-122 is expressed at low levels, was reported to lose miR-122 binding sites during its replication (70). Additionally, adaptive mutations were found in the miR-122 binding sites of HCV RNA during replication in miR-122-deficient cells, resulting in the establishment of miR-122-independent viral replication (45). Several reports noted the involvement of miR-122 in the enhancement of IRES activity and RNA stability (50, 51, 53). Our data showed that EHcV domain I enhanced IRES activity and RNA stability (Fig. 7). Scheel et al. reported that miR-122 enhanced the activity of the EHcV IRES (61). However, our findings suggest that the activity of the EHcV IRES is not affected by miR-122 expression (Fig. 7), which is supported by data recently reported by Yu et al. (63). The interaction of the HCV 5′ UTR with PCBP2 facilitates the long-range interaction between the 5′ UTR and the 3′ X tail, resulting in the enhancement of IRES activity (47). Thus, the EHcV 5′ UTR may interact with the 3′ UTR of EHcV, but not that of HCV, for circularization in the presence of miR-122. The interaction of miR-122 with the HCV 5′ UTR inhibited Xrn1-mediated degradation of the viral RNA by sequestering the 5′ end of the viral RNA (48). It is reasonable to postulate that EHcV domain I facilitates replication by blocking the 5′ terminus of the viral RNA from the exoribonuclease, because EHcV domain I is located in the 5′ terminus of the EHcV 5′ UTR. Alternatively, EHcV domain I may interact with one or more host factors other than miR-122 to mediate RNA replication.
In conclusion, our data suggest that the 5′ UTR of hepacivirus is broadly divided into two components: a cross-species element for IRES-dependent translation and a virus-specific cis-acting element upstream of domain II. IRES structure and function are substantially conserved among hepaciviruses, but a virus-specific structure or sequence is likely involved in determining the organ or host tropism of a specific hepacivirus.
MATERIALS AND METHODS
Cells.
Human embryonic kidney 293T, human lung carcinoma A549, human hepatoma Huh7.5.1, miR-122-knockout Huh7 (Huh-122KO) and the miR-122-rescued derivative (Huh-122KOR) (45), canine kidney MDCK, monkey kidney Vero, and rat hepatoma BRL-3A cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (Gibco, Gaithersburg, MD), 0.1 mM sodium pyruvate (Sigma-Aldrich), and nonessential amino acids (Sigma-Aldrich). Horse fibroblasts were isolated from fresh horse liver using liver digestion medium (Invitrogen, Carlsbad, CA), cultured in the culture medium described above, and immortalized by the exogenous expression of human telomerase reverse transcriptase and the E6 and E7 proteins of human papillomavirus type 16. Tissue sampling for fibroblast preparation was approved by the Institutional Committee of Laboratory Animal Experimentation (University of Yamanashi, Yamanashi, Japan) and was performed in accordance with the guidelines for the care and use of laboratory animals (University of Yamanashi). All cell lines were cultured at 37°C in a 5% CO2 humidified atmosphere.
Plasmid construction.
Bicistronic reporter plasmids containing HCV or the EHcV 5′ UTR were constructed as follows. The PCR product encoding RL, which was amplified from pGL4, was fused upstream of an inactive EMCV IRES (ΔEMCV) sequence (71) by overlap PCR. The ΔEMCV sequence is known to prevent IRES-independent reinitiation of the ribosomes in the intercistronic region (72). The overlap PCR product was cloned into the AflII/BamHI site of the pcDNA4h complete HCV 5′ UTR, and the N-terminal 20 amino acids (aa) of the HCV core (nucleotides 1 to 400; GenBank accession no. AB047639.1) was obtained from pSGR-JFH1 (62) by PCR. The DNA fragments corresponding to the complete EHcV 5′ UTR (strain JPN3; accession no. AB863589.1) or the complete EHcV 5′ UTR flanked by the N-terminal 25 or 50 aa of the EHcV core were amplified by PCR from the previously described subcloning vectors of the EHcV genome (10). Subsequently, these DNA fragments containing HCV or the EHcV 5′ UTR were fused in frame upstream of the FL gene by overlap PCR and inserted at the XbaI site of the pcD4-RL vector using an In-Fusion HD cloning kit (TaKaRa Bio, Shiga, Japan) to prepare pcD4-RL-HCV-FL or pcD4-RL-EHcV-FL. We introduced nucleotide deletion or substitution into the intercistronic sequences of pcD4-RL-HCV-FL and pcD4-RL-EHcV-FL using a KOD Plus mutagenesis kit (Toyobo, Osaka, Japan). The fragment corresponding to each stem-loop in the HCV or EHcV 5′ UTR was amplified from the bicistronic reporter plasmid by PCR and inserted into the EHcV or HCV 5′ UTR by overlap PCR to generate a mutant 5′ UTR in which each domain of the EHcV or HCV 5′ UTR was replaced with its counterpart from the EHcV or HCV 5′ UTR. Subsequently, the fragment was fused to FL and inserted into pcD4-RL as described above.
Monocistronic reporter plasmids were prepared as follows. The DNA fragment corresponding to the RL gene or the FL gene flanked by the EHcV or HCV 5′ UTR was amplified from the bicistronic reporter plasmid and inserted into the XhoI/HindIII sites of pBluescript II SK(−). The DNA fragment corresponding to the 3′ UTR of EHcV strain JPN3 or HCV strain JFH1 was amplified from the plasmid including the EHcV genome (10) or pSGR-JFH1. The DNA fragment corresponding to the 3′ UTR of EHcV strain NZP1 was synthesized by Thermo Scientific (Rockford, IL, USA). Then, the fragment containing the EHcV 3′ UTR was inserted into the HindIII site of pBluescript II SK(−). The excess sequence between the T7 promoter and the EHcV 5′ UTR sequence was deleted using a KOD Plus mutagenesis kit. The resulting T7 promoter-driven transcript had a precise EHcV 5′ UTR. Each stem-loop region in the EHcV 3′ UTR was deleted using a KOD Plus mutagenesis kit.
The plasmids carrying mutants of the HCV subgenomic replicons were prepared as follows. pSGR-JFH1 was digested with EcoRI and PmeI to remove the fragment spanning from the T7 promoter to the neomycin resistance gene. The PCR product containing only the 5′ UTR mutant was amplified from the corresponding plasmid and flanked by the T7 promoter using overlap PCR. The resulting PCR product was fused in frame to the PCR-amplified neomycin resistance gene or the fragment spanning from the neomycin resistance gene to the EMCV IRES. Subsequently, the DNA fragments were inserted into the EcoRI/PmeI site of pSGR-JFH1 using an In-Fusion HD cloning kit. The sequences of these constructs were confirmed by DNA sequencing (Fasmac, Kanagawa, Japan).
Transfection and reporter assay.
Each bicistronic reporter plasmid was introduced into the 293T, Huh7.5.1, A549, VeroE6, or BRL-3A cell line using Trans-IT LT1 reagent (Mirus, Madison, WI) or into horse fibroblasts using Lipofectamine LTX with Plus reagent (Life Technologies, Grand Island, NY). The bicistronic reporter plasmid was in vitro transcribed using an mMessage mMachine T7 Ultra kit (Ambion, Austin, TX) to produce a capped and polyadenylated bicistronic reporter RNA. Each bicistronic reporter RNA was introduced into 293T cells using TransIT-mRNA transfection reagent. The pBluescript II SK(−) plasmid carrying the FL gene flanked by the EHcV or HCV UTR was digested using HindIII and in vitro transcribed using a MEGAscript T7 kit (Ambion). The pBluescript II SK(−) plasmid carrying the RL gene was also digested using HindIII and in vitro transcribed using an mMessage mMachine T7 Ultra kit (Ambion) to produce a capped and polyadenylated RL mRNA. These monocistronic RNAs were introduced into 293T cells using TransIT-mRNA transfection reagent. The resulting cells were lysed with passive lysis buffer (Promega, Madison, WI) to evaluate luciferase activities using a dual-luciferase reporter assay kit (Promega). Luminescence was estimated using Luminescencer-Octa (Atto, Tokyo, Japan).
In vitro translation.
The capped and polyadenylated bicistronic reporter RNA was synthesized as described above. The bicistronic RNA (0.5 μg) was applied to a rabbit reticulocyte lysate system (Promega) and incubated at 30°C for 90 min according to the manufacturer's protocol. The lysate was then diluted in passive lysis buffer (1:25) and subjected to dual-luciferase assay.
Northern blotting.
Total RNA was extracted from the cells using an RNeasy minikit (Qiagen, Valencia, CA, USA), and 0.8 μg of RNA was applied in a 1.2% agarose gel containing 18% formalin and subjected to electrophoresis. The resulting separated RNAs in the gel were stained with ethidium bromide and then transferred onto a positively charged nylon membrane (Roche, Mannheim, Germany) by the capillary method. The resulting membrane was incubated with DIG-Easy Hyb solution (Roche) containing 500 ng/ml digoxigenin (DIG)-labeled RNA probe at 68°C overnight, followed by incubation with alkaline phosphatase-conjugated anti-DIG antibody at room temperature for 1 h. The RNA bands were visualized with a CDP-Star (Roche) and analyzed using LAS-4000 Mini (GE Healthcare, Tokyo, Japan). DIG-labeled RNA probes were prepared using a DIG RNA labeling kit (Roche).
Transient replicon assay and colony formation assay.
The plasmid pSGR-JFH1 and its derivatives were digested with XbaI. The linearized plasmids were in vitro transcribed into replicon RNAs using a Megascript T7 kit. The transcribed RNAs were electroporated into Huh7 cells or the derived cell lines according to a previously described method (73). For the transient replicon assay, the transfected cells were seeded in a 12-well plate, washed with phosphate-buffered saline (PBS) at 4 h posttransfection, incubated in culture medium, and harvested at 48 h posttransfection. For the colony formation assay, the culture medium was replaced with medium containing 1 mg/ml G418 (Nacalai Tesque, Tokyo, Japan) at 24 h posttransfection. The cell colonies were fixed with ice-cold methanol at 21 days posttransfection, stained with crystal violet, and counted using ImageJ software.
RT-PCR and quantitative RT-PCR.
Total RNA was extracted from cells using an RNeasy minikit and reverse transcribed using ReverTra Ace qPCR RT master mix reagent (Toyobo, Tokyo, Japan). RT-PCR was carried out using Ex Taq polymerase (TaKaRa Bio). Quantitative real-time RT-PCR was performed using Fast SYBR green master mix (Life Technologies, Tokyo, Japan) and a StepOne Plus real-time PCR system (Life Technologies). The HCV NS3 region was amplified to evaluate the amount of intracellular replicon RNA, which was standardized to the amount of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA. The primer sequences are listed in Table 1. Total RNA was extracted using an RNeasy minikit (Qiagen) and subjected to quantification of miR-122 using a TaqMan microRNA assay kit (Life Technologies) according to the method used by Fukuhara et al. (56). The amount of miR-122 was standardized to the amount of U6 small nuclear RNA.
Western blotting.
The cells were lysed with 50 mM Tris-HCl (pH 7.5) containing 150 mM NaCl and 1% Triton X-100 (lysis buffer) supplemented with a complete protease inhibitor cocktail (Roche) for 15 min on ice. The lysate was subjected to Western blotting as described previously (10). Mouse anti-NS5A monoclonal antibody (Austral Biologicals, San Ramon, CA), mouse anti-β-actin monoclonal antibody (Sigma-Aldrich), rabbit anti-firefly luciferase polyclonal antibody (Abcam, Cambridge, MA), or rabbit anti-EHcV core polyclonal antibody (10) was used as the primary antibody. Horseradish peroxidase-conjugated goat anti-mouse IgG polyclonal antibody or goat anti-rabbit IgG polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used as a secondary antibody. Immunocomplexes were detected using a SuperSignal West Femto maximum-sensitivity substrate (Thermo Scientific, Rockford, IL, USA) and visualized using an LAS-4000 Mini (GE Healthcare, Tokyo, Japan).
RNA decay assay.
Each Huh7-derivative cell line was seeded at 1 × 105 cells in a 12-well plate and incubated at 37°C overnight. We introduced 1 μg of in vitro-transcribed RNA onto the cells using TransIT-mRNA transfection reagent according to the manufacturer's protocol. The cells were washed three times with prewarmed PBS at 37°C and then incubated in culture medium for 1 h. Subsequently, the cells were collected at the indicated time points to extract total RNA using an RNeasy minikit. The total RNA was subjected to Northern blotting as described above.
Statistical analysis.
The data for statistical analyses are representative of the results of three independent experiments. Each experiment was carried out in triplicate, and the data are expressed as the means of the triplicates with standard deviations. Significant differences between two groups were determined by Student's t test. Statistical significance for multiple comparisons was determined by one-way analysis of variance (ANOVA) with the Tukey-Kramer post hoc test. A P value of less than 0.05 was considered statistically significant. A P value of more than 0.05 was considered not significant.
ACKNOWLEDGMENTS
We thank M. Furugori-Mori for her secretarial work and T. Wakita and R. Bartenschlager for providing plasmids and cell lines.
This work was supported by research programs from the Japan Agency for Medical Research and Development (16fk0210109h1301 and 16fk0210106h0001) and by Grants-in-Aid from JSPS Kakenhi (15K18779 and 15K08493) and from a scholarship donation from Yakult Co. Ltd.
We declare that we have no conflicts of interest.
REFERENCES
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Ding Q, von Schaewen M, Ploss A. 2014. The impact of hepatitis C virus entry on viral tropism. Cell Host Microbe 16:562–568. doi: 10.1016/j.chom.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Attar LM, Mitchell JA, Brooks Brownlie H, Priestnall SL, Brownlie J. 2015. Detection of non-primate hepaciviruses in UK dogs. Virology 484:93–102. doi: 10.1016/j.virol.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kapoor A, Simmonds P, Gerold G, Qaisar N, Jain K, Henriquez JA, Firth C, Hirschberg DL, Rice CM, Shields S, Lipkin WI. 2011. Characterization of a canine homolog of hepatitis C virus. Proc Natl Acad Sci U S A 108:11608–11613. doi: 10.1073/pnas.1101794108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu G, Sun L, Xu T, He D, Wang Z, Ou S, Jia K, Yuan L, Li S. 2016. First description of hepacivirus and pegivirus infection in domestic horses in China: a study in Guangdong Province, Heilongjiang Province and Hong Kong District. PLoS One 11:e0155662. doi: 10.1371/journal.pone.0155662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Figueiredo AS, Lampe E, do Espirito-Santo MP, Mello FC, de Almeida FQ, de Lemos ER, Godoi TL, Dimache LA, Dos Santos DR, Villar LM. 2015. Identification of two phylogenetic lineages of equine hepacivirus and high prevalence in Brazil. Vet J 206:414–416. doi: 10.1016/j.tvjl.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 7.Matsuu A, Hobo S, Ando K, Sanekata T, Sato F, Endo Y, Amaya T, Osaki T, Horie M, Masatani T, Ozawa M, Tsukiyama-Kohara K. 2015. Genetic and serological surveillance for non-primate hepacivirus in horses in Japan. Vet Microbiol 179:219–227. doi: 10.1016/j.vetmic.2015.05.028. [DOI] [PubMed] [Google Scholar]
- 8.Burbelo PD, Dubovi EJ, Simmonds P, Medina JL, Henriquez JA, Mishra N, Wagner J, Tokarz R, Cullen JM, Iadarola MJ, Rice CM, Lipkin WI, Kapoor A. 2012. Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J Virol 86:6171–6178. doi: 10.1128/JVI.00250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyons S, Kapoor A, Sharp C, Schneider BS, Wolfe ND, Culshaw G, Corcoran B, McGorum BC, Simmonds P. 2012. Nonprimate hepaciviruses in domestic horses, United Kingdom. Emerg Infect Dis 18:1976–1982. doi: 10.3201/eid1812.120498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka T, Kasai H, Yamashita A, Okuyama-Dobashi K, Yasumoto J, Maekawa S, Enomoto N, Okamoto T, Matsuura Y, Morimatsu M, Manabe N, Ochiai K, Yamashita K, Moriishi K. 2014. Hallmarks of hepatitis C virus in equine hepacivirus. J Virol 88:13352–13366. doi: 10.1128/JVI.02280-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walter S, Rasche A, Moreira-Soto A, Pfaender S, Bletsa M, Corman VM, Aguilar-Setien A, Garcia-Lacy F, Hans A, Todt D, Schuler G, Shnaiderman-Torban A, Steinman A, Roncoroni C, Veneziano V, Rusenova N, Sandev N, Rusenov A, Zapryanova D, Garcia-Bocanegra I, Jores J, Carluccio A, Veronesi MC, Cavalleri JM, Drosten C, Lemey P, Steinmann E, Drexler JF. 2017. Differential infection patterns and recent evolutionary origins of equine hepaciviruses in donkeys. J Virol 91:e01711-. doi: 10.1128/JVI.01711-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quan PL, Firth C, Conte JM, Williams SH, Zambrana-Torrelio CM, Anthony SJ, Ellison JA, Gilbert AT, Kuzmin IV, Niezgoda M, Osinubi MO, Recuenco S, Markotter W, Breiman RF, Kalemba L, Malekani J, Lindblade KA, Rostal MK, Ojeda-Flores R, Suzan G, Davis LB, Blau DM, Ogunkoya AB, Alvarez Castillo DA, Moran D, Ngam S, Akaibe D, Agwanda B, Briese T, Epstein JH, Daszak P, Rupprecht CE, Holmes EC, Lipkin WI. 2013. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc Natl Acad Sci U S A 110:8194–8199. doi: 10.1073/pnas.1303037110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang B, Yang XL, Li W, Zhu Y, Ge XY, Zhang LB, Zhang YZ, Bock CT, Shi ZL. 2017. Detection and genome characterization of four novel bat hepadnaviruses and a hepevirus in China. Virol J 14:40. doi: 10.1186/s12985-017-0706-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drexler JF, Corman VM, Muller MA, Lukashev AN, Gmyl A, Coutard B, Adam A, Ritz D, Leijten LM, van Riel D, Kallies R, Klose SM, Gloza-Rausch F, Binger T, Annan A, Adu-Sarkodie Y, Oppong S, Bourgarel M, Rupp D, Hoffmann B, Schlegel M, Kummerer BM, Kruger DH, Schmidt-Chanasit J, Setien AA, Cottontail VM, Hemachudha T, Wacharapluesadee S, Osterrieder K, Bartenschlager R, Matthee S, Beer M, Kuiken T, Reusken C, Leroy EM, Ulrich RG, Drosten C. 2013. Evidence for novel hepaciviruses in rodents. PLoS Pathog 9:e1003438. doi: 10.1371/journal.ppat.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kapoor A, Simmonds P, Scheel TK, Hjelle B, Cullen JM, Burbelo PD, Chauhan LV, Duraisamy R, Sanchez Leon M, Jain K, Vandegrift KJ, Calisher CH, Rice CM, Lipkin WI. 2013. Identification of rodent homologs of hepatitis C virus and pegiviruses. mBio 4:e00216-. doi: 10.1128/mBio.00216-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baechlein C, Fischer N, Grundhoff A, Alawi M, Indenbirken D, Postel A, Baron AL, Offinger J, Becker K, Beineke A, Rehage J, Becher P. 2015. Identification of a novel hepacivirus in domestic cattle from Germany. J Virol 89:7007–7015. doi: 10.1128/JVI.00534-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corman VM, Grundhoff A, Baechlein C, Fischer N, Gmyl A, Wollny R, Dei D, Ritz D, Binger T, Adankwah E, Marfo KS, Annison L, Annan A, Adu-Sarkodie Y, Oppong S, Becher P, Drosten C, Drexler JF. 2015. Highly divergent hepaciviruses from African cattle. J Virol 89:5876–5882. doi: 10.1128/JVI.00393-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levi JE, Cabral SP, Nishiya A, Ferreira S, Romano CM, Polite MB, Pereira RA, Mota MA, Kutner JM. 2014. Absence of nonprimate hepacivirus-related genomes in blood donors seroreactive for hepatitis C virus displaying indeterminate blot patterns. J Viral Hepat 21:e164-. doi: 10.1111/jvh.12252. [DOI] [PubMed] [Google Scholar]
- 19.Pfaender S, Walter S, Todt D, Behrendt P, Doerrbecker J, Wolk B, Engelmann M, Gravemann U, Seltsam A, Steinmann J, Burbelo PD, Klawonn F, Feige K, Pietschmann T, Cavalleri JM, Steinmann E. 2015. Assessment of cross-species transmission of hepatitis C virus-related non-primate hepacivirus in a population of humans at high risk of exposure. J Gen Virol 96:2636–2642. doi: 10.1099/vir.0.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfaender S, Cavalleri JM, Walter S, Doerrbecker J, Campana B, Brown RJ, Burbelo PD, Postel A, Hahn K, Anggakusuma Riebesehl N, Baumgartner W, Becher P, Heim MH, Pietschmann T, Feige K, Steinmann E. 2015. Clinical course of infection and viral tissue tropism of hepatitis C virus-like nonprimate hepaciviruses in horses. Hepatology 61:447–459. doi: 10.1002/hep.27440. [DOI] [PubMed] [Google Scholar]
- 21.Ramsay JD, Evanoff R, Wilkinson TE Jr, Divers TJ, Knowles DP, Mealey RH. 2015. Experimental transmission of equine hepacivirus in horses as a model for hepatitis C virus. Hepatology 61:1533–1546. doi: 10.1002/hep.27689. [DOI] [PubMed] [Google Scholar]
- 22.Brown EA, Zhang H, Ping LH, Lemon SM. 1992. Secondary structure of the 5′ nontranslated regions of hepatitis C virus and pestivirus genomic RNAs. Nucleic Acids Res 20:5041–5045. doi: 10.1093/nar/20.19.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Le SY, Ali N, Siddiqui A. 1995. An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 5′ noncoding region. RNA 1:526–537. [PMC free article] [PubMed] [Google Scholar]
- 24.Honda M, Beard MR, Ping LH, Lemon SM. 1999. A phylogenetically conserved stem-loop structure at the 5′ border of the internal ribosome entry site of hepatitis C virus is required for cap-independent viral translation. J Virol 73:1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berry KE, Waghray S, Doudna JA. 2010. The HCV IRES pseudoknot positions the initiation codon on the 40S ribosomal subunit. RNA 16:1559–1569. doi: 10.1261/rna.2197210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolupaeva VG, Pestova TV, Hellen CU. 2000. Ribosomal binding to the internal ribosomal entry site of classical swine fever virus. RNA 6:1791–1807. doi: 10.1017/S1355838200000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukavsky PJ, Otto GA, Lancaster AM, Sarnow P, Puglisi JD. 2000. Structures of two RNA domains essential for hepatitis C virus internal ribosome entry site function. Nat Struct Biol 7:1105–1110. doi: 10.1038/81951. [DOI] [PubMed] [Google Scholar]
- 28.Kieft JS, Zhou K, Jubin R, Doudna JA. 2001. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA 7:194–206. doi: 10.1017/S1355838201001790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji H, Fraser CS, Yu Y, Leary J, Doudna JA. 2004. Coordinated assembly of human translation initiation complexes by the hepatitis C virus internal ribosome entry site RNA. Proc Natl Acad Sci U S A 101:16990–16995. doi: 10.1073/pnas.0407402101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otto GA, Puglisi JD. 2004. The pathway of HCV IRES-mediated translation initiation. Cell 119:369–380. doi: 10.1016/j.cell.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 31.Fukushi S, Okada M, Stahl J, Kageyama T, Hoshino FB, Katayama K. 2001. Ribosomal protein S5 interacts with the internal ribosomal entry site of hepatitis C virus. J Biol Chem 276:20824–20826. doi: 10.1074/jbc.C100206200. [DOI] [PubMed] [Google Scholar]
- 32.Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J. 2001. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science 291:1959–1962. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- 33.Filbin ME, Kieft JS. 2011. HCV IRES domain IIb affects the configuration of coding RNA in the 40S subunit's decoding groove. RNA 17:1258–1273. doi: 10.1261/rna.2594011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto H, Collier M, Loerke J, Ismer J, Schmidt A, Hilal T, Sprink T, Yamamoto K, Mielke T, Burger J, Shaikh TR, Dabrowski M, Hildebrand PW, Scheerer P, Spahn CM. 2015. Molecular architecture of the ribosome-bound Hepatitis C virus internal ribosomal entry site RNA. EMBO J 34:3042–3058. doi: 10.15252/embj.201592469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Locker N, Easton LE, Lukavsky PJ. 2007. HCV and CSFV IRES domain II mediate eIF2 release during 80S ribosome assembly. EMBO J 26:795–805. doi: 10.1038/sj.emboj.7601549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friebe P, Lohmann V, Krieger N, Bartenschlager R. 2001. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J Virol 75:12047–12057. doi: 10.1128/JVI.75.24.12047-12057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Jeng KS, Lai MM. 2011. Poly(C)-binding protein 2 interacts with sequences required for viral replication in the hepatitis C virus (HCV) 5′ untranslated region and directs HCV RNA replication through circularizing the viral genome. J Virol 85:7954–7964. doi: 10.1128/JVI.00339-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romero-Lopez C, Berzal-Herranz A. 2009. A long-range RNA-RNA interaction between the 5′ and 3′ ends of the HCV genome. RNA 15:1740–1752. doi: 10.1261/rna.1680809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romero-Lopez C, Barroso-Deljesus A, Garcia-Sacristan A, Briones C, Berzal-Herranz A. 2012. The folding of the hepatitis C virus internal ribosome entry site depends on the 3′-end of the viral genome. Nucleic Acids Res 40:11697–11713. doi: 10.1093/nar/gks927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 41.Jopling CL, Schutz S, Sarnow P. 2008. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4:77–85. doi: 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Machlin ES, Sarnow P, Sagan SM. 2011. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci U S A 108:3193–3198. doi: 10.1073/pnas.1012464108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimakami T, Yamane D, Welsch C, Hensley L, Jangra RK, Lemon SM. 2012. Base pairing between hepatitis C virus RNA and microRNA 122 3′ of its seed sequence is essential for genome stabilization and production of infectious virus. J Virol 86:7372–7383. doi: 10.1128/JVI.00513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thibault PA, Huys A, Amador-Canizares Y, Gailius JE, Pinel DE, Wilson JA. 2015. Regulation of hepatitis C virus genome replication by Xrn1 and microRNA-122 binding to individual sites in the 5′ untranslated region. J Virol 89:6294–6311. doi: 10.1128/JVI.03631-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ono C, Fukuhara T, Motooka D, Nakamura S, Okuzaki D, Yamamoto S, Tamura T, Mori H, Sato A, Uemura K, Fauzyah Y, Kurihara T, Suda T, Nishio A, Hmwe SS, Okamoto T, Tatsumi T, Takehara T, Chayama K, Wakita T, Koike K, Matsuura Y. 2017. Characterization of miR-122-independent propagation of HCV. PLoS Pathog 13:e1006374. doi: 10.1371/journal.ppat.1006374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts AP, Doidge R, Tarr AW, Jopling CL. 2014. The P body protein LSm1 contributes to stimulation of hepatitis C virus translation, but not replication, by microRNA-122. Nucleic Acids Res 42:1257–1269. doi: 10.1093/nar/gkt941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masaki T, Arend KC, Li Y, Yamane D, McGivern DR, Kato T, Wakita T, Moorman NJ, Lemon SM. 2015. miR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe 17:217–228. doi: 10.1016/j.chom.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM. 2013. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci U S A 110:1881–1886. doi: 10.1073/pnas.1213515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson JA, Zhang C, Huys A, Richardson CD. 2011. Human Ago2 is required for efficient microRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J Virol 85:2342–2350. doi: 10.1128/JVI.02046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. 2012. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci U S A 109:941–946. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C, Junemann C, Niepmann M. 2008. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts AP, Lewis AP, Jopling CL. 2011. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res 39:7716–7729. doi: 10.1093/nar/gkr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conrad KD, Giering F, Erfurth C, Neumann A, Fehr C, Meister G, Niepmann M. 2013. microRNA-122 dependent binding of Ago2 protein to hepatitis C virus RNA is associated with enhanced RNA stability and translation stimulation. PLoS One 8:e56272. doi: 10.1371/journal.pone.0056272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jopling C. 2012. Liver-specific microRNA-122: biogenesis and function. RNA Biol 9:137–142. doi: 10.4161/rna.18827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen CL, Huang JY, Wang CH, Tahara SM, Zhou L, Kondo Y, Schechter J, Su L, Lai MM, Wakita T, Cosset FL, Jung JU, Machida K. 2017. Hepatitis C virus has a genetically determined lymphotropism through co-receptor B7.2. Nat Commun 8:13882. doi: 10.1038/ncomms13882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fukuhara T, Kambara H, Shiokawa M, Ono C, Katoh H, Morita E, Okuzaki D, Maehara Y, Koike K, Matsuura Y. 2012. Expression of microRNA miR-122 facilitates an efficient replication in nonhepatic cells upon infection with hepatitis C virus. J Virol 86:7918–7933. doi: 10.1128/JVI.00567-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murayama A, Sugiyama N, Wakita T, Kato T. 2016. Completion of the entire hepatitis C virus life cycle in Vero cells derived from monkey kidney. mBio 7:e00273-. doi: 10.1128/mBio.00273-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stewart H, Walter C, Jones D, Lyons S, Simmonds P, Harris M. 2013. The non-primate hepacivirus 5′ untranslated region possesses internal ribosomal entry site activity. J Gen Virol 94:2657–2663. doi: 10.1099/vir.0.055764-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang TH, Rijnbrand RC, Lemon SM. 2000. Core protein-coding sequence, but not core protein, modulates the efficiency of cap-independent translation directed by the internal ribosome entry site of hepatitis C virus. J Virol 74:11347–11358. doi: 10.1128/JVI.74.23.11347-11358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, Kim MK, Shin BA, Choi SY. 2011. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scheel TK, Kapoor A, Nishiuchi E, Brock KV, Yu Y, Andrus L, Gu M, Renshaw RW, Dubovi EJ, McDonough SP, Van de Walle GR, Lipkin WI, Divers TJ, Tennant BC, Rice CM. 2015. Characterization of nonprimate hepacivirus and construction of a functional molecular clone. Proc Natl Acad Sci U S A 112:2192–2197. doi: 10.1073/pnas.1500265112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 63.Yu Y, Scheel TKH, Luna JM, Chung H, Nishiuchi E, Scull MA, Echeverria N, Ricardo-Lax I, Kapoor A, Lipkin IW, Divers TJ, Antczak DF, Tennant BC, Rice CM. 2017. miRNA independent hepacivirus variants suggest a strong evolutionary pressure to maintain miR-122 dependence. PLoS Pathog 13:e1006694. doi: 10.1371/journal.ppat.1006694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walter S, Bollenbach A, Doerrbecker J, Pfaender S, Brown RJ, Vieyres G, Scott C, Foster R, Kumar A, Zitzmann N, Griffin S, Penin F, Pietschmann T, Steinmann E. 2016. Ion channel function and cross-species determinants in viral assembly of nonprimate hepacivirus p7. J Virol 90:5075–5089. doi: 10.1128/JVI.00132-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anggakusuma Brown RJ, Banda DH, Todt D, Vieyres G, Steinmann E, Pietschmann T. 2016. Hepacivirus NS3/4A proteases interfere with MAVS signaling in both their cognate animal hosts and humans: implications for zoonotic transmission. J Virol 90:10670–10681. doi: 10.1128/JVI.01634-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hashem Y, des Georges A, Dhote V, Langlois R, Liao HY, Grassucci RA, Pestova TV, Hellen CU, Frank J. 2013. Hepatitis-C-virus-like internal ribosome entry sites displace eIF3 to gain access to the 40S subunit. Nature 503:539–543. doi: 10.1038/nature12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rijnbrand R, Abell G, Lemon SM. 2000. Mutational analysis of the GB virus B internal ribosome entry site. J Virol 74:773–783. doi: 10.1128/JVI.74.2.773-783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chard LS, Kaku Y, Jones B, Nayak A, Belsham GJ. 2006. Functional analyses of RNA structures shared between the internal ribosome entry sites of hepatitis C virus and the picornavirus porcine teschovirus 1 Talfan. J Virol 80:1271–1279. doi: 10.1128/JVI.80.3.1271-1279.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Perard J, Leyrat C, Baudin F, Drouet E, Jamin M. 2013. Structure of the full-length HCV IRES in solution. Nat Commun 4:1612. doi: 10.1038/ncomms2611. [DOI] [PubMed] [Google Scholar]
- 70.Sagan SM, Sarnow P, Wilson JA. 2013. Modulation of GB virus B RNA abundance by microRNA-122: dependence on and escape from microRNA-122 restriction. J Virol 87:7338–7347. doi: 10.1128/JVI.00378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen CY, Sarnow P. 1995. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268:415–417. doi: 10.1126/science.7536344. [DOI] [PubMed] [Google Scholar]
- 72.Wilson JE, Powell MJ, Hoover SE, Sarnow P. 2000. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol Cell Biol 20:4990–4999. doi: 10.1128/MCB.20.14.4990-4999.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Okamoto T, Nishimura Y, Ichimura T, Suzuki K, Miyamura T, Suzuki T, Moriishi K, Matsuura Y. 2006. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J 25:5015–5025. doi: 10.1038/sj.emboj.7601367. [DOI] [PMC free article] [PubMed] [Google Scholar]