

ABSTRACT
Although influenza A virus (IAV) evades cellular defense systems to effectively propagate in the host, the viral immune-evasive mechanisms are incompletely understood. Our recent data showed that hemagglutinin (HA) of IAV induces degradation of type I IFN receptor 1 (IFNAR1). Here, we demonstrate that IAV HA induces degradation of type II IFN (IFN-γ) receptor 1 (IFNGR1), as well as IFNAR1, via casein kinase 1α (CK1α), resulting in the impairment of cellular responsiveness to both type I and II IFNs. IAV infection or transient HA expression induced degradation of both IFNGR1 and IFNAR1, whereas HA gene-deficient IAV failed to downregulate the receptors. IAV HA caused the phosphorylation and ubiquitination of IFNGR1, leading to the lysosome-dependent degradation of IFNGR1. Influenza viral HA strongly decreased cellular sensitivity to type II IFNs, as it suppressed the activation of STAT1 and the induction of IFN-γ-stimulated genes in response to exogenously supplied recombinant IFN-γ. Importantly, CK1α, but not p38 MAP kinase or protein kinase D2, was proven to be critical for HA-induced degradation of both IFNGR1 and IFNAR1. Pharmacologic inhibition of CK1α or small interfering RNA (siRNA)-based knockdown of CK1α repressed the degradation processes of both IFNGR1 and IFNAR1 triggered by IAV infection. Further, CK1α was shown to be pivotal for proficient replication of IAV. Collectively, the results suggest that IAV HA induces degradation of IFN receptors via CK1α, creating conditions favorable for viral propagation. Therefore, the study uncovers a new immune-evasive pathway of influenza virus.
IMPORTANCE Influenza A virus (IAV) remains a grave threat to humans, causing seasonal and pandemic influenza. Upon infection, innate and adaptive immunity, such as the interferon (IFN) response, is induced to protect hosts against IAV infection. However, IAV seems to be equipped with tactics to evade the IFN-mediated antiviral responses, although the detailed mechanisms need to be elucidated. In the present study, we show that IAV HA induces the degradation of the type II IFN receptor IFNGR1 and thereby substantially attenuates cellular responses to IFN-γ. Of note, a cellular kinase, casein kinase 1α (CK1α), is crucial for IAV HA-induced degradation of both IFNGR1 and IFNAR1. Accordingly, CK1α is proven to positively regulate IAV propagation. Thus, this study unveils a novel strategy employed by IAV to evade IFN-mediated antiviral activities. These findings may provide new insights into the interplay between IAV and host immunity to impact influenza virus pathogenicity.
KEYWORDS: casein kinase 1α, hemagglutinin, influenza virus, interferon, interferon receptor degradation, type II interferon receptor (IFNGR)
INTRODUCTION
Influenza viruses continue to circulate among humans, causing severe respiratory illness and imposing a significant economic burden (1–3). Influenza viruses belong to the family Orthomyxoviridae of negative-strand RNA viruses and are categorized into types A, B, C, and D (4, 5). The type A influenza viruses (IAVs) are further classified into diverse subtypes, such as H1N1 and H5N1, based on hemagglutinin (HA) and neuraminidase (NA) proteins expressed on the surface of the virus (6). Antiviral drugs are available for treating influenza. However, numerous strains of the influenza A and B viruses have been shown to be resistant to the current drugs, presumably due to the frequent alteration of influenza viral genomic sequences and viral adaptation to the host environment (7–9). Thus, it is important to unveil the detailed mechanisms for influenza viral regulation of host immunity and to project new therapeutic strategies to better control influenza.
To establish a successful infection, influenza viruses must evade or counterattack the host immune responses. Interferons (IFNs) function as a crucial line of defense against viral infection, restricting virus replication and the spread of viruses (10, 11). The type I IFNs, including both IFN-α and IFN-β, markedly inhibit virus replication (12–15). Upon influenza virus infection, type I IFNs are secreted and bind to the cognate receptor, type I IFN receptor (IFNAR), to initiate a signaling cascade involving activation of the JAK family of tyrosine kinases and the STAT1/STAT2 transcription factors. This leads to the transcriptional induction of various IFN-stimulated genes (ISGs) (16–18), several of which have been determined to exert direct anti-influenza virus activities (19, 20).
IFN-γ, which is designated type II IFN, is secreted by specific immune cells, such as activated T cells and natural killer (NK) cells. It binds to the IFN-γ receptor (IFNGR) complex to elicit a signal within the pathogen-infected cells or other immune cells (21). IFNGR is composed of two subunits, IFNGR1 and IFNGR2. The association of IFN-γ with IFNGR triggers activation of JAK1/JAK2 to cause STAT1 phosphorylation, resulting in the expression of IFN-γ-inducible genes (21–23). IFN-γ has been shown to be critical for innate and adaptive immunity against viral and bacterial infections by inducing the expression of a distinct class of genes (24–28). For example, the binding of IFN-γ to IFNGR causes cells to increase the expression of components of the major histocompatibility complex (MHC) class I antigen presentation machinery, including transporter associated with antigen processing 1 (TAP-1) and low-molecular-weight polypeptide 2 (LMP-2) (29, 30). Besides type I and type II IFNs, there is also a recently classified group of type III IFNs that includes IFN-λ1 to -4. These IFNs signal through a receptor complex (type III IFN receptor [IFNLR]) to display certain antiviral effects (31–33). The exact functions of this type of IFN remain to be further investigated.
While it has been well established that IAV has strategies to inhibit the production of type I IFNs (34–37), it remains unknown if IAV regulates type II IFN responses. We have recently reported that HA of IAV induces the degradation of type I IFN receptor 1 (IFNAR1), which consequently attenuates cellular sensitivity to type I IFNs (38). However, it is unknown what cellular protein(s) mediates the degradation process upon infection with IAV or any other virus. Here, we show that influenza virus regulates type II IFN signaling by inducing degradation of the IFN-γ receptor IFNGR1. Importantly, casein kinase 1α (CK1α) was demonstrated to be critical for IAV infection or HA-induced degradation of type I and type II IFN receptors. The results indicate that CK1α enhances IAV propagation by mediating the degradation of IFN receptors during infection.
RESULTS
IAV infection downregulates IFNGR1.
IAV infection has been shown to induce degradation of IFNAR1 (38). Here, we sought to investigate if IAV regulates the expression of IFNGR, the receptor for IFN-γ. For this purpose, the protein levels of IFNGR1 and IFNAR1 in A549 cells were examined by Western blotting upon infection with influenza A/WSN/33 (H1N1) virus. The level of endogenous IFNGR1, as well as that of IFNAR1, was dramatically reduced by IAV infection, indicating that IAV infection downregulates receptors for both type I and type II IFNs (Fig. 1A). Additionally, we assessed the level of type III IFN receptor 1 (IFNLR1), but almost no change was observed (Fig. 1A). Similar results were obtained when HEK293 cells were infected by IAV for detection of these three receptors over time (Fig. 1B). The decrease of IFNGR1 and IFNAR1 expression was associated with an increase in viral protein expression. Also, the 2009 pandemic A/CA/04/09 (H1N1) virus (IAV-pH1N1) diminished the level of IFNGR1 and IFNAR1, but not IFNLR1, during infection of A549 cells, similar to the A/WSN/33 (H1N1) virus (Fig. 1C). Since the surface expression of IFNGR1 is important for binding and transferring the signal of type II IFN, we next determined the expression of IFNGR1 present on the cell surface by flow cytometry. IAV infection substantially decreased the level (mean fluorescence intensity [MFI]) of IFNGR1 on the surfaces of cells (from 117 to 71) (Fig. 1D). IAV-induced reduction of IFNGR1 could be caused by the inhibition of mRNA synthesis or posttranscriptional regulation, such as increased protein degradation. When the mRNA levels of IFNGR1 in mock-infected and IAV-infected cells were compared, no change was noted (Fig. 1E), indicating that reduction of the IFNGR1 level occurred at the posttranscriptional stage.
FIG 1.

IAV infection reduces the level of IFNGR1 protein. (A and B) A549 cells (A) and HEK293 cells (B) were infected with IAV at an MOI of 1. The levels of IFNGR1, IFNAR1, IFNLR1, and viral HA were analyzed by Western blotting at the indicated time points postinfection. The levels of GAPDH were used as an internal loading control. (C) A549 cells were infected with pandemic IAV-pH1N1 at an MOI of 1. Cells were harvested at the indicated time points, and the levels of IFNGR1, IFNAR1, IFNLR1, viral M1, and GAPDH were detected by Western blotting. (D) HEK293 cells were mock infected (Mock) or infected with IAV at an MOI of 1 (IAV). At 24 hpi, cells were left unstained or incubated with antibody against IFNGR1, followed by staining with FITC-conjugated secondary antibody. The surface expression levels of IFNGR1 were assessed by flow cytometric analysis. The MFI of each sample is shown. The experiment was repeated three times. (E) HEK293 cells were left uninfected (Mock) or infected with IAV at an MOI of 1. The relative mRNA levels of IFNGR1 were analyzed by real-time qPCR at 24 hpi. The data represent means and SD calculated from three reactions per sample. The experiment was independently repeated twice with similar results. n.s., not significant. Mass, molecular mass (kilodaltons).
Hemagglutinin of IAV targets IFNGR1 for degradation.
Our previous study showed that HA of IAV induces the degradation of type I IFN receptor 1 (38). To determine if the HA protein of IAV regulates the expression of IFNGR1, as well as IFNAR1, we transiently expressed HA in cells and monitored the endogenous protein levels of IFNGR1 by Western blotting. HA robustly decreased the expression of IFNGR1 and IFNAR1, but not IFNLR1 (Fig. 2A), as observed during influenza virus infection (Fig. 1). However, NA, another glycoprotein of influenza virus, did not alter the expression of IFN receptors despite the higher expression level of Myc-tagged NA than of Myc-tagged HA in the cells. Thus, NA served as a negative control (Fig. 2A). The IFNGR1 downregulation was reliant on the expression level of viral HA (Fig. 2B). Also, IAV HA, but not NA, decreased the level of IFNGR1 expressed on the surfaces of cells (MFI reduced from 153 to 80) when assessed by flow cytometry (Fig. 2C). Quantitative-PCR (qPCR) analysis revealed that HA does not affect the mRNA levels of IFNGR1 (Fig. 2D). To investigate if HA influences the stability of IFNGR1, cells were treated with cycloheximide (CHX), which blocks the protein translation step at 15 h posttransfection with HA plasmid for the following 3 h. The time points were chosen because the HA-mediated decrease of IFNGR1 was weak until 18 h posttransfection (20% decrease) under these experimental conditions. This allowed us to compare the degradation of IFNGR1 with and without CHX treatment. The level of IFNGR1 under treatment with CHX decreased more rapidly in the presence of HA protein (Fig. 2E) than in the untreated control, indicating that HA decreased the stability of the IFNGR1 protein. However, viral NA did not regulate IFNGR1 protein stability in the same experimental setting (Fig. 2E). Next, we determined if HA expression causes degradation of IFNGR1 via proteasome-dependent or lysosome-dependent mechanisms. When cells were exogenously treated with lysosome inhibitor (NH4Cl), but not proteasome inhibitor (MG132), HA-induced IFNGR1 degradation was repressed (Fig. 2F). However, treatment with these inhibitors could partially rescue IFNAR1 levels in HA-expressing cells (Fig. 2F), which is consistent with our previous observation (38). To further confirm lysosome-dependent IFNGR1 degradation by viral HA, two other established lysosomal inhibitors, chloroquine and bafilomycin A1, were tested. Addition of these inhibitors rescued IFNGR1 levels in HA-expressing cells, affirming the result for lysosomal degradation of IFNGR1 (Fig. 2G).
FIG 2.

IAV HA targets IFNGR1 for degradation. (A) HEK293 cells were transfected with a control vector (CTR) or plasmids encoding Myc-tagged HA of influenza A/New Caledonia/20/99 (H1N1) virus or Myc-tagged NA of influenza A/Thailand/1 (KAN-1)/2004 (H5N1) virus. At 24 h posttransfection, the levels of IFNGR1, IFNAR1, IFNLR1, Myc-tagged HA, Myc-tagged NA, and GAPDH were analyzed by Western blotting. (B) HEK293 cells were transfected with a control vector or increasing doses of HA-encoding plasmids (125, 250, or 500 ng/ml). Twenty-four hours posttransfection, Western blot analysis was performed to detect the levels of IFNGR1, HA, and GAPDH. The relative intensity of each band of IFNGR1 was determined by densitometry based on the GAPDH levels and is depicted below each blot. The relative level of IFNGR1 from a vector-transfected sample was set as 1.0. (C) HEK293 cells were transfected with a control plasmid or plasmids encoding HA or NA. At 24 h posttransfection, cells were left unstained or incubated with antibody against IFNGR1, followed by staining with FITC-conjugated secondary antibody. The levels of IFNGR1 on the surfaces of cells were assessed by flow cytometry. The MFIs are shown. (D) HEK293 cells were transfected with a control vector or plasmids encoding HA. The mRNA levels of IFNGR1 were analyzed by real-time qPCR at 24 h posttransfection. The error bars represent SD from three reactions per sample, and the samples were from three independent experiments. n.s., not significant. (E) HEK293 cells were transfected with a control vector, HA, or NA as indicated. At 15 h posttransfection, cells were treated with solvent (−) or CHX (+) at a concentration of 40 μg/ml for 3 h. The levels of IFNGR1, HA, NA, and GAPDH were analyzed by Western blotting. The relative intensity of each band of IFNGR1 was determined by densitometry based on the GAPDH levels and is depicted below each blot. (F) HEK293 cells were transfected with a control vector or HA as indicated. At 18 h posttransfection, cells were treated with proteasome inhibitor (20 μM MG132), lysosome inhibitor (20 mM NH4Cl), or solvent (dimethyl sulfoxide [DMSO]) (−) for an additional 6 h. Western blotting was performed to detect the levels of IFNGR1, IFNAR1, IFNLR1, HA, and GAPDH. (G) HEK293 cells were transfected with control vector or HA. At 18 h posttransfection, cells were treated with DMSO (−), NH4Cl (20 mM), chloroquine (CQ) (40 μM), or bafilomycin A1 (Baf-A1) (100 nM) for 6 h. The levels of IFNGR1, HA, and GAPDH were analyzed by Western blotting. (H) A549 cells were mock infected or infected with ΔHA-rIAV at two different MOIs (0.3 and 1). At 24 hpi, the cells were harvested and the levels of IFNGR1, IFNAR1, GFP, M1, M2, and GAPDH were detected by Western blotting. Mass, molecular mass (kilodaltons).
To further investigate the importance of HA in regulating IFN receptors during IAV infection, we infected A549 cells with an HA gene-deficient recombinant IAV (ΔHA-rIAV) and determined the expression levels of type I and type II IFN receptors (Fig. 2H). ΔHA-rIAV is deficient in HA expression, since HA-encoding viral RNA in the viral genome is replaced by a green fluorescent protein (GFP) gene. However, there are HA proteins on the virion due to the virus being generated using HA-expressing cells (39, 40). Therefore, the infection by ΔHA-rIAV undergoes only one round of replication without production of infectious viruses in normal cells. As shown in Fig. 2H, HA gene-deficient IAV failed to downregulate IFNGR1 or IFNAR1, demonstrating the crucial function of HA expressed in the cells in degrading IFN receptors during infection. Collectively, these results indicate that IAV HA induces degradation of IFNGR1 protein via the lysosomal degradation pathway.
IAV infection or HA impairs cellular sensitivity to type II IFN.
Following IFN-γ–IFNGR binding, STAT1 is phosphorylated by JAKs, which leads to the transcriptional activation of type II ISGs (21–23). Since IAV reduces the levels of IFNGR1, we investigated if IAV infection affects cellular responsiveness to type II IFN by regulating IFNGR1. To this end, the activation status of STAT1 was assessed after recombinant IFN-γ (rIFN-γ) was exogenously added to mock-infected or IAV-infected cells. IAV infection strongly inhibited IFN-γ-induced STAT1 phosphorylation (Fig. 3A), while the level of STAT1 remained constant. The inhibition of STAT1 phosphorylation was correlated with the reduction of IFNGR1 (Fig. 3A). Next, we determined whether IAV infection regulates the expression of ISGs induced by IFN-γ, such as IRF-1, TAP-1, and LMP-2. The accumulation of IRF-1 protein elicited by IFN-γ was markedly suppressed in cells infected with IAV compared to mock-infected cells (Fig. 3B). Also, IAV was shown to inhibit IFN-γ-triggered mRNA expression of TAP-1 and LMP-2 (Fig. 3C and D). To further determine the association between IAV-mediated IFNGR1 degradation and ISG inhibition, cells were treated with NH4Cl, followed by IAV infection. Inhibition of the lysosomal degradation pathway partially recovered the IFNGR1 level and consequently restored the IFN-γ-induced expression of IRF-1 to some extent (Fig. 3E), suggesting that the inhibition of ISGs by IAV is closely correlated with the diminished IFNGR1 level during infection.
FIG 3.
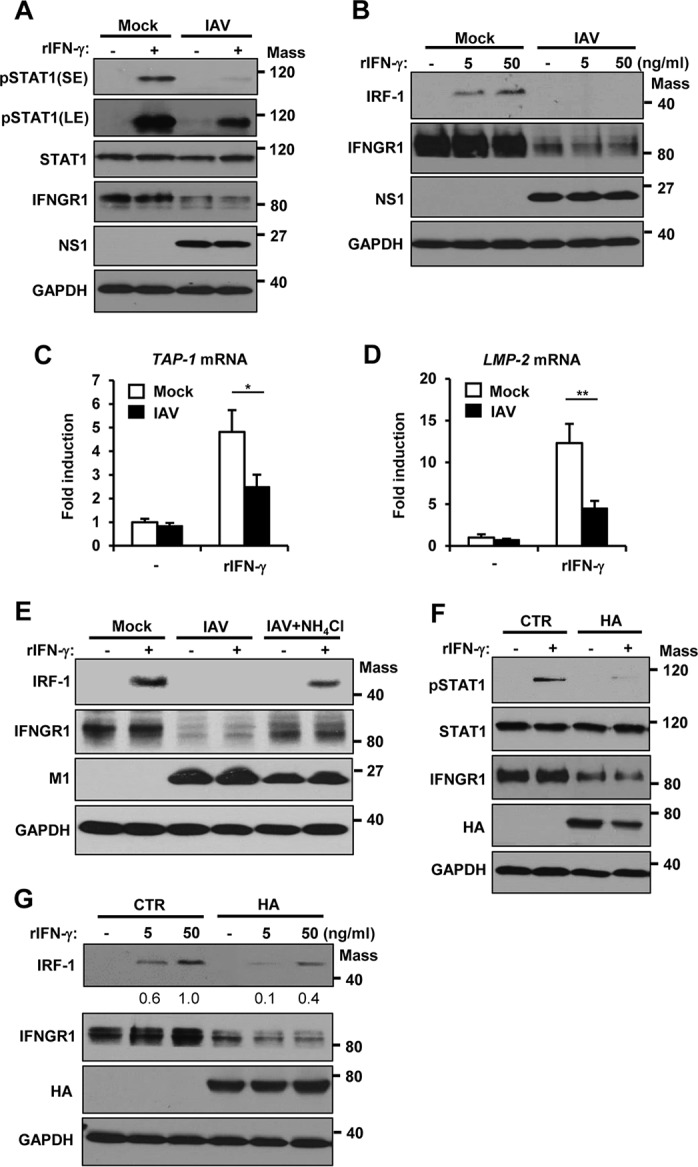
IAV infection or HA renders cells less responsive to recombinant IFN-γ. (A) HEK293 cells were left uninfected (Mock) or infected with IAV at an MOI of 1. At 24 hpi, cells were left untreated (−) or treated with human rIFN-γ (10 ng/ml) for 15 min. The levels of pSTAT1, STAT1, IFNGR1, viral NS1, and GAPDH were determined by Western blotting. The short-exposure (SE) and long-exposure (LE) blots of pSTAT1 are shown. (B) HEK293 cells were infected with IAV at an MOI of 1. At 24 hpi, the cells were treated with rIFN-γ at the indicated concentration. Six hours posttreatment, cells were harvested and Western blotting was performed to detect the levels of IRF-1, IFNGR1, viral NS1, and GAPDH. (C and D) HEK293 cells were left uninfected (Mock) or infected with IAV at an MOI of 1. At 24 hpi, cells were left untreated (−) or treated with rIFN-γ at a concentration of 50 ng/ml. The relative mRNA expression levels of TAP-1 (C) and LMP-2 (D) were analyzed by real-time qPCR at 6 h posttreatment. The error bars represent SD derived from three reactions per sample (*, P ≤ 0.05; **, P ≤ 0.01). The experiment was repeated twice with similar results. (E) HEK293 cells were uninfected (Mock) or infected with IAV at an MOI of 1. At 18 hpi, the cells were treated with NH4Cl (20 mM) for 6 h. At 24 hpi, the cells were treated with rIFN-γ for an additional 6 h. The levels of IRF-1, IFNGR1, viral M1, and GAPDH were analyzed by Western blotting. (F) HEK293 cells were transfected with a control vector or plasmids encoding HA. At 24 h posttransfection, cells were left untreated (−) or treated with rIFN-γ (10 ng/ml) for 15 min. The levels of pSTAT1, STAT1, IFNGR1, HA, and GAPDH were detected. (G) HEK293 cells were transfected with a control vector or HA; 24 h posttransfection, the cells were treated with rIFN-γ at the indicated concentrations for 6 h. Western blotting was performed to detect the levels of IRF-1, IFNGR1, HA, and GAPDH. Mass, molecular mass (kilodaltons).
To establish the role of HA in regulating type II IFN signaling, we next examined the effect of HA expression on IFN-γ-induced STAT1 activation (Fig. 3F) and IRF-1 expression (Fig. 3G). Similar to IAV infection, HA inhibited both STAT1 phosphorylation and IRF-1 expression in response to rIFN-γ, which was related to the downregulation of IFNGR1 (Fig. 3F and G). These results indicate that IAV infection or HA expression substantially diminishes cellular responses to type II IFNs.
IAV or HA induces phosphorylation and polyubiquitination of IFNGR1.
The endocytosis and lysosomal degradation of membrane proteins, such as IFNAR1, is often associated with serine phosphorylation and the subsequent ubiquitination of those proteins (41, 42). Therefore, we determined if IAV infection or HA expression triggers the phosphorylation or ubiquitination of IFNGR1. To this end, FLAG-tagged IFNGR1 was transiently overexpressed in cells, followed by either IAV infection (Fig. 4A) or HA transfection (Fig. 4B). Serine phosphorylation of IFNGR1 was analyzed via denatured immunoprecipitation (IP) and subsequent Western blot analysis. IAV infection (Fig. 4A) or HA expression (Fig. 4B) was shown to enhance the phosphorylation of IFNGR1. Also, we examined the ubiquitination status of immunoprecipitated IFNGR1 during IAV infection or HA expression. Upon infection, an increase of IFNGR1 polyubiquitination was observed (Fig. 4C). Similarly, HA expression promoted the polyubiquitination of IFNGR1 (Fig. 4D).
FIG 4.

IAV infection or HA induces phosphorylation and polyubiquitination of IFNGR1. (A) HEK293 cells were transfected with FLAG-tagged IFNGR1. At 24 h posttransfection, cells were left uninfected or infected with IAV at an MOI of 1, as indicated. The cells were then cultured for an additional 24 h and subjected to a denatured IP experiment. NH4Cl (20 mM) was added to the cell culture 6 h before harvest. The phosphorylation of immunoprecipitated FLAG-IFNGR1 was analyzed by Western blotting (IB) using anti-phosphoserine (p-Serine) antibody. The levels of immunoprecipitated FLAG-IFNGR1, viral M1, and GAPDH in the whole-cell lysates were also detected. (B) HEK293 cells were transfected with FLAG-tagged IFNGR1. At 24 h posttransfection, the cells were transfected with empty-vector control (−) or plasmids encoding HA, as indicated. The cells were cultured for an additional 24 h and subjected to denatured IP. NH4Cl was added to the cell culture 6 h before harvest. The phosphorylation of immunoprecipitated FLAG-IFNGR1 was detected. (C) HEK293 cells were transfected with FLAG-IFNGR1 and/or tHA-Ub. At 24 h posttransfection, cells were left uninfected or infected with IAV at an MOI of 1, as indicated, for an additional 24 h. All of the transfected cells were treated with NH4Cl (20 mM) for 8 h before harvest. The whole-cell lysates were subjected to a denatured IP experiment. The levels of ubiquitination of IFNGR1 were analyzed by Western blotting. (D) HEK293 cells were transfected with FLAG-IFNGR1 and/or HA-tagged ubiquitin, as indicated. At 24 h posttransfection, cells were transfected with a control vector or plasmids encoding HA. The cells were harvested at 18 h posttransfection. The whole-cell lysates were subjected to a denatured IP. The levels of ubiquitination of IFNGR1 were detected by Western blotting. Mass, molecular mass (kilodaltons).
CK1α regulates degradation of IFNGR1 and IFNAR1 upon IAV infection or HA expression.
The degradation of IFNGR1, as well as IFNAR1, caused by influenza virus infection prompted us to investigate the detailed underlying mechanisms. However, little is known about the mechanism by which IFNGR1 is degraded in cells. IFNAR1 can be degraded via phosphorylation by cellular kinases during endoplasmic reticulum (ER) stress response (unfolded-protein response) or upon treatment with high levels of type I IFN (41, 43–45). Therefore, we tested the possible involvement of these kinases in IAV-induced degradation of IFN receptors. The cellular kinase protein kinase D2 (PKD2) is known to phosphorylate IFNAR1 in response to type I IFN treatment, which is crucial for ensuring IFNAR1 ubiquitination and degradation (46, 47). Therefore, we first tested if PKD2 regulates HA-induced degradation of IFNAR1. Treatment of cells with recombinant human IFN-α reduced IFNAR1 levels, and this reduction was inhibited when cells were incubated with a PKD2-specific inhibitor (Fig. 5A). In contrast, treating cells with the PKD2 inhibitor did not alter the HA-induced diminishment of IFNAR1 (Fig. 5A), indicating that PKD2 is not involved in HA-induced IFNAR1 degradation. Similarly, PKD2 inhibition failed to affect the degradation of IFNGR1 caused by influenza viral HA (Fig. 5A).
FIG 5.

Pharmacological inhibition of CK1α led to impaired downregulation of IFN receptors during IAV infection or HA expression. (A) HEK293 cells were pretreated with PKD2 inhibitor (PKD2-I) or solvent (DMSO) for 1 h, followed by transfection with plasmids encoding HA or treatment with human rIFN-α (1,000 U/ml). Cells were harvested for Western blotting at 24 h posttransfection or 6 h after rIFN-α treatment. (B) HEK293 cells were transfected with either an empty vector (CTR) or plasmids encoding HA; 18 h later, cells were treated with either DSMO or SB203580 (p38 MAPK inhibitor) for an additional 6 h. Some of the cells were cotreated with TG (1 μM), as indicated. The levels of IFNGR1, IFNAR1, HA, and GAPDH were detected by Western blotting. (C) HEK293 cells were treated with TG (1 μM) in combination with CK1α-I (5 μM) or solvent (DMSO). At 6 h posttreatment. Western blotting was performed to detect the levels of IFNGR1, IFNAR1, pelF2α, elF2α, and GAPDH. (D) HEK293 cells were pretreated with CK1α-I (5 μM) or solvent (DMSO) for 1 h and then infected with IAV at an MOI of 1. At 24 hpi, the levels of IFNGR1, IFNAR1, viral NS1, and GAPDH were detected by Western blotting. (E) HEK293 cells were pretreated with CK1α-I (5 μM) or solvent (DMSO) for 1 h, followed by transfection with an empty vector (CTR) or HA. Twenty-four hours posttransfection, the levels of IFNGR1, IFNAR1, HA, and GAPDH were analyzed by Western blotting. Mass, molecular mass (kilodaltons).
The PKR-like ER kinase (PERK) pathway of ER stress response induces phosphorylation of elF2α, followed by the activation of p38 MAP kinase (p38 MAPK). In this ER stress response situation, p38 MAPK is known to mediate the priming phosphorylation of IFNAR1, which is required for its ensuing degradation (43, 48, 49). We first sought to determine whether HA-induced IFNGR1 or IFNAR1 degradation is dependent on the activation of p38 MAPK. HA expression or the treatment of cells with thapsigargin (TG), which is an inducer of the ER stress response, decreased the levels of both IFNGR1 and IFNAR1 (Fig. 5B, left). TG (ER stress)-induced IFNAR1 degradation was suppressed when cells were incubated with the inhibitor of p38 MAPK (Fig. 5B), as reported previously (48). However, the HA-induced degradation of both IFNGR1 and IFNAR1 was unaltered by the treatment with p38 MAPK inhibitor (Fig. 5B). This result indicates that HA-mediated degradation of IFN receptors is independent of the activation of p38 MAPK.
Of note, TG-induced IFNGR1 elimination was also independent of p38 MAPK (Fig. 5B), suggesting that IFNGR1 regulation is different from the IFNAR1 degradation process under the ER stress response. In the PERK pathway of ER stress response, the p38 MAPK-mediated phosphorylation of IFNAR1 is thought to be a prerequisite for the subsequent phosphorylation of IFNAR1 by CK1α (48, 50, 51). While p38 MAPK is not involved in TG-induced degradation of IFNGR1, we evaluated the possible role of CK1α in regulating IFNGR1 expression. As shown in Fig. 5C, the treatment of cells with CK1α inhibitor (CK1α-I) markedly inhibited the TG-induced degradation of IFNGR1. This means that CK1α, but not p38 MAPK, is critical for ER stress response-mediated IFNGR1 degradation, while both p38 MAPK and CK1α are important for ER stress response-induced IFNAR1 degradation.
Next, we determined if CK1α plays a role in influenza virus-induced degradation of IFNGR1 or IFNAR1. Cells were pretreated with CK1α-I, followed by IAV infection. The inhibition of CK1α resulted in almost no degradation of IFN receptors during IAV infection, which is associated with the inhibition of virus replication (Fig. 5D). Importantly, the inhibitor of CK1α impaired the decrease of both receptors' expression caused by influenza viral HA (Fig. 5E). To further investigate the regulatory effect of CK1α, we utilized the knockdown approach by using a small interfering RNA (siRNA) specific to CK1α. When CK1α was downregulated by using the siRNA, the reduction of IFNGR1 and IFNAR1 expression was strongly inhibited during infection by IAV (strain WSN) (Fig. 6A) or IAV-pH1N1 (Fig. 6B). This observation is also associated with the repression of IAV replication. Consistent with the result from the IAV infection experiment, HA expression failed to markedly reduce receptor levels in the cells that had decreased expression of CK1α (Fig. 6C). Taken together, these data indicate that CK1α is critical for regulating HA-mediated elimination of type I and type II IFN receptors.
FIG 6.
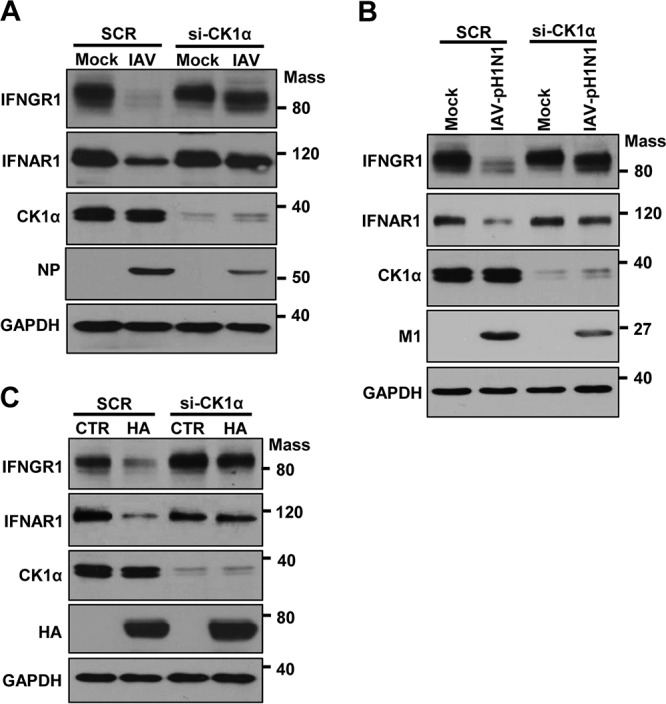
Knockdown of CK1α results in impaired degradation of IFNGR1 and IFNAR1 during IAV infection or HA expression. (A and B) HEK293 cells were transfected with siRNA specific to CK1α (siCK1α) or nonspecific scrambled control siRNA (SCR). At 24 h posttransfection, cells were left uninfected (Mock), infected with IAV (A), or infected with IAV-pH1N1 (B) at an MOI of 1. Twenty-four hours later, the levels of IFNGR1, IFNAR1, CK1α, viral NP, viral M1, and GAPDH were analyzed by Western blotting. (C) HEK293 cells were transfected with siCK1α or SCR. At 24 h posttransfection, cells were transfected with a control vector or plasmids encoding HA. Western blotting was performed 24 h after HA transfection, and the levels of IFNGR1, IFNAR1, CK1α, HA, and GAPDH were detected. Mass, molecular mass (kilodaltons).
CK1α is crucial for IAV-induced phosphorylation and ubiquitination of both IFNGR1 and IFNAR1.
Since CK1α functions as a kinase and the phosphorylation of IFNAR1 is known to be important for its subsequent degradation pathway (50, 51), we investigated if CK1α is critical for IAV-induced phosphorylation and ubiquitination of IFN receptors. To this end, FLAG-tagged IFNGR1 was transiently expressed, and cells were then treated with CK1α inhibitor or solvent, followed by IAV infection. IAV infection led to a robust increase in the phosphorylation of IFNGR1 (Fig. 7A), which was consistent with our data shown in Fig. 4A. However, treatment of cells with the CK1α inhibitor prevented the increase in the phosphorylation level of IFNGR1 caused by IAV infection (Fig. 7A), confirming CK1α-mediated phosphorylation of IFNGR1 upon infection. Furthermore, IAV infection noticeably increased the extent of polyubiquitination of IFNGR1, while treatment with the CK1α inhibitor drastically inhibited infection-induced ubiquitination (Fig. 7B). Similar experiments were performed to determine the inhibitory effect of CK1α inhibitor on IAV-induced phosphorylation or ubiquitination of the receptor for type I IFNs. The increase of phosphorylation (Fig. 7C) and ubiquitination (Fig. 7D) of IFNAR1 caused by IAV infection was impaired when cells were incubated with the CK1α inhibitor. These findings support the conclusion that CK1α is pivotal for the phosphorylation of both IFNAR1 and IFNGR1 and their subsequent degradation process upon influenza virus infection.
FIG 7.

CK1α is crucial for IAV-induced phosphorylation and ubiquitination of IFNGR1 and IFNAR1. (A and B) HEK293 cells were transfected with FLAG-IFNGR1 (A) or cotransfected with FLAG-IFNGR1 and tHA-Ub (B). At 24 h posttransfection, cells were left uninfected or infected with IAV at an MOI of 1 in the presence of CK1α-I (5 μM) or solvent (DMSO). The cells were treated with NH4Cl 6 h before harvest. At 24 hpi, the whole-cell lysates were subjected to a denatured IP experiment. The level of phosphorylation of IFNGR1 (A) or ubiquitination of IFNGR1 (B) was analyzed by Western blotting using the indicated antibodies. (C and D) HEK293 cells were transfected with FLAG-IFNAR1 (C) or cotransfected with FLAG-IFNAR1 and tHA-Ub (D), as indicated. At 24 h posttransfection, the cells were infected with IAV at an MOI of 1. The cells were treated with NH4Cl 6 h before harvest. The phosphorylation of IFNAR1 (C) or ubiquitination of IFNAR1 (D) was assessed as described for panels A and B. Mass, molecular mass (kilodaltons).
CK1α functions as a proviral factor for efficient IAV propagation.
Since IFNAR1 is essential for type I IFN-mediated antiviral responses (43, 52–54), CK1α-regulated degradation of IFNAR1 could be beneficial for IAV replication. To investigate the role of CK1α during IAV replication, A549 cells (Fig. 8A) or HEK293 cells (Fig. 8B) were pretreated with CK1α inhibitor, and then its effect on IAV replication was investigated. IAV decreased the IFNAR1 levels, which were restored following CK1α inhibitor treatment (Fig. 8A and B). The expression levels of viral proteins clearly decreased when both cell types were supplied with the CK1α inhibitor. Further, the addition of CK1α inhibitor on cells also impaired the replication of IAV-pH1N1 at a multiplicity of infection (MOI) of 1 or 3 (Fig. 8C). In support of these inhibitor studies, knockdown of endogenous CK1α by siRNA noticeably inhibited IAV replication in comparison to the scrambled control siRNA-treated condition (Fig. 8D). To further examine the importance of CK1α during IAV replication, CK1α was transiently overexpressed in cells, followed by IAV infection. As shown in Fig. 8E, overexpressed CK1α improved IAV replication, as evidenced by the increased expression of viral proteins, confirming the proviral function of CK1α. To better understand the biological importance of CK1α in IAV propagation, we measured the titers of infectious virus particles released to the supernatant from infected cells with the treatment of CK1α inhibitor or its solvent control at 12, 24, 48, and 72 h postinfection (hpi). The virus titers in the supernatant from cells treated with CK1α inhibitor were strongly reduced, by approximately 100-fold, in comparison to the solvent-treated control cells at 24, 48, or 72 hpi (Fig. 8F). This supports the conclusion that CK1α displays a proviral function during influenza virus replication.
FIG 8.

CK1α regulates IAV propagation. (A and B) A549 cells (A) or HEK293 cells (B) were pretreated with CK1α-I (5 μM) or solvent (DMSO) 1 h before infection with IAV at an MOI of 1. At 24 hpi, the levels of NS1, NP, M1, IFNAR1, and GAPDH were analyzed by Western blotting. (C) HEK293 cells were pretreated with CK1α-I or DMSO for 1 h and then infected with IAV-pH1N1 at the indicated MOIs. The levels of HA, M1, IFNAR1, and GAPDH were detected. (D) HEK293 cells were transfected with siCK1α or SCR. At 24 h posttransfection, the cells were infected with IAV at an MOI of 1. Western blot analysis was performed 24 h postinfection to assess the levels of NP, M1, IFNAR1, CK1α, and GAPDH. (E) HEK293 cells were transfected with a control vector or plasmid encoding Myc-tagged CK1α. At 24 h posttransfection, the cells were infected with IAV at an MOI of 1 for an additional 24 h. The levels of NP, HA, Myc-CK1α, and GAPDH were detected by Western blotting. (F) A549 cells were treated with solvent (DMSO) or CK1α-I and infected with IAV at an MOI of 0.001. At 12, 24, 48, or 72 hpi, the titers of infectious IAV in the supernatants of the cultures were assessed by plaque assay on MDCK cells. Each data point represents the mean of samples generated from three independent experiments. The data represent means ± SD (**, P ≤ 0.01; ***, P ≤ 0.001). Mass, molecular mass (kilodaltons).
Since CK1α may control multiple cellular processes (55–57), we could not exclude the possibility that CK1α enhances influenza virus replication by not only downregulating IFN receptors, but also regulating cellular signaling that is independent of IFN. To investigate this, we utilized IFN production-incompetent Vero cells, which are unable to synthesize type I IFNs, and evaluated the effects of CK1α inhibitor on viral replication. As shown in Fig. 9A, treatment of Vero cells with CK1α inhibitor did not alter the expression levels of viral proteins in the absence of IFN, although it impaired IFNAR1 degradation. This result suggests that the antiviral activity of CK1α inhibitor is reliant on type I IFN signaling. Moreover, cells treated with CK1α inhibitor displayed stronger type I IFN responses during IAV infection, which was evidenced by the increased expression of ISGs, such as ISG56, RIG-I, and OAS1 (Fig. 9B).
FIG 9.

CK1α inhibition increases type I IFN receptor signaling and represses IAV propagation. (A) Vero cells were pretreated with CK1α-I (5 μM) or DMSO for 1 h and then infected with IAV at the indicated MOIs. The levels of NP, NS1, IFNAR1, and GAPDH were analyzed by Western blotting at 24 hpi. (B) A549 cells were pretreated with DMSO (−) or CK1α-I (+) for 1 h, and the cells were then infected with IAV at an MOI of 0.1. At 2 days postinfection, Western blot analysis was performed to detect the levels of ISG56, RIG-I, OAS1, M1, M2, and GAPDH. Mass, molecular mass (kilodaltons). (C) (Left) A549 cells were transfected with siRNA specific to IFNAR1 (si-IFNAR1) or nonspecific scrambled control siRNA (SCR). At 24 h posttransfection, cells were treated with DMSO or CK1α-I (5 μM) and infected with IAV at an MOI of 0.001. The titers of infectious virus in the supernatants of the culture were assessed by plaque assays on MDCK cells at 24, 48, or 72 hpi. Each data point on the curve represents the mean of three independently obtained samples. The data represent means ± SD. P values were calculated between DMSO-treated si-IFNAR1-transfected samples (■) and si-IFNAR1-transfected CK1α inhibitor-treated samples (▲). (Right) A549 cells were transfected with SCR or si-IFNAR1, and at 48 h posttransfection, downregulation of the endogenous IFNAR1 was confirmed by Western blotting. (D) (Left) A549 cells were transfected with siRNA specific to IFNGR1 (si-IFNGR1) or SCR. At 24 h posttransfection, cells were treated with DMSO or CK1α-I (5 μM) and infected with IAV at an MOI of 0.001, and at 48 hpi, viral titers were determined by plaque assay. The data shown are from three independently obtained samples (***, P ≤ 0.001; ****, P ≤ 0.0001; n.s., not significant). (Right) A549 cells were transfected with SCR or si-IFNGR1, and downregulation of IFNGR1 was confirmed 48 h later by Western blotting. Mass, molecular mass (kilodaltons).
To further determine the importance of IFN signaling in CK1α regulation of IAV replication, A549 cells were transfected with either IFNAR1-specific siRNA or a universal scrambled negative-control siRNA duplex. The cells were then infected with IAV in the presence or absence of CK1α inhibitor. When IFNAR1 was downregulated, CK1α inhibitor failed to significantly repress infectious-virus production from A549 cells at 24, 48, and 72 hpi (Fig. 9C). The results indicate that type I IFN receptor signaling is critical for CK1α regulation of IAV replication in A549 cells. However, since IFN-γ is produced only from immune cells, such as NK cells and T cells, there should be no IFN-γ secreted from human epithelial A549 cells. As expected, knockdown of IFNGR1 by using an siRNA did not alter the inhibitory effect of CK1α inhibitor on IAV replication in A549 cells (Fig. 9D). Therefore, the proviral activity of CK1α we observed in our cell culture system is mediated by the regulation of type I IFN signaling.
DISCUSSION
In this study, we demonstrate that influenza virus infection induces the phosphorylation and ubiquitination of the type II IFN receptor, IFNGR1, resulting in the degradation of IFNGR1 and thereby inhibiting cellular responses to IFN-γ. Influenza viral HA was shown to mediate the lysosome-dependent degradation of IFNGR1. Importantly, the cellular kinase CK1α was found to be critical for regulating the IAV-induced degradation of IFNGR1 and IFNAR1 and promoting IAV replication.
IFN-γ is a biologically active 34-kDa homodimer that is secreted from immune cells, such as NK cells and T cells. IFN-γ mediates a wide range of immunoregulatory effects on both innate and acquired immunity (21, 24). In particular, it has been shown to enhance antigen presentation on pathogen-infected cells by increasing the expression of molecules involved in antigen processing and the MHC presentation pathway (29, 30). Also, it could increase the expression of molecules involved in the death receptor pathway of cellular apoptosis in infected cells. These processes may help T or B cells effectively detect and eliminate virus-infected cells, clearing the infection. Our findings indicate that influenza virus induces degradation of IFNGR1 and desensitizes cells to IFN-γ. Thus, elimination of IFNGR by influenza virus should prevent or delay the recognition of infected cells by virus-reactive immune cells. Additionally, IAV-infected monocytes and dendritic cells may not effectively respond to IFN-γ from NK or T cells in producing molecules such as interleukin 12 (IL-12) to promote the host immune response. Taken together, these factors could allow IAV to escape the surveillance of the host immune system. While less well known, IFN-γ may also display antiviral activity by directly inhibiting virus replication in specific cell types (26), which requires further investigation. Future studies should involve developing systems to evaluate the physiological importance of IFNGR1 degradation during influenza.
It has been reported that Kaposi's sarcoma-associated herpesvirus (KSHV) downregulates IFN-γ receptor 1 via viral K3 and K5 proteins. It was shown that the overexpression of viral K3 or K5 induces polyubiquitination of IFNGR1 (58). To our knowledge, this is the only example that illustrates degradation of IFNGR1 caused by virus infection. While hepatitis C virus infection was shown to impair the expression of IFNAR1 and IFNGR1 in primary human hepatocytes, it was unclear if the downregulation occurred by degradation of IFN receptors (59). Since specific phosphorylation is a prerequisite for ubiquitination and subsequent degradation of certain proteins, such as IκBα, β-catenin, and IFNAR1 (41, 60–62), we tested if IFNGR1 is phosphorylated by IAV infection or HA ectopic expression. Indeed, the levels of phosphorylation and ubiquitination of IFNGR1 increased upon IAV infection or HA expression. This suggests that the phosphorylation of IFNGR1 is closely connected with the degradation pathway of IFNGR1 upon infection.
The mechanism of IFNGR1 phosphorylation directing degradation of the receptor is currently unknown, and no kinase(s) has been reported to phosphorylate IFNGR1 for the degradation pathway. However, there are several kinases involved in the process of IFNAR1 phosphorylation. PKD2 mediates the ligand (IFN-α/β)-induced phosphorylation of IFNAR1 (46, 47), while both p38 MAPK and CK1α phosphorylate IFNAR1 upon activation of the ER stress response (48, 50). The PERK pathway of ER stress response activates p38 MAP kinase, which mediates the priming phosphorylation of IFNAR1 at S532. This priming phosphorylation enhances the phosphorylation of IFNAR1 at S535 by CK1α, leading to the degradation of the protein (43, 48, 50, 51). When we investigated the roles of p38 MAPK, PKD2, and CK1α in regulating IFNGR1 or IFNAR1 protein stability during IAV infection, CK1α, but not the others, was identified as a cellular factor critical for regulating IAV-induced degradation of both IFNGR1 and IFNAR1. CK1α belongs to the casein kinase family, which is capable of affecting a multitude of signaling pathways via phosphorylation or can regulate transcription toward specific genes (55–57). Currently, it is unknown how IAV HA induces activation of CK1α and the ensuing degradation process of IFN receptors. Possibly, HA directly or indirectly induces the activation of CK1α to increase association of CK1α with IFN receptors. The detailed molecular interaction and activation are under investigation.
Of note, HA gene-deficient IAV still expressed HA proteins on the virion but failed to downregulate IFN receptors. This suggests that the interaction between HA and the cellular receptor, sialic acid, does not trigger degradation of IFN receptors. It is conceivable that the localization or the local amount of HA protein is critical for IFN receptor degradation; the newly generated HA positioned in a specific intracellular compartment, such as the ER, but not the HA on the virion may activate a signal pathway for receptor degradation. In support of this, the diminishment of IFNGR1 and IFNAR1 levels was correlated with the increased levels of viral proteins newly synthesized in the cells over time (Fig. 1A to C). This line of research requires further exploration.
ER stress response can be caused by three major pathways, PERK, activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1) (63). Among them, PERK-mediated ER stress response is known to cause IFNAR1 degradation (43). However, IAV infection was shown to induce the IRE1-mediated ER stress response but does not cause the PERK-mediated ER stress response that was documented to induce IFNAR1 degradation (63). Consistent with this, IAV HA overexpression does not induce PERK-mediated ER stress responses, either (38, 64). While the PERK-mediated ER stress response requires the activation of both p38 MAPK and CK1α for degradation of IFNAR1, IAV infection needs only CK1α, not p38 MAPK, for IFNAR1 elimination. Therefore, the IAV HA-mediated IFNAR1 degradation process appears to be different from the ER stress response-induced IFNAR1 degradation pathway. However, our results indicate that the degradation of IFNGR1 occurs upon IAV infection or ER stress response, and this degradation is dependent on CK1α but independent of p38 MAPK activity under IAV infection or ER stress response. It will be interesting to further investigate detailed molecular mechanisms to elucidate the commonality and differences between the two pathways (IAV versus ER stress response) for IFN receptor degradation.
Upon IAV infection, IFNAR1 can be degraded via proteasome or lysosomal pathways (38). However, IAV-induced IFNGR1 degradation is solely reliant on the lysosomal degradation pathway and independent of the proteasome pathway. Since both receptors' degradation pathways require CK1α upon IAV infection, it is likely that the two pathways become divergent at or after the ubiquitination step, such as in the ubiquitination type and postubiquitination sorting process. Defining the detailed action mechanisms could enhance our understanding of the commonality and differences between the two pathways.
The levels of IFN receptors present on the surfaces of cells are vital for triggering the IFN-mediated antiviral effects (52). The lack of IFN receptors on the cell surface desensitizes cells to IFNs, forming a state favoring viral propagation. Thus, the molecules involved in the virus-induced IFN receptor degradation pathway are anticipated to function as proviral factors. Indeed, our pharmacologic and genetic studies support the conclusion that CK1α is crucial for receptor degradation and efficient IAV replication. Thus, CK1α may represent a new cellular target for inhibiting influenza virus replication.
Taken together, our findings highlight the function of influenza viral HA as an antagonist of IFN responses to obstruct the host defense system. In particular, this research reveals the importance of a cellular factor, CK1α, in controlling IAV-induced degradation of receptors for both type I and II IFNs and enhancing IAV propagation. This could be another immune-evasive strategy that influenza virus has implemented for enhanced viral spread. Investigation into the comprehensive action modes of IFN receptor elimination by IAV could extend our knowledge of viral pathogenesis and unveil new therapeutic targets for improved containment of influenza.
MATERIALS AND METHODS
Virus and cells.
Influenza A/WSN/33 (H1N1) virus was initially provided by Yoshihiro Kawaoka (University of Wisconsin—Madison). The pandemic influenza A/CA/04/09 (H1N1) virus was used as reported previously (65). The HA gene-deficient recombinant influenza A/Puerto Rico/8/34 (H1N1) virus (ΔHA-rIAV) was provided by Adolfo Garcia-Sastre (Mount Sinai School of Medicine). The recombinant virus lacks the HA-encoding viral RNA but contains GFP instead (39, 40). The wild-type (WT) viruses were amplified on Madin-Darby canine kidney (MDCK) cells as described previously (66–68). The source of MDCK cells was described previously (69). Virus infection and titration were performed as described previously (38–40, 66, 68, 69). For infection, cells were incubated with the indicated virus for 1 h, and then cells infected with the A/WSN/33 (H1N1) virus were incubated with fetal bovine serum (FBS)-containing medium, while cells infected with the A/CA/04/09 (H1N1) virus or ΔHA-rIAV were incubated with FBS-free medium containing 0.3% bovine serum albumin (BSA) and tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (1 μg/ml). The supernatants containing infectious viruses were harvested for titration by plaque assay on MDCK cells. For the plaque assay, using serial dilutions of culture supernatants, viruses were adsorbed onto 2 × 105 MDCK cells/ml for 1 h, and then the cells were incubated with 2× Eagle's minimum essential medium (EMEM) (Gibco) mixed with an equal portion of 1% agarose (SeaKem ME). Human embryonic kidney (HEK) 293 cells; human lung epithelial A549 cells; and Vero cells, which are kidney epithelial cells from African green monkeys, have been described previously (69–71). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) as explained previously (38, 68, 69, 71). The MDCK cells were cultured in minimum essential medium Eagle (MEM) (Mediatech). The cells were cultured in a CO2 incubator at 37°C, and all media were supplemented with 10% FBS (HyClone) and penicillin (100 U/ml)/streptomycin (100 μg/ml) (Invitrogen) unless specifically otherwise indicated.
Reagents and antibodies.
Reagents, including anti-DYKDDDDK (FLAG) G1 affinity resin (GenScript); CHX (Sigma-Aldrich), which was used to block protein synthesis; the protease inhibitor phenylmethylsulfonyl fluoride (PMSF) (Gold Bio); IP lysis buffer (Thermo Scientific); the proteasome inhibitor MG132 (Fisher Scientific); lysosome inhibitors, including NH4Cl (Fisher Scientific), chloroquine (Acros Organics), and bafilomycin A1 (Cayman Chemical); the p38 MAP kinase inhibitor SB203580 (Sigma-Aldrich); the PKD2 inhibitor CID755673 (Cayman); the CK1α inhibitor D4476 (Cayman); TG (Tocris); recombinant human IFN-α (PBL Assay Science); and recombinant human IFN-γ (Gibco), were purchased from the indicated manufacturers. Antibodies against influenza viral NP, viral M1, viral M2, human IFNAR1, and pIFNAR1 (S355/S539) were purchased from Abcam; the antibody against influenza viral NS1 was purchased from Santa Cruz; antibodies against human GAPDH (glyceraldehyde-3-phosphate dehydrogenase), elF2α, pelF2α, STAT1, pSTAT1, IRF-1, CK1α, RIG-I, ISG56, OAS1, FLAG tag, Myc tag, GFP tag, and HA tag were purchased from Cell Signaling Technology; antibodies against IAV HA and NA were purchased from GeneTex; antibodies against human IFNGR1 and IFNLR1 were purchased from R&D Systems; the antibody against phosphoserine was purchased from Millipore; the Alexa Fluor 488 goat anti-mouse antibody was purchased from Invitrogen.
Constructs and transfection.
Mammalian expression plasmids encoding C-terminally 3×FLAG-tagged WT IFNAR1 and Myc-tagged CK1α were provided by Serge Fuchs (University of Pennsylvania) (38, 43, 44). The mammalian expression plasmid encoding 8×HA-tagged WT ubiquitin (tHA-Ub) was provided by Mark Hannink (University of Missouri—Columbia) (72). Plasmids encoding influenza viral HA from A/New Caledonia/20/99 (H1N1) and viral NA from A/Thailand/1(KAN-1)/2004 (H5N1) were provided by NIH/NIAID as described previously (38, 73–75). To generate the Myc-tagged HA, the coding sequence of HA was amplified by PCR from VRC9333, which contains full-length HA of influenza virus A/New Caledonia/20/99 (H1N1), with the primers 5′-CGG AAT TCG GAT GAA GGC CAA ACT GCT G-3′ and 5′-GGG GTA CCT CAG ATA CAG ATC CTG CAC TGC A-3′. The amplified HA-encoding PCR fragment was treated with EcoRI/KpnI and inserted into the EcoRI/KpnI-digested vector pCMV-Myc-N (provided by David Pintel, University of Missouri—Columbia). To generate the Myc-tagged NA, the plasmid NA7708, which encodes NA of influenza virus A/Thailand/1(KAN-1)/2004 (H5N1), was treated with SalI/KpnI, and the NA-encoding fragment was extracted and inserted into the SalI/KpnI-digested vector pCMV-Myc-N. The plasmid encoding FLAG- and Myc-tagged human IFNGR1 was purchased from OriGene. For transient expression, HEK293 cells were transfected with plasmids encoding IAV HA or NA at a concentration of 500 ng/ml by using LipoD293 transfection reagent (SignaGen) and following protocols recommended by the manufacturer. Empty-vector control plasmids were always used to ensure that each transfection sample received the same amounts of total DNA and transfection reagents.
Western blot analysis.
Standard Western blotting was performed as previously described (38, 68, 69, 71). Briefly, cells were lysed in 2× sample buffer containing β-mercaptoethanol and heated at 95°C for 10 min. Equal amounts of protein samples were resolved on a sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred to a nitrocellulose membrane (Bio-Rad). Membrane-bound antibodies were detected using enhanced chemiluminescence substrate (Thermo Scientific). All the data presented were the results of experiments repeated at least twice with independent experimental settings.
Denatured immunoprecipitation assay.
For detection of phosphorylation of IFNGR1 or IFNAR1 during IAV infection, HEK293 cells (1 × 106) were transfected with FLAG-Myc-IFNGR1 (1 μg) or FLAG-IFNAR1 (1 μg). At 24 h posttransfection, the cells were left uninfected or infected with IAV at an MOI of 1 for an additional 24 h. NH4Cl (20 mM) was added to the cell culture for 6 h prior to harvest at 18 h postinfection. For detection of ubiquitination of IFNGR1 or IFNAR1 during IAV infection, HEK293 cells (1 × 106) were transfected with tHA-Ub and either FLAG-Myc-IFNGR1 (1 μg) or FLAG-IFNAR1 (1 μg). At 24 h posttransfection, the cells were mock infected or infected with IAV at an MOI of 1 for an additional 24 h. To increase the amounts of modified IFNAR1 or IFNGR1, NH4Cl (20 mM) was added to the cell culture for 6 h prior to harvest. To assess IFNGR1 ubiquitination upon HA expression, HEK293 cells (1 × 106) were transfected with FLAG-Myc-IFNGR1 (1 μg) and tHA-Ub (0.5 μg). At 24 h posttransfection, the cells were transfected with either a control vector or plasmids encoding HA, as indicated. The cells were lysed in 200 μl of lysis buffer containing 1% SDS and denatured by boiling at 95°C for 5 min. This rapid lysis inactivates cellular ubiquitin hydrolases and therefore preserves Ub-IFNAR1 conjugates present in cells prior to IP. The procedure also disrupts protein-protein interactions, which can affect the results. For IP experiments, the cell lysates were diluted 5-fold (final volume, 1 ml) in regular IP lysis buffer containing protease inhibitor (1 mM PMSF) and incubated with 20 μl of anti-DYKDDDDK G1 affinity resin overnight with rotation at 4°C. The beads were washed three times intensively with IP lysis buffer to remove nonspecific binding. The precipitates were analyzed by Western blotting. Experiments were independently repeated twice with similar results.
Flow cytometric analysis.
For detection of surface levels of IFNGR1 by flow cytometry, HEK293 cells were stained with a mouse monoclonal anti-human IFNGR1 antibody (R&D Systems) for 1 h and then washed three times. The cells were then incubated with a fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse secondary antibody for 30 min, followed by washing 5 times. Data were collected with a CyAn ADP flow cytometer (Beckman Coulter) and analyzed with FlowJo (TreeStar) software (38, 76). Similar results were obtained from three independent experiments.
RNA interference.
siRNA duplexes (27-mer) targeting human casein kinase 1α (siCK1α) and Trilencer-27 universal scrambled negative-control siRNA duplex (SCR) were purchased from OriGene; On-Target Plus human IFNAR1 siRNA (J-020209-05) and On-Target Plus human IFNGR1 siRNA (J-011057-05) were purchased from Dharmacon. All siRNAs were used at a final concentration of 20 nM to transfect HEK293 cells or A549 cells using Lipofectamine RNAiMax transfection reagent according to the manufacturer's instructions. Cells were harvested at 2 days posttransfection, and the knockdown of CK1α, IFNAR1, or IFNGR1 was confirmed by performing Western blot analysis.
Real-time PCR.
Total cellular RNA was purified using Tri Reagent (Sigma-Aldrich) according to the manufacturer's instructions and treated with DNase I (Thermo Scientific) to remove contaminated DNAs. The RNA was reverse transcribed with random primer (Invitrogen), and the resulting cDNA was then analyzed by real-time quantitative PCR (qPCR) using gene-specific primers. Primers for human IFNGR1 (5′-CTT AGC CTG GTA TTC ATC-3′ and 5′-CTC TTC ACA GAC CAC CTC-3′), TAP-1 (5′-TGT GAC AAG GTT CCC ACT GCT TAC-3′ and 5′-GGC TGT GGC CTA TGC AGT CA-3′), and LMP-2 (5′-GCA TAT AAG CCA GGC ATG TCT CC-3′ and 5′-AGC TGT AAT AGT GAC CAG GTA GAT GAC-3′) were used. The qPCRs were performed with SYBR green I chemistry using a Step One Plus real-time PCR instrument. cDNA quantities were normalized to the GAPDH RNA quantities measured in the same samples.
Statistical analysis.
Data were analyzed and compared using a bidirectional, unpaired Student t test. The data represent means ± standard deviations (SD).
ACKNOWLEDGMENTS
We thank Serge Fuchs (University of Pennsylvania), Adolfo García-Sastre's research group (Mount Sinai School of Medicine) and their NIAID Centers of Excellence for Influenza Research and Surveillance program, Yoshihiro Kawaoka (University of Wisconsin—Madison), David Pintel (University of Missouri—Columbia), and Mark Hannink (University of Missouri—Columbia) for their kind provision of research reagents as described in Materials and Methods. Also, we thank the Cell and Immunology Core Facility at the University of Missouri—Columbia.
This work was supported by NIH/NIAID grants R01AI091797 and R21AI127404 (B.H.) and Kansas State University startup funds (W.M.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Claas EC, Osterhaus AD, van Beek R, De Jong JC, Rimmelzwaan GF, Senne DA, Krauss S, Shortridge KF, Webster RG. 1998. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet 351:472–477. doi: 10.1016/S0140-6736(97)11212-0. [DOI] [PubMed] [Google Scholar]
- 2.Chen H, Deng G, Li Z, Tian G, Li Y, Jiao P, Zhang L, Liu Z, Webster RG, Yu K. 2004. The evolution of H5N1 influenza viruses in ducks in southern China. Proc Natl Acad Sci U S A 101:10452–10457. doi: 10.1073/pnas.0403212101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cowling BJ, Jin L, Lau EH, Liao Q, Wu P, Jiang H, Tsang TK, Zheng J, Fang VJ, Chang Z, Ni MY, Zhang Q, Ip DK, Yu J, Li Y, Wang L, Tu W, Meng L, Wu JT, Luo H, Li Q, Shu Y, Li Z, Feng Z, Yang W, Wang Y, Leung GM, Yu H. 2013. Comparative epidemiology of human infections with avian influenza A H7N9 and H5N1 viruses in China: a population-based study of laboratory-confirmed cases. Lancet 382:129–137. doi: 10.1016/S0140-6736(13)61171-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferguson L, Olivier AK, Genova S, Epperson WB, Smith DR, Schneider L, Barton K, McCuan K, Webby RJ, Wan XF. 2016. Pathogenesis of influenza D virus in cattle. J Virol 90:5636–5642. doi: 10.1128/JVI.03122-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foni E, Chiapponi C, Baioni L, Zanni I, Merenda M, Rosignoli C, Kyriakis CS, Luini MV, Mandola ML, Bolzoni L, Nigrelli AD, Faccini S. 2017. Influenza D in Italy: towards a better understanding of an emerging viral infection in swine. Sci Rep 7:11660. doi: 10.1038/s41598-017-12012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouvier NM, Palese P. 2008. The biology of influenza viruses. Vaccine 26(Suppl 4):D49–D53. doi: 10.1016/j.vaccine.2008.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dharan NJ, Gubareva LV, Meyer JJ, Okomo-Adhiambo M, McClinton RC, Marshall SA, St George K, Epperson S, Brammer L, Klimov AI, Bresee JS, Fry AM, Oseltamivir-Resistance Working Group. 2009. Infections with oseltamivir-resistant influenza A(H1N1) virus in the United States. JAMA 301:1034–1041. doi: 10.1001/jama.2009.294. [DOI] [PubMed] [Google Scholar]
- 8.Poland GA, Jacobson RM, Ovsyannikova IG. 2009. Influenza virus resistance to antiviral agents: a plea for rational use. Clin Infect Dis 48:1254–1256. doi: 10.1086/598989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marjuki H, Mishin VP, Chesnokov AP, Jones J, De La Cruz JA, Sleeman K, Tamura D, Nguyen HT, Wu HS, Chang FY, Liu MT, Fry AM, Cox NJ, Villanueva JM, Davis CT, Gubareva LV. 2015. Characterization of drug-resistant influenza A(H7N9) variants isolated from an oseltamivir-treated patient in Taiwan. J Infect Dis 211:249–257. doi: 10.1093/infdis/jiu447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vilcek J. 2006. Fifty years of interferon research: aiming at a moving target. Immunity 25:343–348. doi: 10.1016/j.immuni.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 11.Grandvaux N, ten Oever BR, Servant MJ, Hiscott J. 2002. The interferon antiviral response: from viral invasion to evasion. Curr Opin Infect Dis 15:259–267. doi: 10.1097/00001432-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Seo YJ, Hahm B. 2010. Type I interferon modulates the battle of host immune system against viruses. Adv Appl Microbiol 73:83–101. doi: 10.1016/S0065-2164(10)73004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pitha PM, Kunzi MS. 2007. Type I interferon: the ever unfolding story. Curr Top Microbiol Immunol 316:41–70. [DOI] [PubMed] [Google Scholar]
- 14.Osterlund P, Pirhonen J, Ikonen N, Ronkko E, Strengell M, Makela SM, Broman M, Hamming OJ, Hartmann R, Ziegler T, Julkunen I. 2010. Pandemic H1N1 2009 influenza A virus induces weak cytokine responses in human macrophages and dendritic cells and is highly sensitive to the antiviral actions of interferons. J Virol 84:1414–1422. doi: 10.1128/JVI.01619-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Sastre A. 2011. Induction and evasion of type I interferon responses by influenza viruses. Virus Res 162:12–18. doi: 10.1016/j.virusres.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 17.Darnell JE Jr, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 18.Aaronson DS, Horvath CM. 2002. A road map for those who don't know JAK-STAT. Science 296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 19.Schoggins JW. 2014. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol 6:40–46. doi: 10.1016/j.coviro.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Killip MJ, Fodor E, Randall RE. 2015. Influenza virus activation of the interferon system. Virus Res 209:11–22. doi: 10.1016/j.virusres.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroder K, Hertzog PJ, Ravasi T, Hume DA. 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 22.Greenlund AC, Farrar MA, Viviano BL, Schreiber RD. 1994. Ligand-induced IFN gamma receptor tyrosine phosphorylation couples the receptor to its signal transduction system (p91). EMBO J 13:1591–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan DH, Greenlund AC, Tanner JW, Shaw AS, Schreiber RD. 1996. Identification of an interferon-gamma receptor alpha chain sequence required for JAK-1 binding. J Biol Chem 271:9–12. doi: 10.1074/jbc.271.1.9. [DOI] [PubMed] [Google Scholar]
- 24.Chesler DA, Reiss CS. 2002. The role of IFN-gamma in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev 13:441–454. doi: 10.1016/S1359-6101(02)00044-8. [DOI] [PubMed] [Google Scholar]
- 25.Chaudhary V, Yuen KS, Chan JF, Chan CP, Wang PH, Cai JP, Zhang S, Liang M, Kok KH, Chan CP, Yuen KY, Jin DY. 2017. Selective activation of interferon-gamma signaling by Zika virus NS5 protein. J Virol 91:e00163-17. doi: 10.1128/JVI.00163-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhein BA, Powers LS, Rogers K, Anantpadma M, Singh BK, Sakurai Y, Bair T, Miller-Hunt C, Sinn P, Davey RA, Monick MM, Maury W. 2015. Interferon-gamma inhibits ebola virus infection. PLoS Pathog 11:e1005263. doi: 10.1371/journal.ppat.1005263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A 109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seliger B, Hammers S, Hohne A, Zeidler R, Knuth A, Gerharz CD, Huber C. 1997. IFN-gamma-mediated coordinated transcriptional regulation of the human TAP-1 and LMP-2 genes in human renal cell carcinoma. Clin Cancer Res 3:573–578. [PubMed] [Google Scholar]
- 30.Ferm M, Gronberg A, Tally M. 1996. IFN-gamma treatment increases insulin binding and MHC class I expression in erythroleukemia cells. Immunol Invest 25:37–47. doi: 10.3109/08820139609059289. [DOI] [PubMed] [Google Scholar]
- 31.Syedbasha M, Egli A. 2017. Interferon lambda: modulating immunity in infectious diseases. Front Immunol 8:119. doi: 10.3389/fimmu.2017.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kotenko SV, Durbin JE. 2017. Contribution of type III interferons to antiviral immunity: location, location, location. J Biol Chem 292:7295–7303. doi: 10.1074/jbc.R117.777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donnelly RP, Kotenko SV. 2010. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res 30:555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel D, Schultz LW, Umland TC. 2013. Influenza A polymerase subunit PB2 possesses overlapping binding sites for polymerase subunit PB1 and human MAVS proteins. Virus Res 172:75–80. doi: 10.1016/j.virusres.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Li W, Chen H, Sutton T, Obadan A, Perez DR. 2014. Interactions between the influenza A virus RNA polymerase components and retinoic acid-inducible gene I. J Virol 88:10432–10447. doi: 10.1128/JVI.01383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liedmann S, Hrincius ER, Guy C, Anhlan D, Dierkes R, Carter R, Wu G, Staeheli P, Green DR, Wolff T, McCullers JA, Ludwig S, Ehrhardt C. 2014. Viral suppressors of the RIG-I-mediated interferon response are pre-packaged in influenza virions. Nat Commun 5:5645. doi: 10.1038/ncomms6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A. 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia C, Vijayan M, Pritzl CJ, Fuchs SY, McDermott AB, Hahm B. 2015. Hemagglutinin of influenza A virus antagonizes type I interferon (IFN) responses by inducing degradation of type I IFN receptor 1. J Virol 90:2403–2417. doi: 10.1128/JVI.02749-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marsh GA, Hatami R, Palese P. 2007. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J Virol 81:9727–9736. doi: 10.1128/JVI.01144-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Sobrido L, Cadagan R, Steel J, Basler CF, Palese P, Moran TM, Garcia-Sastre A. 2010. Hemagglutinin-pseudotyped green fluorescent protein-expressing influenza viruses for the detection of influenza virus neutralizing antibodies. J Virol 84:2157–2163. doi: 10.1128/JVI.01433-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar KG, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY. 2003. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J 22:5480–5490. doi: 10.1093/emboj/cdg524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar KG, Krolewski JJ, Fuchs SY. 2004. Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J Biol Chem 279:46614–46620. doi: 10.1074/jbc.M407082200. [DOI] [PubMed] [Google Scholar]
- 43.Liu J, HuangFu WC, Kumar KG, Qian J, Casey JP, Hamanaka RB, Grigoriadou C, Aldabe R, Diehl JA, Fuchs SY. 2009. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 5:72–83. doi: 10.1016/j.chom.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar KG, Barriere H, Carbone CJ, Liu J, Swaminathan G, Xu P, Li Y, Baker DP, Peng J, Lukacs GL, Fuchs SY. 2007. Site-specific ubiquitination exposes a linear motif to promote interferon-alpha receptor endocytosis. J Cell Biol 179:935–950. doi: 10.1083/jcb.200706034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marijanovic Z, Ragimbeau J, Kumar KG, Fuchs SY, Pellegrini S. 2006. TYK2 activity promotes ligand-induced IFNAR1 proteolysis. Biochem J 397:31–38. doi: 10.1042/BJ20060272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng H, Qian J, Baker DP, Fuchs SY. 2011. Tyrosine phosphorylation of protein kinase D2 mediates ligand-inducible elimination of the type 1 interferon receptor. J Biol Chem 286:35733–35741. doi: 10.1074/jbc.M111.263608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng H, Qian J, Varghese B, Baker DP, Fuchs S. 2011. Ligand-stimulated downregulation of the alpha interferon receptor: role of protein kinase D2. Mol Cell Biol 31:710–720. doi: 10.1128/MCB.01154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhattacharya S, Qian J, Tzimas C, Baker DP, Koumenis C, Diehl JA, Fuchs SY. 2011. Role of p38 protein kinase in the ligand-independent ubiquitination and down-regulation of the IFNAR1 chain of type I interferon receptor. J Biol Chem 286:22069–22076. doi: 10.1074/jbc.M111.238766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhattacharya S, HuangFu WC, Dong G, Qian J, Baker DP, Karar J, Koumenis C, Diehl JA, Fuchs SY. 2013. Anti-tumorigenic effects of type 1 interferon are subdued by integrated stress responses. Oncogene 32:4214–4221. doi: 10.1038/onc.2012.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J, Carvalho LP, Bhattacharya S, Carbone CJ, Kumar KG, Leu NA, Yau PM, Donald RG, Weiss MJ, Baker DP, McLaughlin KJ, Scott P, Fuchs SY. 2009. Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol Cell Biol 29:6401–6412. doi: 10.1128/MCB.00478-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhattacharya S, HuangFu WC, Liu J, Veeranki S, Baker DP, Koumenis C, Diehl JA, Fuchs SY. 2010. Inducible priming phosphorylation promotes ligand-independent degradation of the IFNAR1 chain of type I interferon receptor. J Biol Chem 285:2318–2325. doi: 10.1074/jbc.M109.071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuchs SY. 2013. Hope and fear for interferon: the receptor-centric outlook on the future of interferon therapy. J Interferon Cytokine Res 33:211–225. doi: 10.1089/jir.2012.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carbone CJ, Zheng H, Bhattacharya S, Lewis JR, Reiter AM, Henthorn P, Zhang ZY, Baker DP, Ukkiramapandian R, Bence KK, Fuchs SY. 2012. Protein tyrosine phosphatase 1B is a key regulator of IFNAR1 endocytosis and a target for antiviral therapies. Proc Natl Acad Sci U S A 109:19226–19231. doi: 10.1073/pnas.1211491109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho IR, Oh M, Koh SS, Malilas W, Srisuttee R, Jhun BH, Pellegrini S, Fuchs SY, Chung YH. 2012. Hepatitis B virus X protein inhibits extracellular IFN-alpha-mediated signal transduction by downregulation of type I IFN receptor. Int J Mol Med 29:581–586. doi: 10.3892/ijmm.2012.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knippschild U, Kruger M, Richter J, Xu P, Garcia-Reyes B, Peifer C, Halekotte J, Bakulev V, Bischof J. 2014. The CK1 family: contribution to cellular stress response and its role in carcinogenesis. Front Oncol 4:96. doi: 10.3389/fonc.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schittek B, Sinnberg T. 2014. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer 13:231. doi: 10.1186/1476-4598-13-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sinnberg T, Wang J, Sauer B, Schittek B. 2016. Casein kinase 1alpha has a non-redundant and dominant role within the CK1 family in melanoma progression. BMC Cancer 16:594. doi: 10.1186/s12885-016-2643-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Q, Means R, Lang S, Jung JU. 2007. Downregulation of gamma interferon receptor 1 by Kaposi's sarcoma-associated herpesvirus K3 and K5. J Virol 81:2117–2127. doi: 10.1128/JVI.01961-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chandra PK, Gunduz F, Hazari S, Kurt R, Panigrahi R, Poat B, Bruce D, Cohen AJ, Bohorquez HE, Carmody I, Loss G, Balart LA, Wu T, Dash S. 2014. Impaired expression of type I and type II interferon receptors in HCV-associated chronic liver disease and liver cirrhosis. PLoS One 9:e108616. doi: 10.1371/journal.pone.0108616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fuchs SY, Chen A, Xiong Y, Pan ZQ, Ronai Z. 1999. HOS, a human homolog of Slimb, forms an SCF complex with Skp1 and Cullin1 and targets the phosphorylation-dependent degradation of IkappaB and beta-catenin. Oncogene 18:2039–2046. doi: 10.1038/sj.onc.1202760. [DOI] [PubMed] [Google Scholar]
- 61.Tan P, Fuchs SY, Chen A, Wu K, Gomez C, Ronai Z, Pan ZQ. 1999. Recruitment of a ROC1-CUL1 ubiquitin ligase by Skp1 and HOS to catalyze the ubiquitination of I kappa B alpha. Mol Cell 3:527–533. doi: 10.1016/S1097-2765(00)80481-5. [DOI] [PubMed] [Google Scholar]
- 62.Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW. 1997. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J Biol Chem 272:24735–24738. doi: 10.1074/jbc.272.40.24735. [DOI] [PubMed] [Google Scholar]
- 63.Hassan IH, Zhang MS, Powers LS, Shao JQ, Baltrusaitis J, Rutkowski DT, Legge K, Monick MM. 2012. Influenza A viral replication is blocked by inhibition of the IRE1 stress pathway. J Biol Chem 287:4679–4689. doi: 10.1074/jbc.M111.284695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Braakman I, Hoover-Litty H, Wagner KR, Helenius A. 1991. Folding of influenza hemagglutinin in the endoplasmic reticulum. J Cell Biol 114:401–411. doi: 10.1083/jcb.114.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma W, Belisle SE, Mosier D, Li X, Stigger-Rosser E, Liu Q, Qiao C, Elder J, Webby R, Katze MG, Richt JA. 2011. 2009 pandemic H1N1 influenza virus causes disease and upregulation of genes related to inflammatory and immune responses, cell death, and lipid metabolism in pigs. J Virol 85:11626–11637. doi: 10.1128/JVI.05705-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A 96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varble A, Albrecht RA, Backes S, Crumiller M, Bouvier NM, Sachs D, Garcia-Sastre A, ten Oever BR. 2014. Influenza A virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe 16:691–700. doi: 10.1016/j.chom.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seo YJ, Blake C, Alexander S, Hahm B. 2010. Sphingosine 1-phosphate-metabolizing enzymes control influenza virus propagation and viral cytopathogenicity. J Virol 84:8124–8131. doi: 10.1128/JVI.00510-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seo YJ, Pritzl CJ, Vijayan M, Bomb K, McClain ME, Alexander S, Hahm B. 2013. Sphingosine kinase 1 serves as a pro-viral factor by regulating viral RNA synthesis and nuclear export of viral ribonucleoprotein complex upon influenza virus infection. PLoS One 8:e75005. doi: 10.1371/journal.pone.0075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Min J, Mesika A, Sivaguru M, Van Veldhoven PP, Alexander H, Futerman AH, Alexander S. 2007. (Dihydro)ceramide synthase 1 regulated sensitivity to cisplatin is associated with the activation of p38 mitogen-activated protein kinase and is abrogated by sphingosine kinase 1. Mol Cancer Res 5:801–812. doi: 10.1158/1541-7786.MCR-07-0100. [DOI] [PubMed] [Google Scholar]
- 71.Vijayan M, Seo YJ, Pritzl CJ, Squires SA, Alexander S, Hahm B. 2014. Sphingosine kinase 1 regulates measles virus replication. Virology 450-451:55–63. doi: 10.1016/j.virol.2013.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roberts D, Pedmale UV, Morrow J, Sachdev S, Lechner E, Tang X, Zheng N, Hannink M, Genschik P, Liscum E. 2011. Modulation of phototropic responsiveness in Arabidopsis through ubiquitination of phototropin 1 by the CUL3-Ring E3 ubiquitin ligase CRL3(NPH3). Plant Cell 23:3627–3640. doi: 10.1105/tpc.111.087999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei CJ, Xu L, Kong WP, Shi W, Canis K, Stevens J, Yang ZY, Dell A, Haslam SM, Wilson IA, Nabel GJ. 2008. Comparative efficacy of neutralizing antibodies elicited by recombinant hemagglutinin proteins from avian H5N1 influenza virus. J Virol 82:6200–6208. doi: 10.1128/JVI.00187-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang ZY, Wei CJ, Kong WP, Wu L, Xu L, Smith DF, Nabel GJ. 2007. Immunization by avian H5 influenza hemagglutinin mutants with altered receptor binding specificity. Science 317:825–828. doi: 10.1126/science.1135165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wei CJ, Boyington JC, McTamney PM, Kong WP, Pearce MB, Xu L, Andersen H, Rao S, Tumpey TM, Yang ZY, Nabel GJ. 2010. Induction of broadly neutralizing H1N1 influenza antibodies by vaccination. Science 329:1060–1064. doi: 10.1126/science.1192517. [DOI] [PubMed] [Google Scholar]
- 76.Pritzl CJ, Seo YJ, Xia C, Vijayan M, Stokes ZD, Hahm B. 2015. A ceramide analogue stimulates dendritic cells to promote T cell responses upon virus infections. J Immunol 194:4339–4349. doi: 10.4049/jimmunol.1402672. [DOI] [PMC free article] [PubMed] [Google Scholar]