

ABSTRACT
The human JC polyomavirus (JCPyV) infects the majority of the population worldwide and presents as an asymptomatic, persistent infection in the kidneys. In individuals who are immunocompromised, JCPyV can become reactivated and cause a lytic infection in the central nervous system resulting in the fatal, demyelinating disease progressive multifocal leukoencephalopathy (PML). Infection is initiated by interactions between the capsid protein viral protein 1 (VP1) and the α2,6-linked sialic acid on lactoseries tetrasaccharide c (LSTc), while JCPyV internalization is facilitated by 5-hydroxytryptamine 2 receptors (5-HT2Rs). The mechanisms by which the serotonin receptors mediate virus entry and the signaling cascades required to drive viral infection remain poorly understood. JCPyV was previously shown to induce phosphorylation of extracellular signal-regulated kinase (ERK), a downstream target of the mitogen-activated protein kinase (MAPK) pathway, upon virus entry. However, it remained unclear whether ERK activation was required for JCPyV infection. Both ERK-specific small interfering RNA (siRNA) and ERK inhibitor treatments resulted in significantly diminished JCPyV infection in both kidney and glial cells yet had no effect on the infectivity of the polyomavirus simian virus 40 (SV40). Experiments characterizing the role of ERK during steps in the viral life cycle indicate that ERK activation is required for viral transcription, as demonstrated by a significant reduction in production of large T antigen (TAg), a key viral protein associated with the initiation of viral transcription and viral replication. These findings delineate the role of the MAPK-ERK signaling pathway in JCPyV infection, elucidating how the virus reprograms the host cell to promote viral pathogenesis.
IMPORTANCE Viral infection is dependent upon host cell factors, including the activation of cellular signaling pathways. These interactions between viruses and host cells are necessary for infection and play an important role in viral disease outcomes. The focus of this study was to determine how the human JC polyomavirus (JCPyV), a virus that resides in the kidney of the majority of the population and can cause the fatal, demyelinating disease progressive multifocal leukoencephalopathy (PML) in the brains of immunosuppressed individuals, usurps a cellular signaling pathway to promote its own infectious life cycle. We demonstrated that the activation of extracellular signal-regulated kinase (ERK), a component of the mitogen-activated protein kinase (MAPK) pathway, promotes JCPyV transcription, which is required for viral infection. Our findings demonstrate that the MAPK-ERK signaling pathway is a key determinant of JCPyV infection, elucidating new information regarding the signal reprogramming of host cells by a pathogenic virus.
KEYWORDS: ERK, JC polyomavirus, MAPK, PML, T antigen, polyomavirus, transcription
INTRODUCTION
The human JC polyomavirus (JCPyV) is a member of the Polyomaviridae family, which also includes simian virus 40 (SV40) and BK polyomavirus (BKPyV). JCPyV infects 50 to 80% of healthy individuals and causes an asymptomatic, lifelong, persistent infection in the kidneys (1, 2). In immunosuppressed individuals, JCPyV can become reactivated in the central nervous system (CNS) (3, 4) and cause an uncontrolled lytic infection in glial cells, astrocytes and oligodendroctyes (5–7). JCPyV infection and subsequent cytolytic destruction of oligodendrocytes, the myelin-producing cells of the CNS, cause the demyelinating disease progressive multifocal leukoencephalopathy (PML) (8, 9). PML occurs in approximately 5% of individuals with HIV-1, yet highly active antiretroviral therapies (HAART) have reduced the incidence rate in the HIV-1 population significantly (10, 11). PML is also associated with the use of immunomodulatory therapies such as natalizumab or dimethyl fumarate and other fumaric acid ester-containing drugs prescribed for immune-mediated diseases such as multiple sclerosis (MS) (11–15). PML has a high mortality rate, proving fatal within months if left completely untreated. However, treatment of the underlying immunosuppression increases life expectancy to 2 years for ∼50% of HIV-1-positive individuals and ∼77% for individuals receiving natalizumab (11). There are few effective treatment options available for PML, with the exception of immune reconstitution therapy, which can cause immune reconstruction inflammatory syndrome (IRIS), an intense inflammatory response that can be fatal or lead to severe neurological deterioration (10). Therefore, there is a critical need for an improved understanding of the mechanisms driving JCPyV infection and viral pathogenesis.
JCPyV infection is initiated via attachment to α2,6-linked sialic acid on lactoseries tetrasaccharide c (LSTc) through viral protein 1 (VP1), the major viral capsid protein (16–18). While the α2,6-linked sialic acid receptor could be expressed on either a glycolipid or glycoprotein, the sialic acid receptor has been demonstrated to specifically mediate viral attachment (16–18). JCPyV requires the 5-hydroxytryptamine 2 family of serotonin receptors (5-HT2Rs) for viral entry, which is thought to occur through clathrin-dependent endocytosis (19). Following endocytosis, JCPyV traffics through the endocytic compartment (20–22) to the endoplasmic reticulum (ER), where it undergoes partial disassembly (22) before deposition in the nucleus, where the double-stranded DNA (dsDNA) genome is transcribed and replicated.
Like for many other small DNA viruses, the JCPyV transcriptional program is bidirectional. With this replication strategy, JCPyV first initiates transcription of viral early genes, including large tumor antigen (T antigen [TAg]). The accumulation of TAg drives replication of viral DNA and then leads to the transcription of the late viral genes that encode the structural capsid proteins VP1, VP2, and VP3 (9). With the accumulation of viral gene copies within the host, viral gene transcripts overwhelm normal host-cell protein production capabilities, forcing the cell to preferentially synthesize viral proteins associated with capsid formation for eventual viral assembly and egress (23).
The cellular signaling events activated in host cells in response to JCPyV infection and how they regulate viral infection and pathogenesis are poorly understood. Querbes et al. demonstrated that JCPyV requires tyrosine kinase activity for infection, as genistein, a broad-spectrum tyrosine kinase inhibitor, blocks JCPyV infection. Furthermore, the mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase (ERK) is phosphorylated 15 min following infection (20), a time that is consistent with viral attachment and entry events (22). ERK phosphorylation was shown to be sustained for 6 h following infection (20). This indicates that ERK might impact multiple steps in the virus life cycle, including entry and trafficking, as JCPyV is localized at 6 h postinfection in the ER, where it is thought to undergo partial uncoating (20, 22). These findings suggest that JCPyV may usurp MAPK-ERK pathway functions to drive the infectious life cycle, yet the direct impact of the MAPK pathway on the infectious life cycle of JCPyV has not been fully characterized.
The MAPK signaling pathway (Raf/MEK/ERK) induces cellular reprogramming in response to extracellular stimuli, such as growth factors or neurotransmitters (24, 25), through a series of phosphorylation events driven by the MAPK kinases (MAPKKs or MEKs). The small GTPase Ras binds to the serine/threonine kinase Raf (MAPKKK), which phosphorylates the MAPKKs MEK1 and MEK2 (26–29). The dual-specificity kinases MEK1 and MEK2 then phosphorylate ERK1/2 (30–32). Phosphorylated ERK activates downstream signaling targets, becomes dephosphorylated by protein phosphatases, or translocates to the nucleus to regulate transcription factors that function in cell proliferation, differentiation, and cell death (32).
Both DNA and RNA viruses have been reported to activate the Raf/MEK/ERK signal transduction pathway and in some cases utilize it for infectivity (33), including members of the Polyomaviridae family (34–36). Activation of the MAPK pathway by BK polyomavirus (BKPyV) enhances viral replication (35), but BKPyV does not activate the MAPK pathway directly (37). Conversely, mouse polyomavirus (MPyV) activates the Raf/MEK/ERK pathway (34, 36) but does not require activation of the MAPK pathway for infection (36). Furthermore, SV40 polyomavirus activates a MAPK-ERK-independent signaling pathway that induces Ca2+ mobilization and tyrosine kinase activity of protein kinase C (PKC) (34, 38). The MAPK-ERK pathway has been proposed to play a role in JCPyV infection (20, 39), yet the impact of MAPK activation on the infectious viral life cycle remained unclear, as polyomavirus activation of the MAPK pathway is multifactorial and may play critical roles in viral infectivity or could be dispensable.
The goal of this study was to define whether activation of the MAPK-ERK pathway is required for JCPyV infection and to define the steps in the virus life cycle that are impacted by ERK activation. JCPyV infection was significantly reduced in cells expressing ERK1/2-specific small interfering RNA (siRNA) and in cells treated with inhibitors of MEK, which prevent ERK phosphorylation, yet had no impact on SV40 infection. JCPyV induced ERK activation immediately upon infection, and a MEK inhibitor reduced JCPyV infection within 0 to 4 h postinfection. However, inhibition of ERK had no impact on the early steps in the viral life cycle, including attachment or entry, yet resulted in reduced activity of the viral early and late promoters and reduced T-antigen gene expression. These findings demonstrate that activation of the Raf/MEK/ERK signaling pathway is required for driving JCPyV infection and that this pathway is used to reprogram host cells for viral transcription.
RESULTS
Inhibition of ERK blocks JCPyV infection in glial cells.
To determine whether ERK is necessary for JCPyV infection, SVG-A cells were transfected with siRNA targeted toward ERK1 and ERK2 to reduce ERK1/2 protein levels in cells (Fig. 1A) and then infected with JCPyV or the control SV40 polyomavirus (Fig. 1B). Cells expressing the ERK1/2 siRNA demonstrated a 97% decrease in JCPyV infection compared to cells expressing a control siRNA, while no significant reduction in SV40 infection was observed (Fig. 1B). These data suggest that ERK is required for JCPyV infection, yet ERK activity is not required for infection of the closely related polyomavirus SV40. To confirm that JCPyV infection of glial cells is dependent on the activation of MAPK-ERK, cells were treated with ERK inhibitors PD98059 (2′-amino-3′-methoxyflavone) and U0126 (1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio] butadiene), which both bind to MEK1 and MEK2, preventing ERK phosphorylation (40, 41). SVG-A cells treated with PD98059 demonstrated an ∼75% reduction in JCPyV infection in comparison to control dimethyl sulfoxide (DMSO)-treated cells (Fig. 2A). However, PD98059 had no effect on SV40 infection in SVG-A cells (Fig. 2B), indicating that the inhibition of this pathway is specific for JCPyV. Treatment with ERK inhibitor U0126 also reduced JCPyV infection by ∼75% (Fig. 2C) yet had no effect on SV40 infection (Fig. 2D). The efficacy of the MEK inhibitors PD98059 and U0126 in SVG-A cells was analyzed through Western blot analysis of cellular levels of phosphorylated ERK (pERK) in comparison to those in DMSO-treated cells (data not shown). Treatment of cells with PD98059 decreased levels of pERK, yet U0126 resulted in a more significant decrease, suggesting that U0126 is a more effective inhibitor of ERK phosphorylation in SVG-A cells. Due to these results, in addition to U0126 being reported as a more potent inhibitor with a greater 50% inhibitory concentration (IC50) (41), subsequent experiments were performed using U0126 at a concentration of 10 μM to effectively inhibit ERK phosphorylation. The indicated concentrations of inhibitors did not affect cell viability as measured by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay and flow cytometric analysis of propidium iodide-stained cells (data not shown).
FIG 1.
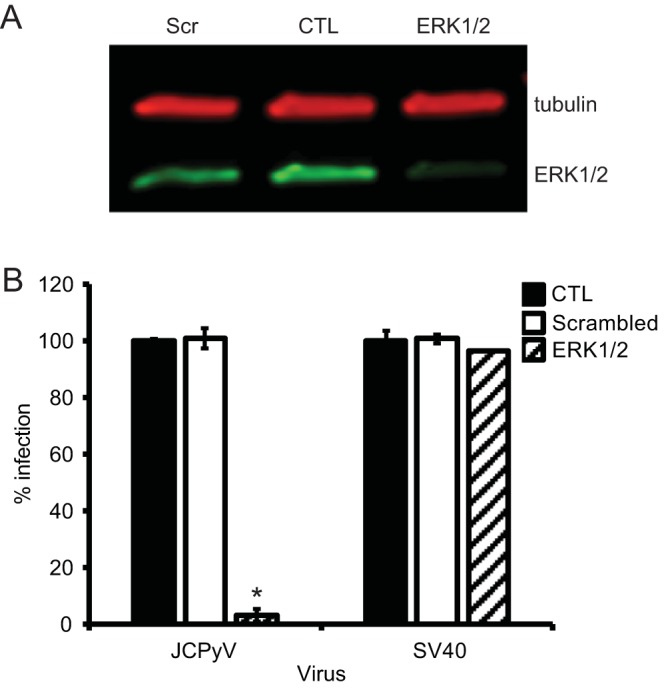
Gene silencing of ERK reduces JCPyV infection in SVG-A cells. SVG-A cells were transfected with siRNAs targeted to ERK1/2, scrambled, or EGFR (control [CTL]) siRNA. At 48 h posttransfection, cells were either harvested for analysis of ERK protein expression (A) or infected (B). (A) Cell lysates of siRNA-treated cells were resolved by SDS-PAGE and analyzed via Western blotting to confirm ERK-specific knockdown using antibodies specific for tubulin (red) and ERK1/2 (green). The blot is representative of results from three independent experiments. (B) SVG-A cells transfected with ERK or control siRNAs were infected with JCPyV (MOI = 0.1 FFU/cell) or SV40 (MOI = 0.001 FFU/cell). Infected cells were scored by indirect immunofluorescence using a VP1-specific antibody that recognizes both JCPyV and SV40 VP1. Data represent the percentage of JCPyV or SV40-infected VP1+ cells per visual field normalized to the number of DAPI+ cells per field for five 10× fields of view for triplicate samples (all samples normalized to EGFR control siRNA-treated cells [100%]). Data are representative of results from 3 independent experiments. Error bars indicate SDs. Student's t test was used to determine statistical significance. *, P < 0.001.
FIG 2.

Inhibition of ERK phosphorylation decreases JCPyV infection in glial cells. SVG-A cells were pretreated with the MEK inhibitor PD98059 (A and B) or U0126 (C and D) at the indicated concentrations or DMSO at an equivalent volume (of maximum inhibitor concentration) and then infected with JCPyV (MOI = 0.1 FFU/cell) (A and C) or SV40 (MOI = 0.001 FFU/cell) (B and D) at 37°C for 1 h. Cells were incubated in media containing inhibitors at the indicated concentrations for 72 h and then fixed and stained by indirect immunofluorescence using a VP1-specific antibody that recognizes both JCPyV and SV40 VP1. Infectivity was scored by quantitating nuclear VP1 expression. Data represent the average number of infected cells per visual field for five 10× fields of view for triplicate samples. Data are representative of results from 3 independent experiments. Error bars indicate SDs. Student's t test was used to determine statistical significance. *, P < 0.05.
JCPyV induces multiphasic ERK activation upon infection.
JCPyV infection has been previously shown to induce ERK phosphorylation as early as 15 min postinfection, with decreasing activation through 9 h postinfection (20), suggesting that phosphorylation of ERK is required for viral entry and trafficking (20, 22). However, ERK activation has not been characterized throughout the JCPyV life cycle. To determine the timing of ERK activation following JCPyV challenge, SVG-A cells were infected with JCPyV or mock infected, harvested at selected time points, and analyzed for ERK phosphorylation by quantitative Western blot analysis (Fig. 3A). A rapid ERK phosphorylation was induced within 10 min following JCPyV challenge and activation was sustained in a multiphasic pattern for the first ∼6 to 12 h in comparison to mock-infected cells. However, the phosphorylated ERK signal waned after 12 h, with only a minor increase in the pERK signal at 72 h postinfection in comparison to that in mock-infected cells (Fig. 3A). These data suggest that ERK is activated early during infection, which corroborates previously published findings (20) and suggests that the rapid ERK activation is not sustained for later steps in the viral life cycle.
FIG 3.

JCPyV induces multiphasic ERK activation upon infection. (A) SVG-A cells were infected with JCPyV (MOI = 1 FFU/cell) for the specified duration and were analyzed via Western blotting using antibodies specific for phosphorylated ERK (pERK) and total ERK (protein control). The percentage of pERK was determined using phosphorylated ERK and total ERK band intensities, determined by using the equation [pERK/(pERK + total ERK)] × 100. The percentage of pERK for JCPyV-infected samples was normalized to the value for mock-infected samples at each time point (100%; dashed line). Percentages of phosphorylated ERK for each time point were plotted via box-and-whisker plot and are representative of results from three independent experiments. (B) SVG-A cells were either pretreated (PT) for 1 h prior to JCPyV infection with U0126 (10 μM) or DMSO (1:1,000) or incubated in complete medium. Cells were then infected with JCPyV (MOI = 0.1 FFU/cell) at 4°C for 1 h. Following infection, cells were washed with medium and then incubated in media with inhibitors for the indicated times. At the specified time point, medium was removed, cells were washed, complete medium was added, and cells were incubated at 37°C for 72 h. Cells were fixed and stained by indirect immunofluorescence using a VP1-specific antibody, and infectivity was scored by quantitating nuclear VP1 expression. Data are representative of the average number of infected cells per visual field for five 20× fields of view for triplicate samples. Data are representative of results from three independent experiments. Error bars indicate SDs. Student's t test was used to determine statistical significance. *, P < 0.05.
To initially characterize how ERK activation (Fig. 3A) or inhibition (Fig. 1 and 2) impacts the JCPyV life cycle, SVG-A cells were treated with the ERK inhibitor U0126 under various treatment regimens (Fig. 3B). As demonstrated in Fig. 2, ERK inhibition led to an ∼75% decrease when cells were pretreated with the U0126 for 1 h prior to infection and the inhibitor was added back to the media for the duration of the infection (Fig. 2 and 3). In order to determine how ERK inhibition impacts the JCPyV life cycle, SVG-A cells were pretreated with U0126 and subsequently incubated with treated media for the first 1 to 4 h following infection, which resulted in an ∼40% decrease in infection (Fig. 3B). This treatment is consistent with the time frame in which the most robust ERK phosphorylation is noted (0 to 2 h postinfection [h.p.i.]), leading to the hypothesis that ERK activation affects the initial events in the viral life cycle, including attachment and entry (22).
Inhibition of ERK does not affect JCPyV attachment.
JCPyV-induced ERK activation is coincident with the timing of viral attachment and entry (Fig. 3) (20, 22). Further, ligand binding to the 5-HT2Rs, which are required for JCPyV entry, can activate the Raf/MEK/ERK pathway, which regulates receptor cell surface expression, endocytosis, and activation of other signaling pathways (42). Therefore, the effect of ERK inhibition on viral attachment to host cells was analyzed. SVG-A cells were pretreated with U0126 or DMSO and then incubated with Alexa Fluor 488-labeled JCPyV (JCPyV-488) virions on ice to allow for viral attachment (Fig. 4), and JCPyV-488 binding to SVG-A cells was subsequently measured by flow cytometry. Treatment of cells with U0126 had no effect on viral attachment in comparison to that with DMSO-treated SVG-A cells, suggesting that ERK inhibition does not affect virus binding to the cell surface.
FIG 4.

JCPyV attachment to SVG-A cells is not altered by ERK inhibition. SVG-A cells in 6-well plates were treated with U0126 (10 μM) or DMSO (1:1,000) at 37°C for 1 h, then washed with PBS, and removed from plates using Cellstripper. Cells were washed and incubated with JCPyV-488 on ice for 2 h and washed with PBS, and binding was analyzed by flow cytometry. Histograms represent the fluorescence intensity of JCPyV-488 for DMSO- and U0126-treated cells for 10,000 events. The shaded histogram represents cells that were not incubated with JCPyV (PBS alone). Data are representative of results from >3 independent experiments.
JCPyV internalization is not affected by ERK inhibition.
JCPyV induces ERK activation within 10 to 15 min of viral infection (Fig. 3) (20), a time consistent with viral entry (22). To determine if ERK activation impacts virus entry, a trypan blue (TB) quenching assay was utilized to measure viral internalization by flow cytometry (22) (Fig. 5). In brief, trypan blue quenches Alexa Fluor 488 fluorescence of plasma membrane-associated labeled virus, but internalized fluorescently labeled virus is protected from TB treatment. SVG-A cells were pretreated with DMSO or U0126, incubated with JCPyV-488 on ice at 4°C to allow for viral attachment for 90 min, and then either fixed or incubated at 37°C for an additional 90 min to allow for entry. Fixed samples were then treated or not with TB (Fig. 5A). Trypan blue treatment resulted in an ∼1-log decrease in JCPyV-488 fluorescence of attached virus as measured by flow cytometry. Additionally, an increase in protected fluorescence in samples incubated at 37°C was observed, indicating that the virus was internalized at 90 min postinfection and was protected from TB treatment (Fig. 5A). The extent of protected fluorescence is consistent with published results of JCPyV entry analyzed via a confocal TB assay (22). Quantitative analysis of flow cytometry results demonstrated that treatment of cells with U0126 had no effect on virus attachment or entry as assessed from equivalent levels of virus binding and entry in U0126- and DMSO-treated cells (Fig. 5B). Taken together, these data indicate that ERK inhibition does not affect JCPyV attachment or entry.
FIG 5.

Entry of JCPyV does not require ERK activation. (A) SVG-A cells treated with either U0126 (10 μM) or DMSO (1:1,000) were incubated with JCPyV-488 on ice at 4°C to promote viral attachment. Cells were fixed (attachment) or transferred to 37°C for 90 min to promote viral internalization (entry). Quenched samples (trypan blue [TB] treated [blue]) and unquenched samples (gray) were then assayed by flow cytometry. Samples were gated to remove cellular debris. Histograms are representative of the mean fluorescence intensity from one replicate for each treatment (n = 10,000 events). (B) The percent protected fluorescence was calculated with the equation (FITC with TB/FITC without TB) × 100 for three individual experiments (each with 10,000 events). Data are the averages from three independent experiments (n = 30,000 events). Error bars indicate SDs.
ERK inhibition affects the JCPyV infectious cycle at a posttrafficking step.
To define whether entry and trafficking of JCPyV are impacted by ERK inhibition, SVG-A cells were pretreated with U0126 or DMSO and then transiently transfected with an infectious clone of JCPyV or SV40 to bypass attachment to viral receptors, endocytosis, and trafficking through the endocytic compartment (Fig. 6). Cells were fixed and stained for VP1 expression at days 4 and 7 following transfection. JCPyV VP1 expression was decreased in U0126-treated cells in comparison to that in DMSO-treated cells (Fig. 6A), yet SV40 VP1 expression was not altered in response to U0126 treatment (Fig. 6B). These data indicate that inhibition of ERK blocks a step in the JCPyV life cycle that occurs after trafficking.
FIG 6.
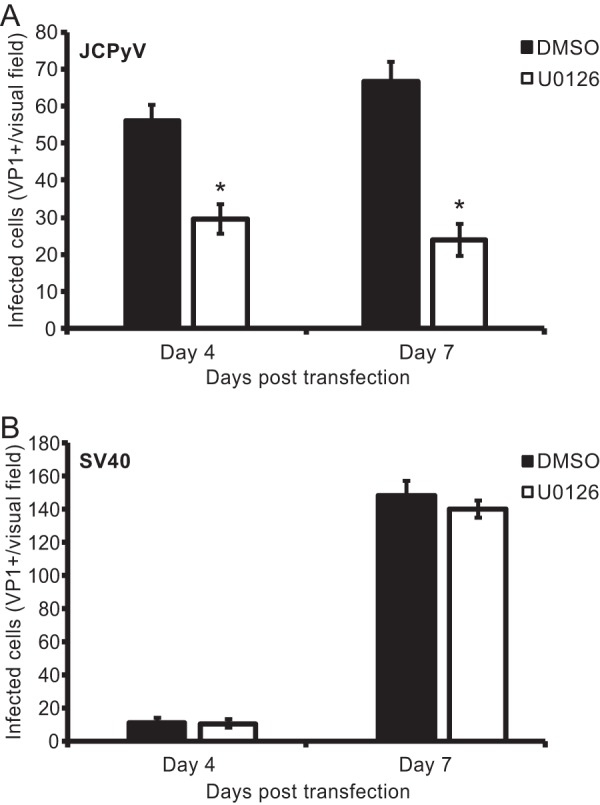
ERK inhibition impacts a postentry step in infection. SVG-A cells were treated with either U0126 (10 μM) or DMSO (1:1,000) for 1 h and then transfected with an infectious clone of JCPyV (A) or SV40 (B). At either 4 or 7 days posttransfection, cells were fixed and stained using a VP1-specific antibody and analyzed by indirect immunofluorescence for infectivity. Data are representative of the number of infected cells per visual field for five 10× fields of view for triplicate samples. Data are representative of results from three independent experiments. Error bars indicate SDs. *, P < 0.05.
ERK inhibition decreases JCPyV early and late promoter activity.
Activation of the Raf/MEK/ERK pathway can lead to phosphorylation of ERK and its subsequent translocation to the nucleus, where it can activate transcription factors, including transcription factors known to be involved in JCPyV infection, such as NFAT4 (43) and SMADs 2 and 4 (39). Polyomaviruses have a bidirectional transcription program, in which the viral early genes are transcribed first, including that for large T antigen, followed by genome replication and then transcription of the late viral genes that encode the structural capsid proteins, including VP1 (9). To analyze potential effects of ERK inhibition on viral transcription, viral promoter activity was measured in SVG-A cells treated with the ERK inhibitor U0126. Cells were treated with DMSO or U0126 and then transfected with an early or late viral promoter construct containing a luciferase reporter (43), and luciferase production was measured at 48 h posttransfection (Fig. 7). ERK inhibition resulted in a significant decrease in both early and late promoter activities as measured by luciferase production, indicating that inhibition of ERK reduces viral promoter activity and may therefore directly impact the transcription of early and late viral genes in SVG-A cells.
FIG 7.
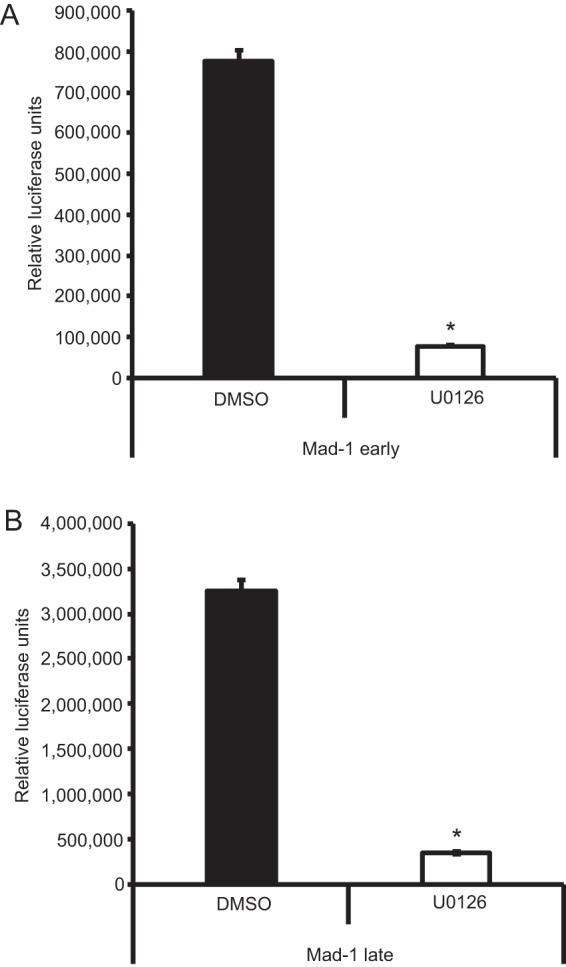
ERK inhibition blocks JCPyV promoter activity. SVG-A cells were pretreated with U0126 (10 μM) or DMSO (1:1,000) for 4 h and subsequently transfected with luciferase-expressing plasmids containing the JCPyV Mad-1 prototype strain early (A) or late (B) promoters and a thymidine kinase (TK) Renilla luciferase plasmid as a control. At 4 h posttransfection, medium containing U0126 (10 μM) or DMSO (1:1,000) was added back to cells. At 48 h posttransfection, cells were assayed for luciferase production, indicative of viral promoter activity, by luminometry. Data represent the average relative luciferase units produced from triplicate samples normalized to TK Renilla luciferase expression. These data are representative of results from three independent experiments. Error bars indicate SDs. *, P < 0.05.
Inhibition of ERK blocks JCPyV infection in kidney cells.
To determine whether ERK is required for JCPyV infection of kidney cells, primary renal proximal tubule epithelial cells (RPTECs) and HEK293A cells stably expressing 5-HT2AR–yellow fluorescent protein (YFP) (HEK293A-5-HT2AR) were analyzed. HEK293A cells are poorly permissive for JCPyV infection unless transfected with 5-HT2Rs (19). RPTECs and HEK293A-5-HT2AR cells were pretreated with either DMSO or U0126 for 1 h, infected with JCPyV (Fig. 8A and B) or SV40 (Fig. 8C), and then incubated with either DMSO or U0126 for 48 h. HEK293A-5-HT2AR cells treated with U0126 demonstrated an ∼50% decrease in JCPyV infection compared to that of cells treated with DMSO, suggesting that ERK is required for JCPyV infection in kidney cells (Fig. 8B) but not required for SV40 infection (Fig. 8C). To ensure that the transformation of the HEK293A-5-HT2AR cells with the adenovirus E1 gene (44) did not influence the impact of ERK inhibition on JCPyV infectivity, primary RPTECs were infected with JCPyV in the presence of U0126 (as described above) (Fig. 8A). Inhibition of ERK in RPTECs resulted in an ∼40% decrease in JCPyV infectivity, suggesting that ERK activity is also required for JCPyV infection in primary kidney cell lines. SV40 does not productively infect RPTECs (45) and thus could not be utilized as a control. Together, these data suggest that the MAPK-ERK cascade is required for JCPyV infection of both kidney and glial cell types.
FIG 8.

Inhibition of ERK phosphorylation decreases JCPyV infection in kidney cells. RPTEC (A) and HEK293A-5-HT2AR (B and C) cells were pretreated with U0126 (10 μM) or DMSO (1:1,000) at 37°C for 1 h and then infected with JCPyV (MOI = 0.5 FFU/cell) (A and B) or SV40 (MOI = 0.001 FFU/cell) (C) at 37°C for 1 h. Cells were incubated in media containing inhibitors at 37°C for 48 h and then fixed and stained by indirect immunofluorescence using JCPyV or SV40 T-antigen-specific antibodies. Infectivity was scored by quantitating nuclear T-antigen expression. Data represent the percentage of JCPyV- or SV40-infected cells per visual field normalized to the number of DAPI+ cells per field for five 20× fields of view for triplicate samples from three independent experiments. Samples were normalized to the DMSO-treated control cells (100%). Error bars indicate SDs. Student's t test was used to determine statistical significance. *, P < 0.05.
T-antigen expression is reduced in kidney and glial cell types when ERK is inhibited.
To determine whether the activation of the MAPK-ERK pathway is necessary for expression of the viral early gene encoding T antigen, SVG-A cells were transfected with an siRNA to reduce ERK1/2 protein levels and then infected with JCPyV, and T-antigen production was measured by indirect immunofluorescence (Fig. 9A). Cells expressing an ERK1/2 siRNA demonstrated an 85% decrease in T-antigen expression compared to that in cells expressing a control siRNA. SVG-A cells are transformed with an origin-defective SV40 T antigen (46), and thus SV40 T-antigen expression cannot be measured in SVG-A cells as a control. To ensure that the effect of ERK inhibition was not affected by T-antigen transformation of the SVG-A cells, HEK293A-5-HT2AR cells were also treated with the ERK1/2 siRNA and infected with JCPyV or SV40 as a control (Fig. 9B and C). JCPyV T-antigen expression was reduced by 79% in HEK293A-5-HT2AR cells treated with the ERK1/2 siRNA (Fig. 9B), while SV40 T-antigen expression was unaffected by ERK inhibition (Fig. 9C). Additionally, RPTECs treated with the ERK inhibitor U0126 demonstrated a significant decrease in T-antigen production as measured by indirect immunofluorescence (Fig. 8A). These data suggest that inhibition of ERK activation impacts the production of T antigen and possibly the initiation of viral JCPyV transcription.
FIG 9.
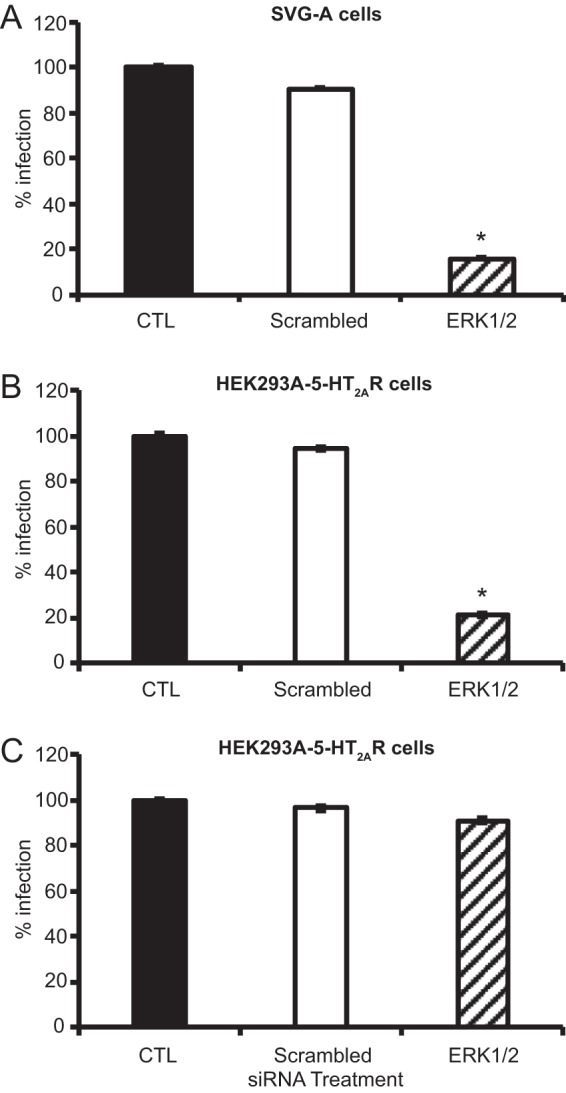
ERK activation is required for viral early gene expression. SVG-A (A) and HEK293A-5-HT2AR (B and C) cells transfected with ERK1/2 or control siRNAs were infected with JCPyV (MOI = 1 FFU/cell) (A and B) or SV40 (MOI = 0.001 FFU/cell) (C). Infected cells were scored by indirect immunofluorescence using JCPyV or SV40 T-antigen-specific antibodies. Data represent the percentage of JCPyV- or SV40-infected cells per visual field normalized to the number of DAPI+ cells per field for five 10× fields of view for triplicate samples (all samples normalized to EGFR control siRNA-treated cells [100%]). Data are representative of results from three independent experiments. Error bars indicate SDs. Student's t test was used to determine statistical significance. *, P < 0.001.
ERK inhibition decreases JCPyV viral gene expression.
ERK inhibition decreased both the activity of viral early and late promoters (Fig. 7) and the production of the viral early gene product T antigen (Fig. 8 and 9), indicating that activation of the MAPK-ERK pathway may impact JCPyV transcription. Further, phosphorylated ERK can initiate transcription and subsequent gene expression (47). As successful JCPyV infection is necessitated by ERK activity, it is possible that JCPyV utilization of the MAPK-ERK pathway provides a means to reprogram host cell transcription patterns for viral transcription. To determine if ERK activity plays a direct role in promoting viral gene expression, SVG-A and HEK293A-5-HT2AR cells were treated with ERK1/2 siRNA or control siRNA and then infected with JCPyV for 48 h. RNA was extracted from cells, converted to cDNA, and analyzed by quantitative PCR (qPCR) to determine relative gene expression of TAg (Fig. 10). ERK silencing in SVG-A and HEK293A-5-HT2AR cells resulted in 74- and 51-fold decreases in viral gene expression, respectively, compared to that in control siRNA-treated cells. These data demonstrate that ERK activation is a positive regulator of JCPyV gene expression in both kidney and glial cell types.
FIG 10.
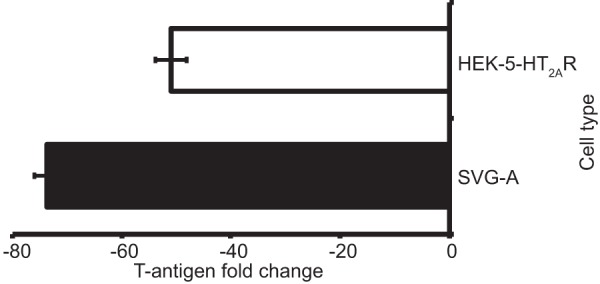
JCPyV T-antigen transcript levels are reduced by ERK inhibition. SVG-A and HEK293A-5-HT2AR cells were treated with either an ERK1/2 or EGFR (CTL) siRNA for 48 h and then infected with JCPyV (MOI = 1 FFU/cell), fed with appropriate media, and incubated at 37°C for 48 h. Cells were harvested and cDNA was prepared from individual triplicate samples, and each sample was analyzed by qPCR to determine viral gene expression. The average fold change in TAg expression in ERK1/2 siRNA-treated cells was calculated per 2–ΔΔCT. Data are representative of the average fold change calculated from triplicate samples from three independent experiments. Error bars represent SDs for three independent experiments.
DISCUSSION
JCPyV infection was previously shown to require the activity of tyrosine kinases (20) and to induce the activation of MAPK-ERK at times consistent with virus entry and trafficking (20, 22). Our results corroborated previously published findings and further revealed that while JCPyV infection results in a robust ERK activation immediately upon infection, there is a multiphasic pattern of ERK phosphorylation throughout the viral life cycle (Fig. 3). However, whether the MAPK-ERK signaling pathway was necessary for JCPyV infection and how the activation of these signaling events affected JCPyV infection remained uncharacterized. Here we demonstrate that JCPyV infection is dependent upon ERK activation, as siRNA directed toward ERK1/2 significantly reduces JCPyV infection (Fig. 1). Further, inhibition of MEK, which blocks phosphorylation of ERK, significantly reduces JCPyV infection (Fig. 2), indicating that the Raf/MEK/ERK pathway is required for JCPyV infection. While ERK activation is induced immediately upon viral infection and inhibition of ERK within the first 4 h of viral infection resulted in a significant decrease in infection (Fig. 3), inhibition of ERK activation had no effect on viral attachment, entry, or trafficking (Fig. 4, 5, and 6). Interestingly, ERK inhibition resulted in significantly reduced viral promoter activity (Fig. 7) and expression of the viral early gene for T antigen in both kidney and glial cells (Fig. 8, 9, and 10), indicating that ERK is required for JCPyV transcription. These findings indicate that JCPyV induces ERK activation upon viral infection to influence downstream steps in the viral life cycle.
Currently, the mechanisms by which JCPyV alters host cell signaling pathways in order to promote viral pathogenesis are poorly understood. We have determined that the activation of ERK plays a critical role in regulating JCPyV gene transcription (Fig. 10), potentially through the activation of crucial viral transcription targets necessary for infection. Viral TAg expression was significantly decreased in both SVG-A and HEK293A-5-HT2AR cells treated with an ERK siRNA (Fig. 9) and in SVG-A (data not shown), HEK293A-5-HT2AR, and RPTEC cells treated with an ERK inhibitor (Fig. 8). These changes in early gene expression suggest that ERK is a positive regulator of JCPyV transcription in both kidney and glial cell types. MAPK-ERK signaling controls the activation of transcription factors necessary for cellular growth, proliferation, and cell death (48); it is seemingly this functionality that is usurped by JCPyV during infection to promote viral propagation. JCPyV infection was previously shown to be enhanced in progenitor-derived astrocytes (PDAs) stimulated with transforming growth factor β1 (TGF-β1) through a mechanism involving the upregulation of MAPK-ERK activity (39). The TGF-β1-stimulated PDAs also had increased expression of the DNA-binding proteins SMADs 2 and 4 (39). Our data build upon these findings by showing that the ERK activation in SVG-A cells, the model cell line for JCPyV studies, is necessary for JCPyV infection and expression of key viral genes independent of TGF-β1 stimulation. Furthermore, while SVG-A cells are transformed with large T antigen, HEK293A-5-HT2AR cells are stably transformed with the adenovirus E1 gene (49) and RPTECs are a nontransformed primary cell type (ATCC), suggesting that the expression of the SV40 T antigen does not impact the effect of ERK inhibition on JCPyV infection. Future studies will focus on defining downstream targets of JCPyV-induced MAPK activation, such as the transcription factor NFAT4, which is crucial for JCPyV infection (43) and can be activated through ERK activity (50).
ERK activation is initiated early during JCPyV infection (Fig. 3) (20), consistent with viral attachment, entry, and trafficking (22). The 5-HT2Rs are required for JCPyV entry (19), and ligand activation of 5-HT2Rs and subsequent endocytosis can activate the MAPK-ERK pathway (51), suggesting that ERK activation may be linked to viral association with the 5-HT2Rs. The 5-HT2Rs induce MAPK-ERK signaling through the action of tyrosine kinases (31, 52, 53) which activate protein kinase C (PKC) (54), stimulating GTPase Ras binding to the serine/threonine kinase Raf and subsequent MEK activation and ERK phosphorylation (30, 55, 56). Our data demonstrate that JCPyV infection activates the MAPK-ERK signaling pathway through a mechanism involving MEK and ERK. Additionally, an inhibitor of Raf, the kinase directly upstream of MEK, significantly reduces JCPyV infection (data not shown), suggesting that JCPyV activates ERK through Raf and MEK. The endocytosis of 5-HT2Rs can lead to the activation of the MAPK-ERK pathway (57, 58), and interestingly, ERK is activated almost immediately following JCPyV challenge (Fig. 3) (20), suggesting that signaling through MAPK-ERK may be activated upon engagement of viral receptors or during cellular entry (22). However, future studies are required to determine the mechanisms by which JCPyV activates ERK. Additionally, the role of attachment receptor motif LSTc in JCPyV-mediated ERK activation cannot be discounted, as sialic acid-containing receptors such as gangliosides can also lead to the activation of the MAPK pathway and activation of downstream transcription factors, like NF-κB (36, 59, 60). Interestingly, JCPyV has been demonstrated to bind to multiple sialylated gangliosides, including GM1 and GM2, albeit with reduced affinity in comparison to that of LSTc, yet gangliosides cannot support JCPyV infection (18). Thus, binding to sialic acid receptors could also activate the MAPK pathway, especially given that MPyV-ganglioside interactions activate MAPK (36). However, inhibition of ERK does not alter virus binding or entry (Fig. 4 and 5), suggesting that while the signal is activated during the initial steps of the virus life cycle (Fig. 3), signaling through this pathway is not required for attachment or entry.
Several members of the Polyomaviridae have been shown to directly or indirectly activate MAPK-ERK, yet the downstream consequences of this activation differ among the polyomaviruses. MPyV activates the MAPK-ERK pathway (34, 36), but MAPK pathway activation is not required for infection (36). BKPyV does not directly activate ERK (35, 37), yet conditions that activate the MAPK-ERK pathway enhance BKPyV replication (35). However, BK polyomavirus infection does activate the AKT-mammalian target of rapamycin (mTOR)-S6 kinase upon infection, likely through receptor interactions, and this signaling pathway enhances BKPyV replication (37). Given that the phosphatidylinositol 3-kinase (PI3K)-mTORC1 pathway can cross-activate or cross-inhibit the ERK pathway (61), it is possible that while BKPyV does not directly activate MAPK-ERK, ERK activation does enhance the mTORC1 signaling pathway, thereby contributing to increased BKPyV replication (37). Activation of mTORC1 and MAPK pathways also regulates translation (61), and therefore, it is possible that inhibition of these pathways could limit translation of transcription factors necessary for viral transcription. Furthermore, SV40 polyomavirus has not been reported to directly activate the Raf/MEK/ERK pathway (34) but rather activates a MAPK-ERK-independent signaling pathway that induces Ca2+ mobilization and tyrosine kinase activity of PKC, without downstream activation of Raf or MAPK-ERK (38). Interestingly, polyomavirus T-antigen proteins, including those of JCPyV, MPyV, and SV40, interact with the MAPK pathway through binding to protein phosphatase 2 (PP2A). The small T-antigen protein of JCPyV binds to PP2A and influences cell cycle progression and viral replication (62). However, the fact that ERK is activated immediately upon JCPyV infection (Fig. 3) (20) suggests that activation of this signaling cascade precedes T-antigen expression. Therefore, it remains possible that JCPyV T-antigen binding to PP2A plays a regulatory role to halt transcription at a critical point in T-antigen production. Thus, while other polyomaviruses can differentially activate the MAPK signaling cascade, it is dispensable for the infectious life cycle of SV40 and MPyV, yet BKPyV is enhanced by ERK activation and JCPyV-induced MAPK activation is necessary for infection specifically by promoting viral transcription. Moreover, as other polyomaviruses have demonstrated the ability to induce biphasic signaling of particular pathways (35, 36, 63), JCPyV may act to alter normal ERK phosphorylation patterns to promote infection. Analysis of ERK phosphorylation patterns following viral challenge indicated that there is a rapid and robust activation of ERK immediately following infection at times consistent with viral attachment, entry, and trafficking (Fig. 3) (16, 22). Our results expand upon previously published findings (20), demonstrating that ERK phosphorylation continues through 12 h.p.i. in a multiphasic pattern that diminishes and eventually stabilizes over the course of infection, with a slight increase in ERK phosphorylation at 72 h.p.i. (Fig. 3). These data may suggest that JCPyV-induced ERK activation is multiphasic in nature and that ERK is required early in the infectious process to accumulate the transcription factors necessary for viral transcription and replication.
A number of other viruses activate the Raf/MEK/ERK signal transduction pathway, including hepatitis C virus, HIV-1, influenza virus (IAV), severe acute respiratory syndrome coronavirus (SARS-CoV), Kaposi's sarcoma-associated herpesvirus (KSHV), and Epstein-Barr virus (EBV), which co-opt various cellular functions of MAPK pathway for infectivity and replication (33, 64). For example, HIV-1 activates the MAPK pathway through GP120 binding to the CD4 and CXCR4 receptors, leading to the activation of ERK and cytoskeletal rearrangements necessary for viral replication and infection (65). Coxsackievirus B3 (CVB3) activates the MAPK pathway, resulting in decreased coxsackievirus and adenovirus receptor (CAR) binding, but reactivation later in the viral replication cycle leads to caspase 3 induction and cell lysis. Moreover, CVB3-induced cell lysis is blocked by the MEK inhibitor U0126, resulting in a reduction in virus release (66). SARS-CoV and mouse hepatitis virus (MHV) also activate the Raf/MEK/ERK pathway, and inhibition of ERK leads to a reduction of viral progeny, thereby limiting viral spread (67–69). Therefore, viruses usurp the Raf/MEK/ERK signaling pathway to drive various aspects of the virus life cycle through unique mechanisms that ultimately impact infection and pathogenesis. While the impact of cellular reprogramming by the MAPK pathway during JCPyV infection affects transcriptional regulation, additional effects on the infectious life cycle should be further elucidated given the range of effects discovered with other viruses.
Taken together, the data presented here demonstrate that JCPyV activates the MAPK-ERK pathway in order to promote viral transcription. ERK is phosphorylated almost immediately following JCPyV infection, yet inhibition of ERK activation through inhibitors and gene silencing abolishes JCPyV infection. Inhibition of ERK does not alter virus attachment, entry, or potential trafficking of the virus. However, viral transcription is significantly decreased when ERK is inhibited, as demonstrated through reduced promoter activity and a significant reduction in the early gene T-antigen production. Infection with the related polyomavirus SV40 was unaffected by the inhibition of ERK, demonstrating that the activation of the MAPK-ERK pathway in glial and kidney cells was specific for JCPyV. Future studies will focus on defining the mechanism by which JCPyV activates the MAPK-ERK pathway and the downstream consequences of ERK activation on cellular transcriptional control.
MATERIALS AND METHODS
Cells, viruses, antibodies, and reagents.
SVG-A cells (46) were cultured in minimum essential medium (MEM; Corning) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (P/S) (Mediatech, Inc.), and 0.2% Plasmocin prophylactic (InvivoGen) (complete medium). HEK293A-5-HT2AR cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/liter of glucose, l-glutamine, and sodium pyruvate (Corning) supplemented with 10% FBS, 1% P/S (Mediatech, Inc.), and G418 sulfate (500 μg/ml) (Corning) (complete medium). Normal human renal proximal tubule epithelial cells (RPTECs) were cultured in renal epithelial cell basal medium (REBM) supplemented with 1% P/S and a renal epithelial cell growth kit as suggested by the manufacturer (ATCC). All cells were grown in a humidified incubator at 37°C with 5% CO2. SVG-A cells, RPTECs (70), and HEK293A-5-HT2AR cells (19) were generously provided by the Atwood laboratory (Brown University) and authenticated by the ATCC.
Generation and propagation of the virus strain Mad-1/SVEΔ and SV40 strain 777 (provided from the Atwood laboratory [Brown University]) were previously described (71, 72). Mad-1/SVEΔ was previously generated through insertion of the SV40 regulatory region (RR) into the Mad-1 RR (Mad-1/SVE) and propagation in SVG-A cells, which yielded strain Mad-1/SVEΔ with a rearranged RR with the JC origin of replication, TATA box, and 78 bp of the 98-bp repeat and an SV40 72-bp repeat with the deletion of most other SV40 72-bp repeats (71). JCPyV was labeled with Alexa Fluor 488 (Thermo Fisher Scientific) according to the manufacturer's instructions, as previously described (73). The infectious viral clones used for transfection were genomic JCPyV DNA of strain JC12, a subclone of Mad-1/SVEΔ (71) subcloned into pUC19 at BamHI sites as previously described (74), and genomic SV40 of strain 776 subcloned into pUC19 at EcoRI sites (provided by the Atwood laboratory, Brown University).
MEK inhibitor U0126 (Cell Signaling Technology) and MEK inhibitor PD98059 (Cell Signaling Technology) were reconstituted in DMSO and used at the desired concentrations prior to or following infection. Antibodies used for indirect immunofluorescence assays include PAB597, a hybridoma supernatant that produces a monoclonal antibody against JCPyV VP1 (generously provided by Ed Harlow) (17) that also cross-reacts with SV40 VP1, PAB962, a JCPyV-specific T-antigen antibody that does not cross-react with SV40 T antigen (generously provided by the Tevethia laboratory [Penn State University]) (75), or AB2 (Calbiochem) to detect SV40 large T antigen (75) and secondary anti-mouse Alexa Fluor 488 and 594 polyclonal antibodies (Thermo Fisher Scientific). Antibodies used for Western blot analysis were a rabbit-specific antibody specific for total ERK (Cell Signaling Technology; no. 4695) and polyclonal antibody for tubulin (loading control; Abcam) and corresponding secondary anti-mouse 680 antibody (LICOR or Invitrogen) or goat anti-rabbit 800 antibody (LICOR). Antibodies used for pERK Western blot analysis were a rabbit-specific phosphorylated ERK antibody (Cell Signaling Technology; no. 9101) and mouse-specific total ERK antibody (Cell Signaling Technology; no. 4696) and corresponding secondary anti-mouse 680 antibody (LICOR) and goat anti-rabbit 800 antibody (LICOR).
siRNA treatment.
SVG-A and HEK293A-5-HT2AR cells were plated to ∼50% confluence in 12-well plates (Grenier Bio-One) and transfected with siRNAs specific for ERK1/2, scrambled control, or epidermal growth factor receptor (EGFR) control (Cell Signaling Technology) (7.5-pmol final concentration of siRNA per well) using RNAiMax transfection reagent (Invitrogen) by mixing transfection complexes in serum- and antibiotic-free MEM or DMEM (Corning) and incubating them at room temperature (RT) for 10 min. Complexes were added to cells and incubated at 37°C for 48 h. Transfection efficiency was monitored using Block-iT Alexa Fluor Red control oligonucleotide (Life Technologies), and optimum ERK protein reduction was confirmed by protein quantitation through Western blot analysis. At 48 h posttransfection, cells were infected with JCPyV or SV40.
siRNA Western blot analysis.
To analyze siRNA knockdown, cells were washed in phosphate-buffered saline (PBS), scraped from wells, washed in PBS, and pelleted by centrifugation at 376 × g at 4°C for 5 min. Cell pellets were resuspended in 30 μl of Tris-HCl lysis buffer containing protease and phosphatase inhibitors and incubated on ice for 20 min, and then insoluble material was removed by centrifugation at 18,600 × g at 4°C for 10 min. Samples were mixed with Laemmli sample buffer (Bio-Rad) and resolved by SDS-PAGE using a 4 to 15% gel (Bio-Rad). Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane using a Transblot Turbo transfer system (Bio-Rad) at 10 V for 30 min. The membrane was blocked with 5% nonfat dry milk in PBS-T (1× PBS–0.05% Tween 20) overnight (O/N), then washed in PBS-T at RT extensively, and incubated with an antibody specific for total ERK (1:500; Cell Signaling Technology) and polyclonal antibody for tubulin (loading control; 1:450; Abcam) in 5% bovine serum albumin (BSA)–PBS-T O/N at 4°C, with rocking. Membranes were washed in PBS-T at RT and then incubated with a secondary anti-rabbit 800 antibody (1:5,000; LICOR) or anti-mouse 680 antibody (1:1,000; Invitrogen) or at RT for 1 h in 5% milk in PBS-T. Membranes were washed in PBS-T and PBS and then imaged using a LICOR Odyssey CLx.
JCPyV and SV40 infection.
Following treatments with inhibitors, vehicle controls, or transfection with siRNAs, medium was removed and cells were infected with JCPyV or SV40 (multiplicities of infection [MOIs] indicated in figure legends). SVG-A cells were infected in MEM containing either 2% or 10% FBS at ∼70% confluence. HEK293A-5-HT2AR cells were infected in complete DMEM containing 10% FBS at ∼50% confluence. Infection of RPTECs was completed in REBM at ∼75% confluence. Prior to RPTEC experiments, 96-well plates were pretreated with laminin from human fibroblasts (Sigma-Aldrich) at 1 μg/cm2 at 37°C for 1 h. Viral infections were completed in volumes of either 200 μl (24 well plate) or 40 μl (96 well plate) and incubated at 37°C for 1 h, and then treated or untreated medium was added and cells were incubated at 37°C for 48 h (T antigen) or 72 h (VP1). Cells were then fixed and stained by indirect immunofluorescence.
Variable timing of ERK inhibition during JCPyV infection.
SVG-A cells were pretreated for 1 h prior to JCPyV infection with U0126 (10 μM) or DMSO (1:1,000) or incubated in complete medium. Cells were then infected with JCPyV at 4°C for 1 h. Following infection, cells were washed in medium and then incubated in medium containing either U0126 or DMSO for the desired duration. For 1-h and 4-h samples, treatment medium was removed at the desired time point, cells were washed in medium, complete medium was added, and cells were incubated at 37°C for the duration of the infection (72 h). Cells were fixed and stained by indirect immunofluorescence using a specific antibody that recognizes JCPyV VP1. Infectivity was scored by quantitating nuclear VP1 expression.
Western blot quantitation of ERK phosphorylation during JCPyV infection.
SVG-A cells at 95% confluence (6 well plates) were either infected with JCPyV (MOI = 1 FFU/cell) or mock infected (complete medium) and incubated at 37°C for the desired times or immediately harvested (0 min). For time points exceeding 1 h, both mock-treated and infected cells were fed with 1 ml of complete medium after a 1-h infection and incubated at 37°C. At the specified time point, cells were washed in 1× PBS, scraped from wells on ice, and pelleted by centrifugation at 376 × g at 4°C for 5 min. Cell pellets were resuspended in 50 μl of Tris-HCl lysis buffer containing protease and phosphatase inhibitors and incubated on ice for 15 min, and insoluble material was removed by centrifugation at 18,600 × g at 4°C for 10 min. Samples were mixed with Laemmli sample buffer (Bio-Rad), incubated at 95°C for 5 min, and then resolved by SDS-PAGE using a 4 to 15% Mini TGX gel (Bio-Rad). Proteins were transferred to nitrocellulose membranes using a Transblot Turbo system (Bio-Rad) at 25 V for 3 min. The membrane was blocked with 5% nonfat dry milk in TBS-T (1× Tris-buffered saline [TBS], 0.05% Tween 20) with rocking at RT for 1 h, then washed in TBS-T at RT, and incubated with a rabbit-specific antibody specific for phosphorylated ERK (1:1,000; Cell Signaling Technology; no. 9101) and mouse-specific antibody targeting total ERK (1:1,000; total ERK protein control; Cell Signaling Technology; no. 4696) in 5% BSA–TBS-T with rocking at 4°C O/N. Membranes were then washed in TBS-T at RT and incubated with both secondary anti-mouse 680 antibody (1:15,000; LICOR) and goat anti-rabbit 800 antibody (1:15,000; LICOR) with rocking at RT for 1 h in 5% nonfat dry milk–TBS-T. Membranes were washed in TBS and then imaged using a LICOR Odyssey CLx. Quantitation of phosphorylated ERK and total ERK expression was performed via analysis of LICOR 680 and 800 intensities corresponding to the total and pERK antibodies using LICOR Odyssey CLx Image Studio software version 5.2. The percentage of phosphorylated ERK was then quantitated using the following equation: [phosphorylated ERK/(phosphorylated ERK + total ERK)] × 100. JCPyV-infected samples for each time point were normalized to mock-infected control samples (100%). Average normalized percentages of phosphorylated ERK for each treatment were plotted via box-and-whisker plot, representing the composite of three independent experiments. Data are representative of the maximum (upper whisker), median, and minimum (lower whisker) of percent phosphorylated ERK from JCPyV-infected samples.
Transfection of JCPyV and SV40 infectious clones.
SVG-A cells were plated to ∼50% confluence in 24-well plates (Grenier Bio-One). Cells were pretreated for 1 h with medium containing either DMSO (1:1,000) or U0126 (10 μM). Cells in medium without antibiotics were transfected with 1 μg of JCPyV-pUC19 or SV40-pUC19 plasmid DNA that had been linearized through digestion with BamHI (JCPyV-pUC19) (Promega) or EcoRI (SV40-pUC19) (Promega) at 37°C for 2 h using Fugene (Roche) at a 1.5:1 ratio (Fugene to DNA) (17) and incubated at 37°C. At 24 h posttransfection, cells were fed with 500 μl of complete medium containing 1% amphotericin B and either DMSO (1:1,000) or U0126 (10 μM) and incubated at 37°C. Cells were fixed and stained by indirect immunofluorescence using a VP1-specific antibody at days 4 and 7 posttransfection.
Indirect immunofluorescence quantitation of viral infectivity.
Following infection, SVG-A cells were washed in PBS, fixed in ice-cold methanol, and incubated at −20°C for >10 min. RPTEC and HEK293A-5-HT2AR cells were washed in PBS and fixed with 4% paraformaldehyde (PFA) at RT for 10 min. Fixed cells were washed with PBS three times for 10 min and then incubated with PBS–0.5% Triton X-100 for permeabilization. RPTEC and HEK293A-5-HT2AR cells were blocked in 10% goat serum (Sigma-Aldrich) at RT for 1 h prior to staining. Cells were then stained using PAB597 (1:10) (17), PAB962 (1:2), or AB2 (Calbiochem; 1:50) (75) and a secondary anti-mouse Alexa Fluor 488 or 594 polyclonal antibody (Thermo Fisher Scientific). Cell nuclei were counterstained using 4′,6-diamidino-2-phenylindole (DAPI). The number of infected cells per visual field was assessed by analysis of nuclear VP1 or TAg staining under a 10× objective using a Nikon Eclipse Ti epifluorescence microscope (Micro Video Instruments, Inc.). At least 5 visual fields per well were quantitated for triplicate wells for experiments performed at least 3 times. Percent infection was determined by determining the number of TAg-positive nuclei divided by the number of DAPI-positive nuclei under a 20× objective. The number of DAPI-positive nuclei was determined utilizing Nikon NIS-Elements Basic Research software (version 4.5). A binary was generated to calculate the number of cells per field, determined by the number of DAPI-positive nuclei based on threshold fluorescence. The binary controlled for nuclei of equal diameter and circularity to distinguish between cells in the same field. The average percent infection was then normalized to the DMSO or siRNA control.
Flow cytometry to measure viral attachment.
SVG-A cells at 100% confluence in 6-well plates (1e6) (Grenier Bio-One) were treated with DMSO (1:1,000) or U0126 (10 μm) at 37°C for 1 h. Cells were removed from 6-well plates by washing with PBS and incubated in Cellstripper (Corning). Cells were pelleted at 376 × g at 4°C for 5 min and washed with PBS. Cells were incubated with Alexa Fluor 488-labeled JCPyV (JCPyV-488) in PBS (100 μl total volume) on ice for 2 h, with agitation every 15 min. Cells were washed, pelleted by centrifugation, and resuspended in 500 μl of PBS. Cells were then analyzed for virus binding using a BD FACSCanto (BD Biosciences) equipped with a 488-nm laser excitation line (Becton, Dickinson, and Company). Data were analyzed using BD FacsDIVA (Becton, Dickinson, and Company) and FlowJo software (Tree Star, Inc.).
Trypan blue quenching assay of viral internalization.
SVG-A cells were plated to ∼95% confluence in 6-well plates (Grenier Bio-One) and pretreated with complete medium containing either DMSO (1:1,000) or U0126 (10 μm) at 37°C for 1 h. Cells were removed from plates using Cellstripper (Corning), pelleted at 376 × g at 4°C for 5 min, and washed with PBS. Cells were then resuspended with cold phenol-free MEM containing 10% FBS and 1% P/S and chilled at 4°C for 30 min. Cells were pelleted and then incubated with JCPyV-488 at 4°C on ice for 1.5 h, with agitation every 15 min to allow for viral attachment. Suspensions were pelleted at 376 × g at 4°C for 5 min and washed with PBS. Cell pellets were then resuspended with cold phenol-free medium containing 10% FBS and 1% P/S containing either DMSO (1:1,000) or U0126 (10 μm). Samples were either fixed (attachment) or placed directly into a 37°C water bath for 90 min (entry). At 0 or 90 min after incubation at 37°C, cells were pelleted at 376 × g at 4°C for 5 min and resuspended in 4% PFA on ice for 10 min. Cells were pelleted, washed, and resuspended in a final volume of 1× PBS or 1× PBS containing trypan blue (0.016%). Samples were analyzed using a BD LSRII equipped with a 488-nm laser line. Data were analyzed using BD FACSDIVA (Becton, Dickinson, and Company) and FlowJo software (Tree Star, Inc.). Quenched (trypan blue addition) and nonquenched samples (without trypan blue) from U0126- and DMSO-treated samples were assessed by flow cytometry (adapted from reference 22) to determine impact of ERK inhibition on viral attachment (viral binding only) and viral internalization (binding and entry through 37°C incubation). Using the vehicle control as a reference, sample events were gated to remove cellular debris using FlowJo software. Percent protected fluorescence of averaged fluorescein isothiocyanate (FITC) readings from quenched (trypan blue addition) and nonquenched samples, determined using the equation (quenched sample/nonquenched sample) × 100, was calculated from three independent experiments.
Viral early/late promoter transfections.
SVG-A cells were plated to ∼60% confluence in 24-well plates (Grenier Bio-One). Cells were pretreated for 4 h with medium containing either DMSO (1:1,000) or U0126 (10 μM). Cells in medium without antibiotics were transfected with 1.8 μg of either JCPyV-Mad-1 early or JCPyV-Mad-1 late plasmid DNA containing a firefly luciferase reporter (generously provided by the Atwood lab [Brown University]) in addition to 0.2 μg of a Renilla luciferase control plasmid pRL-TK (Promega) using a 3:1 ratio (Fugene to DNA) and incubated at 37°C as previously described (43). At 4 h posttransfection, transfection medium was removed and cells were fed with complete medium containing vehicle control DMSO (1:1,000) or U0126 (10 μM) and incubated at 37°C. At 48 h posttransfection, cells were analyzed for viral promoter activity with a dual-luciferase reporter assay system (Promega) per the manufacturer's protocol. Firefly luciferase readings were normalized to the Renilla control (firefly − Renilla = promoter activity), performed with triplicate samples from three independent experiments.
qPCR determination of TAg and VP1 transcript levels.
SVG-A and HEK293A-5-HT2AR cells were plated to ∼70% confluence in 12-well plates (Greiner Bio-One). Cells were transfected with siRNAs specific for ERK1/2 or EGF receptor control (Cell Signaling Technology) (7.5-pmol final concentration of siRNA per well) using RNAiMax transfection reagent (Invitrogen) by mixing transfection complexes in serum- and antibiotic-free MEM or DMEM (Corning) and incubating them at RT for 10 min. Complexes were added to cells and incubated at 37°C for 48 h. Transfection efficiency was monitored using Block-iT Alexa Fluor Red control oligonucleotide (Life Technologies). At 48 h posttransfection, growth medium was removed and cells were washed with PBS, infected with JCPyV at an MOI of 1 FFU/cell in a volume of 350 μl, and incubated at 37°C for 1 h. Cells were fed with complete MEM or DMEM and incubated at 37°C for 48 h. At 48 h postinfection, RNA was extracted from each sample with a SingleShot cell lysis kit (Bio-Rad) per the manufacturer's protocol. For each sample, collected RNA was converted to cDNA with iScript Reverse-Transcription Supermix (Bio-Rad) using 1 μg of RNA per reaction. Analysis of viral gene expression was performed via qPCR using TAg and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; housekeeping gene) primers as previously described (76) (IDT). Using Hard-Shell 96-well PCR plates (Bio-Rad), each well was prepared with 150 nM concentrations of both forward and reverse primers (GAPDH and TAg run individually for each sample), 100 ng of sample cDNA, 10 μl of iQ SYBR green Supermix (Bio-Rad), and nuclease-free water to a total volume of 20 μl per well. The plate was sealed with Microseal ‘B’ seal Seals (Bio-Rad) and centrifuged at 168 × g for 1 min. qPCR analysis was performed on a Bio-Rad CFX96 real-time system, using Bio-Rad CFX Manager software 3.1. Analysis of gene expression was performed with SYBR green within each sample per running conditions previously described (76). Changes in viral gene expression of TAg were determined by qPCR using triplicate samples from three independent experiments. Calculation of change in threshold cycle (ΔCT) for TAg was determined using the following equation: (TAg CT − GAPDH CT). The difference in ΔCT for TAg between ERK siRNA-treated samples and EGFR siRNA-treated samples (ΔΔCT) was calculated using the equation ERK siRNA ΔCT − EGFR siRNA ΔCT. The average fold change of TAg expression between EGFR siRNA- and ERK siRNA-treated cells was calculated using 2−ΔΔCT of average TAg ΔΔCT values.
Statistical analysis.
Student's t test (Microsoft Excel) was used to compare means for at least triplicate samples. P values of <0.05 were considered statistically significant in our statistical analyses.
ACKNOWLEDGMENTS
We thank members of the Atwood laboratory for cell lines, reagents, support, and critical discussion. We thank Pranav Danthi for critical review of the manuscript. We are grateful to the Maine Regional Flow Cytometry Consortium and the Gosse, Kim, Sullivan, and Wheeler laboratories for sharing essential equipment and advice.
This research was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103423 (M.S.M.) and The University of Maine MEIF (M.S.M.).
REFERENCES
- 1.Kean JM, Rao S, Wang M, Garcea RL. 2009. Seroepidemiology of human polyomaviruses. PLoS Pathog 5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, Gosert R, Hirsch HH. 2009. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis 199:837–846. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- 3.Dubois V, Dutronc H, Lafon ME, Poinsot V, Pellegrin JL, Ragnaud JM, Ferrer AM, Fleury HJ. 1997. Latency and reactivation of JC virus in peripheral blood of human immunodeficiency virus type 1-infected patients. J Clin Microbiol 35:2288–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorelik L, Reid C, Testa M, Brickelmaier M, Bossolasco S, Pazzi A, Besets A, Carmillo P, Wilson E, McAuliffe M, Tonkin C, Carulli JP, Lugovskoy A, Lazzarin A, Sunyaev S, Simon K, Cinque P. 2011. Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change its receptor specificity. J Infect Dis 204:103–114. doi: 10.1093/infdis/jir198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverman L, Rubinstein LJ. 1965. Electron microscopic observations on a case of progressive multifocal leukoencephalopathy. Acta Neuropathol 5:215–224. doi: 10.1007/BF00686519. [DOI] [PubMed] [Google Scholar]
- 6.Zurhein G, Chou SM. 1965. Particles resembling papova viruses in hum cerebral demyelinating disease. Science 148:1477–1479. [DOI] [PubMed] [Google Scholar]
- 7.Kondo Y, Windrem MS, Zou L, Chandler-Militello D, Schanz SJ, Auvergne RM, Betstadt SJ, Harrington AR, Johnson M, Kazarov A, Gorelik L, Goldman SA. 2014. Human glial chimeric mice reveal astrocytic dependence of JC virus infection. J Clin Invest 124:5323–5336. doi: 10.1172/JCI76629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Astrom KE, Mancall EL, Richardson EP Jr. 1958. Progressive multifocal leuko-encephalopathy; a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin's disease. Brain 81:93–111. doi: 10.1093/brain/81.1.93. [DOI] [PubMed] [Google Scholar]
- 9.Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO. 2012. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev 25:471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirsch HH, Kardas P, Kranz D, Leboeuf C. 2013. The human JC polyomavirus (JCPyV): virological background and clinical implications. APMIS 121:685–727. doi: 10.1111/apm.12128. [DOI] [PubMed] [Google Scholar]
- 11.Pavlovic D, Patera AC, Nyberg F, Gerber M, Liu M, Progressive Multifocal Leukeoncephalopathy Consortium. 2015. Progressive multifocal leukoencephalopathy: current treatment options and future perspectives. Ther Adv Neurol Disord 8:255–273. doi: 10.1177/1756285615602832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ermis U, Weis J, Schulz JB. 2013. Case reports of PML in patients treated for psoriasis. N Engl J Med 369:1081. [DOI] [PubMed] [Google Scholar]
- 13.Ermis U, Weis J, Schulz JB. 2013. PML in a patient treated with fumaric acid. N Engl J Med 368:1657–1658. doi: 10.1056/NEJMc1211805. [DOI] [PubMed] [Google Scholar]
- 14.Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, Bennett CL. 2009. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol 10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 15.Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, Lee S, Plavina T, Scanlon JV, Sandrock A, Bozic C. 2012. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med 366:1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- 16.Neu U, Maginnis MS, Palma AS, Stroh LJ, Nelson CD, Feizi T, Atwood WJ, Stehle T. 2010. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 8:309–319. doi: 10.1016/j.chom.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maginnis MS, Stroh LJ, Gee GV, O'Hara BA, Derdowski A, Stehle T, Atwood WJ. 2013. Progressive multifocal leukoencephalopathy-associated mutations in the JC polyomavirus capsid disrupt lactoseries tetrasaccharide c binding. mBio 4(3):e00247–13. doi: 10.1128/mBio.00247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ströh LJ, Maginnis MS, Blaum BS, Nelson CD, Neu U, Gee GV, O'Hara BA, Motamedi N, DiMaio D, Atwood WJ, Stehle T. 2015. The greater affinity of JC polyomavirus capsid for alpha2,6-linked lactoseries tetrasaccharide c than for other sialylated glycans is a major determinant of infectivity. J Virol 89:6364–6375. doi: 10.1128/JVI.00489-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Assetta B, Maginnis MS, Gracia Ahufinger I, Haley SA, Gee GV, Nelson CD, O'Hara BA, Allen Ramdial SA, Atwood WJ. 2013. 5-HT2 receptors facilitate JC polyomavirus entry. J Virol 87:13490–13498. doi: 10.1128/JVI.02252-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Querbes W, Benmerah A, Tosoni D, Di Fiore PP, Atwood WJ. 2004. A JC virus-induced signal is required for infection of glial cells by a clathrin- and eps15-dependent pathway. J Virol 78:250–256. doi: 10.1128/JVI.78.1.250-256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Querbes W, O'Hara B, Williams G, Atwood W. 2006. Invasion of host cells by JC virus identifies a novel role for caveolae in endosomal sorting of noncaveolar ligands. J Virol 80:9402–9413. doi: 10.1128/JVI.01086-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson C, Derdowski A, Maginnis M, O'Hara B, Atwood W. 2012. The VP1 subunit of JC polyomavirus recapitulates early events in viral trafficking and is a novel tool to study polyomavirus entry. Virology 428:30–40. doi: 10.1016/j.virol.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whelan S. 2013. Viral replication, p 125–126 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 24.Katz M, Amit I, Yarden Y. 2007. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta 1773:1161–1176. doi: 10.1016/j.bbamcr.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth BL. 2011. Irving Page Lecture: 5-HT(2A) serotonin receptor biology: interacting proteins, kinases and paradoxical regulation. Neuropharmacology 61:348–354. doi: 10.1016/j.neuropharm.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. 2001. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 27.Agell N, Bachs O, Rocamora N, Villalonga P. 2002. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal 14:649–654. doi: 10.1016/S0898-6568(02)00007-4. [DOI] [PubMed] [Google Scholar]
- 28.Shaul YD, Seger R. 2007. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Roskoski R., Jr 2012. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Watts SW. 1998. Activation of the mitogen-activated protein kinase pathway via the 5-HT2A receptor. Ann N Y Acad Sci 861:162–168. doi: 10.1111/j.1749-6632.1998.tb10187.x. [DOI] [PubMed] [Google Scholar]
- 31.Florian JA, Watts SW. 1998. Integration of mitogen-activated protein kinase kinase activation in vascular 5-hydroxytryptamine 2A receptor signal transduction. J Pharmacol Exp Ther 284:346–355. [PubMed] [Google Scholar]
- 32.Lewis TS, Shapiro PS, Ahn NG. 1998. Signal transduction through MAP kinase cascades. Adv Cancer Res 74:49–139. doi: 10.1016/S0065-230X(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 33.Pleschka S. 2008. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol Chem 389:1273–1282. doi: 10.1515/BC.2008.145. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-Viciana P, Collins C, Fried M. 2006. Polyoma and SV40 proteins differentially regulate PP2A to activate distinct cellular signaling pathways involved in growth control. Proc Natl Acad Sci U S A 103:19290–19295. doi: 10.1073/pnas.0609343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seamone ME, Wang W, Acott P, Beck PL, Tibbles LA, Muruve DA. 2010. MAP kinase activation increases BK polyomavirus replication and facilitates viral propagation in vitro. J Virol Methods 170:21–29. doi: 10.1016/j.jviromet.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 36.O'Hara SD, Garcea RL. 2016. Murine polyomavirus cell surface receptors activate distinct signaling pathways required for infection. mBio 7(6):e01836–16. doi: 10.1128/mBio.01836-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirsch HH, Yakhontova K, Lu M, Manzetti J. 2016. BK polyomavirus replication in renal tubular epithelial cells is inhibited by sirolimus, but activated by tacrolimus through a pathway involving FKBP-12. Am J Transplant 16:821–832. doi: 10.1111/ajt.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dangoria NS, Breau WC, Anderson HA, Cishek DM, Norkin LC. 1996. Extracellular simian virus 40 induces an ERK/MAP kinase-independent signalling pathway that activates primary response genes and promotes virus entry. J Gen Virol 77(Part 9):2173–2182. [DOI] [PubMed] [Google Scholar]
- 39.Ravichandran V, Jensen PN, Major EO. 2007. MEK1/2 inhibitors block basal and transforming growth factor 1beta1-stimulated JC virus multiplication. J Virol 81:6412–6418. doi: 10.1128/JVI.02658-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. 1995. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A 92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 42.Raote I, Bhattacharya A, Panicker MM. 2007. Chapter 6. Serotonin 2A (5-HT2A) receptor function: ligand-dependent mechanisms and pathways. In Chattopadhyay A. (ed), Serotonin receptors in neurobiology (electronic). CRC Press, Boca Raton, FL. [PubMed] [Google Scholar]
- 43.Manley K, O'Hara BA, Gee GV, Simkevich CP, Sedivy JM, Atwood WJ. 2006. NFAT4 is required for JC virus infection of glial cells. J Virol 80:12079–12085. doi: 10.1128/JVI.01456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 45.Low J, Humes HD, Szczypka M, Imperiale M. 2004. BKV and SV40 infection of human kidney tubular epithelial cells in vitro. Virology 323:182–188. doi: 10.1016/j.virol.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 46.Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. 1985. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Natl Acad Sci U S A 82:1257–1261. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wortzel I, Seger R. 2011. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer 2:195–209. doi: 10.1177/1947601911407328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoon S, Seger R. 2006. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 49.Shaw G, Morse S, Ararat M, Graham FL. 2002. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 16:869–871. [DOI] [PubMed] [Google Scholar]
- 50.Yang TT, Xiong Q, Graef IA, Crabtree GR, Chow CW. 2005. Recruitment of the extracellular signal-regulated kinase/ribosomal S6 kinase signaling pathway to the NFATc4 transcription activation complex. Mol Cell Biol 25:907–920. doi: 10.1128/MCB.25.3.907-920.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allen JA, Yadav PN, Roth BL. 2008. Insights into the regulation of 5-HT2A serotonin receptors by scaffolding proteins and kinases. Neuropharmacology 55:961–968. doi: 10.1016/j.neuropharm.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berg KA, Stout BD, Maayani S, Clarke WP. 2001. Differences in rapid desensitization of 5-hydroxytryptamine 2A and 5-hydroxytryptamine 2C receptor-mediated phospholipase C activation. J Pharmacol Exp Ther 299:593–602. [PubMed] [Google Scholar]
- 53.Quinn JC, Johnson-Farley NN, Yoon J, Cowen DS. 2002. Activation of extracellular-regulated kinase by 5-hydroxytryptamine (2A) receptors in PC12 cells is protein kinase C-independent and requires calmodulin and tyrosine kinases. J Pharmacol Exp Ther 303:746–752. doi: 10.1124/jpet.102.038083. [DOI] [PubMed] [Google Scholar]
- 54.Berg KA, Clarke WP, Chen Y, Ebersole BJ, McKay RD, Maayani S. 1994. 5-Hydroxytryptamine type 2A receptors regulate cyclic AMP accumulation in a neuronal cell line by protein kinase C-dependent and calcium/calmodulin-dependent mechanisms. Mol Pharmacol 45:826–836. [PubMed] [Google Scholar]
- 55.Watts SW. 1996. Serotonin activates the mitogen-activated protein kinase pathway in vascular smooth muscle: use of the mitogen-activated protein kinase kinase inhibitor PD098059. J Pharmacol Exp Ther 279:1541–1550. [PubMed] [Google Scholar]
- 56.Greene EL, Houghton O, Collinsworth G, Garnovskaya MN, Nagai T, Sajjad T, Bheemanathini V, Grewal JS, Paul RV, Raymond JR. 2000. 5-HT(2A) receptors stimulate mitogen-activated protein kinase via H(2)O(2) generation in rat renal mesangial cells. Am J Physiol Renal Physiol 278:F650–F658. doi: 10.1152/ajprenal.2000.278.4.F650. [DOI] [PubMed] [Google Scholar]
- 57.Kolch W. 2005. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 58.Franklin JM, Carrasco GA. 2013. G-protein receptor kinase 5 regulates the cannabinoid receptor 2-induced up-regulation of serotonin 2A receptors. J Biol Chem 288:15712–15724. doi: 10.1074/jbc.M113.454843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim OS, Park EJ, Joe EH, Jou I. 2002. JAK-STAT signaling mediates gangliosides-induced inflammatory responses in brain microglial cells. J Biol Chem 277:40594–40601. doi: 10.1074/jbc.M203885200. [DOI] [PubMed] [Google Scholar]
- 60.Gouni-Berthold I, Seul C, Ko Y, Hescheler J, Sachinidis A. 2001. Gangliosides GM1 and GM2 induce vascular smooth muscle cell proliferation via extracellular signal-regulated kinase 1/2 pathway. Hypertension 38:1030–1037. doi: 10.1161/hy1101.093104. [DOI] [PubMed] [Google Scholar]
- 61.Mendoza MC, Er EE, Blenis J. 2011. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 36:320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bollag B, Hofstetter CA, Reviriego-Mendoza MM, Frisque RJ. 2010. JC virus small T antigen binds phosphatase PP2A and Rb family proteins and is required for efficient viral DNA replication activity. PLoS One 5(5):e10606. doi: 10.1371/journal.pone.0010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zullo J, Stiles CD, Garcea RL. 1987. Regulation of c-myc and c-fos mRNA levels by polyomavirus: distinct roles for the capsid protein VP1 and the viral early proteins. Proc Natl Acad Sci U S A 84:1210–1214. doi: 10.1073/pnas.84.5.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dawson CW, Laverick L, Morris MA, Tramoutanis G, Young LS. 2008. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J Virol 82:3654–3664. doi: 10.1128/JVI.01888-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balabanian K, Harriague J, Decrion C, Lagane B, Shorte S, Baleux F, Virelizier JL, Arenzana-Seisdedos F, Chakrabarti LA. 2004. CXCR4-tropic HIV-1 envelope glycoprotein functions as a viral chemokine in unstimulated primary CD4+ T lymphocytes. J Immunol 173:7150–7160. doi: 10.4049/jimmunol.173.12.7150. [DOI] [PubMed] [Google Scholar]
- 66.Luo H, Yanagawa B, Zhang J, Luo Z, Zhang M, Esfandiarei M, Carthy C, Wilson JE, Yang D, McManus BM. 2002. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J Virol 76:3365–3373. doi: 10.1128/JVI.76.7.3365-3373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cai Y, Liu Y, Zhang X. 2007. Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J Virol 81:446–456. doi: 10.1128/JVI.01705-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu M, Yang Y, Gu C, Yue Y, Wu KK, Wu J, Zhu Y. 2007. Spike protein of SARS-CoV stimulates cyclooxygenase-2 expression via both calcium-dependent and calcium-independent protein kinase C pathways. FASEB J 21:1586–1596. doi: 10.1096/fj.06-6589com. [DOI] [PubMed] [Google Scholar]
- 69.Varshney B, Lal SK. 2011. SARS-CoV accessory protein 3b induces AP-1 transcriptional activity through activation of JNK and ERK pathways. Biochemistry 50:5419–5425. doi: 10.1021/bi200303r. [DOI] [PubMed] [Google Scholar]
- 70.Assetta B, De Cecco M, O'Hara B, Atwood WJ. 2016. JC polyomavirus infection of primary human renal epithelial cells is controlled by a type I IFN-induced response. mBio 7(4):e00903–16. doi: 10.1128/mBio.00903-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vacante DA, Traub R, Major EO. 1989. Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology 170:353–361. doi: 10.1016/0042-6822(89)90425-X. [DOI] [PubMed] [Google Scholar]
- 72.Nelson C, Carney D, Derdowski A, Lipovsky A, Gee G, O'Hara B, Williard P, DiMaio D, Sello J, Atwood W. 2013. A retrograde trafficking inhibitor of ricin and Shiga-like toxins inhibits infection of cells by human and monkey polyomaviruses. mBio 4(6):e00729–13. doi: 10.1128/mBio.00729-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dugan AS, Gasparovic ML, Atwood WJ. 2008. Direct correlation between sialic acid binding and infection of cells by two human polyomaviruses (JC virus and BK virus). J Virol 82:2560–2564. doi: 10.1128/JVI.02123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gee GV, Tsomaia N, Mierke DF, Atwood WJ. 2004. Modeling a sialic acid binding pocket in the external loops of JC virus VP1. J Biol Chem 279:49172–49176. doi: 10.1074/jbc.M409326200. [DOI] [PubMed] [Google Scholar]
- 75.Maginnis MS, Haley SA, Gee GV, Atwood WJ. 2010. Role of N-linked glycosylation of the 5-HT2A receptor in JC virus infection. J Virol 84:9677–9684. doi: 10.1128/JVI.00978-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chapagain ML, Verma S, Mercier F, Yanagihara R, Nerurkar VR. 2007. Polyomavirus JC infects human brain microvascular endothelial cells independent of serotonin receptor 2A. Virology 364:55–63. doi: 10.1016/j.virol.2007.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]