

ABSTRACT
Reovirus nonstructural protein σ1s is required for the establishment of viremia and hematogenous viral dissemination. However, the function of σ1s during the reovirus replication cycle is not known. In this study, we found that σ1s was required for efficient reovirus replication in simian virus 40 (SV40)-immortalized endothelial cells (SVECs), mouse embryonic fibroblasts, human umbilical vein endothelial cells (HUVECs), and T84 human colonic epithelial cells. In each of these cell lines, wild-type reovirus produced substantially higher viral titers than a σ1s-deficient mutant. The σ1s protein was not required for early events in reovirus infection, as evidenced by the fact that no difference in infectivity between the wild-type and σ1s-null viruses was observed. However, the wild-type virus produced markedly higher viral protein levels than the σ1s-deficient strain. The disparity in viral replication did not result from differences in viral transcription or protein stability. We further found that the σ1s protein was dispensable for cell killing and the induction of type I interferon responses. In the absence of σ1s, viral factory (VF) maturation was impaired but sufficient to support low levels of reovirus replication. Together, our results indicate that σ1s is not absolutely essential for viral protein production but rather potentiates reovirus protein expression to facilitate reovirus replication. Our findings suggest that σ1s enables hematogenous reovirus dissemination by promoting efficient viral protein synthesis, and thereby reovirus replication, in cells that are required for reovirus spread to the blood.
IMPORTANCE Hematogenous dissemination is a critical step in the pathogenesis of many viruses. For reovirus, nonstructural protein σ1s is required for viral spread via the blood. However, the mechanism by which σ1s promotes reovirus dissemination is unknown. In this study, we identified σ1s as a viral mediator of reovirus protein expression. We found several cultured cell lines in which σ1s is required for efficient reovirus replication. In these cells, wild-type virus produced substantially higher levels of viral protein than a σ1s-deficient mutant. The σ1s protein was not required for viral mRNA transcription or viral protein stability. Since reduced levels of viral protein were synthesized in the absence of σ1s, the maturation of viral factories was impaired, and significantly fewer viral progeny were produced. Taken together, our findings indicate that σ1s is required for optimal reovirus protein production, and thereby viral replication, in cells required for hematogenous reovirus dissemination.
KEYWORDS: dissemination, protein synthesis, reovirus, replication
INTRODUCTION
Synthesis of new viral proteins is required for the majority of steps in viral replication, including the formation of viral replicase complexes, modulation of host responses to infection, and assembly of progeny virions. Because viruses do not encode their own ribosomes, they must appropriate cellular translational machinery for the purpose of making viral proteins. Since all viruses usurp host translational processes, cells deploy an assortment of mechanisms to prevent viruses from commandeering protein synthesis machinery, including global inhibition of cellular protein synthesis (1–4). Consequently, viruses must have mechanisms to redirect cellular translational activity and overcome host responses so as to ensure sufficient viral protein synthesis to enable viral replication. The strategies used by some viruses to facilitate viral protein synthesis are well documented (5, 6). For other viruses, including mammalian orthoreoviruses (referred to below as reoviruses), many questions remain surrounding the details of viral protein synthesis.
Reoviruses are nonenveloped icosahedral viruses with a segmented, double-stranded RNA (dsRNA) genome (7). Reovirus virions consist of two concentric protein shells, the outer capsid and the core. The core houses the 10 segments of viral genomic dsRNA. The reovirus entry process culminates with deposition of the transcriptionally active viral core into the cytoplasm (8). Negative-sense RNAs within the entering core particle serve as templates for the synthesis of new viral mRNAs that are extruded from channels at the icosahedral vertices formed by the λ2 protein (9–11). Reoviruses produce noncanonical mRNAs that contain a 5′ cap, as well as short 5′ and 3′ untranslated regions (UTRs), and lack a 3′-polyadenylated tail (12, 13). Although atypical, reovirus mRNAs are efficiently translated by host ribosomes to produce viral proteins. As infection proceeds, reovirus structural proteins, nonstructural proteins, and mRNAs are organized into structures termed viral factories (VFs). VF formation is coordinated by nonstructural proteins μNS and σNS (14–18). Within the VF, viral structural proteins and mRNAs coalesce to form new viral cores. Negative-sense viral RNAs are synthesized within the nascent viral cores using encapsidated positive-sense RNAs as templates (19). New cores serve as secondary transcriptase particles that amplify viral transcription, and thereby reovirus protein production, in the late stages of replication. In many cells, late transcripts produced by secondary transcriptase particles lack 5′-cap structures and are also efficiently translated (20–22). As replication proceeds, viral transcription, translation, genome replication, and virion assembly all continue within the VF (7). Eventually, outer capsid proteins are loaded onto nascent cores to stop the transcriptional activity of the particle (23), and assembled progeny virions are released from the cell by an unknown mechanism.
Host ribosomes and translation factors are recruited to the VF, and active translation of reovirus mRNAs takes place at the margins of VF structures (24). In the current model of reovirus translation, mRNAs produced from reovirus cores are released within the VF, where they associate with cellular translation factors, including the eukaryotic initiation factor 4F (eIF4F) cap-binding complex and the 43S preinitiation complex. Ribosomal subunits are subsequently recruited, along with other host translation initiation and elongation factors, to facilitate protein synthesis (24). This proposed mechanism compartmentalizes reovirus translation to specific sites within the cell. Sequestering active viral translation may aid in the recruitment of viral mRNAs and host translation factors or may enable viral evasion of host responses that impair protein synthesis. However, many questions remain with respect to the mechanisms that underlie reovirus protein synthesis.
Reovirus nonstructural protein σ1s is a 14-kDa protein encoded by the S1 gene segment (25–27). The σ1s open reading frame (ORF) is nested within the coding sequence of reovirus attachment protein σ1 but lies in a different reading frame (25–29). In infected cells, σ1s is found throughout the cytoplasm and nucleus. However, the subcellular localization required for σ1s function is unknown (30–32). In infected mice, σ1s is required for hematogenous reovirus dissemination (33–35). Following oral infection, wild-type reovirus replicates in the intestine and traffics to the bloodstream. The establishment of viremia enables the virus to spread to every organ system in the body, including the central nervous system (CNS). In the absence of σ1s, reovirus replicates efficiently in the intestine (34). However, σ1s-deficient reovirus fails to reach the bloodstream, preventing dissemination throughout the host. For neurotropic strains of reovirus, hematogenous spread is required for full neurovirulence (33). Although σ1s is critical for reovirus pathogenesis, the mechanism by which σ1s promotes spread via the blood is not known.
Previous studies indicate that σ1s is dispensable for reovirus replication in cultured cells, such as L929 and HeLa cells (32–34). In this study, we discovered that σ1s is required for efficient reovirus replication in several cell lines, including mouse simian virus 40 (SV40)-immortalized endothelial cells (SVECs), mouse embryonic fibroblasts (MEFs), human umbilical vein endothelial cells (HUVECs), and T84 human intestinal epithelial cells. In these cell lines, a lack of σ1s led to a marked reduction in viral protein production. Viral transcription and viral protein stability were comparable between wild-type and σ1s-deficient viruses, suggesting that σ1s facilitates reovirus protein synthesis. The reduced viral protein production in the absence of σ1s ultimately leads to impaired viral replication. Together, these findings identify a new role for nonstructural protein σ1s in promoting reovirus protein expression.
RESULTS
The σ1s protein is required for efficient reovirus replication in multiple cell lines.
The σ1s protein is dispensable for reovirus replication in L929 fibroblasts and HeLa cells (32–34). To determine whether σ1s is required for reovirus replication in other cell lines, we quantified viral progeny yields from SV40-immortalized endothelial cells (SVECs), embryonic fibroblasts from C57BL/6 mice (MEFs), and T84 human epithelial cells infected with a wild-type reovirus (rsT1L) or its σ1s-deficient mutant (rsT1L σ1s-null) (Fig. 1A). We found that rsT1L produced significantly higher titers than rsT1L σ1s-null in SVECs at each time point tested (Fig. 1A). By 96 h, rsT1L produced approximately 1,000-fold-higher yields than rsT1L σ1s-null. To determine whether the multiplicity of infection (MOI) influences the requirement for σ1s during reovirus replication in SVECs, we initiated infection with increasing doses of rsT1L or rsT1L σ1s-null (Fig. 1B). As shown in Fig. 1A, rsT1L produced approximately 10-fold more progeny at 24 h than rsT1L σ1s-null at an MOI of 1 PFU/cell. At an MOI of 10 or 100 PFU/cell, the yield differential between rsT1L and rsT1L σ1s-null decreased to 6.5- or 4.4-fold, respectively (Fig. 1B). Similar results were obtained when SVECs were infected with infectious subvirion particles (ISVPs), a reovirus entry intermediate that is more infectious than virions (Fig. 1C) (7). At an MOI of 1 PFU/cell, rsT1L ISVPs exhibited a ∼10-fold-higher progeny yield than rsT1L σ1s-null ISVPs. When the MOI was decreased to 0.1 PFU/cell, rsT1L produced 76-fold more virus than rsT1L σ1s-null. These data indicate that the requirement for σ1s in SVECs cannot be completely overcome by increasing the MOI. However, these results suggest that the requirement for σ1s is greater when infection is initiated at low multiplicities. We also found that rsT1L replicated more efficiently than rsT1L σ1s-null in MEFs (Fig. 1D) and T84 cells (Fig. 1E). At each time point assessed, rsT1L produced at least 10-fold more viral progeny than rsT1L σ1s-null in both cell types. Because endothelial cells are a target for reovirus infection in vivo (36, 37), we surveyed the requirement for σ1s for reovirus replication in additional endothelial cell lines. We found that σ1s was required for efficient reovirus replication in human telomerase reverse transcriptase (hTERT)-immortalized HUVECs (Fig. 1F) but not in 2H11 (mouse lymphatic) or TX-111 (human brain) endothelial cells (data not shown). These data indicate that σ1s is not required for reovirus replication specifically in endothelial cells. Rather, σ1s promotes reovirus replication in a cell line-specific manner. Together, these findings indicate that although not strictly required for reovirus replication in many cell lines, σ1s is required for optimal viral replication in SVECs, MEFs, HUVECs, and T84 cells.
FIG 1.

Nonstructural protein σ1s is required for efficient reovirus replication in multiple cell lines. (A and B) SVECs were infected with rsT1L or rsT1L σ1s-null at an MOI of 1 PFU/cell (A) or 10 or 100 PFU/cell (B). (C) SVECs were infected with rsT1L or rsT1L σ1s-null ISVPs at an MOI of 1 or 0.1 PFU/cell. (D through F) MEFs (D), T84 cells (E), or hTERT-immortalized HUVECs (F) were infected with rsT1L or rsT1L σ1s-null at an MOI of 1 PFU/cell. For all experiments, viral titers were determined at the indicated time points by plaque assays. Results are presented as mean viral yields from three independent experiments. Error bars represent standard deviations. *, P < 0.05 (as determined by Student's t test).
Because the magnitude of the replication difference between rsT1L and rsT1L σ1s-null was greater in SVECs than in MEFs, HUVECs, or T84 cells, we used SVECs to determine how σ1s functions to promote reovirus replication. To confirm that impaired replication of rsT1L σ1s-null results from the absence of the σ1s protein, we assessed viral replication in SVECs that stably express T1L σ1s (Fig. 2). As in untransduced cells (Fig. 1A), rsT1L produced ∼10-fold-higher yields than rsT1L σ1s-null at 24 h in SVECs that stably express green fluorescent protein (GFP). However, rsT1L and rsT1L σ1s-null produced equivalent yields in cells expressing T1L σ1s. This finding indicates that the replication defect for rsT1L σ1s-null in SVECs is due to a lack of σ1s expression.
FIG 2.
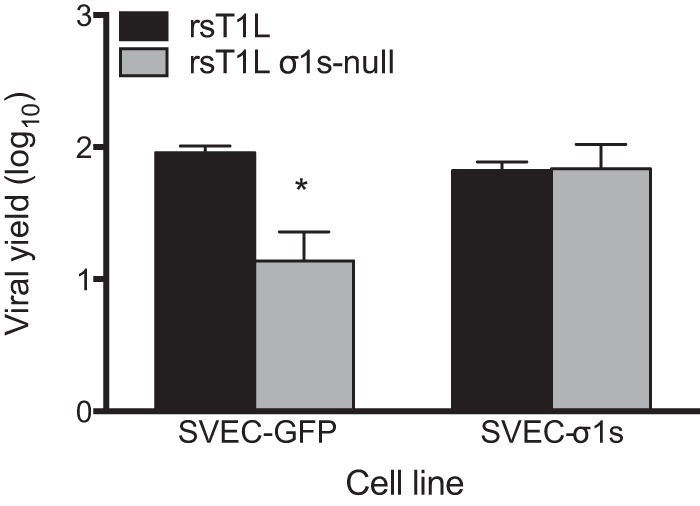
Ectopic σ1s protein expression rescues the replication of σ1s-deficient reovirus in SVECs. SVECs transduced with a retrovirus expressing GFP or σ1s were infected with rsT1L or rsT1L σ1s-null at an MOI of 1 PFU/cell. Viral titers were determined by plaque assays at 0 and 24 h. Results are presented as mean viral yields from two independent experiments. Error bars represent standard deviations. *, P < 0.05 (as determined by Student's t test).
The σ1s protein does not inhibit cell death, apoptosis, or type I interferon (IFN-I) responses.
To determine whether differences in replication between rsT1L and rsT1L σ1s-null in SVECs resulted from differential cell killing or apoptosis induction, we measured cell viability and the activities of caspases 3 and 7 (caspase-3/7) in SVECs following rsT1L or rsT1L σ1s-null infection across a range of MOIs. Cell viabilities were comparable for rsT1L and rsT1L σ1s-null infections at each MOI tested. This result indicates that σ1s does not block reovirus-mediated killing of SVECs (Fig. 3A). Consistent with previous findings that T1 reoviruses do not induce apoptosis (7), rsT1L and rsT1L σ1s-null did not induce appreciable caspase-3/7 activity at any MOI tested (Fig. 3B). Moreover, no difference in caspase-3/7 activity was observed between rsT1L and rsT1L σ1s-null. Together, these data indicate that replication differences between rsT1L and rsT1L σ1s-null do not result from a blockade of cell killing by the σ1s protein.
FIG 3.

The σ1s protein does not modulate cell death or type I interferon responses in SVECs. (A and B) SVECs were either mock infected or infected with rsT1L or rsT1L σ1s-null at an MOI of 1, 10, 100, or 1,000 PFU/cell. (A) At 24 h, cell viability was determined by trypan blue staining. Results are presented as mean viabilities from three independent experiments. Error bars indicate standard deviations. (B) At 24 h, caspase-3/7 activity was measured by a caspase-3/7 Glo assay. TNF-α and cycloheximide (CHX) were included as a positive control for caspase-3/7 activation. Results are presented as mean luminescence values from three independent experiments. (C through E) SVECs were infected with rsT1L or rsT1L σ1s-null at an MOI of 100 PFU/cell. (C) IFN-β levels in the supernatants were measured by ELISA at 24 h. Results are presented as mean IFN-β concentrations from three independent experiments. (D) At 6, 12, and 18 h, whole-cell lysates were prepared, and proteins were separated by SDS-PAGE. Immunoblot analysis was performed for phosphorylated STAT1 (p-STAT1), total STAT1, and β-actin (left panels) or p-STAT2, total STAT2, and β-actin (right panels). (E) At 24 h, IFIT1 mRNA levels were quantified by RT-qPCR. Results are presented as IFIT1 expression relative to that at 0 h postinfection.
To determine whether σ1s functions as an IFN-I antagonist, we compared IFN-I responses in SVECs infected with rsT1L or rsT1L σ1s-null. We found no difference in beta interferon (IFN-β) production (Fig. 3C), STAT1 or STAT2 phosphorylation (Fig. 3D), or interferon-stimulated gene (ISG) induction (Fig. 3E) following infection with rsT1L or rsT1L σ1s-null. These findings indicate that the difference in replication between rsT1L and rsT1L σ1s-null in SVECs does not result from σ1s functioning to antagonize IFN-I responses.
The σ1s protein is not required for reovirus infectivity.
To determine whether σ1s is required for reovirus infectivity, we performed a fluorescent focus assay and quantified the percentage of SVECs infected with rsT1L or rsT1L σ1s-null (Fig. 4). Very few infected cells were detected (∼1% or less) following infection with rsT1L or rsT1L σ1s-null at each MOI tested (Fig. 4A and B). Although the percentages of infected cells were low, comparable proportions of cells were infected by rsT1L and rsT1L σ1s-null at each MOI (Fig. 4B). Similar results were obtained when infection was initiated with ISVPs (Fig. 4C). We noted that rsT1L-infected cells stained brighter than cells infected with rsT1L σ1s-null. This observation suggests that rsT1L produced more viral antigen in infected cells than rsT1L σ1s-null. Although cells infected with rsT1L σ1s-null could still be identified, the difference in staining intensity could complicate the quantification of rsT1L σ1s-null-infected cells. To definitively quantify the cells infected with rsT1L and rsT1L σ1s-null, we performed an infectious center assay (Fig. 4D). SVECs were adsorbed with rsT1L or rsT1L σ1s-null, washed thoroughly, collected by trypsinization, diluted, and seeded onto monolayers of L929 cells. The L929 monolayers were then overlaid with agar for plaque assays. Although σ1s enhances reovirus replication in SVECs, viral progeny are still produced in the absence of the σ1s protein. In contrast, σ1s is not required for reovirus replication in L929 cells (33, 34). Thus, although the level of progeny produced by rsT1L σ1s-null from SVECs is lower than that produced by rsT1L, any virus that replicates in SVECs will form plaques in the L929 cell monolayer. In agreement with the results of our fluorescent focus assay, we found that rsT1L and rsT1L σ1s-null produced comparable numbers of infectious centers from SVECs (Fig. 4D). Together, these data indicate that σ1s does not facilitate reovirus replication by modulating reovirus entry. Rather, our results suggest that the σ1s protein is required for a postentry step in the reovirus replication cycle. Our findings further indicate that reovirus does not infect SVECs as efficiently as other cell lines, such as L929 or HeLa cells. Attempts to plaque reovirus on SVECs were unsuccessful (data not shown). Consequently, we used plaque titers determined on L929 cells to standardize infection for the remainder of the study.
FIG 4.

The σ1s protein is dispensable for reovirus infectivity. (A) SVECs were infected with rsT1L or rsT1L σ1s-null at an MOI of 10 or 100 PFU/cell. At 6 h, cells were treated with 10 mM ammonium chloride to prevent secondary rounds of infection. At 24 h, viral protein was visualized by immunofluorescence staining for reovirus antigen. Nuclei were stained with DAPI. (B) The percentages of infected cells in panel A were quantified, and the results are presented as the mean percentage of infected cells from three independent experiments for each condition. Error bars indicate standard deviations. (C) SVECs were infected with rsT1L or rsT1L σ1s-null ISVPs at the indicated MOIs and were processed as described for panel A. The percentages of infected cells were determined, and the results are presented as the mean percentage of infected cells from three independent experiments for each condition. (D) SVECs were infected with rsT1L or rsT1L σ1s-null at the indicated MOIs. After 1 h of adsorption, the SVECs were washed, trypsinized, and counted, and 1,000 cells per well were first plated on monolayers of L929 fibroblasts and then processed for plaque assays. Results are presented as the mean number of infectious centers per 1,000 cells from three independent experiments for each condition.
The σ1s protein is dispensable for reovirus transcription in SVECs.
To determine whether σ1s is required for reovirus mRNA synthesis, we quantified S4 positive- and negative-sense RNA levels produced by rsT1L or rsT1L σ1s-null in SVECs over an 18-h time course (Fig. 5). We found that rsT1L and rsT1L σ1s-null produced positive-sense (Fig. 5A) and negative-sense (Fig. 5B) RNAs with similar kinetics and to comparable magnitudes. We quantified mRNA levels for the remaining nine reovirus gene segments at 12 and 18 h by subtracting negative-sense RNA levels (the indicator of genomic dsRNA) from positive-sense RNA levels (Fig. 5C). Comparable mRNA levels were detected for the majority of the gene segments, with the exception of L1 and M1. We detected approximately 8-fold more L1 and M1 mRNA for rsT1L than for rsT1L σ1s-null at 18 h postinfection. However, only the difference for the L1 segment reached statistical significance. It is possible that transcription of the L1 gene segment, as the largest viral RNA, is more susceptible to differences in viral protein production between rsT1L and rsT1L σ1s-null (see below). Although less L1 and M1 mRNA was produced by rsT1L σ1s-null than by rsT1L in SVECs, substantial amounts of L1 and M1 mRNA were generated by rsT1L σ1s-null; L1 and M1 mRNA levels increased 24-fold and 97-fold over the levels at the 0-h time point, respectively. Together, these data indicate that σ1s is not required for reovirus mRNA synthesis.
FIG 5.

The σ1s protein is dispensable for reovirus mRNA synthesis. SVECs were infected with rsT1L or rsT1L σ1s-null at an MOI of 10 PFU/cell. Total RNA was purified at the indicated times postinfection. (A and B) The relative quantity (RQ) of reovirus S4 positive-sense (A) or negative-sense (B) RNA compared to the quantity at the 0-h time point was determined by RT-qPCR using a TaqMan assay. (C) mRNA levels for the L1, L2, L3, M1, M2, M3, S1, S2, and S3 genes. Positive- and negative-sense RNA levels for these gene segments were determined by RT-qPCR using SYBR green. The RQ of positive- or negative-sense RNA was determined with reference to the quantity at the 0-h time point. The RQ of reovirus mRNA was determined by subtracting the RQ of negative-sense viral RNA (representing genomic RNA production) from the RQ of positive-sense RNA. Data are presented as the mean log2 RQ from three independent experiments. Error bars indicate standard deviations. *, P < 0.05 (as determined by Student's t test).
The σ1s protein is required for optimal reovirus protein expression in SVECs.
To determine whether σ1s is required for reovirus protein synthesis in SVECs, we analyzed viral protein levels in cells infected with rsT1L or rsT1L σ1s-null. Reovirus outer capsid proteins μ1 and σ3 were readily detected in rsT1L-infected cells. However, markedly less μ1 and σ3 were produced at each time point by rsT1L σ1s-null (Fig. 6A). As with μ1 and σ3, rsT1L produced substantially more nonstructural protein σNS than rsT1L σ1s-null (Fig. 6B). Infection with rsT1L or rsT1L σ1s-null ISVPs produced similar results (Fig. 6C). It is notable that in SVECs, rsT1L and rsT1L σ1s-null produced equivalent levels of M2, S4, and S3 mRNAs, which encode μ1, σ3, and σNS, respectively (Fig. 5). This finding indicates that the lack of protein production by rsT1L σ1s-null does not result from a deficiency in viral mRNA. Rather, the discrepancy in viral protein levels between rsT1L and rsT1L σ1s-null likely results from either reduced protein synthesis or protein stability in cells infected with rsT1L σ1s-null. The difference in viral protein synthesis between rsT1L and rsT1L σ1s-null was not restricted to SVECs; rsT1L also produced substantially more viral protein in MEFs than did rsT1L σ1s-null (Fig. 6D). Together, these data indicate that σ1s is required for optimal reovirus protein expression.
FIG 6.

The σ1s protein is required for optimal reovirus protein expression in SVECs and MEFs. (A and B) SVECs were infected with rsT1L or rsT1L σ1s-null virions at an MOI of 10 PFU/cell. (C) rsT1L or rsT1L σ1s-null ISVPs at an MOI of 1 or 0.1 PFU/cell. (D) MEFs were infected with rsT1L or rsT1L σ1s-null at an MOI of 1 PFU/cell. Cell lysates were collected at the indicated times, and proteins were separated by SDS-PAGE. Reovirus proteins were detected by immunoblotting using a reovirus-specific (A, C, and D) or σNS-specific (B) polyclonal antiserum. β-Actin was detected as a loading control.
The σ1s protein does not affect reovirus protein stability.
To determine whether σ1s modulates reovirus protein stability, we performed steady-state analysis of viral protein levels (Fig. 7). SVECs infected with rsT1L or rsT1L σ1s-null ISVPs were treated with cycloheximide (CHX) at 13 h postinfection to halt new protein synthesis. The levels of μ1 and σNS were quantified over the subsequent 8 h. Cells were infected with ISVPs to ensure that sufficient viral protein would be produced in rsT1L σ1s-null-infected cells to be quantified. Cyclin D1 was used as a positive control for CHX activity due to its short half-life (<2 h) (38). Following CHX treatment, we observed comparable degradation patterns for μ1 following rsT1L and rsT1L σ1s-null infections (Fig. 7A and B). Using densitometry and linear regression analysis, we found that the rates of μ1 loss in rsT1L- and rsT1L σ1s-null-infected cells did not differ significantly (y, −0.05325 for rsT1L and −0.04372 for rsT1L σ1s-null) (Fig. 7C). Similar results were obtained for reovirus nonstructural protein σNS (y, −0.02449 for rsT1L and −0.008924 for rsT1L σ1s-null) (Fig. 7D to F). Together, these data indicate that σ1s does not affect reovirus protein stability.
FIG 7.

The σ1s protein does not affect reovirus protein stability. SVECs were either mock infected or infected with rsT1L or rsT1L σ1s-null ISVPs at an MOI of 1 PFU/cell. At 13 h, either dimethyl sulfoxide (used as a vehicle control [VC]) or cycloheximide (CHX; 25 μg/ml; used to halt protein synthesis) was added to the culture medium. Cell lysates were collected at the indicated times posttreatment, and proteins were separated by SDS-PAGE. (A, B, D, and E) Reovirus outer capsid protein μ1 (A and B) or nonstructural protein σNS (D and E) was detected by immunoblotting. Upper bands in panels A and B represent full-length μ1 protein. (C and F) Densitometry was performed on the full-length μ1 protein (C) and the σNS protein (F) to determine the rate of protein degradation by linear regression analysis. Results are presented as the average relative quantity of protein from two independent experiments. Error bars indicate standard deviations.
σ1s influences viral factory morphology in SVECs.
Reovirus replication, including viral positive- and negative-sense RNA synthesis, viral protein synthesis, and virion assembly, occurs within VFs (16, 18, 23, 24, 39). The capacity of rsT1L σ1-null to produce low levels of viral progeny suggests that VFs can form, even though rsT1L σ1s-null synthesizes substantially less viral protein than rsT1L. To determine whether σ1s is required for VF formation, we stained for reovirus nonstructural protein σNS, as a marker of VFs, at 6, 12, 18, and 24 h after the infection of SVECs with rsT1L or rsT1L σ1s-null (Fig. 8A). Over time, rsT1L VFs progressed from punctate (6 h) to globular (12 h) to large, filamentous structures characteristic of mature VFs (18 h). In contrast, punctate structures were not observed in rsT1L σ1s-null-infected SVECs until 12 h. Late in infection, most rsT1L σ1s-null VFs were small and globular or remained as punctate structures. Very few progressed to a large, filamentous morphology. At 24 h, we quantified the VFs in four different morphological categories: (i) large/filamentous, (ii) small/globular, (iii) punctate, and (iv) diffuse (Fig. 8B and C). A single filamentous VF that encircled, or nearly encircled, the nucleus was categorized as large/filamentous. Multiple smaller, rounded VFs throughout the cytoplasm, with a discernible margin and interior, were categorized as small/globular. For infected cells that contained only small inclusions with no discernible margin or interior, the VF morphology was classified as punctate. Finally, when σNS staining was observed throughout the cytoplasm, with no or very few punctae (<20/cell), the VF morphology was categorized as diffuse. Large/filamentous VFs surrounding the nucleus were observed in the majority of rsT1L-infected cells (70%), with fewer cells containing small/globular VFs (26%) or punctate structures (4%). In contrast, very few rsT1L σ1s-null-infected cells contained large/filamentous VFs (4%). However, we observed larger populations of small/globular VFs (36%) and punctate structures (20%). Interestingly, the largest percentage of rsT1L σ1s-null-infected cells (40%) exhibited the diffuse σNS staining pattern, which was not observed in rsT1L-infected cells. The diffuse staining pattern suggests a defect in VF nucleation or maturation that results in free σNS in the cytoplasm. These data indicate that while VF structures can form in the absence of σ1s, the σ1s protein promotes efficient VF development or maturation, possibly by enhancing new viral protein synthesis, which is necessary for VF enlargement.
FIG 8.

The σ1s protein influences VF morphology in SVECs. (A) SVECs were infected with rsT1L or rsT1L σ1s-null at an MOI of 100 PFU/cell. At the indicated times, cells were fixed and were stained for reovirus nonstructural protein σNS (green) and nuclear DNA (DAPI) (blue). (B) Colored arrows indicate the four staining patterns identified: large/filamentous (blue), small/globular (red), punctate (green), and diffuse (purple). (C) At least 100 infected cells were counted and were classified by the staining patterns of the VFs. Data are presented as the average percentage of cells exhibiting each indicated staining pattern from three independent experiments with standard deviations.
DISCUSSION
Like all viruses, reoviruses must synthesize viral proteins in order to replicate efficiently. However, the viral and cellular factors that regulate reovirus protein production are not defined. In this study, we identified nonstructural protein σ1s as a viral mediator of optimal reovirus protein expression, at least in some cell types. We found that wild-type virus (rsT1L) generated substantially more viral protein than a σ1s-deficient mutant (rsT1L σ1s-null) in SVECs and MEFs (Fig. 6) and that diminished viral protein expression restricts reovirus replication (Fig. 1). Impaired protein production by rsT1L σ1s-null does not result from reduced viral transcription, as evidenced by the fact that viral mRNA levels are comparable in rsT1L- and rsT1L σ1s-null-infected cells (Fig. 5). In addition, the rates of viral protein degradation do not differ between rsT1L and rsT1L σ1s-null, indicating that the σ1s protein does not function to stabilize viral proteins (Fig. 7). From these findings, we hypothesize that σ1s functions to facilitate reovirus protein expression and thereby reovirus replication.
Although the general steps in reovirus translation are known, the cellular and molecular events that govern reovirus protein synthesis are only partially understood. In the current model of reovirus translation, capped viral mRNAs produced by incoming virions are used as templates for the initial rounds of viral protein synthesis by cellular ribosomes (7). New viral structural and nonstructural proteins accumulate to form VFs, which recruit viral positive-sense RNAs that become incorporated into new core particles and serve as templates for negative-sense RNA synthesis (7). Nascent viral cores function as secondary transcriptase particles that continue to synthesize positive-sense RNAs (7). The guanylyltransferase and methyltransferase activities in secondary cores are inhibited, and viral mRNAs synthesized from these particles are uncapped (20). Late in infection, uncapped reovirus mRNAs are translated with much greater efficiency than capped viral mRNAs (20–22). As replication proceeds, VFs enlarge as additional viral RNAs and proteins are produced and incorporated into the structure. The addition of heterohexamers of outer capsid proteins σ3 and μ1 to the nascent core represses the transcriptional activity of new cores, and the insertion of σ1 trimers into the RNA exit channel in λ2 completes virion assembly.
We propose three hypotheses for the mechanism by which σ1s might promote optimal reovirus protein expression. First, σ1s might directly enhance reovirus protein production by associating with viral mRNAs or host translation factors. To efficiently synthesize viral proteins, reovirus RNAs must engage the host cell translation machinery. This is accomplished spatially by the recruitment of ribosomes and cellular translation factors (initiation, elongation, termination, and recycling) to the VF margin, likely via interaction with reovirus nonstructural proteins σNS and μNS (24). The localization of translation factors and ribosomes to the VF margin is hypothesized to place the cellular translation machinery and nascent reovirus mRNAs in close proximity to facilitate the loading of ribosomes onto the viral mRNAs (24). However, reovirus mRNAs differ from highly translated cellular mRNAs in that they lack 3′ poly(A) tails, contain short 3′ and 5′ UTRs, and can be uncapped during the late stages of infection. Despite these differences, reovirus mRNAs are highly translated in infected cells (7).
Many viruses encode proteins that engage the host translation machinery to facilitate preferential translation of viral RNAs (6). Nonstructural protein 3 (NSP3) from rotavirus, also a member of the Reoviridae family of viruses, binds the tetranucleotide sequence common to the 3′ termini of all rotavirus mRNAs in a manner analogous to the engagement of the poly(A) tail on cellular mRNAs by poly(A)-binding protein (PABP) (40). NSP3 also engages the scaffolding protein eIF4G, which, in turn, interacts with the cap binding protein eIF4E (41). In this way, rotavirus recapitulates the mRNA circularization that enables efficient translation (42–46). Like rotavirus mRNAs, reovirus mRNAs contain a conserved tetranucleotide sequence (CATC) at their 3′ termini (7). However, it is not known how noncanonical reovirus mRNAs engage host translation machinery.
The σ1s protein also shares similarities with rotavirus NSP6. The 12-kDa protein NSP6 is encoded by ORF3, which is nested within the gene for nonstructural protein 5 (47), an arrangement analogous to the placement of the σ1s coding sequence within the σ1 ORF. Like σ1s, NSP6 is not absolutely essential for viral replication but does promote efficient viral growth in cultured cells (48). The function of NSP6 is not fully defined, but NSP6 has the capacity to bind both single-stranded RNA (ssRNA) and dsRNA (49). It is possible that σ1s also binds viral RNA, possibly through the conserved N-terminal basic region. Binding to viral RNA might allow σ1s to serve as an adaptor between reovirus mRNAs and host factors, such as translation factors. Alternatively, σ1s could act as a viral translation factor that diverts cellular translational activity to favor reovirus protein synthesis.
Second, σ1s might function to allow reovirus to overcome host translational shutoff (2–4). Reovirus activates protein kinase R (PKR) and RNase L, induces eIF2α phosphorylation, and elicits the formation of stress granules, all of which impair normal translational processes (50–52). To replicate, reovirus must have the capacity to translate viral proteins when host protein synthesis is inhibited. Moreover, reovirus replication is impaired in MEFs expressing a nonphosphorylatable form of eIF2α (53), suggesting that host protein shutoff may favor reovirus replication. A number of viruses encode proteins that either prevent the induction of host translation shutoff or allow the virus to circumvent the translational block (5, 6). Reovirus outer capsid protein σ3 has the capacity to bind dsRNA and prevent PKR activation (54–58). However, reovirus likely has other mechanisms to overcome translational arrest, and σ1s might function to allow reovirus to bypass global translation inhibition. Our data indicate that σ1s does not function as a direct antagonist of IFN-I responses (Fig. 3). However, σ1s could function downstream of IFN-I signaling to counteract specific ISGs or other cell responses to infection.
Third, and finally, σ1s might facilitate the assembly and expansion of VFs, which are the platform for viral translation. Active reovirus translation occurs at the VF margin, and enlargement of the VF could recruit increasing amounts of cellular translation machinery to boost the level of viral translation over the course of infection. The absence of σ1s could perturb VF expansion and limit viral protein production. Although ectopic expression of σNS and μNS is sufficient to form VF-like structures in cells (14, 15), σ1s could promote VF maturation during infection. These three possibilities are not mutually exclusive; σ1s could perform multiple functions simultaneously.
Our data indicate that σ1s potentiates reovirus protein expression in SVECs and MEFs. However, reovirus must have the capacity to produce protein in the absence of σ1s, since early viral translation occurs prior to the synthesis of σ1s itself. It is likely that σ1s-independent protein production can occur throughout the replication cycle, because although rsT1L σ1s-null produced significantly less viral protein in SVECs and MEFs than rsT1L, it still produced sufficient viral protein to support low levels of VF formation and progeny production. We hypothesize that σ1s functions to facilitate reovirus protein synthesis at later stages of viral replication, when large amounts of viral protein are required for VF enlargement and virion assembly.
In this study, we characterized the requirement for σ1s in facilitating reovirus protein expression and replication in SVECs, a mouse endothelial cell line. Endothelial cells are targets for reovirus infection in vivo (36, 37). Although σ1s is also required for reovirus replication in HUVECs (Fig. 1F), it is dispensable for viral replication in 2H11 or TX-111 endothelial cells (data not shown). Together, these findings indicate that σ1s is not an endothelial-cell-specific reovirus replication factor. However, further work will be required to determine whether σ1s functions to promote reovirus protein expression and replication in vivo. In addition to 2H11 and TX-111 cells, σ1s is not required for replication in L929 and HeLa cells or in the intestines and CNS of infected mice (30, 32–34). A similar cell-type-specific requirement is observed for the virion host shutoff (vhs) protein from herpes simplex virus 2 (HSV-2). HSV-2 lacking the vhs protein is severely attenuated in the host (59). However, vhs-deficient HSV-2 replicates comparably to wild-type HSV-2 in certain cells, including Vero cells, but is significantly attenuated in others, such as HeLa cells (60). In HeLa cells, the replication of the vhs-deficient mutant is diminished due to decreased viral translation, whereas no difference in viral protein synthesis is observed in Vero cells (60–63). The cell-type-specific differences that dictate the requirement for σ1s in mediating reovirus protein expression remain to be determined.
In vivo, σ1s is required for hematogenous reovirus spread (33, 34). Although σ1s is dispensable for reovirus replication in the intestine and CNS, viruses lacking σ1s fail to reach the bloodstream (33, 34). Because σ1s is a nonstructural protein, this observation suggests that σ1s is required for viral replication in one or more cell types that are necessary to traffic reovirus to the blood. By facilitating viral protein synthesis in cells that are required for hematogenous dissemination, σ1s might promote efficient reovirus replication and thereby systemic spread.
MATERIALS AND METHODS
Cells and viruses.
Murine L929 fibroblasts were maintained in Joklik's modified Eagle medium (JMEM; Sigma) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen), 1% l-glutamine (Invitrogen), 1% penicillin-streptomycin (Invitrogen), and 0.1% amphotericin B (Sigma). SV40-immortalized endothelial cells (SVEC4-10 [SVECs]; ATCC), C57BL/6 murine embryonic fibroblasts (MEFs), 2H11 cells, TX-111 cells, and EcoPack cells were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented to contain 10% heat-inactivated FBS and 1% l-glutamine. Human telomerase reverse transcriptase (hTERT)-immortalized human umbilical vein endothelial cells (HUVECs) were maintained in endothelial basal medium supplemented with the SingleQuots kit (Lonza). T84 human epithelial cells were maintained in a 1:1 mixture of Ham's F-12 medium and DMEM (Invitrogen) supplemented to contain 10% heat-inactivated FBS and 1% l-glutamine.
All reovirus strains were generated using plasmid-based reverse genetics (64, 65). The T1L S1 allele containing the point mutation to disrupt the σ1s translational start site (σ1s-null) was constructed previously (30, 33, 34). The σ1s truncation mutants were generated using QuikChange (Stratagene) primer-based mutagenesis. Primer sequences are available on request. Purified reovirus virions were generated using second- or third-passage L929 cell lysates from twice-plaque-purified reovirus (66). Viral particles were extracted with Vertrel (MicroCare), centrifuged on a CsCl density gradient, and exhaustively dialyzed in virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.8]). Viral titers were determined by plaque assays on L929 fibroblasts (67).
Infectious subvirion particles (ISVPs) were generated by treating 2 × 1011 reovirus particles with chymotrypsin (200 μg/ml) for 1 h at 37°C. Chymotrypsin activity was halted by the addition of 2 mM phenylmethylsulfonyl fluoride (PMSF; Sigma). ISVP conversion was confirmed by separating particles (2 × 105 particles) via SDS-PAGE and staining with Coomassie blue.
Viral replication assays.
Monolayers of cells in 24-well plates (1 × 105 cells/well) were either mock infected or infected as indicated in Fig. 1 at 4°C for 1 h, washed twice with phosphate-buffered saline (PBS), and overlaid with 1 ml of fresh medium. Infected cells were freeze-thawed twice at various times postinfection. The viral titer in each sample was determined by a plaque assay on L929 cells (67). Viral yields were calculated using the equation log10(yieldtx) = log10(PFU/ml)tx − log10(PFU/ml)t0 where t0 is the time of infection and tx is the time postinfection.
Retrovirus transduction of SVECs.
An N-terminally FLAG-tagged T1L σ1s construct was excised from pcDNA3.1-FLAG T1L σ1s using BamHI for transfer into the BglII site in pMSCV-puro (BamHI and BglII have compatible cohesive ends). The insertion was confirmed by sequencing. Retrovirus was generated by transfecting pMSCV-puro-GFP or pMSCV-puro-FLAG σ1s into EcoPack cells by use of Lipofectamine LTX with Plus reagent (Invitrogen) according to the manufacturer's instructions. Supernatants from transfected EcoPack cells were collected at 3 days posttransfection, clarified through a 0.45-μm syringe filter, and applied to SVECs in 6-well dishes (∼50% confluence) with 4 μg/ml Polybrene (Santa Cruz). At 24 h, transduced SVECs were selected in a medium containing 2 μg/ml puromycin (Life Technologies).
Cell viability.
Monolayers of SVECs in 24-well plates (5 × 104 cells/well) were either mock infected or infected with rsT1L or rsT1L σ1s-null at an MOI of 1,000, 100, 10, or 1 PFU/cell in triplicate. The numbers of live and dead cells were determined at 24 h by trypan blue exclusion assays. Data are presented as the percentages of viable cells.
Caspase activity assays.
Monolayers of SVECs in 96-well plates (5,000 cells/well) were either mock infected or infected with rsT1L or rsT1L σ1s-null at an MOI of 1,000, 100, 10, or 1 PFU/cell in triplicate. Uninfected cells were treated with 25 μg/ml cycloheximide (CHX) plus 25 ng/ml tumor necrosis factor alpha (TNF-α) as a control for caspase-3/7 activity. Caspase-3/7 activity was measured at 24 h postinfection by use of Caspase-Glo 3/7 (Promega) according to the manufacturer's instructions.
IFN-β ELISA.
Monolayers of SVECs (1 × 105/well) in 24-well plates (Corning) were either mock infected or adsorbed in duplicate with rsT1L or rsT1L σ1s-null at an MOI of 100 PFU/cell at room temperature for 1 h. At 6, 12, 18, and 24 h postinfection, mouse IFN-β in cell culture medium was measured by use of the VeriKine mouse IFN-β enzyme-linked immunosorbent assay (ELISA) kit (PBL) according to the manufacturer's instructions.
Immunofluorescence.
Monolayers of SVECs in clear-bottom 96-well plates (1 × 104 cells/well) or 8-well chamber slides (5 × 104 cells/well) were either mock infected or adsorbed with the viruses indicated at 4°C for 1 h, washed twice with PBS, and overlaid with fresh medium. At the times indicated, infected cells were fixed by use of 4% paraformaldehyde for 30 min. To stain for reovirus proteins, cells were permeabilized with 2% Triton X-100 (TX-100) in PBS for 10 min and were incubated in blocking buffer (2% normal goat serum plus 1% TX-100 in PBS) for 30 min. Cells were incubated in blocking buffer containing primary antibodies as indicated in Fig. 2 and 8 for 1 h. Following three washes with PBS containing 1% TX-100, the cells were incubated in blocking buffer containing a secondary antibody for 1 h and were then washed three times with PBS. 4′,6-Diamidino-2-phenylindole (DAPI; 1:1,000; Sigma) was included with the secondary-antibody incubation to stain nuclear DNA. Incubations with fluorescent secondary antibodies and DAPI were performed with protection from light. Images for quantification were taken using an EVOS FL Auto cell imaging system (Invitrogen). Representative images were taken using a Nikon Eclipse Ti-U microscope (Nikon). Images were deconvoluted using identical parameters with NIS-Elements Viewer software (Nikon). The primary antibodies used were a reovirus-specific rabbit polyclonal antiserum (dilution, 1:1,000) and a σNS-specific guinea pig polyclonal antiserum (dilution, 1:1,000). The secondary antibodies used were an Alexa Fluor 488-conjugated goat anti-rabbit antibody (dilution, 1:1,000; Life Technologies) and an Alexa Fluor 488-conjugated goat anti-guinea pig antibody (dilution, 1:1,000; AbCam).
Infectious center assay.
Monolayers of SVECs (1 × 106/well in a 6-well plate) were prechilled at 4°C for 1 h and were then adsorbed with rsT1L or rsT1L σ1s-null at an MOI of 1, 10, or 100 PFU/cell at 4°C. The cells were washed three times with cold PBS and were collected by trypsinization. The cells were counted, and equivalent numbers of cells (1,000, 100, or 10) were plated on monolayers of L929 fibroblasts (2 × 106 cells/well in a 6-well plate) for 2 h. The monolayers were then overlaid for plaque assays (67).
Reverse transcription-quantitative PCR (RT-qPCR) assays.
Monolayers of SVECs in 6-well plates (1 × 106 cells/well) were either mock infected or infected with rsT1L or rsT1L σ1s-null at an MOI of 10 PFU/cell. Total RNA was collected using the RNeasy Plus kit (Qiagen) at the times indicated in Fig. 5. Reovirus RNA in 500 ng of total RNA was quantified using TaqMan Fast Virus 1-Step master mix (Applied Biosystems) or a SuperScript III Platinum SYBR green One-Step qRT-PCR kit (Invitrogen). The RNA was heated at 95°C for 3 min and then placed on ice prior to addition to 96-well PCR plates for TaqMan or SYBR green assays.
For TaqMan assays to detect the S4 gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was detected as an endogenous control using the Pre-Developed TaqMan Assay Reagents kit for mouse GAPDH (TaqMan assays; Applied Biosystems). PCRs were prepared according to the manufacturer's specifications for fast RT-qPCRs, with minor modifications. The reverse transcription step was performed as described in the manufacturer's protocol (50°C for 3 min; 95°C for 30 s) using only the reverse S4 primer to synthesize cDNA from positive-sense viral RNA or only the forward S4 primer to synthesize cDNA from negative-sense viral RNA. Primer and probe sequences are available on request. After reverse transcription and heat inactivation of the reverse transcriptase, the opposing primer and probe were added, and qPCR was performed as described in the manufacturer's protocol (40 cycles of 95°C for 3 s and 60°C for 30 s). The reverse transcription step was performed using an Eppendorf Mastercycler pro system, and qPCR was performed using the Applied Biosystems StepOne Plus system.
Expression of the ISG IFIT1 was quantified following rsT1L or rsT1L infection of SVECs at an MOI of 10 PFU/cell using the RNA purification and TaqMan protocols described above without modification from the manufacturer's protocol. IFIT1 mRNA expression was measured using the IFIT1 gene expression assay from Applied Biosystems.
For SYBR green assays, PCRs were prepared according to the manufacturer's specifications for standard RT-qPCRs, with minor modifications. As described above, reverse transcription (60°C for 3 min; 95°C for 5 min) was performed using a single primer, and the opposing primer was added prior to qPCR detection (40 cycles of 95°C for 15 s and 60°C for 30 s). Primer sequences are available on request. Melting curve analysis was performed for SYBR assays to ensure primer specificity. The reverse transcription step was performed using an Eppendorf Mastercycler pro system, and qPCR was performed using the StepOne Plus system.
For both assays, the relative quantity (RQ) of reovirus positive- or negative-sense RNA at the indicated times was determined using the sample at 0 h postinfection as the reference sample. ΔΔCT was calculated for each sample as (unknown CTtx − GAPDH CTtx) − (unknown CTt0 − GAPDH CTt0), where CT is the threshold cycle, t0 is the time of infection, and tx is the time postinfection. ΔΔCT was then used to calculate the RQ using the equation RQ = 2˄−ΔΔCT. Finally, the RQ of viral mRNA was determined by subtracting the RQ of negative-sense RNA from the RQ of positive-sense RNA.
Immunoblotting.
Monolayers of cells in 6-well plates (1 × 106 cells/well) were either mock infected or infected with rsT1L or rsT1L σ1s-null at the MOIs indicated on Fig. 3 and in the legend to Fig. 6. Whole-cell lysates were collected at various times postinfection in radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% sodium dodecyl sulfate, 1% desoxycholate, 1/100 IGEPAL [NP-40]). Total protein in each sample was quantified using the DC protein assay (Bio-Rad), and equivalent amounts of protein (25 μg) were separated by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane and were incubated in blocking buffer (5% milk in 1× Tris-buffered saline with 0.05% Tween 20 [TBS-T]) with rocking for 1 h. Membranes were incubated in blocking buffer containing either a reovirus-specific rabbit polyclonal antiserum (dilution, 1:2,000), a σNS-specific guinea pig antiserum (dilution, 1:2,000), rabbit polyclonal anti-STAT1 (dilution, 1:1,000; Cell Signaling, Danvers, MA), rabbit monoclonal anti-phospho-STAT1 (Tyr701) (dilution, 1:1,000; Cell Signaling, Danvers, MA), rabbit polyclonal anti-STAT2 (dilution, 1:100; Cell Signaling, Danvers, MA), or rabbit polyclonal anti-STAT2 (phospho Y690) (dilution, 1:1,000; Abcam, Cambridge, United Kingdom) at 4°C overnight. Following three TBS-T washes, membranes were incubated in blocking buffer containing horseradish peroxidase-conjugated goat anti-rabbit IgG (dilution, 1:2,000; Jackson Immunolabs) or peroxidase-conjugated goat anti-guinea pig IgG (dilution, 1:2,000; Cell Signaling) for 1 h. Membranes were washed three times with TBS-T, and proteins were detected using SuperSignal West chemiluminescent substrate (Thermo Fisher). Blots were stripped using Restore Western blot stripping buffer (Thermo Scientific) for 15 min at room temperature, blocked as described above, and blotted for β-actin using a mouse β-actin-specific monoclonal antibody (dilution, 1:10,000; Sigma) and peroxidase-conjugated goat anti-mouse IgG (dilution, 1:2,000; Jackson Immunolabs). Fluorescent immunoblots were prepared as described above with minor changes. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes presoaked in methanol. Primary-antibody incubation was performed with a reovirus rabbit polyclonal antiserum (dilution, 1:1,000) and a β-actin-specific monoclonal antibody simultaneously. The secondary antibodies used were Alexa Fluor 488-conjugated goat anti-rabbit (dilution, 1:2,000; Life Technologies) and Alexa Fluor 546-conjugated goat anti-mouse (dilution, 1:2,000; Life Technologies) antibodies and were incubated simultaneously. Proteins were detected as described above and were imaged using the ChemiDoc MP imaging system (Bio-Rad).
Steady-state protein analysis.
Monolayers of SVECS in 6-well plates (1 × 106 cells/well) were infected with rsT1L or rsT1L σ1s-null ISVPs at an MOI of 10 PFU/cell. Infected cells were treated with 25 μg/ml cycloheximide at 13 h postinfection to halt new protein synthesis. Cell lysates were collected at the times indicated on Fig. 7. Lysates were prepared, and μ1, σNS, or β-actin was detected as described above. Cyclin D1 was detected using a rabbit monoclonal antibody against cyclin D1 (dilution, 1:1,000; Cell Signaling) incubated in TBS-T plus 5% bovine serum albumin (BSA), followed by peroxidase-conjugated goat anti-rabbit IgG. Densitometry was performed on the upper μ1 band (full-length μ1) or the σNS band to determine the quantity relative to that at 0 h posttreatment. All μ1 and σNS levels were normalized to relative quantities of β-actin. Densitometric analysis was performed using the Image Lab program (version 4.1; Bio-Rad). Linear regression analysis was performed using Prism (version 6.04; GraphPad, Inc.) to measure the rate of protein degradation.
Statistics.
Differences in reovirus replication, RNA synthesis, and VF morphology were evaluated using an unpaired Student t test. Viral protein stability was evaluated by regression analysis. Statistical analysis was performed using Prism software (GraphPad Software, Inc., La Jolla, CA, USA). P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
We thank Joseph Koon and Morgan Howells for experimental assistance. We thank Pranav Danthi for reading the manuscript. Blossom Damania provided hTERT-immortalized HUVECs, and Ruud Dings provided 2H11 mouse lymphatic endothelial cells. The pMSCV-puro plasmid and EcoPack cells were provided by Craig Forrest (UAMS).
This research was supported by Public Health Service grants K22 AI94079 and R01 AI118801 (both to K.W.B.). J.D.S. was supported by the UAMS Initiative for Maximizing Student Diversity (R25 GM83247). Additional support was provided by the Center for Microbial Pathogenesis and Host Inflammatory Responses (P20 GM103625).
REFERENCES
- 1.Levy DE, Marié IJ, Durbin JE. 2011. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol 1:476–486. doi: 10.1016/j.coviro.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalet A, Gatti E, Pierre P. 2015. Integration of PKR-dependent translation inhibition with innate immunity is required for a coordinated anti-viral response. FEBS Lett 589:1539–1545. doi: 10.1016/j.febslet.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Poblete-Durán N, Prades-Perez Y, Vera-Otarola J, Soto-Rifo R, Valiente-Echeverria F. 2016. Who regulates whom? An overview of RNA granules and viral infections. Viruses 8:E180. doi: 10.3390/v8070180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohr I. 2006. Phosphorylation and dephosphorylation events that regulate viral mRNA translation. Virus Res 119:89–99. doi: 10.1016/j.virusres.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Walsh D, Mathews MB, Mohr I. 2013. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol 5:a012351. doi: 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh D, Mohr I. 2011. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol 9:860–875. doi: 10.1038/nrmicro2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dermody TS, Parker JSL, Sherry B. 2013. Orthoreovirus, p 1304–1346. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 8.Chandran K, Nibert ML. 2003. Animal cell invasion by a large nonenveloped virus: reovirus delivers the goods. Trends Microbiol 11:374–382. doi: 10.1016/S0966-842X(03)00178-1. [DOI] [PubMed] [Google Scholar]
- 9.Cleveland DR, Zarbl H, Millward S. 1986. Reovirus guanylyltransferase is L2 gene product λ2. J Virol 60:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reinisch KM, Nibert ML, Harrison SC. 2000. Structure of the reovirus core at 3.6 Å resolution. Nature 404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- 11.White CK, Zweerink HJ. 1976. Studies on the structure of reovirus cores: selective removal of polypeptide λ2. Virology 70:171–180. doi: 10.1016/0042-6822(76)90247-6. [DOI] [PubMed] [Google Scholar]
- 12.Kozak M, Shatkin AJ. 1977. Sequences of two 5′-terminal ribosome-protected fragments from reovirus messenger RNAs. J Mol Biol 112:75–96. doi: 10.1016/S0022-2836(77)80157-5. [DOI] [PubMed] [Google Scholar]
- 13.Kozak M, Shatkin AJ. 1978. Identification of features in 5′ terminal fragments from reovirus mRNA which are important for ribosome binding. Cell 13:201–212. doi: 10.1016/0092-8674(78)90150-2. [DOI] [PubMed] [Google Scholar]
- 14.Broering TJ, Parker JS, Joyce PL, Kim J, Nibert ML. 2002. Mammalian reovirus nonstructural protein μNS forms large inclusions and colocalizes with reovirus microtubule-associated protein μ2 in transfected cells. J Virol 76:8285–8297. doi: 10.1128/JVI.76.16.8285-8297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broering TJ, Arnold MM, Miller CL, Hurt JA, Joyce PL, Nibert ML. 2005. Carboxyl-proximal regions of reovirus nonstructural protein μNS necessary and sufficient for forming factory-like inclusions. J Virol 79:6194–6206. doi: 10.1128/JVI.79.10.6194-6206.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller CL, Arnold MM, Broering TJ, Hastings CE, Nibert ML. 2010. Localization of mammalian orthoreovirus proteins to cytoplasmic factory-like structures via nonoverlapping regions of μNS. J Virol 84:867–882. doi: 10.1128/JVI.01571-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller CL, Broering TJ, Parker JS, Arnold MM, Nibert ML. 2003. Reovirus σNS protein localizes to inclusions through an association requiring the μNS amino terminus. J Virol 77:4566–4576. doi: 10.1128/JVI.77.8.4566-4576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broering TJ, Kim J, Miller CL, Piggott CD, Dinoso JB, Nibert ML, Parker JS. 2004. Reovirus nonstructural protein μNS recruits viral core surface proteins and entering core particles to factory-like inclusions. J Virol 78:1882–1892. doi: 10.1128/JVI.78.4.1882-1892.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antczak JB, Joklik WK. 1992. Reovirus genome segment assortment into progeny genomes studied by the use of monoclonal antibodies directed against reovirus proteins. Virology 187:760–776. doi: 10.1016/0042-6822(92)90478-8. [DOI] [PubMed] [Google Scholar]
- 20.Skup D, Millward S. 1980. mRNA capping enzymes are masked in reovirus progeny subviral particles. J Virol 34:490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skup D, Millward S. 1980. Reovirus-induced modification of cap-dependent translation in infected L cells. Proc Natl Acad Sci U S A 77:152–156. doi: 10.1073/pnas.77.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zarbl H, Skup D, Millward S. 1980. Reovirus progeny subviral particles synthesize uncapped mRNA. J Virol 34:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farsetta DL, Chandran K, Nibert ML. 2000. Transcriptional activities of reovirus RNA polymerase in recoated cores. Initiation and elongation are regulated by separate mechanisms. J Biol Chem 275:39693–39701. [DOI] [PubMed] [Google Scholar]
- 24.Desmet EA, Anguish LJ, Parker JS. 2014. Virus-mediated compartmentalization of the host translational machinery. mBio 5:e01463-14. doi: 10.1128/mBio.01463-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkar G, Pelletier J, Bassel-Duby R, Jayasuriya A, Fields BN, Sonenberg N. 1985. Identification of a new polypeptide coded by reovirus gene S1. J Virol 54:720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cashdollar LW, Chmelo RA, Wiener JR, Joklik WK. 1985. Sequences of the S1 genes of the three serotypes of reovirus. Proc Natl Acad Sci U S A 82:24–28. doi: 10.1073/pnas.82.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ernst H, Shatkin AJ. 1985. Reovirus hemagglutinin mRNA codes for two polypeptides in overlapping reading frames. Proc Natl Acad Sci U S A 82:48–52. doi: 10.1073/pnas.82.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dermody TS, Nibert ML, Bassel-Duby R, Fields BN. 1990. Sequence diversity in S1 genes and S1 translation products of 11 serotype 3 reovirus strains. J Virol 64:4842–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cenatiempo Y, Twardowski T, Shoeman R, Ernst H, Brot N, Weissbach H, Shatkin AJ. 1984. Two initiation sites detected in the small S1 species of reovirus mRNA by dipeptide synthesis in vitro. Proc Natl Acad Sci U S A 81:1084–1088. doi: 10.1073/pnas.81.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boehme KW, Hammer K, Tollefson WC, Konopka-Anstadt JL, Kobayashi T, Dermody TS. 2013. Nonstructural protein σ1s mediates reovirus-induced cell cycle arrest and apoptosis. J Virol 87:12967–12979. doi: 10.1128/JVI.02080-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoyt CC, Bouchard RJ, Tyler KL. 2004. Novel nuclear herniations induced by nuclear localization of a viral protein. J Virol 78:6360–6369. doi: 10.1128/JVI.78.12.6360-6369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodgers SE, Connolly JL, Chappell JD, Dermody TS. 1998. Reovirus growth in cell culture does not require the full complement of viral proteins: identification of a σ1s-null mutant. J Virol 72:8597–8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boehme KW, Frierson JM, Konopka JL, Kobayashi T, Dermody TS. 2011. The reovirus σ1s protein is a determinant of hematogenous but not neural virus dissemination in mice. J Virol 85:11781–11790. doi: 10.1128/JVI.02289-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boehme KW, Guglielmi KM, Dermody TS. 2009. Reovirus nonstructural protein σ1s is required for establishment of viremia and systemic dissemination. Proc Natl Acad Sci U S A 106:19986–19991. doi: 10.1073/pnas.0907412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boehme KW, Lai CM, Dermody TS. 2013. Mechanisms of reovirus bloodstream dissemination. Adv Virus Res 87:1–35. doi: 10.1016/B978-0-12-407698-3.00001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71. doi: 10.1016/j.chom.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai CM, Boehme KW, Pruijssers AJ, Parekh VV, Van Kaer L, Parkos CA, Dermody TS. 2015. Endothelial JAM-A promotes reovirus viremia and bloodstream dissemination. J Infect Dis 211:383–393. doi: 10.1093/infdis/jiu476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanie T, Onoyama I, Matsumoto A, Yamada M, Nakatsumi H, Tateishi Y, Yamamura S, Tsunematsu R, Matsumoto M, Nakayama KI. 2012. Genetic reevaluation of the role of F-box proteins in cyclin D1 degradation. Mol Cell Biol 32:590–605. doi: 10.1128/MCB.06570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Broering TJ, McCutcheon AM, Centonze VE, Nibert ML. 2000. Reovirus nonstructural protein μNS binds to core particles but does not inhibit their transcription and capping activities. J Virol 74:5516–5524. doi: 10.1128/JVI.74.12.5516-5524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poncet D, Laurent S, Cohen J. 1994. Four nucleotides are the minimal requirement for RNA recognition by rotavirus non-structural protein NSP3. EMBO J 13:4165–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piron M, Vende P, Cohen J, Poncet D. 1998. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J 17:5811–5821. doi: 10.1093/emboj/17.19.5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Groft CM, Burley SK. 2002. Recognition of eIF4G by rotavirus NSP3 reveals a basis for mRNA circularization. Mol Cell 9:1273–1283. doi: 10.1016/S1097-2765(02)00555-5. [DOI] [PubMed] [Google Scholar]
- 43.Deo RC, Groft CM, Rajashankar KR, Burley SK. 2002. Recognition of the rotavirus mRNA 3′ consensus by an asymmetric NSP3 homodimer. Cell 108:71–81. doi: 10.1016/S0092-8674(01)00632-8. [DOI] [PubMed] [Google Scholar]
- 44.Gratia M, Sarot E, Vende P, Charpilienne A, Baron CH, Duarte M, Pyronnet S, Poncet D. 2015. Rotavirus NSP3 is a translational surrogate of the poly(A) binding protein-poly(A) complex. J Virol 89:8773–8782. doi: 10.1128/JVI.01402-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gratia M, Vende P, Charpilienne A, Baron HC, Laroche C, Sarot E, Pyronnet S, Duarte M, Poncet D. 2016. Challenging the roles of NSP3 and untranslated regions in rotavirus mRNA translation. PLoS One 11:e0145998. doi: 10.1371/journal.pone.0145998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gajardo R, Vende P, Poncet D, Cohen J. 1997. Two proline residues are essential in the calcium-binding activity of rotavirus VP7 outer capsid protein. J Virol 71:2211–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattion NM, Mitchell DB, Both GW, Estes MK. 1991. Expression of rotavirus proteins encoded by alternative open reading frames of genome segment 11. Virology 181:295–304. doi: 10.1016/0042-6822(91)90495-W. [DOI] [PubMed] [Google Scholar]
- 48.Komoto S, Kanai Y, Fukuda S, Kugita M, Kawagishi T, Ito N, Sugiyama M, Matsuura Y, Kobayashi T, Taniguchi K. 2017. Reverse genetics system demonstrates that rotavirus nonstructural protein NSP6 is not essential for viral replication in cell culture. J Virol 91:e00695-17. doi: 10.1128/JVI.00695-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rainsford EW, McCrae MA. 2007. Characterization of the NSP6 protein product of rotavirus gene 11. Virus Res 130:193–201. doi: 10.1016/j.virusres.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 50.Qin Q, Carroll K, Hastings C, Miller CL. 2011. Mammalian orthoreovirus escape from host translational shutoff correlates with stress granule disruption and is independent of eIF2α phosphorylation and PKR. J Virol 85:8798–8810. doi: 10.1128/JVI.01831-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin Q, Hastings C, Miller CL. 2009. Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection. J Virol 83:11090–11101. doi: 10.1128/JVI.01239-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith JA, Schmechel SC, Raghavan A, Abelson M, Reilly C, Katze MG, Kaufman RJ, Bohjanen PR, Schiff LA. 2006. Reovirus induces and benefits from an integrated cellular stress response. J Virol 80:2019–2033. doi: 10.1128/JVI.80.4.2019-2033.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith JA, Schmechel SC, Williams BR, Silverman RH, Schiff LA. 2005. Involvement of the interferon-regulated antiviral proteins PKR and RNase L in reovirus-induced shutoff of cellular translation. J Virol 79:2240–2250. doi: 10.1128/JVI.79.4.2240-2250.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Denzler KL, Jacobs BL. 1994. Site-directed mutagenic analysis of reovirus σ3 protein binding to dsRNA. Virology 204:190–199. doi: 10.1006/viro.1994.1523. [DOI] [PubMed] [Google Scholar]
- 55.Olland AM, Jané-Valbuena J, Schiff LA, Nibert ML, Harrison SC. 2001. Structure of the reovirus outer capsid and dsRNA-binding protein σ3 at 1.8 Å resolution. EMBO J 20:979–989. doi: 10.1093/emboj/20.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schiff LA, Nibert ML, Co MS, Brown EG, Fields BN. 1988. Distinct binding sites for zinc and double-stranded RNA in the reovirus outer capsid protein σ3. Mol Cell Biol 8:273–283. doi: 10.1128/MCB.8.1.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bergeron J, Mabrouk T, Garzon S, Lemay G. 1998. Characterization of the thermosensitive ts453 reovirus mutant: increased dsRNA binding of σ3 protein correlates with interferon resistance. Virology 246:199–210. doi: 10.1006/viro.1998.9188. [DOI] [PubMed] [Google Scholar]
- 58.Wang Q, Bergeron J, Mabrouk T, Lemay G. 1996. Site-directed mutagenesis of the double-stranded RNA binding domain of bacterially-expressed σ3 reovirus protein. Virus Res 41:141–151. doi: 10.1016/0168-1702(96)01281-6. [DOI] [PubMed] [Google Scholar]
- 59.Murphy JA, Duerst RJ, Smith TJ, Morrison LA. 2003. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J Virol 77:9337–9345. doi: 10.1128/JVI.77.17.9337-9345.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duerst RJ, Morrison LA. 2004. Herpes simplex virus 2 virion host shutoff protein interferes with type I interferon production and responsiveness. Virology 322:158–167. doi: 10.1016/j.virol.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 61.Dauber B, Pelletier J, Smiley JR. 2011. The herpes simplex virus 1 vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J Virol 85:5363–5373. doi: 10.1128/JVI.00115-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dauber B, Poon D, Dos Santos T, Duguay BA, Mehta N, Saffran HA, Smiley JR. 2016. The herpes simplex virus virion host shutoff protein enhances translation of viral true late mRNAs independently of suppressing protein kinase R and stress granule formation. J Virol 90:6049–6057. doi: 10.1128/JVI.03180-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dauber B, Saffran HA, Smiley JR. 2014. The herpes simplex virus 1 virion host shutoff protein enhances translation of viral late mRNAs by preventing mRNA overload. J Virol 88:9624–9632. doi: 10.1128/JVI.01350-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boehme KW, Ikizler M, Kobayashi T, Dermody TS. 2011. Reverse genetics for mammalian reovirus. Methods 55:109–113. doi: 10.1016/j.ymeth.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kobayashi T, Antar AA, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Furlong DB, Nibert ML, Fields BN. 1988. σ1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Virgin HW IV, Bassel-Duby R, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol 62:4594–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]