

ABSTRACT
SC29EK is an electronically constrained α-helical peptide HIV-1 fusion inhibitor that is highly effective against both wild-type and enfuvirtide (T20)-resistant viruses. In this study, we focused on investigating the mechanism of HIV-1 resistance to SC29EK by two approaches. First, SC29EK-escaping HIV-1 variants were selected and characterized. Three mutant viruses, which possessed two (N43K/E49A) or three (Q39R/N43K/N126K and N43K/E49A/N126K) amino acid substitutions in the N- and C-terminal repeat regions of gp41 were identified as conferring high resistance to SC29EK and cross-resistance to the first-generation (T20 and C34) and newly designed (sifuvirtide, MT-SC29EK, and 2P23) fusion inhibitors. The resistance mutations could reduce the binding stability of SC29EK, impair viral Env-mediated cell fusion and entry, and change the conformation of the gp41 core structure. Further, we determined the crystal structure of SC29EK in complex with a target mimic peptide, which revealed the critical intra- and interhelical interactions underlying the mode of action of SC29EK and the genetic pathway to HIV-1 resistance. Taken together, the present data provide new insights into the structure and function of gp41 and the structure-activity relationship (SAR) of viral fusion inhibitors.
IMPORTANCE T20 is the only membrane fusion inhibitor available for treatment of viral infection, but it has relatively low anti-HIV activity and genetic barriers for resistance, thus calling for new drugs blocking the viral fusion process. As an electronically constrained α-helical peptide, SC29EK is highly potent against both wild-type and T20-resistant HIV-1 strains. Here, we report the characterization of HIV-1 variants resistant to SC29EK and the crystal structure of SC29EK. The key mutations mediating high resistance to SC29EK and cross-resistance to the first and new generations of fusion inhibitors as well as the underlying mechanisms were identified. The crystal structure of SC29EK bound to a target mimic peptide further revealed its action mode and genetic pathway to inducing resistance. Hence, our data have shed new lights on the mechanisms of HIV-1 fusion and its inhibition.
KEYWORDS: HIV-1, fusion inhibitor, SC29EK, six-helix bundle, resistance
INTRODUCTION
Entry of human immunodeficiency virus type 1 (HIV-1) into target cells requires membrane fusion, which is mediated by the trimeric viral envelop (Env) glycoprotein gp120/gp41 complex (1). Sequential binding of the surface subunit gp120 to cell receptor CD4 and a coreceptor (CCR5 or CXCR4) brings the Env with a cascade of structural rearrangements, enabling the hydrophobic fusion peptide located at the N terminus of gp41 to be released and inserted into the cell membrane. Subsequently, three C-terminal heptad repeats (CHR) of gp41 fold antiparallelly onto the central hydrophobic grooves created by the trimeric N-terminal heptad repeat (NHR) coiled coil, leading to a stable six-helix bundle (6-HB) structure that pulls the viral and cellular membranes in close apposition for fusion. The crystal structures of diverse 6-HBs have universally revealed a deep hydrophobic pocket on the C-terminal portion of NHR helices, which is docked by hydrophobic residues from the N-terminal pocket-binding domain (PBD) of the CHR helix (2–4). We and others have recently identified a subpocket located immediately downstream of the deep pocket, which further stabilizes the interactions of the NHR and CHR helices (5–7). Both pockets offer ideal targets for developing anti-HIV therapeutics.
Peptides derived from the NHR or CHR sequence of gp41 possess potent anti-HIV activity and act as competitors by binding the prehairpin intermediate of gp41 to prevent 6-HB formation, thereby inhibiting viral fusion and entry in a dominant negative fashion (8–10). The CHR-derived 36-residue peptide T20 (enfuvirtide/fuzeon) was approved for clinical use in 2003, creating the first member of a new class of anti-HIV drugs: HIV entry inhibitors (11, 12). T20 has demonstrated effectiveness as a salvage therapy for HIV/AIDS patients who failed to respond to antiretroviral therapeutics that include reverse transcriptase inhibitors (RTIs) and protease inhibitors (PIs); however, it has relatively weak anti-HIV activity, thus a requiring frequent high dosage, and a low genetic barrier to inducing drug resistance. The mutations mediating T20 resistance are mapped primarily to the inhibitor-binding sites in the NHR region, with the amino acid stretch Gly36 to Leu45 particularly being a hot spot (13–18). To overcome the drawbacks of T20, there have been tremendous efforts to develop new-generation fusion inhibitors with improved pharmaceutical profiles. C34, a 34-residue CHR-derived peptide, has been widely applied for inhibitor design, as its N terminus contains the pocket-binding domain (PBD) and shows higher anti-HIV potency (8, 9). SC34EK was originally designed by introducing the glutamate (E) and lysine (K) substitutions into the solvent-accessible site of C34, which promoted the formation of intrahelical salt bridges, thus stabilizing the α-helicity of the inhibitor (19, 20). Impressively, its truncated form, SC29EK (29 residues), maintained the highly potent antiviral activity against both wild-type and T20-resistant strains, highlighting the potential for clinical development (19). A schematic illustration of HIV-1 gp41 and its peptide derivatives is shown in Fig. 1.
FIG 1.

Schematic illustration of HIV-1 gp41 and its peptide derivatives. FP, fusion peptide; NHR, N-terminal heptad repeat; CHR, C-terminal heptad repeat; TM, transmembrane domain; CT, cytoplasmic tail. The gp41 numbering of HIV-1HXB2 is used. The sequences corresponding to the T20-resistant site and the pocket-forming site are marked in purple and blue, respectively, on the NHR-derived peptide N36, with the SC29EK-induced mutations highlighted in red. The sequences corresponding to the pocket-binding domain (PBD) are marked in deep red on the CHR-derived peptides, while the position and sequence of the M-T hook structure are shown in green. The negatively and positively charged residues introduced for potential formation of salt bridges in inhibitors are indicated with solid black lines.
In this study, we focused on characterizing SC29EK-induced HIV-1 variants in order to gain novel insights into the genetic pathways and mechanisms of HIV-1 resistance to the electronically constrained α-helical peptides. The presented results benefit our understanding of the structure-function relationship of HIV-1 Env and the structure-activity relationship (SAR) of gp41-dependent membrane fusion inhibitors. As the electronically constrained α-helical peptides have widely been considered a vital strategy for inhibitor design, our data would also help in developing therapeutics against other enveloped viruses with class I membrane fusion glycoproteins.
RESULTS
Sequence characterization of SC29EK-resistant HIV-1 variants.
To characterize the structure-activity relationship of newly designed HIV-1 fusion inhibitors, we previously conducted the in vitro selection of escape HIV-1 variants to SC29EK, in which the drug concentration was raised from 0.8 to 2,650 nM after 35 generations of virus passage over 7 months, thus implying the emergence of mutant viruses with high resistance to SC29EK (21). To determine the underlying mechanisms, the entire env genes of HIV-1 variants were amplified by PCR and cloned for DNA sequencing. Finally, three dominating mutant viruses were identified (Fig. 2), including one mutant with two amino acid substitutions (N43K/E49A) and two mutants with three amino acid substitutions (Q39R/N43K/N126K and N43K/E49A/N126K). Obviously, the Q39R, N43K, and E49A substitutions located within the inhibitor-binding site of gp41 might serve as primary mutations for resistance, while the N126K substitution in the CHR has been recognized as a secondary mutation readily emerging both in vitro and in vivo (22). Among a number of cloned Envs, no consistent substitutions were observed in the other sites of gp41 or in gp120 sequence.
FIG 2.

SC29EK-induced mutations in the NHR and CHR sites of HIV-1NL4-3 gp41. The amino acid sequences of wild-type (WT) and mutant viruses are aligned. The positions of selected mutations are in bold, and numbering is according to that of HIV-1HXB2 gp41. The pocket-forming sequence in NHR and the pocket-binding domain in CHR with the M-T hook residues are underlined.
Profile of resistance of HIV-1 mutants to SC29EK.
To clarify the mutations critically determining the phenotype of SC29EK-induced resistance, we generated a panel of HIV-1NL4-3 Env mutants carrying the characterized amino acid substitutions either singly or in combination (Table 1). The corresponding pseudoviruses then were generated, and the inhibitory activity of SC29EK was determined by a single-cycle infection assay. As shown in Table 1, three mutant viruses, N43K/E49A, Q39R/N43K/N126K, and N43K/E49A/N126K, were confirmed to possess high resistance to SC29EK, with changes relative to the resistance of the wild-type virus at 654.75-, 497.75-, and 707.31-fold, respectively. Compared to single mutations of Q39R and E49A, the single N43K mutation conferred a 113.2-fold change in resistance, indicating its dominating role in the three resistant mutants. Obviously, the secondary mutation N126K could boost the resistance level of the Q39R/N43K mutant, but it played little role in resistance of the N43K/E49A virus.
TABLE 1.
Resistance of HIV-1 mutants to SC29EK and the first-generation fusion inhibitors
| HIV-1NL4-3 | Resistance to: |
|||||
|---|---|---|---|---|---|---|
| SC29EK |
T20 |
C34 |
||||
| IC50 (nM)a | Fold changeb | IC50 (nM) | Fold change | IC50 (nM) | Fold change | |
| Wild type | 3.46 ± 1.28 | 1 | 83.24 ± 16.07 | 1 | 1.77 ± 0.13 | 1 |
| Q39R | 6.72 ± 1.3 | 1.94 | 24.67 ± 11.73 | 0.3 | 1.31 ± 0.38 | 0.74 |
| N43K | 391.67 ± 141.38 | 113.2 | 937.11 ± 200.8 | 11.26 | 30.87 ± 2.49 | 17.44 |
| E49A | 19.7 ± 4.84 | 5.69 | 174.49 ± 37.43 | 2.1 | 4.91 ± 1 | 2.78 |
| N126K | 4.86 ± 0.52 | 1.4 | 131.5 ± 12.5 | 1.62 | 5 ± 0.54 | 2.82 |
| Q39R/N43K | 458.47 ± 43.06 | 132.51 | 785.39 ± 294.31 | 9.44 | 12.93 ± 0.65 | 7.3 |
| N43K/E49A | 2,265.44 ± 390.31 | 654.75 | 1,222.25 ± 349.41 | 14.68 | 64.78 ± 15.76 | 36.6 |
| Q39R/N43K/N126K | 1,722.22 ± 143.36 | 497.75 | 1,248.6 ± 198.27 | 15 | 31.01 ± 6.17 | 17.52 |
| N43K/E49A/N126K | 2,447.29 ± 552.89 | 707.31 | 1,595.03 ± 518.88 | 19.16 | 81.23 ± 18.88 | 45.89 |
The experiment was performed in triplicate and repeated three times. Data are expressed as means ± standard deviations. IC50, 50% inhibitory concentration.
The fold change in the IC50 was determined relative to the wild-type level.
Cross-resistance to the first-generation fusion inhibitors T20 and C34.
We then determined the cross-resistance levels of SC29EK-induced mutants to the drug T20 and the template peptide C34. As shown in Table 1, three mutant viruses (N43K/E49A, Q39R/N43K/N126K, and N43K/E49A/N126K) also displayed a considerable degree of resistance to the two inhibitors: the N43K/E49A mutant showed resistance to T20 and C34 with increases of 14.68- and 36.6-fold, respectively; the Q39R/N43K/N126K mutant showed increases in resistance to T20 and C34 of 15- and 17.52-fold, respectively; and the N43K/E49A/N126K mutant had increases in resistance to T20 and C34 of 19.16- and 45.89-fold, respectively. As expected, the single N43K mutation played major roles in the cross-resistance profiles, with changes of 11.26-fold for T20 and 17.44-fold for C34, while the E49A mutation also contributed a modest resistance phenotype. Interestingly, it was found that the single Q39R mutant displayed increased susceptibility to T20.
Cross-resistance to newly designed fusion inhibitors.
Sifuvirtide (SFT) is also an electronically constrained C34-based HIV-1 fusion inhibitor, and it has been approved for phase III clinical trials in China (23). Here, we were interested in determining the cross-resistance of SC29EK-induced mutants to SFT. As shown in Table 2, the N43K/E49A, Q39R/N43K/N126K, and N43K/E49A/N126K mutants had increased resistance of 32.6-, 14.87-, and 39.95-fold, respectively, while the N43K mutant had a resistance of 21.99-fold, suggesting a resistance profile similar to that for C34.
TABLE 2.
Resistance of SC29EK-induced HIV-1 mutants to three newly designed fusion inhibitors
| HIV-1NL4-3 | Resistance to: |
|||||
|---|---|---|---|---|---|---|
| SFT |
MT-SC29EK |
2P23 |
||||
| IC50 (nM)a | Fold changeb | IC50 (nM) | Fold change | IC50 (nM) | Fold change | |
| Wild type | 1.36 ± 0.6 | 1 | 0.58 ± 0.21 | 1 | 0.67 ± 0.19 | 1 |
| Q39R | 1.19 ± 0.22 | 0.88 | 0.94 ± 0.25 | 1.62 | 0.76 ± 0.15 | 1.14 |
| N43K | 29.90 ± 2.48 | 21.99 | 6.45 ± 1.21 | 11.12 | 0.66 ± 0.1 | 0.99 |
| E49A | 2.58 ± 0.37 | 1.89 | 1.02 ± 0.48 | 1.76 | 3.76 ± 0.43 | 5.61 |
| N126K | 2.91 ± 0.24 | 2.14 | 0.9 ± 0.15 | 1.55 | 1.53 ± 0.72 | 2.28 |
| Q39R/N43K | 13.15 ± 3.38 | 9.67 | 10.7 ± 0.6 | 18.45 | 1.28 ± 0.26 | 1.92 |
| N43K/E49A | 44.33 ± 5.27 | 32.6 | 26.55 ± 5.17 | 45.78 | 4.80 ± 0.41 | 6.23 |
| Q39R/N43K/N126K | 20.23 ± 3.35 | 14.87 | 32.68 ± 10.43 | 56.35 | 3.30 ± 0.51 | 4.92 |
| N43K/E49A/N126K | 54.33 ± 14.22 | 39.95 | 42.56 ± 8.66 | 73.38 | 10.32 ± 1.55 | 15.41 |
The experiment was performed in triplicate and repeated three times. Data are expressed as means ± standard deviations. IC50, 50% inhibitory concentration.
The fold change in the IC50 was determined relative to the wild-type level.
We previously showed that the M-T hook structure-modified SC29EK (MT-SC29EK) had significantly improved antiviral activity and genetic barriers to inducing resistance (21). Thus, we sought to compare the inhibitory activities and resistance profiles of SC29EK and MT-SC29EK. The results revealed that MT-SC29EK exhibited greatly increased potency in inhibiting both wild-type and SC29EK-resistant mutants (Table 2). For example, while SC29EK inhibited the N43K/E49A, Q39R/N43K/N126K, and N43K/E49A/N126K mutants with mean 50% inhibitory concentrations (IC50s) of 2,265.44, 1,722.22, and 2,447.29 nM, respectively, MT-SC29EK inhibited the three mutants with mean IC50s of 26.55, 32.68, and 42.56 nM, respectively, which indicated that MT-SC29EK was 85.33-, 52.7-, and 57.5-fold more potent than the template SC29EK. In comparison, MT-SC29EK exhibited markedly decreased resistance changes relative to SC29EK. As shown, the three mutant viruses had resistance changes of 45.78-, 56.35-, and 73.38-fold, respectively, and the E43K mutant had a change of 11.12-fold (Table 2). Therefore, the data verified the importance of the M-T hook structure for the design of HIV-1 fusion inhibitor peptides.
On the basis of the M-T hook structure, we previously developed the helical short-peptide fusion inhibitor 2P23, which targeted mainly the gp41 pocket site and showed highly potent activities against HIV-1, HIV-2, and simian immunodeficiency virus (SIV) (24). Here, we sought to characterize the cross-resistance of SC29EK-induced mutants to 2P23. Interestingly, the single N43K mutation did not confer resistance to 2P23, but the single E49A mutation conferred moderate resistance (Table 2). The N43K/E49A, Q39R/N43K/N126K, and N43K/E49A/N126K mutants did exhibit cross-resistance to 2P23, but the fold changes in resistance were dramatically reduced compared to those for the other inhibitors. Indeed, 2P23 was the most potent inhibitor against SC29EK-induced mutant viruses and exhibited the lowest fold changes in resistance among all the characterized fusion inhibitors.
The resistance mutations dramatically reduce the binding stability of SC29EK.
To explore the mechanisms underlying the resistance phenotypes, we first characterized the effects of the resistance mutations on the binding stability of SC29EK. To this end, the NHR-derived peptide N36 with a wild-type sequence (N36wt) or its mutants with single or double amino acid substitutions (N36Q39R, N36N43K, N36E49A, N36Q39R/N43K, and N36N43K/N49A) were synthesized and used as target surrogates. SC29EK and N36 were mixed at equal molar concentrations and incubated at 37°C for 30 min, and the α-helicity and thermostability of each 6-HB complex were determined by circular dichroism (CD) spectroscopy. As shown in Fig. 3A, the CD spectra of all peptide pairs displayed typical double minima at 208 and 222 nm, which indicated that SC29EK could interact with both the wild-type and mutant N36 peptides to form secondary structures with high α-helical contents; however, the thermostability of 6-HBs, defined as the midpoint of the thermal unfolding transition (Tm) value, demonstrated significantly reduced binding stability of SC29EK with N36 mutants (Fig. 3B). While the complex SC29EK+N36wt showed a Tm of 67°C, the complexes with single mutations, SC29EK+N36Q39R and SC29EK+N36N43K, had Tms of 64 and 57°C, respectively, and the complexes with combined mutations, SC29EK/N36Q39R/N43K and SC29EK+N36N43K/E49A, had Tms of 55 and 56°C, respectively. Therefore, SC29EK-induced mutations, alone or in combination, resulted in significantly reduced binding stability of the inhibitor with the target site thus contributing the resistance phenotypes.
FIG 3.

Interactions of SC29EK with target mimic peptides determined by CD spectroscopy. The α-helicity (A) and thermostability (B) of SC29EK in complexes with the NHR-derived peptide N36 or its mutants were measured. The final concentration of each peptide in PBS was 10 μM. The experiments were performed 2 times, and representative data are shown.
Effects of SC29EK-induced mutations on the functionality of HIV-1 Env.
We next focused on investigating the effects of SC29EK-induced resistance mutations on the functionality of viral Env. First, the cell entry efficiency of HIV-1NL4-3 pseudoviruses with single or combined mutations was determined by a single-cycle infection assay, in which the infectivity of wild-type (WT) virus was normalized to 100% and the relative infectivities of other mutants were calculated accordingly. It was found that the single Q39R, N43K, or N126K mutation had a minor effect on the Env-mediated virus entry, but the single E49A mutation resulted in a dramatic reduction (Fig. 4A). Regardless, all three selected SC29EK-resistant viruses with combined mutations (N43K/E49A, Q39R/N43K/N126K, and N43K/E49A/N126K) showed dramatically decreased infectivity. Further, we measured the kinetics of diverse Env-mediated cell-cell fusion by a dual split-protein (DSP)-based fusion assay. As shown in Fig. 4B, the single Q39R or N126 mutation resulted in an Env with a modestly increased fusion activity, whereas the single N43K or E49A mutation caused its significant decrease. Thus, the Q39R/N43K combination seemingly had compensatory effects on the functionality of viral Env, whereas the N43K/E49A combination exhibited synergistic effects, thus resulting in an Env with the lowest fusion activity. In addition, N126K could improve the fusion kinetics of the Env with N43K/E49A or Q39R/N43K, suggesting its compensatory role as a secondary mutation.
FIG 4.

Effects of SC29EK-induced mutations on the functionality of HIV-1 Env. (A) Relative infectivity of HIV-1NL4-3 and its mutants determined by a single-cycle infection assay. The wild-type (WT) and mutant pseudoviruses were normalized to a fixed amount by p24 antigen, and viral entry was tested in TZM-bl cells. The luciferase activity was measured and corrected for background, the luciferase activity of wild-type HIV-1NL4-3 was treated as 100%, and the relative activities of other mutant viruses were calculated accordingly. A t test was performed to judge the significance of the difference between the WT and mutants. * and **, P < 0.01 and P < 0.05, respectively. (B) Kinetics of HIV-1 Env-mediated cell-cell fusion determined by a dual split-protein assay (DSP). For both viral entry and fusion, data were derived from the results of three independent experiments and are expressed as means and standard deviations.
We also examined whether the introduced mutations affected the expression and secretion of the Env glycoproteins in transfected cells. First, the human anti-gp120 antibody VRC01 and anti-gp41 antibody 10E8 were applied in capture enzyme-linked immunosorbent assay (ELISA)-based assays, as both antibodies target highly conserved epitopes and possess broadly reactive neutralizing activity (25, 26). As shown in Fig. 5, no significant changes were found to be associated with the observed phenotypes. To further analyze the processing of the gp160 precursor, the human anti-HIV polyclonal antibody HIV-IG was used in a Western blotting assay. The results verified that the Q39R, N43K, and E49A mutants had no obvious changes in the protein expression, while the N126K mutation resulted in the loss of glycosylation in gp41, as expected (Fig. 6).
FIG 5.

Determination of HIV-1 Env expression by capture ELISA. HEK293T cells were transfected with plasmids expressing wild-type (WT) or mutant HIV-1NL4-3 Envs. The glycoproteins (gp120/gp41) in the lysates of transfected cells (A) or culture supernatant (B) were captured by sheep anti-gp120 antibody D7324 and detected with human MAbs VRC01 and 10E8. The experiments were conducted in duplicate and repeated 2 times.
FIG 6.

Determination of HIV-1 Env processing by Western blotting. The viral Env glycoproteins in the lysates of expression plasmid-transfected cells were probed with human anti-HIV polyclonal antibody (HIV-IG). The reaction bands corresponding to gp160, gp120, and gp41 are marked.
The resistance mutations cause conformational changes in gp41 6-HB core structure.
The 6-HB conformation formed by the NHR-derived peptide N36 and the CHR-derived peptide C34 has been considered a core structure of gp41, which plays an essential role in viral fusion and entry (4). By using three conformation-specific monoclonal antibodies (MAbs) (NC-1, 17C8, and 2G8), we previously developed ELISA-based assays to observe the effects of the selected mutations on the 6-HB structure, which could reveal new insights into the mechanisms of resistance, 6-HB stability, and viral infectivity (27, 28). Here, we detected the conformational changes of 6-HBs formed by the wild-type and mutant N36 and C34 peptides. As shown in Fig. 7, the Q39R mutation resulted in 6-HBs with significantly increased reactivity with all three antibodies, whereas the N43K mutation caused reduced reactivity. Differently, the E49A mutation decreased the reactivity of NC-1 but increased the reactivity of 17C8 and 2G8. While the N43K/E49A combination severely impaired the reactivities of the three antibodies, the Q39R/N43K combination enhanced the reactivities of NC-1 and 17C8 while reducing the recognition by 2G8. Combined, the reaction patterns of diverse 6-HBs with the three antibodies implied the increased or decreased exposure of antigenic epitopes, which might correlate with the structural rearrangements of the 6-HB core, thus determining the fusion activity of gp41 and the binding affinity of inhibitors.
FIG 7.

Effects of SC29EK-induced mutations on the conformation of the viral 6-HB structure. The reactivities of 6-HBs formed by C34 and N36 or its mutants with conformation-dependent MAbs NC-1 (A), 17C8 (B), and 2G8 (C) were tested by ELISA. The peptide mixture was used to coat the plate wells at 10 μg/ml, and the final concentration of a tested MAb was 5 μg/ml. Data were derived from three independent experiments and are expressed as means and standard deviations. A t test was performed to judge the significance of the difference between the WT and mutants. * and **, P < 0.01 and P < 0.05, respectively.
Crystal structure of SC29EK bound to the target mimic peptide N36.
To reveal the structural basis underlying the activity of SC29EK and its resistance profile, we determined the crystal structure of SC29EK in complex with its NHR counterpart N36 (Protein Data Bank [PDB] accession number 5Z0W). The crystal of the SC29EK/N36 complex diffracted the X ray to a resolution limit of 1.9 Å. The crystals belonged to the space group of R3 and contained one SC29EK peptide and one N36 peptide in an asymmetric unit, representing one-third of an intact 6-HB. We could locate all residues of SC29EK/N36 peptides in the final electron density map. The final atomic model has excellent refinement statistics and stereochemistry quality. The statistics for data collection, structure refinement, and structure validation are summarized in Table 3. The overall structure of SC29EK/N36 exhibits a canonical 6-HB conformation. Briefly, three N36 helices formed an NHR trimeric coiled coil with three hydrophobic grooves between adjacent helices, whereas three SC29EK helices are accommodated in these grooves in an antiparallel orientation (Fig. 8). The pocket-binding domain (PBD) of SC29EK, comprising the three hydrophobic residues Trp-117, Trp-120, and Ile-124, occupies the deep hydrophobic pockets at the C-terminal portion of the N36 trimer. The extensive hydrophobic interactions between PBD and the pocket stabilize the conformation of the 6-HB.
TABLE 3.
Crystallographic data collection and refinement statistics
| Parameter | Valuea |
|---|---|
| Data collection statistics | |
| Beamline | SSRF BL19U |
| Wavelength (Å) | 0.977750 |
| Resolution rage | 50.0–1.90 (2.01–1.90) |
| Space group | R3 |
| Cell dimensions | |
| a, b, c (Å) | 56.925, 56.925, 51.454 |
| α, β, γ (°) | 90, 90, 120 |
| Redundancy | 4.53 |
| Total no of reflections | 22,345 |
| No. of unique reflections | 4,928 |
| Rmerge (%)b | 7.3 (84.5) |
| I/σI | 11.21 (1.68) |
| Completeness (%) | 99.5 (97.3) |
| Refinement statistics | |
| Resolution range (Å) | 35.60–1.90 (2.34–1.90) |
| No. of reflections | 4,897 (245) |
| Rwork/Rfreec | 0.2057/0.2570 |
| No. of atoms | |
| Protein | 553 |
| Water | 23 |
| B factors | |
| Protein | 39.81 |
| Water | 47.21 |
| Root mean square deviations | |
| Bond length (Å) | 0.008 |
| Bond angle (°) | 0.810 |
| Ramachandran plot (%) | |
| Favored regions | 100 |
| Allowed regions | 0 |
| Disallowed regions | 0 |
Values in parentheses are those for the highest-resolution shell.
Rmerge = ∑hkl∑j|Ihkl,j − Ihkl|/∑hkl ∑jIhkl,j, where Ihkl is the average of symmetry-related observations of a unique reflection.
Rwork = ∑hkl||Fobs| − |Fcalc||/∑hkl|Fobs|, where h, k, and l are the indices of the reflections and Fobs and Fcalc are the observed and calculated structure factors deduced from the model, respectively. Rfree is defined as a cross-validation R factor for 5% of reflections against which the model was not refined.
FIG 8.
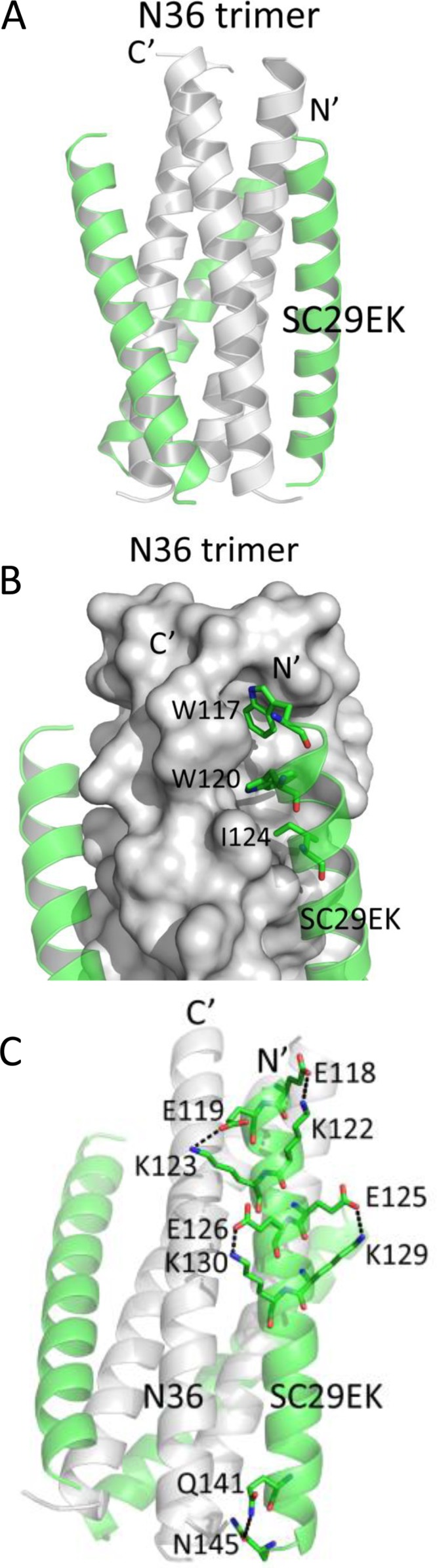
Crystal structure of the 6-HB formed by SC29EK and N36. (A) Ribbon model of the SC29EK/N36-based 6-HB structure. The N36 peptides are colored in gray; SC29EK inhibitors are colored in green. The N terminus of SC29EK and the C terminus of N36 are labeled. (B) Binding of the N-terminal pocket-binding domain (PBD) of SC29EK with the pocket site of the N36 trimer. The N36 trimer is shown as surfaces and colored in gray; the residues Trp-117,Trp-120, and Ile-124 from the PBD of SC29EK are shown as sticks. (C) Intrahelical salt bridges and hydrogen bond of SC29EK in the 6-HB structure. Residues related to the salt bridges or hydrogen bond on SC29EK are shown as stick models with labels.
In the original peptide design, multiple oppositely charged amino acid pairs (glutamate and lysine at positions i and i + 4) were introduced into the N-terminal portion of SC29EK with the purpose of promoting the formation of intrahelical salt bridges Glu-118/Lys-122, Glu-119/Lys-123, Glu-125/Lys-129 and Glu-126/Lys-130. We were able to observe all the designed salt bridges in the crystal structure of SC29EK/N36, which indicates that the salt bridges stabilize the helical conformation of SC29EK. In the C-terminal portion of the inhibitor, we did not observe the possible salt bridges; instead, a hydrogen bond between the side chains of Gln-141 and Asn-145 was identified (Fig. 8), which also stabilizes the helical conformation of SC29EK.
Importantly, a number of interpeptide hydrogen bonds existed between SC29EK and N36 in the crystal structure of SC29EK/N36, which contributed significantly to the binding affinity of SC29EK and the stabilization of 6-HB conformation. As illustrated in Fig. 9, we found three hydrogen bonds between Trp-120 and Leu-57, Tyr-127 and His-53, and Thr-128 and Gln-56 which tethered the middle portion of the inhibitor to the N36 trimer. We identified five more hydrogen bonds, Ser-138 and Gln-40, Gln-141 and Ser-35, Gln-142 and Val-38, Gln-142 and Gln-40, and Asn-145 and Gln-41, which anchored the C-terminal portion of SC29EK to the N36 trimer. It is worth noting that Gln-142 interacts with Val-38 and Gln-40 from two adjacent N36 helices simultaneously. Additionally, Gln-40 of N36 also hydrogen bonds with Ser-138 of SC29EK. Collectively, the extensive hydrophobic interactions, salt bridge, and hydrogen bond network between SC29EK and N36 contribute to the binding affinity of SC29EK.
FIG 9.

Interhelical interactions of the SC29EK/N36-based 6-HB structure. (A) Binding of the N-terminal portion of SC29EK with the N36 trimer in a ribbon model. The N36 peptides are colored in gray, and SC29EK inhibitors are colored in green. Residues related to hydrogen bonds within the 6-HB are shown as stick models with labels. The dashed lines indicate hydrogen bonds between SC29EK and the N36 trimer. (B) Binding of the C-terminal portion of SC29EK with the N36 trimer in a ribbon model. The same representation and color scheme as in the crystal structure in panel A are used. The C terminus of SC29EK and the N terminus of N36 are labeled. (C) Sequence map of a single SC29EK interacting with two N36 helices. The dashed black lines indicate hydrogen bonds between SC29EK and N36. The C and N termini of the sequences are labeled.
Structural insights into the mechanism of SC29EK-induced resistance.
By combining the crystal structure of the SC29EK/N36 complex with the virological results, we further analyzed the mechanisms of HIV-1 resistance to SC29EK. As shown in Fig. 10, the side chain of Gln-39 from N36 formed two hydrogen bonds with the main-chain NH group and the side chain OH group of Ser-35 from N36, and an ordered water molecule connected the side chains of Gln-39 and Asn-43 from N36 and the side chain of Gln-141 from SC29EK. Therefore, HIV-1 bearing the Q39R mutation could disrupt the Gln-39-centered hydrogen bond network, thus hampering the binding of SC29EK. In addition, the bulky side chain of arginine might also generate steric obstruction to destabilize the conformation of 6-HB.
FIG 10.

Structural basis of HIV-1 resistance to SC29EK. (A) Gln-39 and Asn-43 are involved in an extensive hydrogen bond network formed by a cluster of residues and two water molecules (stick model), which mediate the complicated interactions between SC29EK and the N36 trimer. Dashed lines indicate the hydrogen bonds. (B) Gln-40 is a key member of a remarkable glutamine-rich polar layer of the 6-HB. Dashed lines indicate the hydrogen bonds. (C) The position and conformation of Glu-49 are shown in a stick model.
The structure of the SC29EK/N36 complex shows that residue Asn-43 of N36 is located at the interface between NHR and CHR and is deeply buried in the core of 6-HB, and it is involved an extensive hydrogen bond network constituted by the highly conserved residues Q40 of N36 and S138 and Q142 of SC29EK. In detail, the side chain amine group of Asn-43 donated a hydrogen bond to the side chain carboxyl group of Gln-40; the side chain of Gln-40 also formed hydrogen bonds with the side chains of Ser-138 and Gln-142 on SC29EK, and it combined to two water molecules: while the first water molecule kept the network with Gln-39 and Gln-141 described above, the second water molecule maintained the connections with Asn-42, Arg-46 on N36, and Lys-137 on SC29EK. Therefore, the Asn-43-centered hydrogen bond network is critical to the interactions between the N36 and SC29EK helices, and thus the N43K mutation not only disrupts the critical hydrogen bond network between N36 and SC29EK but also introduces a steric obstruction at the core of 6-HB. Additionally, the N43K mutation could repulse the positively charged Lys-137 from SC29EK, which also reduced the binding of the inhibitor.
In the crystal structure of the 6-HB structure-containing N36/SC29EK, the negatively charged residue Glu-49 is exposed between two SC29EK helices, and we did not detect specific interaction between Glu-49 and SC29EK. Thus, the E49A mutation might not affect the binding of SC29EK, which is consistent with its effect on the Tm value determined by CD spectroscopy.
DISCUSSION
In the present study, we focused on exploring the mechanism of HIV-1 resistance to the electronically constrained α-helical peptide fusion inhibitor SC29EK. Three mutant viruses, E43K/E49A, Q39R/N43K/N126K, and N43K/E49A/N126K, were identified as conferring high resistance to SC29EK and cross-resistance to the first-generation and newly designed fusion inhibitors. It was found that the resistance mutations markedly reduced the binding stability of SC29EK, impairing viral Env-mediated cell fusion and entry. We further determined the crystal structure of SC29EK bound to a target mimic peptide, which revealed their detailed interactions underlying the mode of action of SC29EK and the genetic pathway to HIV-1 resistance. We believe that the presented data have provided important information about the structure-function relationship of gp41 and the structure-activity relationship (SAR) of viral fusion inhibitors, which definitely help our understanding of the mechanisms of HIV-1 entry and its inhibition.
Discovery of the peptide T20 and its successful development as the first drug blocking viral membrane fusion triggered tremendous efforts to exploit the mechanisms of viral entry and inhibition; however, the emergence and spread of T20-resistant HIV-1 variants have largely limited its clinical application and necessitated the design of novel fusion inhibitors. As a leading strategy, introducing positively and negatively charged residues into the solvent-accessible site of a peptide has been applied, resulting in a group of electronically constrained α-helical peptides with significantly improved pharmaceutical profiles, such as SC34EK (20), SC29EK (19), sifuvirtide (23), T2635 (29), and CP32M (30). Meanwhile, there were considerable efforts dedicated to elucidate the mechanism of HIV-1 resistance to the first-generation and newly designed fusion inhibitors (22, 27, 28, 31–36). It was found that escaping HIV-1 variants of the optimized α-helical peptide inhibitors usually require multiple mutations in gp41, thus suggesting a markedly increased genetic barrier for resistance (22, 33–36). Importantly, the N43K mutation in the inhibitor-binding site of the NHR helices was frequently selected as a primary mutation, which critically determined the resistance phenotype alone or in combination (33–36). In this study, we found that a single N43K mutation could confer high-level resistance to SC29EK, with a change in resistance of greater than 100-fold. The N43K also mediated considerable cross-resistance to the first-generation fusion inhibitors T20 (11.26-fold) and C34 (17.44-fold) as well as the newly designed fusion inhibitors sifuvirtide (21.99-fold) and MT-SC29EK (11.12-fold), indicating that this substitution plays a central role in SC29EK-induced resistance. Again, the results verified that the N43K-mediated resistance could be overcome by the electronically constrained short-peptide inhibitor 2P23, which targets mainly the gp41 pocket site rather than the N43K-containing region. In contrast to N43K, the Q39R and E49A mutations were rarely identified in the previous in vitro selection of escaping HIV-1 mutants. Here, both mutations were found to render a mild resistance to the inducer SC29EK, but they were able to greatly boost the N43K-mediated resistance in the presence or absence of a compensatory N126K mutation (Table 1), hence resulting in the emergence of three highly resistant HIV-1 mutants (E43K/E49A, Q39R/N43K/N126K, and N43K/E49A/N126K). It is worth noting that the selection and characterization of HIV-1 mutants resistant to SC22EK, a further truncated form of SC29EK, identified a single E49K substitution which conferred high resistance to the short-peptide inhibitors targeting the gp41 pocket and modest cross-resistance to the longer peptides T20, C34, and sifuvirtide (28). Taken together, these data highlight the importance of the negatively charged residue Glu-49 for action of the electronically constrained peptide inhibitors, such as SC29EK and SC22EK.
On the basis of mutational analysis and molecular docking, Eggink et al. described several pathways of resistance to peptide-based HIV-1 fusion inhibitors, including small-amino-acid-mediated reduced contact, large-amino-acid-mediated steric obstruction, acidic-amino-acid-mediated electrostatic repulsion, and basic-amino-acid-mediated electrostatic attraction (37). Based on the crystal structure of the fusion inhibitor sifuvirtide and its resistance mutations, we also proposed that the disruption of hydrogen bonds and hydrophobic contacts would impair the binding of inhibitors, thus critically determining the resistance (38). It is conceivable that the virus with multiple mutations would render the resistance through different molecular mechanisms described above, resulting in significantly reduced binding affinity of the inhibitors and dramatically damaged functionality of the viral Env complex (27, 28). In this study, the crystal structure of SC29EK bound to the target mimic peptide N36 did reveal the critical intra- and interhelical interactions underlying the mechanism of action of SC29EK and its resistance pathway. It was verified that the Q39R and N43K mutations conferred the resistance by destroying the hydrogen bond networks that were essential for binding stability of the inhibitor and simultaneously generating unfavorable repulsive charge-charge interactions. The structure also supported that the E49A mutation had no effect on the binding stability of SC29EK as determined by CD spectroscopy (Fig. 3), demonstrating that the E49A-mediated resistance might be dominated by a detrimental viral Env as shown in cell fusion and virus entry assays (Fig. 4). More ideally, a crystal structure of SC29EK in complex with a target mimic peptide bearing the single or combined resistant mutations would facilitate elucidation of the resistance mechanisms in more detail.
MATERIALS AND METHODS
Cell lines, plasmids, and antibodies.
TZM-bl indicator cells stably expressing CD4 and CCR5 along with endogenously expressed CXCR4 and the plasmid pSG3Δenv, which encodes an Env-defective, luciferase-expressing HIV-1 genome, were obtained from John C. Kappes and Xiaoyun Wu through the AIDS Reagent Program, Division of AIDS, NIAID, NIH (17). The plasmids DSP1-7 and DSP8-11 for the DSP-based fusion assay were kindly provided by Zene Matsuda at the Institute of Medical Science of the University of Tokyo (Tokyo, Japan) (39, 40). The monoclonal antibody NC-1 was kindly provided by Shibo Jiang at the Lindsley F. Kimball Research Institute of the New York Blood Center (New York, NY) (41). The monoclonal antibodies 17C8 and 2G8 were kindly provided by Yinghua Chen at the School of Life Sciences of Tsinghua University (Beijing, China) (42). HEK293T cells were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). Both cell lines were cultured in complete growth medium that consisted of Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml of penicillin-streptomycin, 2 mM l-glutamine, 1 mM sodium pyruvate, and 1× MEM nonessential amino acids (Gibco/Invitrogen, USA) and were maintained at 37°C in 5% CO2.
Peptide synthesis.
A panel of inhibitor peptides (SC29EK, T20, C34, SFT, MT-SC29EK, and 2P23) and the target mimic peptides (N36, N36Q39R, N36N43K, N36E49A, N36Q39R/N43K, and N36N43K/E49A) were synthesized with a standard solid-phase 9-fluorenylmethoxy carbonyl (FMOC) method as described previously (43). All peptides were protected by N-terminal acetylation and C-terminal amidation. They were purified to a purity of >95% by reversed-phase high-performance liquid chromatography (HPLC) and characterized for correct amino acid composition by mass spectrometry. Concentrations of the peptides were measured by UV absorbance and a theoretically calculated molar extinction coefficient based on tryptophan and tyrosine residues.
Site-directed mutagenesis.
HIV-1NL4-3 Env mutants were generated by site-directed mutagenesis as described previously (44). Briefly, two primers contained the desired mutation and occupied the same starting and ending positions on opposite strands of the plasmid. DNA synthesis was performed by PCR in a 50-μl reaction volume using 1 ng of denatured plasmid template, 50 pM concentrations of the upper and lower primers, and 5 U of the high-fidelity polymerase PrimeSTAR (TaKaRa, Dalian, China). PCR amplification was performed for one cycle of denaturation at 98°C for 5 min, followed by 18 cycles of 98°C for 15 s and 68°C for 15 min, with a final extension at 72°C for 10 min. The amplicons were digested with DpnI at 37°C for 1 h, and DpnI-resistant molecules were recovered by transforming Escherichia coli strain DH5α to antibiotic resistance. The introduced mutations were confirmed by DNA sequencing.
Single-cycle infection assay.
HIV-1 pseudoviruses were generated as described previously (44). In brief, 293T cells were cotransfected with an Env-expressing plasmid and a backbone plasmid (pSG3Δenv) that carries an Env-defective, luciferase-expressing HIV-1 genome. Supernatants were harvested 48 h after transfection, and 50% tissue culture infectious doses (TCID50) were determined in TZM-bl cells. Peptides were prepared in graded concentrations, mixed with 100 TCID50 of viruses, and then incubated 1 h at room temperature. The mixture was added to TZM-bl cells (104/well) containing DEAE (final concentration, 15 μl/ml) and incubated for 48 h at 37°C. The luciferase activity was measured using luciferase assay reagents and a luminescence counter (Promega, Madison, WI, USA).
DSP-based cell-cell fusion assay.
A dual split-protein (DSP)-based cell-cell fusion assay was adapted to determine the Env fusion activities of HIVNL4-3 and its mutants (27). Briefly, a total of 1.5 × 104 293T cells (effector cells) were seeded on a 96-well plate, and a total of 8 × 104 U87-CXCR4 cells (target cells) were seeded on a 24-well plate. On the following day, 293T cells were transfected with a mixture of an Env-expressing plasmid and a DSP1-7 plasmid, and U87-CXCR4 cells were transfected with a DSP8-11 plasmid. At 24 h posttransfection, the target cells were resuspended in 300 μl prewarmed culture medium containing EnduRen live-cell substrate (Promega) at a final concentration of 17 ng/μl and then transferred to each well of the effector cells with equal volumes. The cell mixture was spun down, and luciferase activity was measured with a luminescence counter (Promega).
ELISA.
To determine the effects of introduced mutations on Env expression, a capture enzyme-linked immunosorbent assay (ELISA) was conducted as described previously (28). Briefly, the wells of an ELISA plate were coated with a sheep anti-gp120 antibody (D7324) at 10 μg/ml and blocked with 3% bovine serum albumin (BSA). Cell lysates or culture supernatants (50 μl) of Env-transfected cells were added to the wells and incubated at 37°C for 1 h. After four washes, 50 μl of human anti-gp120 monoclonal antibody (MAb) VRC01 or anti-gp41 MAb 10E8 diluted 10 μg/ml was added and incubated at 37°C for 1 h. The bound antibodies were detected with horseradish peroxidase (HRP)-conjugated goat anti-human IgG (Sigma). The reaction was visualized by addition of 3,3,5,5-tetramethylbenzidine, and the A450 was measured. To detect the 6-HBs formed by N36 and C34 peptides, three 6-HB conformation-specific MAbs (NC-1, 17C8, and 2G8) were applied in an ELISA as described previously (27). Briefly, the isolated or mixed peptides were applied to the ELISA wells at 10 μg/ml and blocked with 3% BSA. After washing, the anti-6-HB MAb diluted at 5 μg/ml was added to the wells and incubated at 37°C for 1 h. After three washes, the bound antibodies were detected with HRP-conjugated anti-mouse IgG (Sigma). Similarly, the reaction was visualized by addition of 3,3,5,5-tetramethylbenzidine, and the A450 was measured.
Western blotting.
Cell lysates were centrifuged at 20,000 × g at 4°C for 15 min to remove insoluble materials. Equal amounts of total proteins were separated by 10% SDS-PAGE and then transferred to a nitrocellulose membrane. After blocking with 5% nonfat dry milk solution in Tris-buffered saline (TBS) (pH 7.4) at room temperature for 1 h, the membrane was incubated with the human anti-HIV polyclonal antibody HIV-IG (obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH) overnight at 4°C. On the following day, the membrane was washed three times in TBS-Tween 20 (TBS-T) and then incubated with IRDye 800CW goat anti-human IgG at room temperature for 2 h. The membrane was then scanned using the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE, USA).
CD spectroscopy.
Circular dichroism (CD) spectroscopy was conducted according to our protocol described previously (30). In brief, SC29EK was incubated with an equal molar concentration of the wild-type or mutant NHR peptide N36 at 37°C for 30 min in phosphate-buffered saline (PBS) (pH 7.2). CD spectra were acquired on a Jasco spectropolarimeter (model J-815) using a 1-nm bandwidth with a 1-nm step resolution from 195 to 260 nm at room temperature and corrected by subtraction of a solvent blank. The α-helical content was calculated from the CD signal by dividing the mean residue ellipticity (θ) at 222 nm by the value expected for 100% helix formation (−33,000 degrees cm−2 dmol−1). Thermal denaturation was performed by monitoring the ellipticity change at 222 nm from 20°C to 98°C at a rate of 1.2°C/min.
Crystallization, data collection, and structure determination.
To assemble the SC29EK/N36, two peptides were dissolved in denaturing buffer (100 mM NaH2PO4, 10 mM Tris-HCl, pH 8.0, 8 M urea) at an equal molar ratio and then dialyzed against buffer containing 50 mM Tris-HCl (pH 8.0) and 100 mM NaCl at 4°C overnight for refolding. The complex was subjected to size exclusion chromatography (Superdex 75 10/300 GL; GE Healthcare), and a peak elution corresponding to the size of a 6-HB was collected. After being concentrated, the SC29EK/N36 complex was crystallized by mixing equal volumes (1:1) of purified sample (20 mg/ml) and reservoir solution containing 0.1 M Tris-HCl (pH 8.0) and 28% (wt/vol) polyethylene glycol (PEG) 4000 in a hanging-drop vapor diffusion system at 22°C. The formed crystals were soaked in reservoir solution containing 15% glycerol, followed by 30 to 60 s of flash-freezing in liquid nitrogen. The diffraction data sets for SC29EK/N36 were collected at beamline BL-19U1 at the Shanghai Synchrotron Radiation Facility, China. The phasing problem was solved by the molecular replacement method using the software CCP4i with a crystal structure of HIV gp41 core (PDB accession number 5H0N) as a searching model (45, 46). The final model was manually adjusted in COOT based on the electron density map and refined with the program PHENIX (47, 48). The data collection and structure refinement statistics are shown in Table 3. The structure was validated by MolProbity analysis (49). The MolProbity score was 1.29, rating as the 99th percentile among structures of comparable resolution, and the Ramachandran plot found all the residues in the favored area. All structural figures were generated with the program PyMOL.
ACKNOWLEDGMENTS
This work was supported by grants from the Natural Science Foundation of China (81630061, 81473255, and 81673484) and the CAMS Innovation Fund for Medical Sciences (2017-I2M-1-014).
REFERENCES
- 1.Eckert DM, Kim PS. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem 70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 2.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 3.Tan K, Liu J, Wang J, Shen S, Lu M. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A 94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. doi: 10.1016/S0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 5.Qiu Z, Chong H, Yao X, Su Y, Cui S, He Y. 2015. Identification and characterization of a subpocket on the N-trimer of HIV-1 Gp41: implication for viral entry and drug target. AIDS 29:1015–1024. doi: 10.1097/QAD.0000000000000683. [DOI] [PubMed] [Google Scholar]
- 6.Crespillo S, Camara-Artigas A, Casares S, Morel B, Cobos ES, Mateo PL, Mouz N, Martin CE, Roger MG, El Habib R, Su B, Moog C, Conejero-Lara F. 2014. Single-chain protein mimetics of the N-terminal heptad-repeat region of gp41 with potential as anti-HIV-1 drugs. Proc Natl Acad Sci U S A 111:18207–18212. doi: 10.1073/pnas.1413592112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu S, Gochin M. 2013. Identification of fragments targeting an alternative pocket on HIV-1 gp41 by NMR screening and similarity searching. Bioorg Med Chem Lett 23:5114–5118. doi: 10.1016/j.bmcl.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He Y. 2013. Synthesized peptide inhibitors of HIV-1 gp41-dependent membrane fusion. Curr Pharm Des 19:1800–1809. doi: 10.2174/1381612811319100004. [DOI] [PubMed] [Google Scholar]
- 9.Steffen I, Pohlmann S. 2010. Peptide-based inhibitors of the HIV envelope protein and other class I viral fusion proteins. Curr Pharm Des 16:1143–1158. doi: 10.2174/138161210790963751. [DOI] [PubMed] [Google Scholar]
- 10.Eggink D, Berkhout B, Sanders RW. 2010. Inhibition of HIV-1 by fusion inhibitors. Curr Pharm Des 16:3716–3728. doi: 10.2174/138161210794079218. [DOI] [PubMed] [Google Scholar]
- 11.Lalezari JP, Henry K, O'Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M, TORO 1 Study Group . 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med 348:2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 12.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med 4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 13.Sista PR, Melby T, Davison D, Jin L, Mosier S, Mink M, Nelson EL, DeMasi R, Cammack N, Salgo MP, Matthews TJ, Greenberg ML. 2004. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS 18:1787–1794. doi: 10.1097/00002030-200409030-00007. [DOI] [PubMed] [Google Scholar]
- 14.Poveda E, Rodes B, Labernardiere JL, Benito JM, Toro C, Gonzalez-Lahoz J, Faudon JL, Clavel F, Schapiro J, Soriano V. 2004. Evolution of genotypic and phenotypic resistance to enfuvirtide in HIV-infected patients experiencing prolonged virologic failure. J Med Virol 74:21–28. doi: 10.1002/jmv.20141. [DOI] [PubMed] [Google Scholar]
- 15.Greenberg ML, Cammack N. 2004. Resistance to enfuvirtide, the first HIV fusion inhibitor. J Antimicrob Chemother 54:333–340. doi: 10.1093/jac/dkh330. [DOI] [PubMed] [Google Scholar]
- 16.Baldwin CE, Sanders RW, Deng Y, Jurriaans S, Lange JM, Lu M, Berkhout B. 2004. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J Virol 78:12428–12437. doi: 10.1128/JVI.78.22.12428-12437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rimsky LT, Shugars DC, Matthews TJ. 1998. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J Virol 72:986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naito T, Izumi K, Kodama E, Sakagami Y, Kajiwara K, Nishikawa H, Watanabe K, Sarafianos SG, Oishi S, Fujii N, Matsuoka M. 2009. SC29EK, a peptide fusion inhibitor with enhanced alpha-helicity, inhibits replication of human immunodeficiency virus type 1 mutants resistant to enfuvirtide. Antimicrob Agents Chemother 53:1013–1018. doi: 10.1128/AAC.01211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otaka A, Nakamura M, Nameki D, Kodama E, Uchiyama S, Nakamura S, Nakano H, Tamamura H, Kobayashi Y, Matsuoka M, Fujii N. 2002. Remodeling of gp41-C34 peptide leads to highly effective inhibitors of the fusion of HIV-1 with target cells. Angew Chem Int Ed Engl 41:2937–2940. doi:. [DOI] [PubMed] [Google Scholar]
- 21.Chong H, Qiu Z, Sun J, Qiao Y, Li X, He Y. 2014. Two M-T hook residues greatly improve the antiviral activity and resistance profile of the HIV-1 fusion inhibitor SC29EK. Retrovirology 11:40. doi: 10.1186/1742-4690-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eggink D, Bontjer I, Langedijk JP, Berkhout B, Sanders RW. 2011. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J Virol 85:10785–10797. doi: 10.1128/JVI.05331-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Y, Xiao Y, Song H, Liang Q, Ju D, Chen X, Lu H, Jing W, Jiang S, Zhang L. 2008. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem 283:11126–11134. doi: 10.1074/jbc.M800200200. [DOI] [PubMed] [Google Scholar]
- 24.Xiong S, Borrego P, Ding X, Zhu Y, Martins A, Chong H, Taveira N, He Y. 2017. A helical short-peptide fusion inhibitor with highly potent activity against human immunodeficiency virus type 1 (HIV-1), HIV-2, and simian immunodeficiency virus. J Virol 91:e01839-16. doi: 10.1128/JVI.01839-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su Y, Chong H, Xiong S, Qiao Y, Qiu Z, He Y. 2015. Genetic pathway of HIV-1 resistance to novel fusion inhibitors targeting the gp41 pocket. J Virol 89:12467–12479. doi: 10.1128/JVI.01741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su Y, Chong H, Qiu Z, Xiong S, He Y. 2015. Mechanism of HIV-1 resistance to short-peptide fusion inhibitors targeting the gp41 pocket. J Virol 89:5801–5811. doi: 10.1128/JVI.00373-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. 2007. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc Natl Acad Sci U S A 104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He Y, Cheng J, Lu H, Li J, Hu J, Qi Z, Liu Z, Jiang S, Dai Q. 2008. Potent HIV fusion inhibitors against enfuvirtide-resistant HIV-1 strains. Proc Natl Acad Sci U S A 105:16332–16337. doi: 10.1073/pnas.0807335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eggink D, de Taeye SW, Bontjer I, Klasse PJ, Langedijk JP, Berkhout B, Sanders RW. 2016. HIV-1 escape from a peptidic anchor inhibitor through stabilization of the envelope glycoprotein spike. J Virol 90:10587–10599. doi: 10.1128/JVI.01616-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Feo CJ, Wang W, Hsieh ML, Zhuang M, Vassell R, Weiss CD. 2014. Resistance to N-peptide fusion inhibitors correlates with thermodynamic stability of the gp41 six-helix bundle but not HIV entry kinetics. Retrovirology 11:86. doi: 10.1186/s12977-014-0086-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimane K, Kawaji K, Miyamoto F, Oishi S, Watanabe K, Sakagami Y, Fujii N, Shimura K, Matsuoka M, Kaku M, Sarafianos SG, Kodama EN. 2013. HIV-1 resistance mechanism to an electrostatically constrained peptide fusion inhibitor that is active against T-20-resistant strains. Antimicrob Agents Chemother 57:4035–4038. doi: 10.1128/AAC.00237-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z, Shan M, Li L, Lu L, Meng S, Chen C, He Y, Jiang S, Zhang L. 2011. In vitro selection and characterization of HIV-1 variants with increased resistance to sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem 286:3277–3287. doi: 10.1074/jbc.M110.199323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimura K, Nameki D, Kajiwara K, Watanabe K, Sakagami Y, Oishi S, Fujii N, Matsuoka M, Sarafianos SG, Kodama EN. 2010. Resistance profiles of novel electrostatically constrained HIV-1 fusion inhibitors. J Biol Chem 285:39471–39480. doi: 10.1074/jbc.M110.145789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eggink D, Baldwin CE, Deng Y, Langedijk JP, Lu M, Sanders RW, Berkhout B. 2008. Selection of T1249-resistant human immunodeficiency virus type 1 variants. J Virol 82:6678–6688. doi: 10.1128/JVI.00352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eggink D, Langedijk JP, Bonvin AM, Deng Y, Lu M, Berkhout B, Sanders RW. 2009. Detailed mechanistic insights into HIV-1 sensitivity to three generations of fusion inhibitors. J Biol Chem 284:26941–26950. doi: 10.1074/jbc.M109.004416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao X, Chong H, Zhang C, Waltersperger S, Wang M, Cui S, He Y. 2012. Broad antiviral activity and crystal structure of HIV-1 fusion inhibitor sifuvirtide. J Biol Chem 287:6788–6796. doi: 10.1074/jbc.M111.317883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kondo N, Miyauchi K, Meng F, Iwamoto A, Matsuda Z. 2010. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J Biol Chem 285:14681–14688. doi: 10.1074/jbc.M109.067090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel 25:813–820. doi: 10.1093/protein/gzs051. [DOI] [PubMed] [Google Scholar]
- 41.Jiang S, Lin K, Lu M. 1998. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J Virol 72:10213–10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Chen X, Huang J, Jiang S, Chen YH. 2009. Identification of critical antibody-binding sites in the HIV-1 gp41 six-helix bundle core as potential targets for HIV-1 fusion inhibitors. Immunobiology 214:51–60. doi: 10.1016/j.imbio.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 43.Chong H, Yao X, Sun J, Qiu Z, Zhang M, Waltersperger S, Wang M, Cui S, He Y. 2012. The M-T hook structure is critical for design of HIV-1 fusion inhibitors. J Biol Chem 287:34558–34568. doi: 10.1074/jbc.M112.390393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chong H, Yao X, Zhang C, Cai L, Cui S, Wang Y, He Y. 2012. Biophysical property and broad anti-HIV activity of albuvirtide, a 3-maleimimidopropionic acid-modified peptide fusion inhibitor. PLoS One 7:e32599. doi: 10.1371/journal.pone.0032599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su S, Zhu Y, Ye S, Qi Q, Xia S, Ma Z, Yu F, Wang Q, Zhang R, Jiang S, Lu L. 2017. Creating an artificial tail anchor as a novel strategy to enhance the potency of peptide-based HIV fusion inhibitors. J Virol 91:e01445-16. doi: 10.1128/JVI.01445-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948–1954. doi: 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- 49.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]