

ABSTRACT
Genome uncoating is essential for replication of most viruses. For poxviruses, the process is divided into two stages: removal of the envelope, allowing early gene expression, and breaching of the core wall, allowing DNA release, replication, and late gene expression. Subsequent studies showed that the host proteasome and the viral D5 protein, which has an essential role in DNA replication, are required for vaccinia virus (VACV) genome uncoating. In a search for additional VACV uncoating proteins, we noted a report that described a defect in DNA replication and late expression when the gene encoding a 68-kDa ankyrin repeat/F-box protein (68k-ank), associated with the cellular SCF (Skp1, cullin1, F-box-containing complex) ubiquitin ligase complex, was deleted from the attenuated modified vaccinia virus Ankara (MVA). Here we showed that the 68k-ank deletion mutant exhibited diminished genome uncoating, formation of DNA prereplication sites, and degradation of viral cores as well as an additional, independent defect in DNA synthesis. Deletion of the 68k-ank homolog of VACV strain WR, however, was without effect, suggesting the existence of compensating genes. By inserting VACV genes into an MVA 68k-ank deletion mutant, we discovered that M2, a member of the poxvirus immune evasion (PIE) domain superfamily and a regulator of NF-κB, and C5, a member of the BTB/Kelch superfamily associated with cullin-3-based ligase complexes, independently rescued the 68k-ank deletion phenotype. Thus, poxvirus uncoating and DNA replication are intertwined processes involving at least three viral proteins with mutually redundant functions in addition to D5.
IMPORTANCE Poxviruses comprise a family of large DNA viruses that infect vertebrates and invertebrates and cause diseases of medical and zoological importance. Poxviruses, unlike most other DNA viruses, replicate in the cytoplasm, and their large genomes usually encode 200 or more proteins with diverse functions. About 90 genes may be essential for chordopoxvirus replication based either on their conservation or individual gene deletion studies. However, this number may underestimate the true number of essential functions because of redundancy. Here we show that any one of three seemingly unrelated and individually nonessential proteins is required for the incompletely understood processes of genome uncoating and DNA replication, an example of synthetic lethality. Thus, poxviruses appear to have a complex genetic interaction network that has not been fully appreciated and which will require multifactor deletion screens to assess.
KEYWORDS: gene redundancy, genome replication, genome uncoating, poxvirus replication, recombinant DNA technology, synthetic lethality, vaccinia virus
INTRODUCTION
Genome uncoating is a prerequisite for the replication of most viruses (1, 2). Uncoating may be divided into two stages for poxviruses, which are large DNA viruses that replicate in the cytoplasm and are comprised of a nucleoprotein core surrounded by a lipoprotein envelope (3). The first stage of uncoating commences almost immediately upon infection and consists of removal of the envelope and liberation of cores into the cytoplasm (4). This step, involving fusion of the viral envelope with the host plasma or endosomal membrane, has been reviewed elsewhere (5, 6). Less is known about the second stage of uncoating, which is usually referred to simply as uncoating and consists of rupture of the core wall and liberation of the genome. This stage begins after a lag and was originally defined experimentally as the sensitization of the viral DNA to DNase (4). Further studies indicated that the latter step requires RNA and protein synthesis (7). Although it was initially proposed that the protein synthesis requirement was mediated by activation of host gene expression, the subsequent finding that intact cytoplasmic cores synthesize early mRNAs (8–11) suggested that uncoating may be mediated by a viral protein. Further insights came from studies with inhibitors and human genome-wide RNA interference (RNAi) screens, which indicated a role for the ubiquitin-proteasome system in both uncoating and DNA replication (12–15). It was not until a recent small interfering RNA (siRNA) screen of VACV conserved early genes that a viral protein required for uncoating was identified by Kilcher and coworkers (16). In an analysis with 81 siRNAs, D5 was the only protein whose depletion prevented genome uncoating, the formation of DNA prereplication sites, and degradation of viral cores. D5 is a highly conserved protein that is directly involved in DNA replication (17) and has essential ATPase (18) and primase (19) activities. Mutagenesis studies suggested that only the ATPase domain is required for uncoating (16). Whether additional viral proteins, not revealed by the siRNA screen, are involved in uncoating is an open question. In particular, a screen carried out with single siRNAs would not detect two or more genes that have mutually redundant functions.
We were intrigued by the report of Sperling and coworkers (20) that deletion of open reading frame (ORF) 186, encoding a 68-kDa ankyrin repeat protein (68k-ank) of modified vaccinia virus (VACV) Ankara (MVA), led to a defect in DNA replication and late gene expression in mammalian cells without affecting replication in chicken embryo fibroblasts (CEF), indicating a host-specific defect. MVA was generated by serial passages in CEF, during which a substantial amount of genetic information, including all other ankyrin repeat-encoding genes was lost (21–23). Although MVA containing the 68k-ank has a host range defect in most mammalian cells, viral protein synthesis occurs at a high level, and the block is at a virion assembly step (24). In addition to four 33-residue ankyrin repeats, the 68k-ank protein contains a C-terminal F-box-like domain, which is found in cellular adapter proteins involved in the ubiquitin-dependent proteolysis pathway, and the 68k-ank protein was shown to interact with the SCF (Skp1, cullin1, F-box-containing complex) ubiquitin ligase complex (25). The known role of the proteasome in VACV replication and the above-described properties of the 68k-ank protein led us to investigate whether the defect of the MVA deletion mutant might be based in part on a failure of genome uncoating, although Sperling and coworkers (20) had found that the F-box domain is not essential.
The current study confirmed that a deletion of the 68k-ank ORF from MVA resulted in a block in postreplicative gene expression in several mammalian cell lines. Furthermore, we provide evidence for a defect in uncoating as well as an independent impairment of DNA replication. In contrast to the results obtained with MVA, deletion of the ORF encoding the 68k-ank homolog of VACV strain WR did not adversely affect replication, suggesting the existence of compensating or redundant genes. Two such VACV genes, M2L and C5L, were identified by reversing the block in postreplicative gene expression of an MVA 68k-ank deletion mutant. Uncoating, DNA replication, and postreplicative gene expression of MVA in mammalian cells were enabled by independent expression of either the 68k-ank, M2, or C5 protein. Thus, poxvirus uncoating is a complex process involving the proteasome and multiple viral proteins in addition to D5.
RESULTS
The 68k-ank protein is required for MVA DNA replication and postreplicative gene expression in a variety of mammalian cell lines.
Sperling et al. (20) previously reported that deletion of the 68k-ank gene from MVA abolished late gene expression in HaCaT human skin keratinocytes and NIH 3T3 murine embryonic fibroblasts but not in permissive CEF. We constructed a new 68k-ank deletion mutant (MVA-Δ186) by replacing ORF 186 with an ORF encoding enhanced green fluorescent protein (GFP) regulated by a VACV late promoter (Fig. 1A). The absence of ORF 186 was verified by PCR and sequencing. The previous finding regarding a defect in late gene expression was confirmed and extended by comparing the syntheses of the intermediate/late A3 protein by MVA-Δ186, the parental MVA, and MVA-GFP (encoding GFP at an irrelevant site) in a variety of cell lines. Synthesis of the A3 protein by MVA-Δ186 was specifically diminished in human lung carcinoma A549, human cervical carcinoma epithelial HeLa, human embryonic kidney 293-T, and hamster fibroblast BHK-21 cells but not in CEF (Fig. 1B). Nevertheless, similar amounts of the early I3 protein were produced by parental MVA, MVA-GFP, and MVA-Δ186 in each of the cell lines. Thus, the 68k-ank protein plays a critical role in MVA postreplicative gene expression in several different mammalian cell lines.
FIG 1.
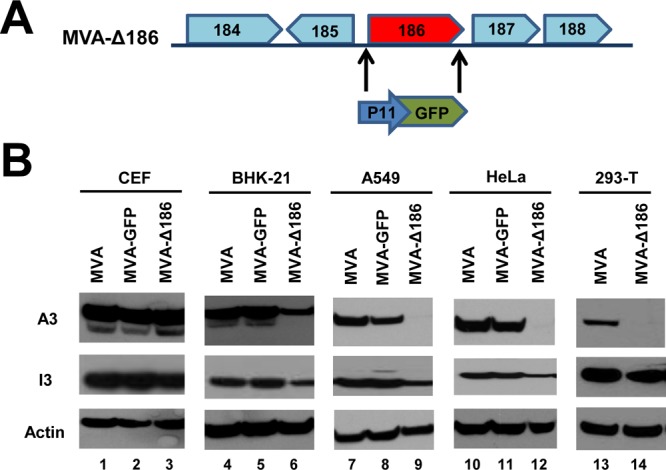
Deletion of MVA ORF 186 encoding the 68k-ank protein reduces postreplicative gene expression in mammalian cells. (A) Diagram showing replacement of MVA ORF 186 with the GFP gene regulated by the VACV P11 late promoter for construction of MVA-Δ186. (B) The indicated cells were infected for 16 h with 0.5 PFU/cell of parental MVA, MVA expressing GFP regulated by the P11 promoter (MVA-GFP), and MVA-Δ186. Proteins were resolved by SDS-PAGE, transferred to membranes, and probed with antibodies to VACV intermediate/late A3 protein (upper panels) and early I3 protein (middle panels). Actin (lower panels) served as a loading control.
Sperling et al. (20) further demonstrated by dot blot analysis that deletion of the 68k-ank gene of MVA led to decreased viral genome replication, which is a prerequisite for late mRNA and protein synthesis. The incorporation of the clickable nucleoside 5-ethynyl-2′-deoxyuridine (EdU) offers an advantageous way of localizing nascent DNA by fluorescence microscopy in individual cells (26). In human A549 cells infected with the WR strain of VACV, Senkevich et al. (27) combined EdU incorporation, immunostaining of the early I3 single-stranded DNA binding protein, and staining with 4′,6-diamidino-2-phenylindole (DAPI) to visualize a dramatic decrease in host nuclear DNA synthesis and the de novo synthesis of viral DNA in cytoplasmic factories. As shown in Fig. 2, nuclear EdU fluorescence was bright in mock-infected HeLa cells but dim or absent by 3 h after infection with either MVA or MVA-Δ186. Distinct cytoplasmic DNA factories were evident by bright EdU labeling in the cells infected with MVA at each of the 3-, 4-, and 5-h time points; I3 was present throughout the cytoplasm but was highly concentrated in the replication sites (Fig. 2). In the cells infected with MVA-Δ186, small EdU-labeled viral factories were seen only occasionally, and there was always diffuse cytoplasmic I3 staining (Fig. 2). These data confirmed the deficiency in viral DNA replication in human cells infected with MVA-Δ186, which would account for the defect in postreplicative protein synthesis. Interestingly, the data also show that viral DNA replication was not necessary for the decrease in nuclear DNA synthesis.
FIG 2.
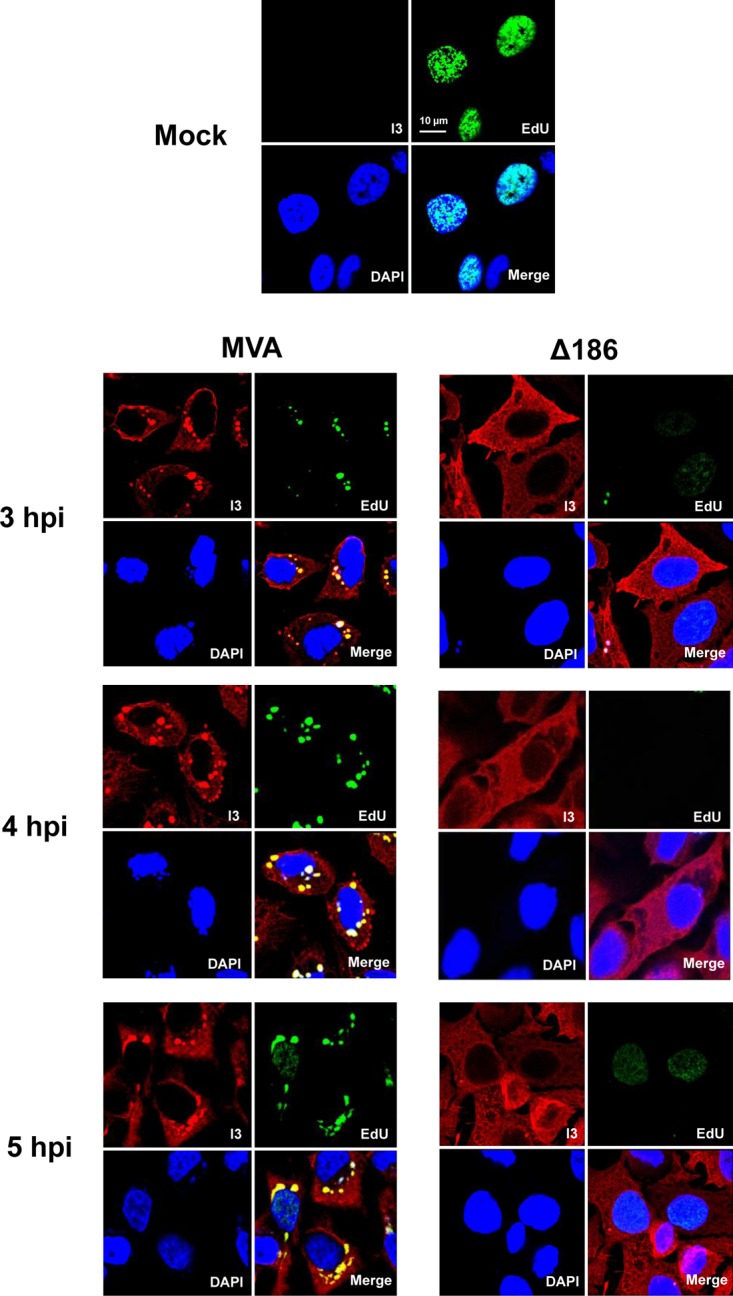
Deletion of MVA ORF 186 encoding the 68k-ank protein prevents viral genome replication. HeLa cells were mock infected or infected with 5 PFU/cell of MVA or MVA-Δ186 (Δ186) and incubated with EdU (10 μM) for 1-h periods at 3, 4, and 5 h after infection. Following each incubation time, the cells were fixed, permeabilized, and reacted with Alexa Fluor 647 azide. The I3 single-stranded DNA binding protein was visualized by staining with a specific MAb, followed by an anti-mouse secondary antibody conjugated to Alexa Fluor 568 and DAPI to stain total DNA. Images were captured with a confocal microscope. Each panel is divided into quadrants: upper left, I3 (red); upper right, EdU (green); lower left, DAPI (blue); and lower right, merge. The scale bar represents 10 μm. hpi, hours postinfection.
68k-ank contributes to MVA genome uncoating in human cells.
Since uncoating of the viral genome precedes DNA replication, it was important to determine whether the defect in DNA synthesis might be a secondary effect. We addressed this question by quantification of punctate viral cores, which are destabilized after uncoating and can be detected by immunostaining with antibody to the A4 core protein (12, 14) and by detection of viral genomes which are accessible to the early I3 single-stranded DNA binding protein only after uncoating (14, 28, 29). In the experiment depicted in Fig. 3, HeLa cells were infected with MVA or MVA-Δ186 in the absence of drugs or in the presence of the protein synthesis inhibitor cycloheximide (CHX) or the DNA synthesis inhibitor cytosine arabinoside (AraC). At 1 and 4 h after infection, the cells were fixed and stained with DAPI and antibodies to the A4 and I3 proteins. During the first hour of a normal VACV infection, the numbers of cores represent a balance due to entry and degradation. Without inhibitors, low numbers of cores that stained with A4 antibody were present in cells infected with MVA or MVA-Δ186 at 1 h (Fig. 3A and B). The numbers of cores decreased by 4 h in MVA-infected cells due to uncoating and degradation, but they increased markedly in cells infected with MVA-Δ186, suggesting core stabilization under the latter conditions. As expected, the large difference between MVA and MVA-Δ186 also occurred in the presence of AraC but not in the presence of CHX, which prevents uncoating and allows cores to accumulate during the 4 h of infection with both viruses.
FIG 3.

Deletion of MVA ORF 186 encoding the 68k-ank protein prevents viral genome uncoating. HeLa cells were untreated or treated with 100 μg/ml of cycloheximide (CHX) or 44 μg/ml of cytosine arabinoside (AraC) and infected with 5 PFU/cell of MVA or MVA-Δ186. After 1 h of adsorption at 4°C, the cells were washed and incubated at 37°C for 1 or 4 h. Cells were then fixed, permeabilized, and immunostained with mouse anti-I3 MAb and rabbit anti-A4 polyclonal antibody, followed by Alexa Fluor 568 anti-mouse IgG and Alexa Fluor 647 anti-rabbit IgG, respectively. Nuclei were stained blue with DAPI. (A) The subcellular localizations of I3 (red), A4 (green), and DAPI (blue) were determined by confocal microscopy, with the scale bar representing 10 μm. (B) Viral cores that stained with anti-A4 antibody and cells that stained with DAPI were enumerated from 40 to 120 cells, and the average numbers of viral cores per cell were plotted. Standard deviations were calculated from the numbers obtained in three random fields.
In the untreated MVA-infected cells, I3 produced at 1 h by intact cores appeared diffuse, whereas at 4 h considerable I3 protein localized with DNA in viral factories (Fig. 3A). In contrast, the I3 staining remained diffuse in MVA-Δ186-infected cells at both times (Fig. 3A), consistent with the data in Fig. 2. Importantly, in the presence of AraC, punctate I3 staining of genomes released from cores could be seen at 4 h after infection with MVA but not after infection with MVA-Δ186 (Fig. 3A). Of course, no I3 was made in the CHX control. Thus, uncoating as assessed by both core destabilization and release of genomic DNA was inhibited in the absence of the 68k-ank protein.
68k-ank is required for DNA synthesis in addition to uncoating.
Although a reduction in genome replication and postreplicative gene expression could be explained entirely by an uncoating defect, this does not preclude an additional direct block in DNA synthesis. To test the latter idea, we developed a droplet digital PCR (ddPCR) plasmid replication assay (Fig. 4A) based on previous findings that VACV can replicate any circular DNA (30, 31). This assay allowed us to examine the ability of VACV to replicate DNA while circumventing the genome uncoating step. HeLa cells were transfected with pUC19 plasmid and 24 h later infected with MVA or MVA-Δ186. The cells were lysed at 6 and 12 h after infection, and the total DNA was extracted and incubated with DpnI enzyme to digest the methylated input plasmid and with BamHI to cleave the replicated DNA concatemers into unit-length segments. Primers for ddPCR were designed to flank a region of the plasmid with a cluster of GATC DpnI recognition sites (Fig. 4B) so that only replicated unmethylated DNA would remain intact and be amplified. The number of DNA copies determined by ddPCR was much higher (P < 0.01) for MVA than for MVA-Δ186 (Fig. 4C). We concluded that the 68k-ank protein has roles in promoting both uncoating and DNA synthesis.
FIG 4.

68k-ank is required for replication of uncoated DNA. (A) Overview of plasmid replication assay. HeLa cells were transfected with pUC19 and infected with VACV 24 h later. The methylated template plasmids and unmethylated replicated DNA are represented by gray and red circles, respectively. Total DNA was extracted and digested with BamHI to cut DNA into unit-length segments and DpnI to cleave methylated template plasmids. After the digestion, droplet digital PCR (ddPCR) was performed to detect newly synthesized DNA. (B) The single BamHI and multiple DpnI restriction sites in pUC19 are shown. The ddPCR primers (P+ and P−) flanked a region with multiple DpnI sites. (C) The number of copies of replicated plasmid DNA was determined at 6 and 12 h after infection of HeLa cells with 5 PFU/cell of MVA or MVA-Δ186. **, P < 0.01.
Other VACV genes can compensate for loss of a 68k-ank homolog.
Because the MVA 68k-ank gene is highly conserved in orthopoxviruses, we were curious to determine the effects of deletion of the homologous gene of the well-characterized VACV WR strain. Accordingly, we deleted WR ORF 199 (B18R according to the Copenhagen nomenclature) by replacing it with the GFP gene (Fig. 5A). After verification of the deletion by PCR and DNA sequencing, we compared the syntheses of the intermediate/late A3 and early I3 proteins by the parent VACV WR and the WR deletion mutant by Western blotting. No reduction in gene expression was observed (Fig. 5B), suggesting that one or more genes in VACV WR compensated for the loss of the 68k-ank homolog and that those genes had been lost by MVA during the extensive passages in CEF. We considered two approaches that might help us identify the putative compensating genes. One was to make additional deletions in VACV WR. However, this approach might fail if more than one compensating gene was present. Furthermore, we knew that VACV WR remained replication competent after deletion of a large DNA fragment from the left end of the genome that included 68k-ank gene homolog as well as other genes (32). The second tactic was to insert additional genes into MVA-Δ186. This method had advantages and was facilitated by the availability of a panel of recombinant MVAs (rMVAs) that had up to 80,000 bp of DNA inserted into the left end of the genome (33). To implement this method, the 68k-ank ORF was deleted from three such MVA recombinants (rMVA 51.1, rMVA 44.2, and rMVA 44/47), and their ability to express the intermediate/late A3 protein was evaluated in several cell lines. Interestingly, each recombinant MVA expressed A3 despite deletion of the 68k-ank gene (Fig. 5C). (We noted that the protein expression defect was less pronounced in BS-C-1 cells infected with MVA-Δ186 than in the other mammalian cells tested.) Further studies were carried out with rMVA 51.1 because it appeared to have a smaller DNA insert than the other recombinant viruses tested here (33).
FIG 5.

Deletion of VACV WR 199 (B18R), the homolog of the 68k-ank gene, does not impair postreplicative gene expression. (A) Diagram showing replacement of VACV ORF B18R with the GFP gene regulated by the VACV P11 late promoter. (B) The indicated cells were infected with 0.5 PFU/cell of parental VACV WR strain (WR) or VACV WR ΔB18R (ΔB18R) for 16 h. The proteins were resolved by SDS-PAGE, transferred to membranes, and probed with antibodies to A3 intermediate/late protein (upper row) and early I3 protein (middle row). Actin (lower row) served as a loading control. (C) The indicated cells were infected with 0.5 PFU/cell of MVA, MVA-GFP, MVA-Δ186, and recombinant rMVAs 44/47.1, 44.2, 51.1, 44/47.1-Δ186, 44.2-Δ186, and 51.1-Δ186 for 16 h. Proteins were resolved by SDS-PAGE, transferred to membranes, and probed with antibodies to the A3 protein and actin.
The terminal regions of VACV contain numerous short ORFs representing pseudogenes in addition to full-length protein-coding ORFs. Whole-genome sequencing of rMVA 51.1 revealed insertions within the left end of MVA, which corresponded to the region contained in cosmid 51 used for recombination (Fig. 6). The insertions restored DNA of MVA deletions I, V, and II. Based on these data, we focused on ORFs C17-C12, C5-C1, and M1-K1, which were absent from the parental MVA (Fig. 7A). Segments of DNA containing these ORFs were PCR amplified from the genome of rMVA 51.1 and inserted into a noncoding region (deletion III) near the right end of MVA-Δ186 (Fig. 7B). A cassette containing the late P11 promoter fused to the mCherry ORF was introduced simultaneously to allow visualization and isolation of fluorescent recombinant plaques. After confirming the insertions by PCR and DNA sequencing, HeLa cells were infected with the new viruses as well as the parental MVA and MVA-Δ186. Lysates were then analyzed for expression of the A3 protein (Fig. 7B). Whereas rMVA-Δ186-C17-C12 failed to show increased A3 gene expression, both rMVA-Δ186-M1-K1 and rMVA-Δ186-C5-C1 did (Fig. 7B). Therefore, at least two compensatory genes were present in rMVA 51.1.
FIG 6.

Comparison of genome sequences of MVA and rMVA 51.1. The left 50,000 bp of the genome of MVA 51.1, corresponding to the DNA segment in cosmid 51, is shown. ORFs of >100 bp and starting with ATG, CTG, or TTG are color coded with arrows pointing in the direction of transcription. Green, identical to MVA; blue, mutated relative to MVA; red, inserted or repaired relative to MVA. The three MVA deletions dI, dV, and dII are indicated. HindIII fragments C, N/M, K, and F are shown at the bottom.
FIG 7.

C5L and M2L ORFs rescue the postreplicative gene expression defect of MVA-Δ186. (A) Diagram representing the MVA-Δ186 genome with position of cosmid 51 and site of GFP replacement of MVA ORF 186. The enlargement shows missing ORFs within deletion I (C17-C12), deletion V (C5-C1), and deletion II (M1-K1). (B) Diagram representing the genome of MVA-Δ186 and DNA containing the mCherry ORF regulated by the P11 promoter, followed by an insert containing C17-C12, C5-C1, or M1-K1 DNA flanked by sequences to enable homologous recombination. The Western blot shows A3 protein and an actin loading control from cells infected for 16 h with 0.5 PFU/cell of MVA-Δ186, rMVA-Δ186-C17-C12, rMVA-Δ186-M1-K1, or rMVA-Δ186-C5-C1. (C) Diagram representing the genome of MVA-Δ186 and DNA containing the mCherry ORF regulated by the P11 promoter, followed by an insert containing an individual ORF (C1, C2, C3, C5, K1, M1, or M2) regulated by the mH5 promoter and flanked by sequences to enable homologous recombination. The Western blot shows A3 protein and an actin loading control from cells infected for 16 h with 0.5 PFU/cell of MVA, MVA-Δ186, or rMVA-Δ186 constructs expressing C1, C2, C3, C5, K1, M1, or M2 protein.
To identify the compensatory genes, the individual ORFs were amplified by PCR from rMVA 51.1. In each case the ORF was regulated by the mH5 promoter (34), since the native promoter sequences of these genes have not been characterized. The genes were then inserted into deletion III of MVA-Δ186 to form the seven recombinants rMVA-Δ186-C1, -C2, -C3, -C5, -K1, -M1, and -M2, and HeLa cells were infected with each. Western blotting showed that only ORFs C5 and M2 compensated for the 68k-ank gene (Fig. 7C). We also confirmed that rMVA-Δ186-C5 and rMVA-Δ186-M2 no longer had a defect in viral DNA replication by assessing EdU incorporation and I3 labeling in HeLa cells (Fig. 8A).
FIG 8.
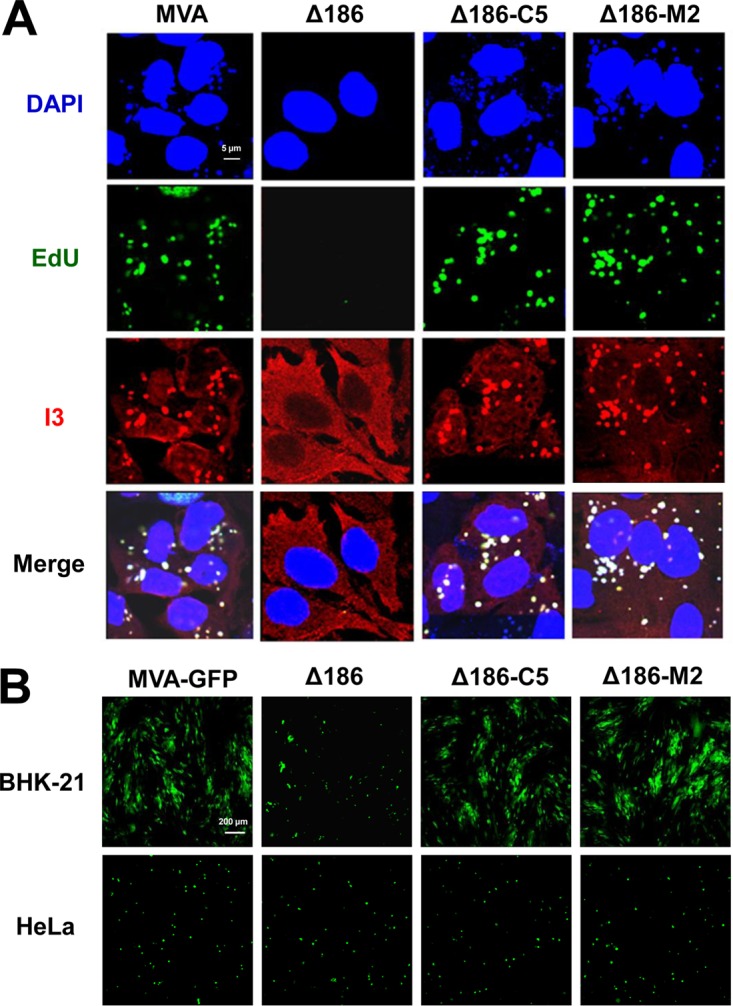
C5 and M2 restore genome replication by MVA-Δ186. (A) HeLa cells were infected with 5 PFU/cell of MVA, MVA-Δ186, rMVA-Δ186-C5, or rMVA-Δ186-M2. At 3 h after infection, the cells were incubated with EdU (10 μM) and then fixed, permeabilized, and reacted with Alexa Fluor 647 azide. The I3 single-stranded DNA binding protein was visualized by staining with a MAb followed by an anti-mouse secondary antibody conjugated to Alexa Fluor 568. DAPI was used to stain total DNA. Images were collected with a confocal microscope. The scale bar represents 5 μm. (B) BHK-21 and HeLa cells were infected with 0.05 PFU/cell of the indicated viruses expressing GFP under the control of the P11 late promoter for 24 h. Virus spread was visualized by confocal microscopy. The presence of similar numbers of cells was confirmed by differential image contrast (DIC) microscopy (not shown). The scale bar represents 200 μm.
The studies described thus far showed that expression of C5 and M2 compensates for the absence of the 68k-ank protein with regard to DNA replication and postreplicative gene expression. However, even without deletion of the 68k-ank gene, MVA cannot produce infectious virus in human cells due to an assembly block. We therefore turned to BHK-21 cells, which are permissive for MVA (35, 36). Unlike MVA, MVA-Δ186 was unable to spread in BHK-21 cells (Fig. 8B). However, the ability to spread was restored by either C5 or M2 expression, indicating that these proteins fully replaced 68k-ank. Although C5 and M2 restored postreplicative gene expression in HeLa cells (Fig. 6C), they did not restore formation of infectious virus as determined by a spread assay (Fig. 8B), indicating persistence of the host range defect.
DISCUSSION
The two major themes of this work are genome uncoating/DNA replication and gene redundancy. It seems apparent that uncoating is a prerequisite for poxvirus DNA replication, as the genome must be accessible to newly synthesized viral proteins. However, there has been increasing evidence that uncoating and DNA synthesis are deeply intertwined, with the ubiquitin-proteosome system playing a key role. Evidence for this has come from studies with proteasome inhibitors and human genome-wide RNAi screens (12–15). Mercer and coworkers (14) provided evidence for a model in which core proteins are ubiquitinated prior to or during virion assembly and suggested that these ubiquitinated proteins become substrates for the proteasome following virus entry and early gene expression. Evidence for a proteasome requirement for viral DNA replication independent of uncoating was also obtained by allowing uncoating to occur in the presence of a reversible DNA replication inhibitor and then inhibiting proteasome activity before reversal (12, 14) and also by inhibiting virus-mediated replication of transfected circular DNA, which does not require uncoating (12). In addition, inhibition of the ubiquitin-activating enzyme E1 by UBEI-41 reduced DNA replication and late gene expression (12, 13) without affecting uncoating (14). Also, siRNA knockdown experiments showed that cullin-3 ubiquitin ligase was needed for DNA replication (14). Nevertheless, how the ubiquitin-proteosome system facilitates uncoating or DNA synthesis has not been elucidated.
Although numerous viral early proteins are required for DNA synthesis (37, 38), so far only one such viral protein, D5, has been shown to be involved in uncoating (16). D5 is a DNA primase and putative helicase that is essential for viral DNA replication but without obvious links to the ubiquitin-proteosome system. Therefore, it seemed likely to us that additional viral proteins are involved in uncoating. Because the phenotype of a mutant virus unable to replicate its genome might have a primary block in uncoating, we were intrigued by a report by Sperling and coworkers (20). They found that the 68k-ank protein of MVA is required for DNA replication and late gene expression in human and murine cells. In the present study, we confirmed their findings by showing that a 68k-ank deletion mutant had defects in genome replication as assessed by diminished EdU incorporation and in postreplicative gene expression as determined by Western blotting. In addition, we found that genome uncoating, formation of DNA prereplication sites, and degradation of viral cores were severely reduced. Although an uncoating defect could be sufficient to explain the inhibition of genome replication, we also obtained evidence for a direct block in DNA synthesis using a ddPCR plasmid replication assay. The assay was based on the remarkable ability of poxviruses to mediate the replication of transfected plasmids and other circular DNAs that requires all known viral replication proteins (30, 31, 39). Since plasmid replication does not require core uncoating but was inhibited in cells infected with the 68k-ank deletion mutant, we concluded that the 68k-ank protein has a dual role in uncoating and DNA synthesis. Western blotting showed that similar amounts of D5 were made in cells infected with MVA and MVA-Δ186 (data not shown), eliminating the possibility that the 68k-ank protein was needed for expression or stability of D5.
The 68k-ank protein encoded by the MVA 186 ORF has homologs in other strains of VACV as well as other orthopoxviruses, but these have not been shown to be essential for replication in cell culture. We considered that the 68k-ank protein might have a unique role for MVA because of the many genes that were disrupted during the extensive passages of this virus in CEF. To test this hypothesis, we deleted the homologous ORF from VACV WR and found no defect in postreplicative gene expression. To follow up the idea of compensatory genes, we took advantage of a panel of MVA recombinants that were made by transfecting cosmids with DNA from the left end of a replication-competent VACV and which recovered the ability to replicate in mammalian cells to various degrees (33). Unlike the case for the parent MVA, deletion of the 68k-ank gene from three of these constructs did not prevent postreplicative gene expression, indicating that one or more of the additional genes had a compensatory function. By whole-genome sequencing of the recombinant virus rMVA 51.1, we found that DNA corresponding to the ORFs missing from three deletions in the MVA genome had been restored. We then used a forward strategy to identify two genes, C5 and M2, that independently enabled DNA replication and postreplicative gene expression by the MVA 68k-ank deletion mutant. These genes were not included in the siRNA screen of early VACV proteins that led to the identification of D5 as an uncoating factor (16).
The structural motifs of 68k-ank (B18), C5, and M2 indicate the presence of protein-interacting domains (Table 1). The 68k-ank protein has four ankyrin repeats near the N terminus and an F-box-like domain that interacts with components of the SCF (Skp1, cullin1, F-box-containing complex) (25). The combination of ankyrin repeats and F-box-like domains (called Ank/PRANC) is found in proteins encoded by nearly all poxvirus genera, and many species have multiple copies (40). Sonnberg and coworkers (41) proposed that these bipartite proteins bind through the F box to Skp1, the adapter subunit of the SCF1 complex, and that additional interactions of the ankyrin repeats provide target specificity. The role of the F-box domain of the MVA 68k-ank protein is uncertain, however, since an F-box-truncated protein still enabled MVA late gene expression (20).
TABLE 1.
VACV proteins with roles in uncoating and DNA synthesis
| Protein, Cop (WR)a | Size (kDa) | Expression | Viruses with homologs | Essential | Domains | Ubiquitin/proteasome interaction |
|---|---|---|---|---|---|---|
| D5 (110) | 90 | Early | All poxviruses | Yes | Helicase, primase | ? |
| B18 (199) | 68 | Early | Orthopoxviruses | No | Ankyrin repeats, F box | Yes |
| C5 (023) | 25 | Early | Orthopoxviruses | No | BTB, Kelch | Yes |
| M2 (031) | 25 | Early | Orthopoxviruses | No | Signal peptide, PIE | ? |
Cop, VACV Copenhagen nomenclature; WR, WR nomenclature.
The C5 protein belongs to a superfamily of poxviral proteins with BTB (POZ) and Kelch domains (42). BTB is a protein interaction domain comprised of a cluster of α-helices flanked by short β-sheets (43). Kelch repeats form β-propeller tertiary structures involved in protein interactions (44). The WR strain of VACV has 4 BTB/Kelch proteins (C2, C5, F3, and A55), none of which is individually essential for replication in cell culture (45–48). Cellular BTB domains have been shown to function as specific adaptors for cullin-3-based ubiquitin ligase to target proteins for ubiquitination, and there is evidence that certain ectromelia BTB/Kelch proteins do so as well (49). The protein-interacting Kelch domain, like the ankyrin repeats, may provide target specificity (42). Since cullin-3 ubiquitin ligase is needed for VACV DNA replication, a role for C5 as a cullin-3 adaptor is an interesting possibility (14).
The M2 protein belongs to the poxvirus immune evasion (PIE) superfamily based on the presence of a conserved β-sandwich fold (50). M2 is nonessential for replication in cell culture (45, 51) and is a glycosylated protein that is expressed early in infection, localizes to the endoplasmic reticulum (ER), and is one of several VACV proteins that can inhibit NF-κB as one of its functions (52, 53). Ligands for 68k-ank, C5, and M2 that could account for their roles in uncoating/DNA replication remain to be determined.
The redundancy of the 68k-ank, C5, and M2 genes is an interesting aspect of this work and is related to synthetic lethality, a well-known genetic phenomenon in which deficiencies in the expression of two or more genes result in cell or organismal death, whereas a deficiency in any one gene does not. Such redundancy may lead to a gross overestimation of the number of nonessential genes. For example, the majority of the 6,000 genes of Saccharomyces cerevisiae are individually dispensable but form a complex interaction network (54, 55). In most cases, nonhomologous genes acting on the same cellular process or pathway, rather than homologous genes, provide functional redundancy. The situation with poxviruses may be similar. A cowpox virus complete single-gene knockout bacterial artificial chromosome library was constructed, and infectious virus was successfully recovered for 109 of the 183 clones, indicating that only 74 ORFs are essential (56). However, Dobson and Tscharke (32) pointed out the difficulty due to redundancy in defining essential and nonessential genes of orthopoxviruses. In their study, VACV with large deletions at either end of the genome exhibited no or only mild host range defects, whereas combining deletions from both ends had a devastating effect. The present study underscores this phenomenon for uncoating and DNA synthesis and suggests that multifactor gene screens are necessary for analyzing the complexity of poxvirus replication and host interactions.
MATERIALS AND METHODS
Cells, viruses, and chemicals.
Cells and viruses were propagated essentially as described previously (57). A panel of human replication-competent recombinant MVAs (rMVAs) (44/47.1, 44.2, and 51.1) with segments of added DNA of various lengths was described previously (33). AraC and CHX were purchased from Sigma-Aldrich.
Library preparation for pyrosequencing.
Libraries for 454 pyrosequencing were made using the Rapid Library Preparation Method Manual (October 2009) GS FLX Titanium Series (Roche, Branford, CT) and the Paired End Library Preparation Method Manual, 3kb Span (October 2009) GS FLX Titanium Series. Each library was processed using the emPCR Method, Manual Lib-L MV (October 2009) in separate emulsion reactions. The paired-end sample was loaded in a single lane, and the fragment sample was loaded in two lanes of an 8-region 454 GS FLX Titanium sequencing run.
Genome assembly and gap closure.
A de novo assembly with several reiterations was initially done. Physical mapping was carried out using an in-house script coupled with Nucmer software (58), and all gaps were closed by PCR and Sanger sequencing. Several reference genomes (MVA, Lister, and Copenhagen genome sequence) were used for verification and comparison purposes. The inverted terminal repetitions were identified and junctions sequenced to generate the final genome drafts. PCR and Sanger sequencing were also done for mutations within coding regions to correct mismatches caused by sequencing errors or homopolymers. Whole-genome alignments were compared to Acam3000 (accession number AY603355). Determination of homology of annotated ORFs was carried out using MyOrfeome script (https://sourceforge.net/projects/myorfeome/) and several other reference genomes, such as VACV-Lister_107 (accession number DQ121394), VACV-CVA (accession number AM501482), Acam2000 (accession number AY313847), Acam3000 (accession number AY603355), and VACV-Cop (accession number M35027).
Construction of recombinant viruses.
Modified viruses were constructed by homologous recombination using fluorescent reporter genes as described previously (59). ORF 186 of MVA and rMVA 44/47.1, 44.2, and 51.1 was deleted by homologous recombination with a PCR product containing the P11 VACV promoter-driven GFP gene flanked by sequences on either side of ORF 186. Fluorescent plaques were identified and cloned by repeated plaque isolation. A similar strategy was adopted to delete the B18R homolog of the 68k-ank gene from VACV strain WR. Similarly, MVA-GFP was generated by inserting P11 promoter-driven GFP into deletion III of MVA. The insertion regions of the recombinant viruses were PCR amplified and sequenced to confirm the identities.
To introduce the C17-C12, C5-C1, or M1-K1 DNA segment into the genome of MVA-Δ186 at the deletion III site, the segments were PCR amplified and inserted downstream of P11 VACV promoter-driven mCherry in a modified pLW44-derived vector (60). This plasmid was transfected into CEF that were infected with MVA-Δ186, and fluorescent foci that expressed both GFP and mCherry were picked and plaque purified. The C5, C3, C2, C1, M1, M2, and K1 ORFs under the control of the mH5 promoter were inserted into MVA-Δ186 in a similar manner. Expression of each gene was confirmed by ddPCR.
EdU assay and immunostaining.
EdU assays were performed according to the Click-it Plus EdU imaging kit protocol (Thermo Fisher) as described previously (27). Briefly, HeLa cells plated on coverslips were mock infected or infected with VACV and at various times were incubated with EdU for 1 h before fixation with 4% paraformaldehyde in phosphate-buffered saline (PBS) and permeabilization with 0.1% Triton X-100. The click reaction was carried out by incubation with Alexa Fluor 647 azide, followed successively by immunostaining with anti-I3 mouse monoclonal antibody (MAb) (61), a secondary anti-mouse antibody conjugated to Alexa Fluor 594, and DAPI. Images were collected with a Leica laser-scanning confocal microscope.
Visualization of intracellular viral cores.
Immunostaining and visualization of intracellular viral cores were as previously described (12). Briefly, HeLa cells plated on glass coverslips were infected with purified virus (5 PFU per cell) at 4°C for 1 h to allow attachment. After removal of unbound virions by washing, the cells were incubated for 1 or 4 h at 37°C in medium without or with CHX (100 μg/ml) or AraC (44 μg/ml). The cells were then fixed, permeabilized, and immunostained as described above. Anti-A4 rabbit polyclonal antibody (62), anti-I3 mouse MAb (61), and appropriate secondary antibodies were used for immunostaining. Images were collected with a Leica laser-scanning confocal microscope. Viral cores that stained with anti-A4 antibody and cells that stained with DAPI were counted using Imaris 8.4 software (Bitplane Scientific Solutions).
Western blotting.
Cells were lysed with radioimmunoprecipitation assay (RIPA) buffer and clarified by low-speed centrifugation. Proteins were resolved by electrophoresis on NuPAGE gels (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membranes using the iBlot system (Invitrogen). Membranes were incubated with primary antibodies in 3% nonfat milk in PBS overnight at 4°C and subsequently with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) and were developed with SuperSignal West Pico, Dura, or Femto chemiluminescent substrate (Pierce, Rockford, IL).
Plasmid replication assay.
HeLa cells were transfected with pUC19 plasmid and 24 h later were infected with VACV. Total DNA was extracted using a QIAamp blood minikit (Qiagen, Valencia, CA) and digested with BamHI and DpnI. Replicated plasmid DNA was quantified by ddPCR using primers 5′-CTTCATTTTTAATTTAAAAGG-3′ and 5′-GAAAAAGAGTTGGTAGCTCTTG-3′.
Statistical analysis.
Data analysis was performed using IBM SPSS 11.5 software. Statistical differences were determined using Student's t test. P values of <0.05 were considered significant. Error bars represent standard deviations.
Accession number(s).
The genome sequence of rMVA 51.1 was deposited under GenBank accession number MG663594.
ACKNOWLEDGMENTS
We thank Catherine Cotter for help with cells and Dan Bruno, Craig Martens, Steve Porcella, and Wei Xiao for genome sequencing. David Evans, Mariano Esteban, and Paula Traktman kindly provided antibodies.
This research was supported by the Division of Intramural Research, NIAID, NIH.
B.L. planned, carried out, and evaluated experiments, made most of the figures, and contributed to writing the paper. D.P. initiated the project and carried out some experiments. J.D.M.-R. sequenced and annotated the recombinant MVA genome and made one figure. S.G. participated in confocal microscopy experiments and analysis of data. L.S.W. made suggestions regarding the use of the MVA recombinants and experimental procedures. B.M. conceived the project, suggested and interpreted experiments, and wrote the paper.
We declare no conflict of interest.
REFERENCES
- 1.Greber UF, Singh I, Helenius A. 1994. Mechanisms of virus uncoating. Trends Microbiol 2:52–56. doi: 10.1016/0966-842X(94)90126-0. [DOI] [PubMed] [Google Scholar]
- 2.Kilcher S, Mercer J. 2015. DNA virus uncoating. Virology 479:578–590. doi: 10.1016/j.virol.2015.01.024. [DOI] [PubMed] [Google Scholar]
- 3.Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res 66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- 4.Joklik WK. 1964. The intracellular uncoating of poxvirus DNA. I. The fate of radioactively-labeled rabbitpox virus. J Mol Biol 8:263–276. [DOI] [PubMed] [Google Scholar]
- 5.Moss B. 2012. Poxvirus cell entry: how many proteins does it take? Viruses 4:688–707. doi: 10.3390/v4050688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt FI, Bleck CK, Mercer J. 2012. Poxvirus host cell entry. Curr Opin Virol 2:20–27. doi: 10.1016/j.coviro.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Joklik WK. 1964. The intracellular uncoating of poxvirus DNA. II. The molecular basis of the uncoating process. J Mol Biol 8:277–288. [DOI] [PubMed] [Google Scholar]
- 8.Kates JR, McAuslan B. 1967. Messenger RNA synthesis by a “coated” viral genome. Proc Natl Acad Sci U S A 57:314–320. doi: 10.1073/pnas.57.2.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kates JR, McAuslan BR. 1967. Poxvirus DNA-dependent RNA polymerase. Proc Natl Acad Sci U S A 58:134–141. doi: 10.1073/pnas.58.1.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munyon WE, Paoletti E, Grace JT Jr. 1967. RNA polymerase activity in purified infectious vaccinia virus. Proc Natl Acad Sci U S A 58:2280–2288. doi: 10.1073/pnas.58.6.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodson B. 1967. Vaccinia mRNA synthesis under conditions which prevent uncoating. Biochem Biophys Res Commun 27:169–175. doi: 10.1016/S0006-291X(67)80057-3. [DOI] [PubMed] [Google Scholar]
- 12.Satheshkumar PS, Anton LC, Sanz P, Moss B. 2009. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J Virol 83:2469–2479. doi: 10.1128/JVI.01986-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teale A, Campbell S, Van Buuren N, Magee WC, Watmough K, Couturier B, Shipclark R, Barry M. 2009. Orthopoxviruses require a functional ubiquitin-proteasome system for productive replication. J Virol 83:2099–2108. doi: 10.1128/JVI.01753-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mercer J, Snijder B, Sacher R, Burkard C, Bleck CK, Stahlberg H, Pelkmans L, Helenius A. 2012. RNAi screening reveals proteasome- and cullin3-dependent stages in vaccinia virus infection. Cell Rep 2:1036–1047. doi: 10.1016/j.celrep.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Sivan G, Martin SE, Myers TG, Buehler E, Szymczyk KH, Ormanoglu P, Moss B. 2013. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc Natl Acad Sci U S A 110:3519–3524. doi: 10.1073/pnas.1300708110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kilcher S, Schmidt FI, Schneider C, Kopf M, Helenius A, Mercer J. 2014. siRNA screen of early poxvirus genes identifies the AAA+ ATPase D5 as the virus genome-uncoating factor. Cell Host Microbe 15:103–112. doi: 10.1016/j.chom.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Evans E, Klemperer N, Ghosh R, Traktman P. 1995. The vaccinia virus D5 protein, which is required for DNA replication, is a nucleic acid-independent nucleotide triphosphatase. J Virol 69:5353–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyle KA, Arps L, Traktman P. 2007. Biochemical and genetic analysis of the vaccinia virus D5 protein: multimerization-dependent ATPase activity is required to support viral DNA replication. J Virol 81:844–859. doi: 10.1128/JVI.02217-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Silva FS, Lewis W, Berglund P, Koonin EV, Moss B. 2007. Poxvirus DNA primase. Proc Natl Acad Sci U S A 104:18724–18729. doi: 10.1073/pnas.0709276104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sperling KM, Schwantes A, Staib C, Schnierle BS, Sutter G. 2009. The orthopoxvirus 68-kilodalton ankyrin-like protein is essential for DNA replication and complete gene expression of modified vaccinia virus Ankara in nonpermissive human and murine cells. J Virol 83:6029–6038. doi: 10.1128/JVI.01628-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayr A, Hochstein-Mintzel V, Stickl H. 1975. Passage history, properties, and applicability of the attenuated vaccinia virus strain MVA. Infection 3:6–14. doi: 10.1007/BF01641272. [DOI] [Google Scholar]
- 22.Antoine G, Scheiflinger F, Dorner F, Falkner FG. 1998. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 244:365–396. doi: 10.1006/viro.1998.9123. [DOI] [PubMed] [Google Scholar]
- 23.Meyer H, Sutter G, Mayr A. 1991. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol 72:1031–1038. doi: 10.1099/0022-1317-72-5-1031. [DOI] [PubMed] [Google Scholar]
- 24.Sutter G, Moss B. 1992. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc Natl Acad Sci U S A 89:10847–10851. doi: 10.1073/pnas.89.22.10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sperling KM, Schwantes A, Schnierle BS, Sutter G. 2008. The highly conserved orthopoxvirus 68k ankyrin-like protein is part of a cellular SCF ubiquitin ligase complex. Virology 374:234–239. doi: 10.1016/j.virol.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 26.Salic A, Mitchison TJ. 2008. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Senkevich TG, Katsafanas G, Weisberg A, Olano LR, Moss B. 2017. Identification of vaccinia virus replisome and transcriptome proteins by iPOND coupled with mass spectrometry. J Virol 91:e01015-17. doi: 10.1128/JVI.01015-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domi A, Beaud G. 2000. The punctate sites of accumulation of vaccinia virus early proteins are precursors of sites of viral DNA synthesis. J Gen Virol 81:1231–1235. doi: 10.1099/0022-1317-81-5-1231. [DOI] [PubMed] [Google Scholar]
- 29.Welsch S, Doglio L, Schleich S, Locker JK. 2003. The vaccinia virus I3L gene product is localized to a complex endoplasmic reticulum-associated structure that contains the viral parental DNA. J Virol 77:6014–6028. doi: 10.1128/JVI.77.10.6014-6028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merchlinsky M, Moss B. 1988. Sequence-independent replication and sequence-specific resolution of plasmids containing the vaccinia virus concatemer junction: requirements for early and late trans-acting factors, p 87–93. In Kelly T, Stillman B (ed), Cancer cells 6/eukaryotic DNA replication. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 31.De Silva FS, Moss B. 2005. Origin-independent plasmid replication occurs in vaccinia virus cytoplasmic factories and requires all five known poxvirus replication factors. Virol J 2:23. doi: 10.1186/1743-422X-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobson BM, Tscharke DC. 2015. Redundancy complicates the definition of essential genes for vaccinia virus. J Gen Virol 96:3326–3337. doi: 10.1099/jgv.0.000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wyatt LS, Carroll MW, Czerny C-P, Merchlinsky M, Sisler JR, Moss B. 1998. Marker rescue of the host range restricted defects of modfied vaccinia virus Ankara. Virology 251:334–342. doi: 10.1006/viro.1998.9397. [DOI] [PubMed] [Google Scholar]
- 34.Wyatt LS, Shors ST, Murphy BR, Moss B. 1996. Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine 14:1451–1458. doi: 10.1016/S0264-410X(96)00072-2. [DOI] [PubMed] [Google Scholar]
- 35.Carroll M, Moss B. 1997. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 238:198–211. doi: 10.1006/viro.1997.8845. [DOI] [PubMed] [Google Scholar]
- 36.Drexler I, Heller K, Wahren B, Erfle V, Sutter G. 1998. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J Gen Virol 79:347–352. doi: 10.1099/0022-1317-79-2-347. [DOI] [PubMed] [Google Scholar]
- 37.Moss B. 2013. Poxvirus DNA replication. Cold Spring Harb Perspect Biol 5:a010199. doi: 10.1101/cshperspect.a010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Czarnecki MW, Traktman P. 2017. The vaccinia virus DNA polymerase and its processivity factor. Virus Res 234:193–206. doi: 10.1016/j.virusres.2017.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeLange AM, McFadden G. 1986. Sequence-nonspecific replication of transfected plasmid DNA in poxvirus-infected cells. Proc Natl Acad Sci U S A 83:614–618. doi: 10.1073/pnas.83.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mercer AA, Fleming SB, Ueda N. 2005. F-box-like domains are present in most poxvirus ankyrin repeat proteins. Virus Genes 31:127–133. doi: 10.1007/s11262-005-1784-z. [DOI] [PubMed] [Google Scholar]
- 41.Sonnberg S, Seet BT, Pawson T, Fleming SB, Mercer AA. 2008. Poxvirus ankyrin repeat proteins are a unique class of F-box proteins that associate with cellular SCF1 ubiquitin ligase complexes. Proc Natl Acad Sci U S A 105:10955–10960. doi: 10.1073/pnas.0802042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barry M, van Buuren N, Burles K, Mottet K, Wang QA, Teale A. 2010. Poxvirus exploitation of the ubiquitin-proteasome system. Viruses (Basel) 2:2356–2380. doi: 10.3390/v2102356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bardwell VJ, Treisman R. 1994. The POZ domain: a conserved protein-protein interaction motif. Genes Dev 8:1664–1677. doi: 10.1101/gad.8.14.1664. [DOI] [PubMed] [Google Scholar]
- 44.Adams J, Kelso R, Cooley L. 2000. The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol 10:17–24. doi: 10.1016/S0962-8924(99)01673-6. [DOI] [PubMed] [Google Scholar]
- 45.Kotwal GJ, Moss B. 1988. Analysis of a large cluster of nonessential genes deleted from a vaccinia virus terminal transposition mutant. Virology 167:524–537. [PubMed] [Google Scholar]
- 46.Pires De Miranda M, Reading PC, Tscharke DC, Murphy BJ, Smith GL. 2003. The vaccinia virus kelch-like protein C2L affects calcium-independent adhesion to the extracellular matrix and inflammation in a murine intradermal model. J Gen Virol 84:2459–2471. doi: 10.1099/vir.0.19292-0. [DOI] [PubMed] [Google Scholar]
- 47.Beard PM, Froggatt GC, Smith GL. 2006. Vaccinia virus keltch protein A55 is a 64 kDa intracellular factor that affects virus-induced cytopathic effect and the outcome of infection in a murine intradermal model. J Gen Virol 87:1521–1529. doi: 10.1099/vir.0.81854-0. [DOI] [PubMed] [Google Scholar]
- 48.Froggatt GC, Smith GL, Beard PM. 2007. Vaccinia virus gene F3L encodes an intracellular protein that affects the innate immune response. J Gen Virol 88:1917–1921. doi: 10.1099/vir.0.82815-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilton BA, Campbell S, Van Buuren N, Garneau R, Furukawa M, Xiong Y, Barry M. 2008. Ectromelia virus BTB/kelch proteins, EVM150 and EVM167, interact with cullin-3-based ubiquitin ligases. Virology 374:82–99. doi: 10.1016/j.virol.2007.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelson CA, Epperson ML, Singh S, Elliott JI, Fremont DH. 2015. Structural conservation and functional diversity of the poxvirus immune evasion (PIE) domain superfamily. Viruses (Basel) 7:4873–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith KA, Stallard V, Roos JM, Hart C, Sormier N, Cohen LK, Roberts BE, Payne LG. 1993. Host range selection of vaccinia recombinants containing insertions of foreign genes into non-coding sequences. Vaccine 11:43–53. doi: 10.1016/0264-410X(93)90338-X. [DOI] [PubMed] [Google Scholar]
- 52.Gedey R, Jin XL, Hinthong O, Shisler JL. 2006. Poxviral regulation of the host NF-kappa B response: the vaccinia virus M2L protein inhibits induction of NF-kappa B activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J Virol 80:8676–8685. doi: 10.1128/JVI.00935-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hinthong O, Jin XL, Shisler JL. 2008. Characterization of wild-type and mutant vaccinia virus M2L proteins' abilities to localize to the endoplasmic reticulum and to inhibit NF-kappa B activation during infection. Virology 373:248–262. doi: 10.1016/j.virol.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C. 2009. Systematic mapping of genetic interaction networks. Annu Rev Genet 43:601–625. doi: 10.1146/annurev.genet.39.073003.114751. [DOI] [PubMed] [Google Scholar]
- 55.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, et al. . 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 56.Xu ZY, Zikos D, Osterrieder N, Tischer BK. 2014. Generation of a complete single-gene knockout bacterial artificial chromosome library of cowpox virus and identification of its essential genes. J Virol 88:490–502. doi: 10.1128/JVI.02385-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cotter CA, Earl PL, Wyatt LS, Moss B. 2017. Preparation of cell cultures and vaccinia virus stocks. Curr Protoc Mol Biol 117:16.16.1–16.16.18. doi: 10.1002/cpmb.33. [DOI] [PubMed] [Google Scholar]
- 58.Delcher AL, Salzberg SL, Phillippy AM. 2003. Using MUMmer to identify similar regions in large sequence sets. Curr Protoc Bioinformatics Chapter 10:Unit 10.3. doi: 10.1002/0471250953.bi1003s00. [DOI] [PubMed] [Google Scholar]
- 59.Wyatt LS, Earl PL, Moss B. 2017. Generation of recombinant vaccinia viruses. Curr Protoc Mol Biol 117:16.17.1–16.17.18. doi: 10.1002/cpmb.32. [DOI] [PubMed] [Google Scholar]
- 60.Bisht H, Roberts A, Vogel L, Bukreyev A, Collins PL, Murphy BR, Subbarao K, Moss B. 2004. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc Natl Acad Sci U S A 101:6641–6646. doi: 10.1073/pnas.0401939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin YC, Li J, Irwin CR, Jenkins H, DeLange L, Evans DH. 2008. Vaccinia virus DNA ligase recruits cellular topoisomerase II to sites of viral replication and assembly. J Virol 82:5922–5932. doi: 10.1128/JVI.02723-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Demkowicz WE, Maa JS, Esteban M. 1992. Identification and characterization of vaccinia virus genes encoding proteins that are highly antigenic in animals and are immunodominant in vaccinated humans. J Virol 66:386–398. [DOI] [PMC free article] [PubMed] [Google Scholar]