

ABSTRACT
Sensory neurons in trigeminal ganglia (TG) of calves latently infected with bovine herpesvirus 1 (BoHV-1) abundantly express latency-related (LR) gene products, including a protein (ORF2) and two micro-RNAs. Recent studies in mouse neuroblastoma cells (Neuro-2A) demonstrated ORF2 interacts with β-catenin and a β-catenin coactivator, high-mobility group AT-hook 1 (HMGA1) protein, which correlates with increased β-catenin-dependent transcription and cell survival. β-Catenin and HMGA1 are readily detected in a subset of latently infected TG neurons but not TG neurons from uninfected calves or reactivation from latency. Consequently, we hypothesized that the Wnt/β-catenin signaling pathway is differentially expressed during the latency and reactivation cycle and an active Wnt pathway promotes latency. RNA-sequencing studies revealed that 102 genes associated with the Wnt/β-catenin signaling pathway were differentially expressed in TG during the latency-reactivation cycle in calves. Wnt agonists were generally expressed at higher levels during latency, but these levels decreased during dexamethasone-induced reactivation. The Wnt agonist bone morphogenetic protein receptor 2 (BMPR2) was intriguing because it encodes a serine/threonine receptor kinase that promotes neuronal differentiation and inhibits cell death. Another differentially expressed gene encodes a protein kinase (Akt3), which is significant because Akt activity enhances cell survival and is linked to herpes simplex virus 1 latency and neuronal survival. Additional studies demonstrated ORF2 increased Akt3 steady-state protein levels and interacted with Akt3 in transfected Neuro-2A cells, which correlated with Akt3 activation. Conversely, expression of Wnt antagonists increased during reactivation from latency. Collectively, these studies suggest Wnt signaling cooperates with LR gene products, in particular ORF2, to promote latency.
IMPORTANCE Lifelong BoHV-1 latency primarily occurs in sensory neurons. The synthetic corticosteroid dexamethasone consistently induces reactivation from latency in calves. RNA sequencing studies revealed 102 genes associated with the Wnt/β-catenin signaling pathway are differentially regulated during the latency-reactivation cycle. Two protein kinases associated with the Wnt pathway, Akt3 and BMPR2, were expressed at higher levels during latency but were repressed during reactivation. Furthermore, five genes encoding soluble Wnt antagonists and β-catenin-dependent transcription inhibitors were induced during reactivation from latency. These findings are important because Wnt, BMPR2, and Akt3 promote neurogenesis and cell survival, processes crucial for lifelong viral latency. In transfected neuroblastoma cells, a viral protein expressed during latency (ORF2) interacts with and enhances Akt3 protein kinase activity. These findings provide insight into how cellular factors associated with the Wnt signaling pathway cooperate with LR gene products to regulate the BoHV-1 latency-reactivation cycle.
KEYWORDS: Akt3, ORF2, Wnt signaling, bovine herpesvirus 1, neuronal survival
INTRODUCTION
Acute infection of cattle with bovine herpesvirus 1 (BoHV-1) can result in upper respiratory tract disease, conjunctivitis, and inflammation (1, 2). Mucosal damage in the respiratory tract and immune suppression also occur after infection, which allows commensal bacteria in the upper respiratory tract to colonize the lung and cause life-threatening pneumonia. In addition to BoHV-1, stress and other viral pathogens can suppress immune responses and cause bacterial pneumonia. This polymicrobial disease is referred to as bovine respiratory disease complex (BRDC), the most important disease in cattle (3, 4). A BoHV-1 entry receptor, poliovirus receptor related 1, is a BRDC susceptibility gene for Holstein calves (5), adding support to the notion that BoHV-1 is an important BRDC cofactor.
BoHV-1 establishes lifelong latency in sensory neurons within trigeminal ganglia (TG) (6, 7). Virus transmission in nature depends on the ability of BoHV-1 to establish, maintain, and reactivate from latency. In contrast to acute infection, the latency-related (LR) gene, which encodes two micro-RNAs and at least two proteins (ORF2 and ORF1), is the only viral gene abundantly expressed during latency (8, 9). An LR mutant virus with three stop codons adjacent to the initiating methionine of ORF2 in the LR gene does not replicate efficiently in the ocular cavity or tonsils (10) or reactivate from latency in calves (11), suggesting ORF2 regulates crucial steps during the latency-reactivation cycle. ORF2 is a multifunctional protein that localizes to the rim of the nucleus (12), inhibits apoptosis (13, 14), interacts with DNA (15), and interacts with Notch family members (12, 16, 17). LR-encoded micro-RNAs interfere with expression of a viral transcriptional regulatory protein (bICP0) (18). Although viral genes likely mediate certain aspects of establishing and maintaining latency, we predict that cellular factors are also crucial.
Reactivation of the virus from the latent state is initiated by external stimuli (e.g., stress and immunosuppression). During reactivation, viral gene expression is stimulated, LR gene expression decreases, and infectious virus is produced and transported back to mucosal surfaces. The ability of BoHV-1 to reactivate from the latent state results in virus transmission to susceptible animals. Regulation of virus-host interactions that control the latency-reactivation cycle is not well understood. BoHV-1 is an excellent model to study these events, because the natural host can be used and the synthetic corticosteroid dexamethasone (DEX) consistently initiates reactivation from latency in infected calves (6, 7, 9, 19, 20). We have used experimentally infected calves treated with DEX to initiate reactivation from latency in order to identify virus-host interactions important for the latency-reactivation cycle. These studies identified host cellular factors and pathways that may be crucial for latency maintenance (21) and reactivation (22). For example, the Wnt/β-catenin signaling pathway is differentially regulated during the latency-reactivation cycle. A cellular transcription factor, β-catenin, and a β-catenin coactivator, high-mobility group AT-hook 1 (HMGA1) protein, are readily detected in TG neurons of latently infected but not uninfected calves or calves latently infected that were treated with DEX to initiate reactivation from latency (21, 23). Many β-catenin+ neurons express ORF2 but not bICP0, which is important for productive infection. Additional studies revealed that the viral protein ORF2 interacts with a complex containing β-catenin and HMGA1 in transfected cells, enhances β-catenin-dependent transcription, and cooperates with β-catenin to promote survival of neuroblastoma cells (23). In contrast, during DEX-induced reactivation from latency, expression of a soluble Wnt antagonist, Dickkopf-1 (DKK1), is induced more than 10-fold (21). DKK-1 is responsible for stress-induced neuronal death (24, 25). These studies suggest that the canonical Wnt/β-catenin signaling pathway regulates the establishment and maintenance of latency because this pathway promotes cell survival and axonal growth and directs axons to their proper synaptic targets (26–30).
In this study, RNA sequencing was performed using TG from calves at very early times after DEX treatment to initiate reactivation from latency. During the latency-reactivation cycle, 102 genes involved with the Wnt/β-catenin signaling pathway were differentially regulated. In agreement with previous microarray studies (21–23), the Wnt/β-catenin signaling pathway was more active in TG during latency versus in uninfected calves or calves undergoing reactivation from latency. The top three Wnt agonists that were differentially expressed during latency are the guanine nucleotide-binding protein alpha Q (GNAQ), Wnt16, and a gene that regulates TG neuronal patterning, bone morphogenetic protein receptor 2 (BMPR2). These studies also identified that a protein kinase associated with cell survival (Akt3) was expressed at higher levels in TG neurons during latency than TG neurons from uninfected calves or 30 min after reactivation from latency was initiated with DEX. ORF2 interacts with Akt3, stimulates Akt3 activity, and alters its localization in mouse neuroblastoma cells (Neuro-2A). In contrast, Akt1 was not expressed at higher levels during latency and does not appear to interact with ORF2. Several Wnt antagonists were stimulated during reactivation from latency. These Wnt antagonists included DKK1, Dickkopf-1-like (DKKL1), two secreted frizzled related proteins (SFRP), and two Sox transcripts that encode proteins that inhibit β-catenin-dependent transcription. Collectively, these studies confirm and extend previous findings that the Wnt/β-catenin signaling pathway is regulated during the latency-reactivation cycle.
RESULTS
Stress-induced reactivation from latency regulates the canonical Wnt signaling pathway.
Previous microarray studies suggested the Wnt/β-catenin signaling pathway is differentially regulated during the BoHV-1 latency-reactivation cycle (21, 23). To confirm and expand these studies, we compared cellular gene expression in TG during latency to that in TG from uninfected calves and at very early times after DEX-induced reactivation from latency (30 min, 1.5 h, and 3 h). RNA sequencing was performed because there are at least 19 related Wnt receptors, 7 Wnt coreceptors, and two families of Wnt antagonists that may not have been accurately identified or were missed by microarray analysis. These studies revealed expression of Wnt/β-catenin-associated genes was significantly different at all times evaluated compared to expression in latently infected TG: uninfected, P = 1.54e−3; 30 min after DEX, P = 7.16e−6; 1.5 h after DEX, P = 3.55e−3; and 3 h after DEX, P = 8.15e−4. Ingenuity pathway analysis (IPA) identified 102 unique genes or gene families involved with the Wnt/β-catenin signaling pathway that were differentially regulated across these time points: 29 in uninfected TG, 72 at 30 min after DEX, 35 at 1.5 h after DEX, and 58 at 3 h after DEX (Fig. 1). Table 1 provides a summary of the statistical analysis of the read data.
FIG 1.

Heat map summarizing differentially expressed genes that are associated with the Wnt/β-catenin signaling pathway in TG during BoHV-1 latency-reactivation cycle. RNA sequencing was performed using TG from calves for each group (one uninfected, three latently infected, and eight after DEX treatment to induce reactivation [two calves at 30 min, three calves at 1.5 h, and three calves at 3 h after DEX treatment]). Genes associated with the Wnt/β-catenin signaling pathway in TG of uninfected calves were compared to those of calves latently infected. Genes associated with the Wnt/β-catenin signaling pathway in TG during early states of reactivation (30 min, 1.5 h, and 3 h after DEX treatment) were compared to those in latency. *, fold change in latently infected TG compared to uninfected TG; **, fold change after DEX treatment compared to latently infected TG. Statistical analyses of the read data are summarized in Table 1.
TABLE 1.
Summary of RNA sequencing of TG from calves used in this studya
| Sampleb | No. of raw reads | No. of reads after trimming | No. of mapped reads | Mapping (%) |
|
|---|---|---|---|---|---|
| Concordant pair | Overall | ||||
| Latency (1) | 87,772,072 | 86,899,588 | 80,341,469 | 84.90 | 91.53 |
| Latency (2) | 113,935,336 | 113,602,597 | 103,298,949 | 83.70 | 90.66 |
| Latency (3) | 103,295,710 | 102,763,023 | 94,102,756 | 84.60 | 91.10 |
| 30 min (1) | 130,998,440 | 130,868,656 | 68,976,317 | 49.80 | 52.65 |
| 30 min (2) | 134,891,362 | 134,664,406 | 46,302,068 | 32.20 | 34.33 |
| 1.5 h (1) | 181,545,774 | 181,349,993 | 97,355,270 | 50.60 | 53.63 |
| 1.5 h (2) | 118,128,452 | 117,968,772 | 109,163,219 | 86.50 | 92.41 |
| 1.5 h (3) | 78,031,304 | 77,912,973 | 71,808,615 | 86.30 | 92.03 |
| 3 h (1) | 20,268,356 | 20,254,807 | 19,210,229 | 90.10 | 94.78 |
| 3 h (2) | 61,588,174 | 61,550,670 | 58,667,117 | 91.00 | 95.26 |
| 3 h (3) | 193,385,058 | 193,130,128 | 181,472,604 | 88.90 | 93.84 |
| Uninfected | 146,102,758 | 145,870,066 | 134,616,584 | 86.20 | 92.14 |
Summary of reads for RNA-sequencing results during latency (3 calves). Reads for latently infected calves treated with DEX for 30 min (2 calves), 1.5 h after DEX treatment (3 calves), and 3 h after DEX treatment (3 calves) are also provided. As a control, RNA sequencing was performed from an uninfected calf.
The numbers in parentheses denote TG from different animals.
When comparing gene expression in TG of latently infected calves relative to that of uninfected calves, the genes most increased during latency included several genes reported to enhance the canonical Wnt/β-catenin signaling pathway (Fig. 1 and 2). The top four differentially expressed genes during latency are shown in Fig. 2A and B. The number 1 gene is the gene encoding guanine nucleotide-binding protein alpha-Q (GNAQ), which enhances canonical Wnt signaling by multiple mechanisms, including β-catenin stabilization (31, 32). The second gene is the Wnt16 gene, a soluble Wnt agonist that promotes cortical bone development (33) and human acute lymphoblastoid leukemia cell growth (34). The third gene is the bone morphogenetic protein receptor 2 (BMPR2) gene, which is regulated by Wnt or regulates the Wnt/β-catenin signaling pathway (35, 36). Interestingly, BMPR2 is preferentially bound by BMP4 (bone morphogenetic protein 4), which is required for dorsoventral patterning of the TG, peripheral innervation, and survival of sensory neurons (37–39). BMPR2 is also essential for the effects of growth differentiation factor 5 (GDF5) on stimulating dendrite growth of hippocampal neurons (40), confirming BMPR2 mediates neuronal differentiation. Within 30 min after DEX treatment, RNA expression of positive Wnt/β-catenin regulators was repressed relative to that of latently infected TG (Fig. 1 and 2A): the level for GNAQ was 42-fold lower, that for Wnt16 was 54-fold lower, and that for BMPR2 was 44-fold lower.
FIG 2.
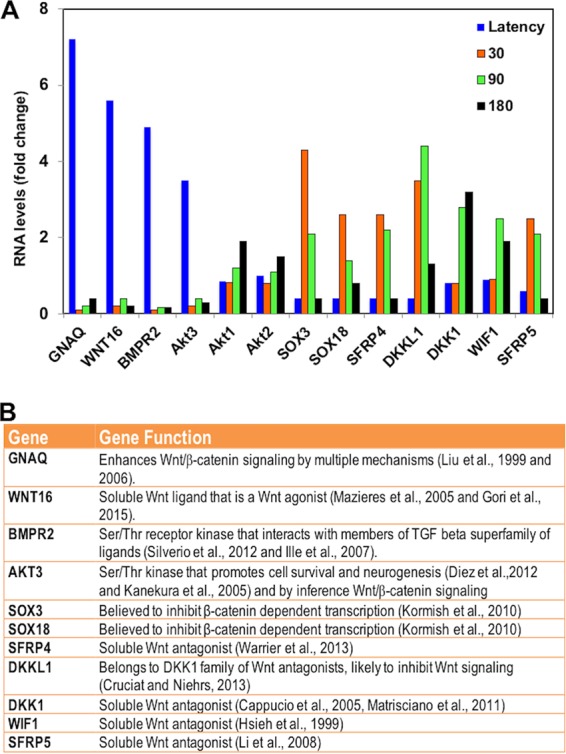
Relative expression of most differentially regulated genes associated with Wnt/β-catenin signaling pathway in TG during latency-reactivation cycle. (A) The top four differentially expressed genes during latency-reactivation cycle were chosen from the data presented in Fig. 1. Akt1 and Ak2 expression were not significantly different at the time points that were analyzed but were included as a comparison to Akt3. (B) List of genes and brief description of known functions.
The fourth most differentially regulated gene during latency is the Akt3 gene (Fig. 2). Numerous studies concluded that the Akt signaling pathway activates the Wnt signaling pathway and vice versa (41–47). The finding that Akt3, but not Akt1 or Akt2, RNA levels were reduced 51-fold during reactivation (Fig. 2A) is intriguing, because Akt3, Akt1, and Ak2 have similar substrates and are primarily regulated by the phosphatidylinositol 3-kinase (PI3K) pathway (48–50). Although published studies are focused primarily on Akt1, many studies did not identify the Akt isoform that was involved, and examination of Akt3 functions has lagged behind that of Akt. However, Akt3 possesses enhanced neuroprotective and axonal growth properties relative to Akt1 (51, 52), functions we deem essential for maintaining lifelong latency. Akt activity is also linked to herpes simplex virus 1 (HSV-1) reactivation from latency and the antiapoptosis functions of latency-associated transcript (LAT) (53–56). The conclusions of the HSV-1 studies relied on an inhibitor (AKT VIII) that interferes with all Akt isoforms. Further evidence suggesting Akt3 is regulated during the BoHV-1 latency-reactivation cycle comes from the finding that Appl1 RNA levels were higher during latency (2.5-fold higher) but reduced 30 min after DEX treatment (23-fold lower) (Fig. 1). Appl1 encodes a protein that interacts with Akt1 and regulates Akt substrate specificity (57, 58). In summary, these observations imply Akt3 signaling is regulated during the BoHV-1 latency-reactivation cycle and further supports the finding that Akt3 is involved in Wnt signaling.
Additional positive regulators of the Wnt/β-catenin signaling pathway were significantly induced during latency. These include the soluble Wnt agonist Wnt3 (2.5-fold), which activates the canonical Wnt pathway and maintains gastrulation in mice (59). Three Wnt coreceptors, frizzled 3 (FZD3), FZD4, and FZD7, are Wnt agonists. FZD3 activates the Wnt pathway in peripheral sensory neurons (60), FZD4 expression promotes β-catenin-dependent transcription (61), and FZD7 expression maintains the pluripotent state in human embryonic cells because it activates the Wnt/β-catenin pathway (62). Furthermore, levels of transcripts for positive Wnt/β-catenin transcriptional regulators TCFL2 (transcription factor 7 like 2) (63), TCF4 (T-cell factor 4) (64), and CREBBP (CREB binding protein) (65) were 2- to 3-fold higher in latently infected TG than uninfected TG. Interestingly, these positive regulators of the Wnt/β-catenin pathway were repressed 3- to 20-fold following DEX treatment.
From 30 min to 1.5 h after latently infected calves were treated with DEX, expression of Wnt/β-catenin antagonists was induced: SOX3 at 4.3-fold, SOX18 at 2.7-fold, Dikkopf-1-like protein (DKKL1) at 4.2-fold, Dikkopf-1 (DKK1) at 3-fold, Wnt inhibitory factor 1 (WIF-1) at 2.7-fold (66), secreted frizzled-related protein 4 (SFRP4) at 2.4-fold (67, 68), and SFRP5 (69) at 2.1-fold (Fig. 2B). There are 20 closely related SOX transcription factors, and several interact with β-catenin and inhibit β-catenin-dependent transcription (70), which implies SOX3 and SOX18 interfere with β-catenin-dependent transcription. DKK1 is firmly established as a secreted Wnt antagonist that induces neuronal death following stress (24, 25, 71–73). DKKL1 is a recently discovered DKK1 family member and thus is likely a Wnt antagonist (74). In general, DEX-induced soluble Wnt antagonists inhibit signaling by binding Wnt proteins (SFRP and WIF1) or the Wnt receptor complex (DKK family members) (67, 75, 76). In conclusion, RNA-sequencing studies revealed that the canonical Wnt/β-catenin pathway was regulated during the BoHV-1 latency-reactivation cycle in calves.
Immunohistochemistry studies confirm certain genes are differentially expressed in TG during latency-reactivation cycle.
Immunohistochemistry (IHC) studies were performed to confirm that certain cellular genes were differentially expressed in TG neurons during the latency-reactivation cycle. We initially analyzed GNAQ because its expression was 7 times greater in TG during latency than in uninfected TG. Furthermore, GNAQ expression was reduced more than 40-fold during DEX-induced reactivation (Fig. 1 and 2A), and GNAQ has several effects on the Wnt signaling pathway, including β-catenin stabilization (31, 32). IHC studies demonstrated GNAQ was detected in significantly more TG neurons during latency than TG from uninfected calves (Fig. 3A, B, and D). Although GNAQ was detected in certain TG neurons at 6 h after latently infected calves were treated with DEX to initiate reactivation from latency (Fig. 3C), the number of GNAQ+ neurons was significantly lower than that of TG from latently infected calves (Fig. 3D).
FIG 3.
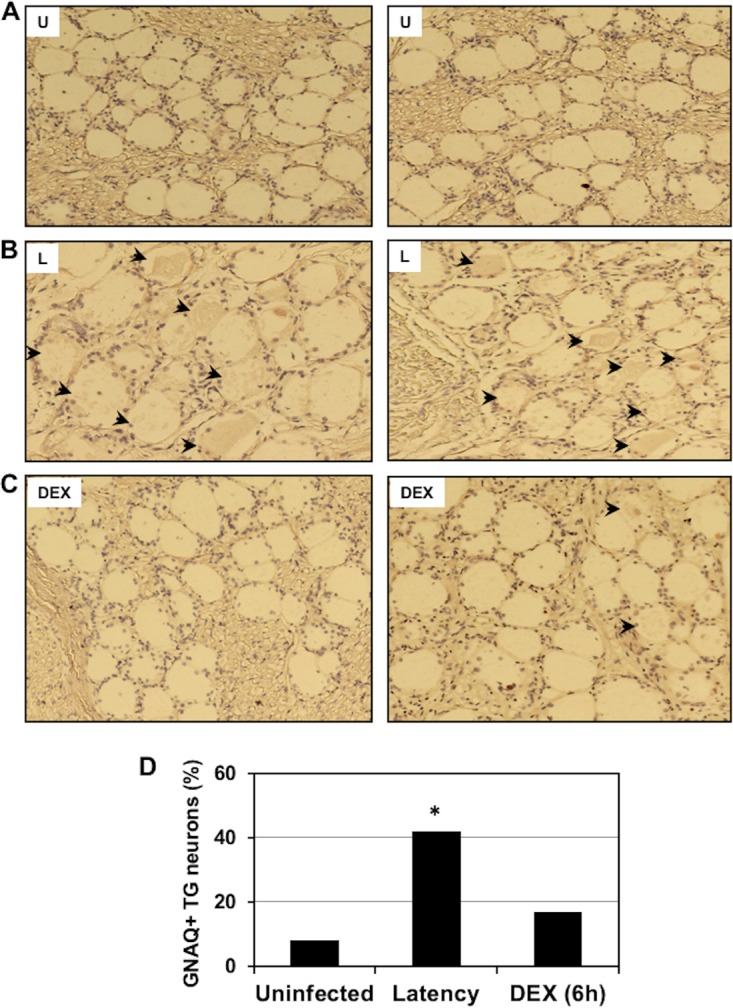
Detection of GNAQ in TG neurons during latency-reactivation cycle. TG were collected from 3 uninfected calves (U) (A), latently infected calves (L) (B), or latently infected calves treated with DEX for 6 h to initiate reactivation from latency (DEX) (C). Thin sections were cut from formalin-fixed, paraffin-embedded TG sections. The GNAQ antibody (ab75825; Abcam) was diluted 1:450. Biotinylated goat anti-rabbit IgG (Vector Laboratories) was used as a secondary antibody. Arrows denote GNAQ+ neurons in the respective samples. (D) The percentage of GNAQ-positive neurons from 296 uninfected neurons, 241 latently infected neurons, and 209 TG neurons at 6 h after latently infected calves were treated with DEX. An asterisk denotes significant differences (P < 0.05) in the numbers of GNAQ+ neurons as determined by a Student t test. Magnification is approximately ×200.
Similarly, we examined BMPR2 expression in TG neurons. BMPR2 was detected in a small subset of TG neurons of uninfected calves (16%); however, the number of BMPR2+ neurons increased significantly in latently infected tissue (75%) (Fig. 4A and B). The time point of 6 h after DEX treatment was used to initiate reactivation from latency and a significant decrease in BMPR2+ neurons was detected (20%), which was similar to that of TG from uninfected calves.
FIG 4.
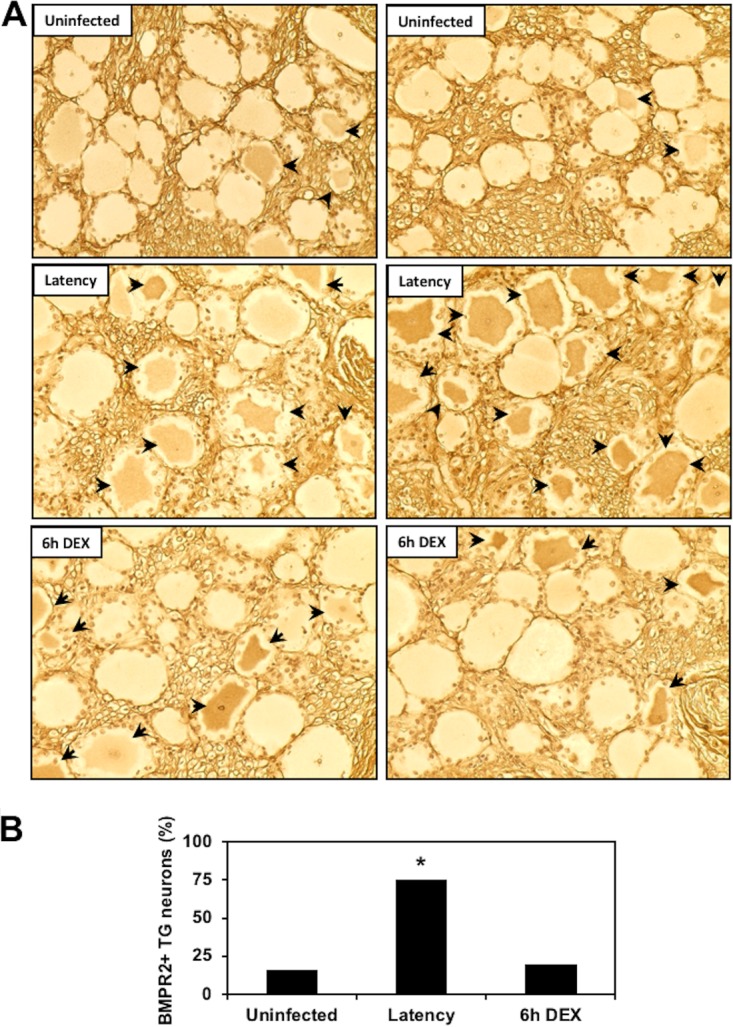
Comparison of BMPR2 expression during the BoHV-1 latency-reactivation cycle. (A) TG were collected from 3 uninfected calves, 3 latently infected calves, or 3 latently infected calves treated with DEX for 6 h to initiate reactivation from latency. IHC was performed as described in Materials and Methods using a BMPR2 monoclonal antibody (MA5-15827; ThermoFisher Scientific); arrows denote BMPR2+ TG neurons. Magnification is approximately ×400, and these sections are representative of many sections that were examined. (B) Quantification of BMPR2+ TG neurons. The percentage of BMPR2+ TG neurons from uninfected calves (429 neurons were counted), latently infected calves (435 neurons), or latently infected calves treated with DEX for 6 h to initiate reactivation (417 neurons) were calculated. The asterisk denotes significant differences (P < 0.05) in the numbers of BMPR2+ TG neurons as determined by a Student t test.
Additional studies indicated DKK1 was not readily detected in TG neurons from latently infected calves (Fig. 5A) or TG of uninfected calves (data not shown). However, DKK1 was detected in a subset of TG neurons from latently infected calves that were treated with DEX for 6 h (Fig. 5B, arrows denote DKK1+ TG neurons). DKK1 staining was also detected outside TG neurons after 6 h of DEX treatment (two areas are denoted by filled circles). Careful examination of TG sections from latently infected calves treated with DEX appeared to have light DKK1 staining between certain neurons (denoted by filled circles). Extracellular DKK1 staining was not readily detected in TG of latently infected calves. As DKK1 is a soluble secreted protein (24, 25, 71–73), staining outside TG neurons or satellite cells was not unexpected. We did not compare the number of TG neurons in latently infected TG to those in uninfected TG or during reactivation because DKK1 is a soluble protein. Wnt16, SFRP4, SFRP5, DKKL1, and WIF1 are also soluble proteins and thus were not examined by IHC.
FIG 5.
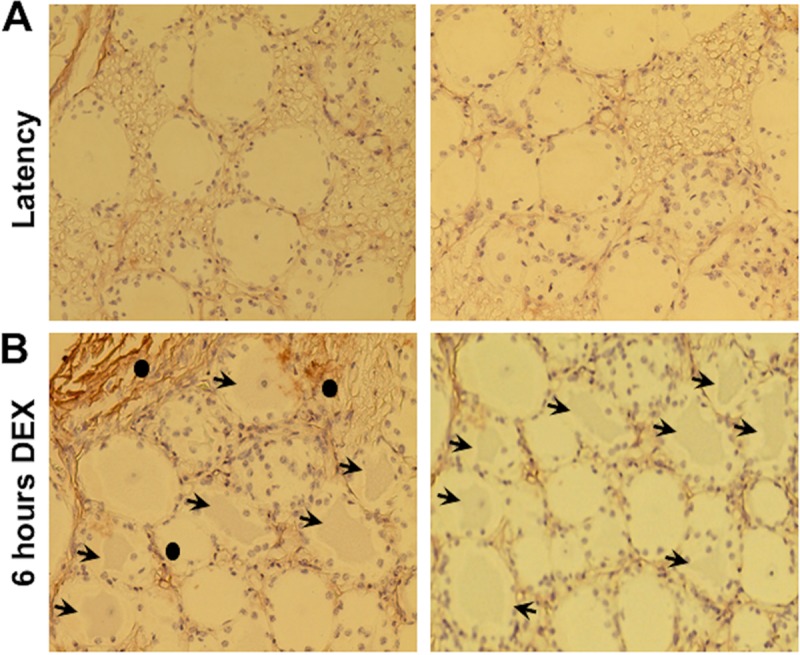
Comparison of DKK1 expression during the BoHV-1 latency-reactivation cycle. TG were collected from 3 latently infected calves (A) or latently infected calves treated with DEX for 6 h to initiate reactivation from latency (B). IHC was performed as described in Materials and Methods using a DKK1-specific antibody (ab188597; Abcam); arrows denote DKK1+ TG neurons, and closed circles denote extracellular DKK1 staining. Magnification is approximately ×400, and these sections are representative of many sections that were examined.
The Akt3 protein is differentially expressed during latency.
Since Akt activity was reported to regulate certain aspects of HSV-1 latency (53–56), we examined Akt3 expression by IHC. At the transcript level, Akt3 was 3.6-fold higher in latently infected TG than in mock-infected animals but was dramatically reduced during reactivation from latency (Fig. 1 and 2A). Certain TG neurons from uninfected calves appeared to be lightly stained by the Akt3 antibody (Fig. 6A), which was expected because Akt3 RNA levels in TG from uninfected calves were detected. Conversely, Akt3+ TG neurons were readily detected during latency (Fig. 6A, blue and black arrows). A subset of Akt3+ TG neurons from latently infected calves also appeared to contain Akt3 in the nucleus (Fig. 6A, blue arrows). By 6 h after DEX treatment of latently infected calves, there were fewer TG neurons from calves stained by the Akt3 antibody and the intensity of staining appeared to be less (denoted by black arrow). Nuclei were readily visible in TG neurons from uninfected calves and latently infected calves treated with DEX for 6 h (marked by closed circles) but generally were not stained by the Akt3 antibody. When Akt3+ neurons from the respective samples were counted, more Akt3+ neurons were detected during latency than TG neurons from uninfected calves and following DEX treatment of latently infected calves for 6 h (Fig. 6B).
FIG 6.
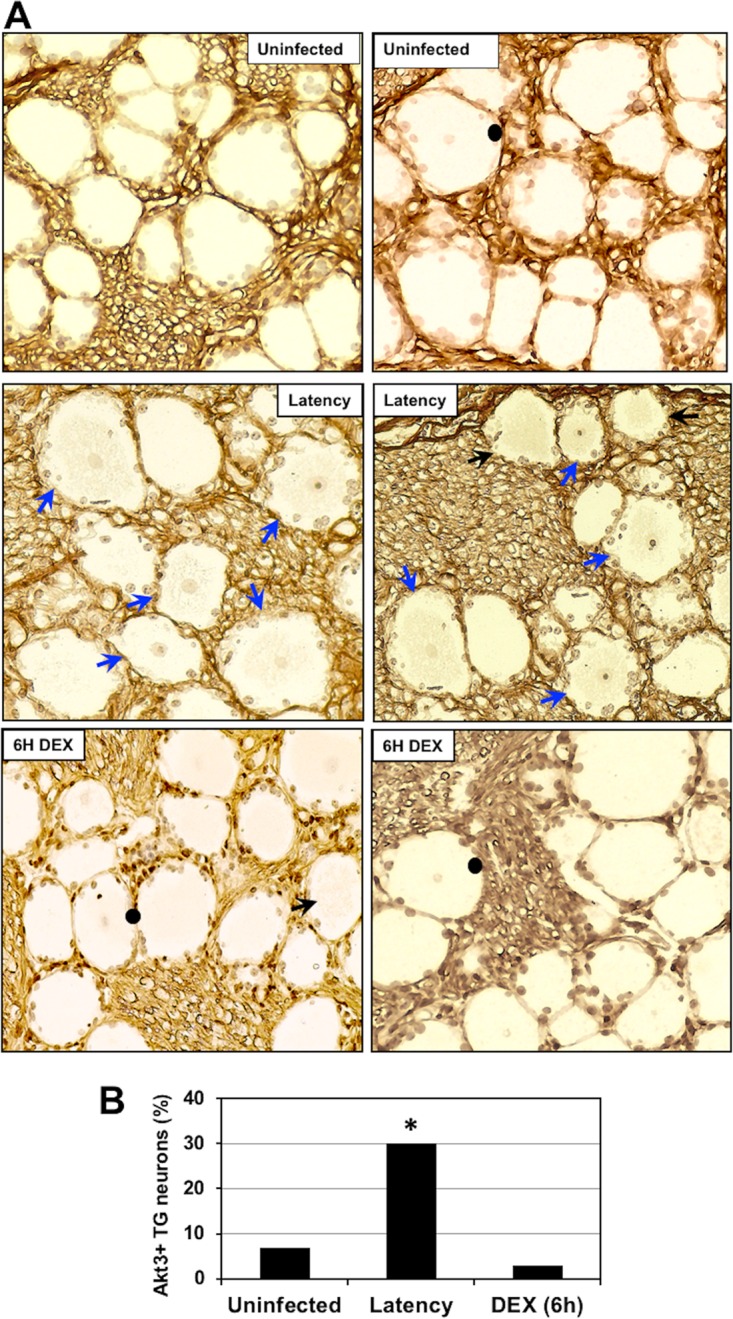
Comparison of Akt3 expression during the BoHV-1 latency-reactivation cycle. (A) TG were collected from 3 uninfected calves, 3 latently infected calves, or 3 latently infected calves treated with DEX for 6 h to initiate reactivation from latency. Thin sections were cut from formalin-fixed paraffin-embedded TG sections. The Akt3 antibody (ab152157; Abcam) was diluted 1:500. Biotinylated goat anti-rabbit IgG (Vector Laboratories) was used as a secondary antibody. Blue arrows denote Akt3-positive TG neurons that contained an Akt3-positive nucleus and a counterstained nucleolus. Black arrows denote TG neurons in which the nucleus was not visible, but they were Akt3+. Closed circles denote TG neurons that contain a nucleus in which the nucleolus is counterstained but was not stained by the Akt3 antibody. These images are representative of many sections stained with the Akt3 antibody. Magnification is approximately 400×. (B) The percentage of Akt3-positive TG neurons from 500 total neurons was estimated from sections derived from 3 latently infected calves, 3 mock-infected calves, and 3 latently infected calves treated with DEX for 6 h. An asterisk denotes significant differences (P < 0.05) in the number of Akt3-positive neurons as determined by a Student t test.
To determine if Akt3 was preferentially expressed in latently infected neurons, consecutive sections were cut from TG of calves latently infected, and one slide was stained with an ORF2 peptide-specific antibody and one with the Akt3 antibody. ORF2 is expressed in a subset of latently infected TG neurons and thus is a marker for certain latently infected neurons (77, 78). ORF2 expression in latently infected TG is not uniform and appears to cluster in certain areas within the TG. Consequently, we identified areas of TG sections that contained several ORF2+ neurons and then compared Akt3 staining in consecutive sections (Fig. 7). The ORF2 antibody stained eight TG neurons in this section obtained from a latently infected calf; the Akt3 antibody stained 4 of these TG neurons (neurons 1, 3, 4, and 5). From additional TG sections from 3 latently infected calves, we quantified the number of ORF2+ TG neurons stained by the Akt3 antibody. Of the 500 TG neurons that were ORF2+, 62% of TG neurons in consecutive sections were Akt3+. In areas of the TG where ORF2 was not detected, we detected 16% that were lightly stained by the Akt3 antibody. In summary, many ORF2+ TG neurons expressed Akt3 in calves latently infected with BoHV-1.
FIG 7.
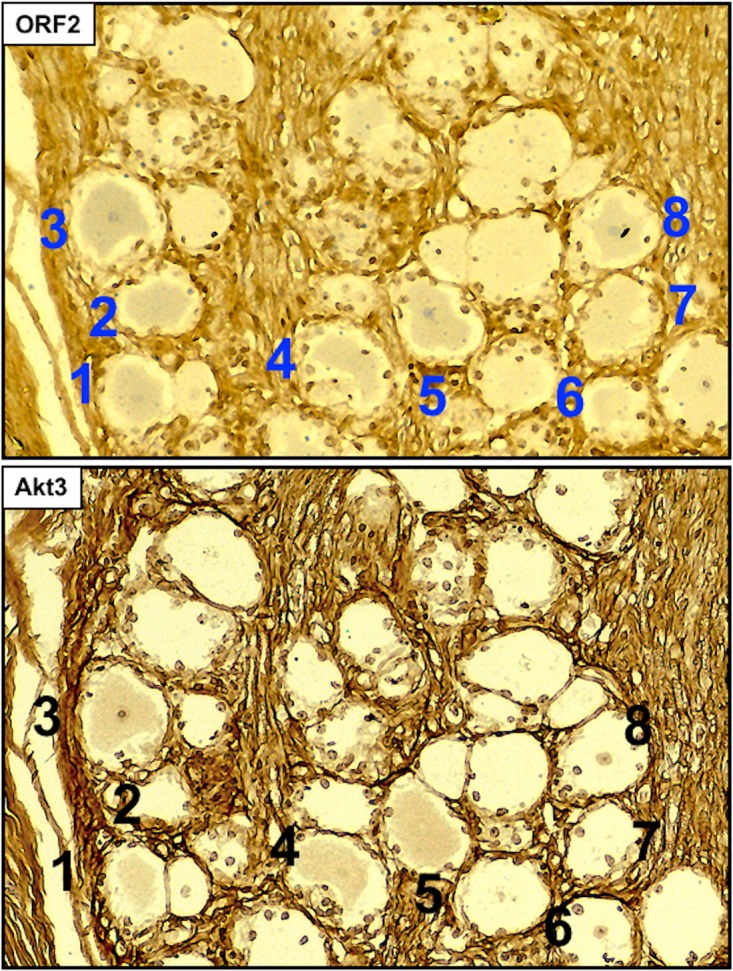
Akt3 is frequently detected in ORF2+ neurons during latency. Consecutive sections from formalin-fixed paraffin-embedded TG sections from latently infected calves were prepared. One section was stained with the Akt3 antibody (ab152157; Abcam) that was diluted 1:500. A consecutive section was stained with a peptide-specific ORF2 antibody (1:500 dilution). Biotinylated goat anti-rabbit IgG (Vector Laboratories) was used as a secondary antibody for the sections. Areas of sections that contained ORF2+ neurons were subsequently examined for Akt3 staining. Numbers denote the ORF2-positive neurons, and neurons 1, 3, 4, and 5 were also Akt3+. Magnification is approximately 400×, and these sections are representative of many sections that were examined.
ORF2 increases Akt3 steady-state levels and alters Akt3 subcellular localization.
We examined the effect that ORF2 may have on Akt3, because Akt3 was expressed at higher levels during latency and the HSV-1 latency-associated transcript (LAT) promotes Akt activity (53, 54). Furthermore, inhibiting PI3K or Akt activity induces HSV-1 reactivation in a neuronal model of latency (55, 56). Akt family members belong to the AGC (cyclic AMP dependent, cGMP, and protein kinase C) family of serine/threonine protein kinases because they have similar catalytic domains and phosphorylate many of the same substrates (79). Neuro-2A cells were used for these studies because they can be readily transfected, and ORF2 is consistently detected in these cells following transfection with an expression plasmid (12, 16). Although Neuro-2A cells are an established neuroblastoma cell line that grows efficiently, they can be differentiated into dopamine-like neurons (80) and consequently have certain neuron-like features. Following cotransfection of Neuro-2A cells with increasing concentrations of a plasmid that expresses ORF2, a dose-dependent increase in endogenous Akt3 levels was observed (Fig. 8A). At the highest concentration of ORF2 used for this study (2 μg DNA), approximately 15-fold higher Akt3 steady-state levels were detected (Fig. 8B). An ORF2 construct that lacked the nuclear localization signal (ORF2ΔNLS) also increased Akt3 steady-state levels (Fig. 8C). In contrast, an ORF2 construct containing stop codons at the amino terminus of ORF2 does not express ORF2 and was unable to increase steady-state levels of Akt3. In contrast to the results obtained with Akt3, ORF2 expression in Neuro-2A cells reduced Akt1 steady-state protein levels approximately 2-fold (Fig. 8D and E).
FIG 8.
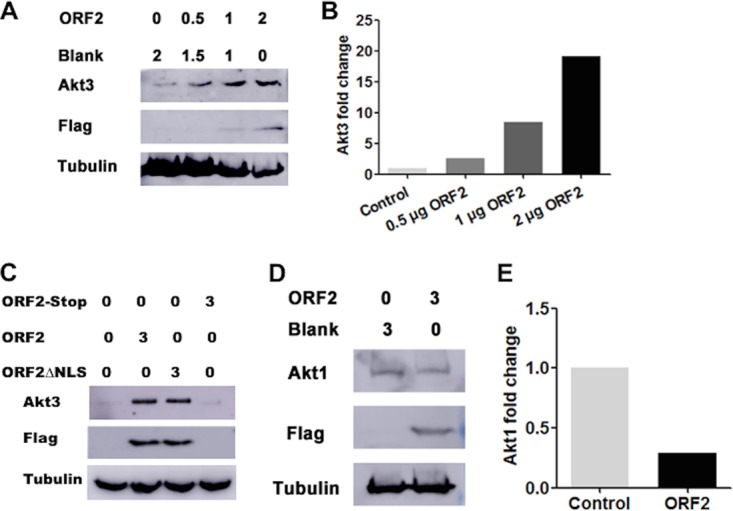
ORF2 stabilizes Akt3 steady-state protein levels in transfected Neuro-2A cells. (A) Neuro-2A cells were transfected with the designated amounts of an ORF2 expression plasmid (μg DNA). Total cell lysate was prepared at 40 h after transfection and Akt3 levels examined by Western blotting using an Akt3-specific antibody (4059; Cell Signaling Technology). ORF2 was detected with a Flag monoclonal antibody (Sigma). Tubulin protein levels were included as a loading control. (B) Quantification of Akt3 and tubulin bands from Western blots shown in panel A were collected using the ImageJ software package. The band intensity of Akt3 was initially normalized to tubulin, and then the fold change after infection was calculated. Akt3 levels in cells transfected with the empty vector were set to 1. (C) Neuro-2A cells were transfected with 3 μg of a plasmid that expresses ORF2, a stop codon ORF2 mutant that cannot express ORF2 (ORF2 stop), or an ORF2 mutant that has a deletion in the nuclear localization signal (ORF2ΔNLS). At 48 h after transfection, Akt3 and ORF2 were examined in transfected Neuro-2A cells by Western blotting. (D) Neuro-2A cells were transfected with 3 μg of a plasmid that expresses ORF2. At 48 h after transfection, Akt1 and ORF2 levels were examined in transfected Neuro-2A cells by Western blotting. (E) Quantification of Akt1 and tubulin bands from Western blots shown in panel D were analyzed as described for panel B. These results are representative of three independent studies.
Given that an increase in nuclear Akt3 was observed in latently infected TG neurons by IHC, confocal microscopy was subsequently performed to test whether ORF2 influenced Akt3 localization in transfected Neuro-2A cells. When Neuro-2A cells were transfected with a plasmid that expresses Akt3, most of the Akt3 staining was present in the cytoplasm or perinuclear region (28 out of 30 Akt3+ cells that were examined) (Fig. 9A). As previously reported (13, 16, 17), ORF2 is primarily localized to the rim area of the nucleus (Fig. 9B). When ORF2 and Akt3 were cotransfected into Neuro-2A cells, ORF2 did not appear to be localized as much to the rim area of the nucleus, and Akt3 was detected in the nucleus of 24 out of 50 cells that expressed both proteins (Fig. 9C shows a representative Neuro-2A cell that was ORF2+ and Akt3+). In some cells in which Akt3 was present in the nucleus of cells expressing ORF2 there was colocalization of ORF2 and Akt3, as indicated by yellow staining. In summary, these studies suggested ORF2 increased steady-state levels of the Akt3 protein and relocalized a subset of Akt3 to the nucleus.
FIG 9.

Localization of Akt3 and ORF2 in transfected Neuro-2A cells. Neuro-2A cells were transfected (Lipofectamine 2000; Invitrogen) with 0.5 μg of a plasmid that expresses Akt3 (green) (A), Flag-tagged ORF2 (red) (B), or Akt3 and ORF2 (C). At 48 h posttransfection, cells were fixed and then stained with antibodies that recognize Akt3 (green-stained cells) (ab1521; Abcam) and/or ORF2 (red-stained cells). Nuclear DNA was stained with DAPI. Images were observed by confocal microscopy. The selected images are representative of 4 experiments (at least 100 stained cells were examined). For panel C, we show ORF2 alone, Akt3 only, and merged images that also show DAPI-stained cells that are ORF2 and Akt3 positive. The scale bar in each panel is equal to 50 μm.
ORF2 interacts with and activates Akt3.
Confocal microscopy suggested ORF2 was partially localized with a subset of Akt3 and ORF2 increased Akt3 nuclear localization (Fig. 8 and 9). To test whether ORF2 was associated with Akt3, coimmunoprecipitation (co-IP) studies were performed in Neuro-2A cells transfected with ORF2. When Akt3 was immunoprecipitated, ORF2 was consistently detected in the immune precipitate (Fig. 10A). IP with the Flag antibody also revealed Akt3 was detected in the immunoprecipitate when ORF2 was included in the transfection (Fig. 10A). In certain experiments, ORF2 migrates as a doublet (Fig. 10A, for example), which may be the result of phosphorylation (12, 15, 16) or proteolysis. As expected, the Flag antibody only detected an ORF2-specific band in Neuro-2A cells transfected with the ORF2 expression plasmid. In contrast to Akt3, Akt1 did not stably interact with ORF2 in transfected Neuro-2A cells (Fig. 10B).
FIG 10.

Analysis of the effect ORF2 has on Akt3 and Akt1. For all studies described below, Neuro-2A cells were transfected with the designated amount of plasmids (μg DNA), and whole-cell extract (WCE) was collected at 48 h after transfection. (A) IP was performed with an Akt3 antibody (4059; Cell Signaling Technology) using WCE (400 μg protein). Immunoprecipitated proteins bound to magnetic protein A beads (S1425S; Invitrogen) were washed extensively, suspended in SDS-PAGE buffer, and separated in a 10% SDS-PAGE gel. After proteins were transferred to a PVDF membrane, the immunoblot (IB) was probed with the Akt3 antibody or anti-Flag monoclonal antibody (F1804; Sigma). (Right) IP was performed with the Flag antibody to IP ORF2 using total cell lysate (400 μg protein). Immunoprecipitated proteins were processed as described here and separated in a 10% SDS-PAGE gel, and the IB was probed with the Flag antibody or Akt3 antibody. (B) IP was performed with the Akt1 antibody (2938; Cell Signaling Technology) or Flag antibody using total cell lysate (400 μg protein). Immunoprecipitated proteins were analyzed by Western blotting and then probed with an Akt1 or anti-Flag monoclonal antibody. Western blot of WCE (50 μg protein) was probed with Flag or Akt1 antibody. (C) IP was performed with the Akt3 antibody, the precipitated proteins separated by 10% SDS-PAGE, and protein transferred to a membrane. The Western blot was then probed with the Akt antibody that recognizes phosphorylated Akt at serine 472 (pSer473; 9271; Cell Signaling Technology) or Akt3 antibody. ORF2 in WCE was detected using the Flag antibody. This was the same image as that shown in panel A, because these studies were performed in the same experiment. Levels of pSer472 Akt3 were quantified using ImageJ. (D) IP was performed with the Akt1 antibody using total cell lysate (400 μg protein). Immunoprecipitated proteins were analyzed in Western blots and then probed with the Akt antibody that recognizes phosphorylated Akt at serine 473 or anti-Akt1 antibody (2938; Cell Signaling Technology). WCE (50 μg protein) was probed with the anti-Flag monoclonal antibody to detect ORF2 (F1804; Sigma). Levels of phosphorylated serine 473 on Akt1 were quantified. (E) IP was performed with the Akt1 or Akt3 antibody using total cell lysate (400 μg protein). As described for earlier panels, immunoprecipitated proteins were examined for Akt1 or Akt3. Akt1 or Akt3 in 50 μg WCE was identified by Western blot analysis as a control. The results shown are representative of 3 independent studies.
Akt activation requires phosphorylation at threonine 308 and serine 473 by two cellular protein kinases, PDK1 (phosphoinositide-dependent kinase) and mTORC2 (mammalian target of rapamycin complex 2) (49, 81, 82). Phosphorylation of serine 473 is required for activation of Akt1, and this site is well conserved in Akt3; however, the site is at position 472 (83). The antibody that recognizes Akt1 phosphorylation at serine 473 (p-Ser473) also recognizes p-Ser472 in Akt3. Consequently, we tested whether ORF2 regulated Akt3 phosphorylation by performing IP with the Akt3 antibody and estimating p-Ser472 Akt3 levels in the immune precipitate with an antibody that recognizes Akt p-Ser473. In cells transfected with ORF2, a 4-fold increase of p-Ser472 was associated with Akt3 (Fig. 10C). In contrast, ORF2 did not increase p-Ser473 levels when the IP was performed with the Akt1-specific antibody (Fig. 10D). Additional studies tested whether the Akt3 antibody cross-reacted with Akt1 and led to IP of Akt1 or vice versa. Following IP with Akt1 or Akt3, we were unable to detect Akt3 or Akt1, respectively, even when the proteins were overexpressed in Neuro-2A cells. As expected, Akt1 or Akt3 was detected in the immunoprecipitate when the IP was performed with the same antibody. In summary, these studies revealed ORF2 specifically interacted with Akt3 but not Akt1 and ORF2 activated Akt3, as judged by increased phosphorylation of Ser472.
DISCUSSION
In this study, we provided evidence that more than 100 genes involved in the Wnt/β-catenin signaling pathway were differentially expressed in TG during the BoHV-1 latency-reactivation cycle. During latency, the Wnt pathway in TG was more active than that in TG from uninfected calves or during DEX-induced reactivation from latency (Fig. 11 depicts a schematic summarizing the findings in this study). The ability of ORF2 to enhance β-catenin-dependent transcription in transfected cells (21) correlates with stabilizing the Wnt/β-catenin signaling pathway during latency. Other viral gene products may also be involved, because two virus-encoded micro-RNAs and ORF1 are expressed in certain latently infected neurons (8), and other herpesvirus-encoded micro-RNAs were proposed to regulate the Wnt signaling pathway (84). Regardless of which LR gene products are involved, Wnt/β-catenin agonists, including GNAQ, Wnt16, and BMPR2, are expressed at higher levels during latency than in uninfected calves or during early stages of reactivation. BMPR2, via its interactions with BMP4 and GDF5, seems to be very important during the latency-reactivation cycle, because this signaling axis is required for dorsoventral patterning of TG, peripheral innervation, and survival of sensory neurons (37–39). Furthermore, BMPR2 was present in approximately 75% of the total neurons that were examined. Since only 15 to 20% of TG neurons are believed to express ORF2 during latency (12, 78, 85), we suggest BMPR2 expression is induced in “bystander” uninfected TG neurons, because several Wnt agonists are soluble secreted factors. The Wnt pathway enhances neurogenesis and neuronal survival (26, 86, 87), suggesting the Wnt/β-catenin signaling pathway promotes the establishment and maintenance of lifelong latency (Fig. 11A).
FIG 11.
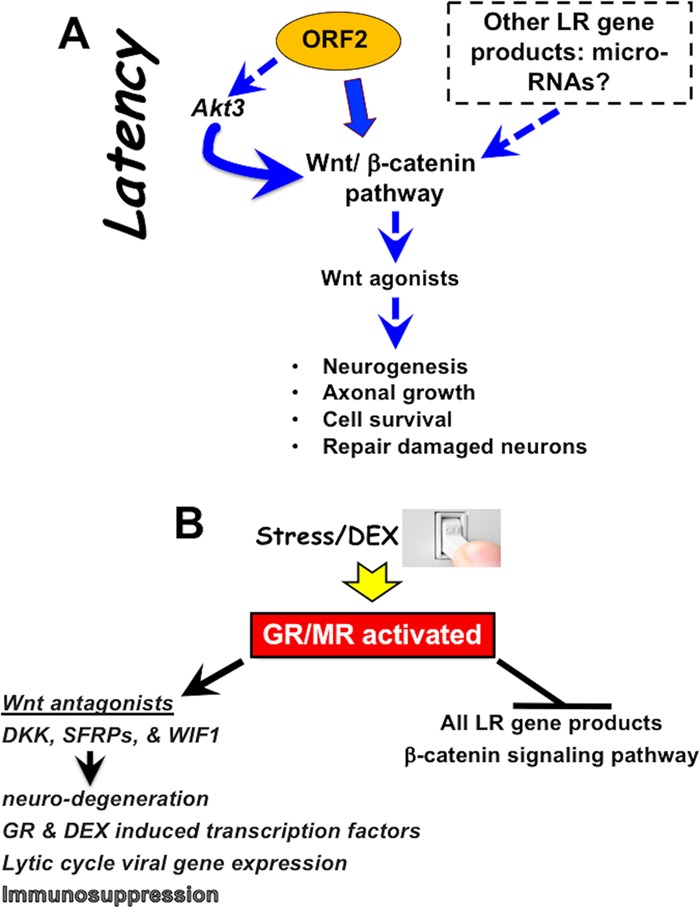
Schematic of viral gene expression and putative role of Wnt signaling pathway during BoHV-1 latency-reactivation cycle. Salient features during DEX-induced reactivation from latency and the role that the Wnt signaling pathway plays. For details, see Discussion.
The ability of ORF2 to interact with and influence Akt3 activation (Fig. 11A) is likely to promote latency, because Akt activity was reported to regulate key events during the HSV-1 latency-reactivation cycle. For example, inhibiting Akt activity with a specific inhibitor (AKT VIII) impairs the ability of HSV-1 LAT to inhibit apoptosis and promote neurite sprouting in mouse neuroblastoma cells (C1300) (53, 54). In an in vitro HSV-1 latency model using primary superior cervical ganglion sympathetic neurons, LAT is readily detected and a quiescent infection can be maintained for weeks at a time if nerve growth factor (NGF) signaling is active (55, 56). When NGF is removed from cultures, lytic-cycle viral gene expression is induced and infectious virus produced, resembling reactivation from latency in vivo. Downstream targets of the NGF receptor include PI3K and AKT, reviewed in reference 88. Latently infected neuronal cultures containing NGF also reactivate from latency when cultures are treated with AKT VIII (56). Several studies also concluded Akt signaling stimulated the Wnt/β-catenin signaling pathway by directly phosphorylating β-catenin on serine 552 and increasing β-catenin-dependent transcription (42, 89). In line with this finding, the PI3K/AKT pathway increases nuclear β-catenin localization (90), increases β-catenin protein levels (43), and thus promotes neuronal survival after traumatic brain injury (91). In a subset of TG neurons from latently infected calves and in transfected Neuro-2A cells, Akt3 was detected in the nucleus, consistent with other studies (83). This is important because nuclear Akt is required for NGF-mediated antiapoptotic signaling in PC12 neuron-like cells (92). Akt3 is more important than Akt1 and Akt2 with respect to preventing stroke-induced neuronal injury (52), inhibiting apoptosis in neurons, and stimulating axonal development (51). In summary, we predict Akt3 stabilizes the Wnt/β-catenin signaling pathway in TG of latently infected calves, which facilitates survival of latently infected neurons.
Stress, as mimicked by DEX, consistently induces BoHV-1 reactivation from latency (11, 22, 93–100) (Fig. 11B depicts a summary of events during early stages of BoHV-1 reactivation). Increased expression of at least seven putative Wnt antagonists (DKK1, DKKL1, Wif-1, SFRP4, SFRP5, SOX3, and SOX18) occurred during early stages of DEX-induced reactivation, suggesting that inhibiting the Wnt signaling pathway promotes reactivation from latency (Fig. 11B). The canonical Wnt/β-catenin signaling pathway is negatively regulated by the DKK family of secreted proteins, which interacts with Wnt coreceptors and low-density lipoprotein receptor-related proteins (LRP) and prevents their interactions with Wnt family members (73, 75, 76). DKK1 induction is required for development of ischemic neuronal death (71, 101) and stress-induced hippocampal damage (24) and is linked to neuronal death in patients with Alzheimer's and Parkinson's disease (27, 72, 86, 102–104). SFRP family members, by virtue of interacting with Wnt family members, also antagonize the Wnt pathway. For example, SFRP4 induces apoptosis (105, 106) by suppressing Akt survival pathways for its proapoptotic properties (107). Induction of apoptosis has been reported to stimulate herpesvirus replication (108) and reactivation from latency (109), suggesting there is a correlation between stress-mediated activation of DKK, SFRP family members, and reactivation from latency.
Increased corticosteroids can stimulate productive infection and directly activate the immediate-early (IE) promoter that drives expression of bICP0 and bICP4, the two most important viral transcriptional regulators encoded by BoHV-1 (110, 111). LR gene products are also reduced by DEX during reactivation from latency (12, 18, 98), which is significant because the two virus-encoded micro-RNAs can reduce bICP0 steady-state protein levels (18). Finally, it is well established that corticosteroids induce immunosuppression (112–114), which enhances the ability of BoHV-1 to successfully reactivate from latency.
In conclusion, these studies provide additional evidence that the Wnt/β-catenin signaling pathway is active during BoHV-1 latency. Conversely, stress antagonizes this signaling pathway, which correlates with induction of lytic-cycle viral gene expression and reactivation from latency. It is currently not known whether the Wnt/β-catenin signaling pathway is regulated during the latency-reactivation cycle of other alphaherpesvirinae subfamily members. Studies to test this prediction are under way.
MATERIALS AND METHODS
Infection of calves.
All TG samples from calves used for this study were previously described (21–23, 115). In brief, BoHV-1-free calves (∼200 kg) were inoculated with 107 PFU of BoHV-1 into ocular and nasal cavities as described previously (10, 11, 116–119). Calves were housed under strict isolation and given antibiotics to prevent bacterial pneumonia. At 60 days postinfection, calves were not shedding virus and were operationally defined as being latently infected. Certain calves were injected intravenously in their jugular vein with 100 mg DEX to initiate reactivation from latency. All calves (latently infected, DEX treated, or uninfected) were transported to the University of Nebraska Veterinary Diagnostic laboratory and then anesthetized with xylazine (Rompun) followed by electrocution. Experiments were performed in accordance with the American Association of Laboratory Animal Care guidelines and University of Nebraska IACUC committee (A3459). Following euthanasia, TG were collected and minced into small pieces, and then a portion of the TG was formalin fixed and paraffin embedded. The remainder of both TG samples was minced into small pieces and placed into a single 50-ml conical tube, and the tube was placed in a dry-ice ethanol bath and then stored at −80°C. After decapitation, it takes 5 to 10 min to collect TG, mince TG, place TG pieces in formalin, or submerge TG pieces in 50-ml conical tubes in a dry-ice ethanol bath.
RNA sequencing.
Total RNA was prepared from approximately 2 g of TG tissue using TRIzol (Life Technologies) according to the manufacturer's instructions. RNA integrity and concentration were quantified with an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). An Illumina TruSeq stranded RNA library preparation kit was used to construct cDNA libraries, which were sequenced as 75-bp paired-end reads on an Illumina NextSeq 500 (Illumina, San Diego, CA).
Read alignment of the RNA-Seq data was carried out as follows. First, the raw paired-end sequence reads in individual fastq files were trimmed to remove adapter sequences and low-quality bases using Trimmomatic software (version 0.35) (120). The remaining reads were mapped to the UMD 3.1 genome assembly with Tophat2 (version 2.1.1) (121) using the National Center for Biotechnology Information (NCBI) Bos taurus reference annotation to guide the alignment. The default parameters for Tophat2 were used.
Cuffdiff software (version 2.2.1) (122) was employed to process the aligned sequence read files and test for differential gene expression. Transcript abundance was estimated for each transcript in the NCBI Bos taurus reference annotation (UMD3.1) for each sample. A gene was defined as being expressed provided it had an average fragment per kilobase of transcript per million mapped reads (FPKM) of ≥1.0 in at least one of the two groups in the comparison. Unexpressed genes were removed from further downstream analysis. Differential gene expression was detected by comparing the log (base 2) ratios of the FPKM values for every gene and transcript. The resulting P values were corrected for multiple testing using Benjamini-Hochberg correction (123). P values were considered statistically significant at a Benjamini-Hochberg adjusted P value of ≤0.05. Statistical analysis of the read data is summarized in Table 1.
Qiagen IPA.
Ingenuity pathway analysis (IPA; Qiagen, Redwood City, CA) was used to deduce direct and indirect molecular relationships among differentially expressed genes. Each of the data sets was imported with a flexible format using gene symbol as the identifier. A core analysis was performed on genes in each set, where a P value for each network is calculated according to the fit of the user set of significant genes and the size of the network. P values were considered statistically significant at a Benjamini-Hochberg adjusted P value of ≤0.05.
IHC analysis.
IHC studies were performed using an ABC kit (Vector Laboratories) according to specifications of the manufacturer as previously described (21, 23, 115, 124). Thin sections (4 to 5 μm) of TG were cut, mounted on glass slides, and processed as described previously (12, 21). Slides were subsequently incubated with Akt3 antibody (1:500 dilution; 152157; Abcam), GNAQ antibody (1:450 dilution; ab75825), BMPR2 (1:400 dilution; MA5-15827; ThermoFisher), or DKK1 antibody (1:500 dilution; ab188597; Abcam) overnight in a humidified chamber at 4°C. The next day, slides were washed in 1× Tris-buffered saline (TBS) and incubated in biotinylated goat anti-rabbit IgG (PK-6101; Vector Laboratories) for 30 min at room temperature in a humidified chamber. Avidin-biotinylated enzyme complex was added to the slides for 30 min of incubation at room temperature. After three washes in 1× TBS, slides were incubated with freshly prepared substrate (SK-4800; Vector Laboratories), rinsed with distilled water, and lightly counterstained with hematoxylin. IHC results were evaluated in a blinded fashion.
Cells, plasmids, and antibodies.
Murine neuroblastoma cells (Neuro-2A; CCL-131) were obtained from the ATCC (Manassas, VA) and grown in minimal essential medium (MEM; Life Technology) supplemented with 10% fetal calf serum (FCS), penicillin (10 U/ml), and streptomycin (100 μg/ml).
The ORF2 expression construct was generated in the pCMV-Tag-2B vector (Stratagene) and was described previously (12, 14, 16). A Flag epitope is present at the N terminus of ORF2, and the human IE cytomegalovirus (CMV) promoter drives its expression. Sequences derived from ORF2 with a nuclear localization signal (NLS) deletion (amino acids [aa] 64 to 70) (ORF2ΔNLS) were synthesized by Integrated DNA Technology (IDT; Coralville, IA) and cloned into the pCMVTag-2B plasmid using BamHI-HindIII restriction enzymes. A plasmid that expresses Akt1 was a gift from Jie Chen (pCDNA3-HA-Akt1; plasmid 73408; Addgene). A plasmid that expresses Akt3 was a gift from William Sellers (1236 pcDNA3 Myr HA Akt3; plasmid 9017; Addgene).
All plasmids were transfected into Neuro-2A cells in 60-mm dishes using Lipofectamine 3000 transfection reagent (L3000075; Invitrogen) according to the manufacturers' instructions. The anti-Flag monoclonal antibody (F1804; Sigma), Akt3 antibody (4059 [Cell Signaling Technology] and ab152157 [Abcam]), phosphorylated Akt at serine residue 473 (9271; Cell Signaling Technology), an Akt1 specific antibody (2938; Cell Signaling Technology), or a peptide-specific ORF2 antibody was used for Western blotting, confocal microscopy, and IP studies.
Co-IP studies and Western blot analysis.
For coimmunoprecipitation (co-IP) studies, Neuro-2A cells grown in 60-mm dishes were transfected with the designated plasmids using Lipofectamine 3000 transfection reagent (L3000075; Invitrogen). At 48 h after transfection, cells were lysed with 1 ml of radioimmunoprecipitation assay (RIPA) buffer (1× PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail (Roche). Cell lysate was clarified by centrifugation at 13,000 rpm for 10 min. The clarified supernatant (approximately 400 μg protein) was incubated with the anti-FLAG M2 affinity gel with gentle rotation using a roller shaker for 2 h at 4°C. The anti-FLAG M2 affinity gel (A2220; Sigma) was prepared according to the manufacturer's specifications. To perform IP with Akt3 or Akt1 antibody, the clarified supernatant (approximately 400 μg protein) was incubated with anti-Akt1 or anti-Akt3 antibody for 2 h at 4°C with rotation and then incubated with Dynabeads (10001D; Life Technologies). After washing three times with 0.5 ml of PBS, beads were boiled in SDS loading buffer and Western blotting performed to detect the designated proteins.
For Western blotting studies, cells were collected, washed once with PBS, and then lysed in RIPA buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) with protease and phosphatase inhibitors (Thermo-Scientific). The respective samples were boiled in Laemmli sample buffer for 5 min, and all samples were separated on an 8% or 10% SDS-polyacrylamide gel. Immunodetection of the respective proteins was performed using the antibodies described above.
Immunofluorescence assay.
Neuro-2A cells seeded into 2-well chamber slides (Nunc, Inc., IL) were transfected with the designated plasmids using Lipofectamine 3000 transfection reagent (L3000075; Invitrogen). At 48 h after transfection, cells were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 10 min at room temperature and permeabilized with 0.25% Triton X-100 in PBS (pH 7.4) for 10 min at room temperature, blocked with 1% bovine serum albumin (BSA) in PBST (PBS plus 0.1% Tween 20) for 30 min, and incubated with anti-Akt3 antibody (1:500 dilution; ab152157; Abcam) or anti-Flag antibody (F1804; Sigma) in 1% BSA in PBST for 12 h. After three washes, cells were incubated with Alexa Fluor 488 goat anti-rabbit IgG (H+L) (1:500 dilution; A-11008; Invitrogen) or Alexa Fluor 633 goat anti-mouse IgG (H+L) (1:500 dilution; A-21050; Invitrogen) for 1 h in the dark. After three washes, DAPI (4′,6-diamidino-2-phenylindole) staining was performed to visualize the nucleus. Coverslips were added to the slides by using Gel Mount aqueous mounting medium (Electron Microscopy Sciences). Images were obtained by confocal microscopy (Leica).
ACKNOWLEDGMENTS
C.J. was supported by grants from the USDA-NIFA Competitive Grants Program (13-01041 and 16-09370), funds derived from the Sitlington Endowment, and funds from the Oklahoma Center for Respiratory and Infectious Diseases (National Institutes of Health Centers for Biomedical Research Excellence grant P20GM103648). A.W. was supported by funds from the Agricultural Research Service (CRIS 3040-32000-031-00D). L.Z. was partially supported by the China Scholarship Council, Chinese National Science Foundation Grants (no. 31472172), and the National Key Research Program (no. 2016YFD0500704).
The use of product and company names is necessary to accurately report the methods and results; however, the USDA neither guarantees nor warrants the standard of the products. The use of names by the USDA implies no approval of the product to the exclusion of others that may also be suitable. The USDA is an equal opportunity provider and employer.
REFERENCES
- 1.Hodgson PD, Aich P, Manuja A, Hokamp K, Roche FM, Brinkman FSL, Potter A, Babiuk LA, Griebel PJ. 2005. Effect of stress on viral-bacterial synergy in bovine respiratory disease: novel mechanisms to regulate inflammation. Comp Funct Genomics 6:244–250. doi: 10.1002/cfg.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones C, Chowdhury S. 2010. Bovine herpesvirus type 1 (BHV-1) is an important cofactor in the bovine respiratory disease complex. Vet Clin North Am Food Anim Pract Bovine Resp Dis 26:303–321. doi: 10.1016/j.cvfa.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Rice JA, Carrasco-Medina L, Hodgins DC, Shewen PE. 2008. Mannheimia haemolytica and bovine respiratory disease. Anim Health Res Rev 8:117–128. doi: 10.1017/S1466252307001375. [DOI] [PubMed] [Google Scholar]
- 4.Srikumaran S, Ambagela A, Kelling CL. 2007. Immune evasion by pathogens of bovine respiratory disease complex. Anim Health Res Rev 8:215–229. doi: 10.1017/S1466252307001326. [DOI] [PubMed] [Google Scholar]
- 5.Neibergs HL, Seabury CM, Wojtowicz AJ, Wang Z, Scraggs E, Kiser JN, Neupane M, Womack JE, Van Eenennaam A, Hagevortm GR, Lehenbauer TW, Aly S, Davis J, Taylor JF, Bovine Respiratory Disease Complex Coordinated Agricultural Research Team. 2014. Susceptibility loci revealed for bovine respiratory disease complex in pre-weaned Holstein calves. BMC Genomics 15:1164. doi: 10.1186/1471-2164-15-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones C. 2003. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin Microbiol Rev 16:79–95. doi: 10.1128/CMR.16.1.79-95.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones C. 2013. Bovine herpes virus 1 (BHV-1) and herpes simplex virus type 1 (HSV-1) promote survival of latently infected sensory neurons, in part by inhibiting apoptosis. J Cell Death 6:1–16. doi: 10.4137/JCD.S10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones C, Geiser V, Henderson G, Jiang Y, Meyer F, Perez S, Zhang Y. 2006. Functional analysis of bovine herpesvirus 1 (BHV-1) genes expressed during latency. Vet Microbiol 113:199–210. doi: 10.1016/j.vetmic.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Jones C, Frizzo da Silva L, Sinani D. 2011. Regulation of the latency-reactivation cycle by products encoded by the bovine herpesvirus 1 (BHV-1) latency-related gene. J Neurovirol 17:535–545. doi: 10.1007/s13365-011-0060-3. [DOI] [PubMed] [Google Scholar]
- 10.Inman M, Lovato L, Doster A, Jones C. 2001. A mutation in the latency-related gene of bovine herpesvirus 1 leads to impaired ocular shedding in acutely infected calves. J Virol 75:8507–8515. doi: 10.1128/JVI.75.18.8507-8515.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inman M, Lovato L, Doster A, Jones C. 2002. A mutation in the latency-related gene of bovine herpesvirus 1 disrupts the latency reactivation cycle in calves. J Virol 76:6771–6779. doi: 10.1128/JVI.76.13.6771-6779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinani D, Frizzo da Silva L, Jones C. 2013. A bovine herpesvirus 1 protein expressed in latently infected neurons (ORF2) promotes neurite sprouting in the presence of activated Notch1 or Notch3. J Virol 87:1183–1192. doi: 10.1128/JVI.02783-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinani D, Jones C. 2011. Localization of sequences in a protein encoded by the latency related gene of bovine herpesvirus 1 (ORF2) that inhibits apoptosis and interferes with Notch1 mediated trans-activation of the bICP0 promoter. J Virol 85:12124–12133. doi: 10.1128/JVI.05478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen W, Jones C. 2008. Open reading frame 2, encoded by the latency-related gene of bovine herpesvirus 1, has antiapoptotic activity in transiently transfected neuroblastoma cells. J Virol 82:10940–10945. doi: 10.1128/JVI.01289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pittayakhajonwut D, Sinani D, Jones C. 2013. A protein (ORF2) encoded by the latency related gene of bovine herpesvirus 1 interacts with DNA. J Virol 87:5943–5501. doi: 10.1128/JVI.00193-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinani D, Liu Y, Jones C. 2014. Analysis of a bovine herpesvirus 1 protein encoded by an alternatively spliced latency related (LR) RNA that is abundantly expressed in latently infected neurons. Virology 464-465:244–252. [DOI] [PubMed] [Google Scholar]
- 17.Workman A, Sinani D, Pittayakhajonwut D, Jones C. 2011. A protein (ORF2) encoded by the latency related gene of bovine herpesvirus 1 interacts with Notch1 and Notch3. J Virol 85:2536–2546. doi: 10.1128/JVI.01937-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaber T, Workman A, Jones C. 2010. Small noncoding RNAs encoded within the bovine herpesvirus 1 latency-related gene can reduce steady-state levels of infected cell protein 0 (bICP0). J Virol 84:6297–6307. doi: 10.1128/JVI.02639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones C. 1998. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv Virus Res 51:81–133. doi: 10.1016/S0065-3527(08)60784-8. [DOI] [PubMed] [Google Scholar]
- 20.Jones C. 2014. Reactivation from latency by alpha-herpesvirinae subfamily members: a stressful situation. Curr Topics Virol 12:99–118. [Google Scholar]
- 21.Liu Y, Hancock M, Workman A, Doster A, Jones C. 2016. Beta-catenin, a transcription factor activated by canonical Wnt signaling, is expressed in sensory neurons of calves latently infected with bovine herpesvirus 1. J Virol 90:3148–3159. doi: 10.1128/JVI.02971-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Workman A, Eudy J, Smith L, Frizzo da Silva L, Sinani D, Bricker H, Cook E, Doster A, Jones C. 2012. Cellular transcription factors induced in trigeminal ganglia during dexamethasone-induced reactivation from latency stimulate bovine herpesvirus 1 productive infection and certain viral promoters. J Virol 86:2459–2473. doi: 10.1128/JVI.06143-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu L, Workman A, Jones C. 2017. A potential role for a beta-catenin coactivator (high mobility group AT-hook 1 protein) during the latency-reactivation cycle of bovine herpesvirus 1. J Virol 91:e02132-. doi: 10.1128/JVI.02132-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matrisciano F, Buscetti CL, Bucci D, Orlando R, Caruso A, Molinaro G, Cappuccion I, Riozzi B, Gradini R, Motolese M, Caraci F, Copani A, Scaccianoce S, Melchiorri D, Bruno V, Battaglia G, Nicoletti F. 2011. Induction of the Wnt antagonist Dickkopf-1 is involved in stress-induced hippocampal damage. PLoS One 6:e16447. doi: 10.1371/journal.pone.0016447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moors M, Bose R, Johansson-Haque K, Edoff K, Okret S, Ceccatelli S. 2012. Dickkopf mediates glucocorticoid-induced changes in human neural progenitor cell proliferation and differentiation. Toxicol Sci 125:488–495. doi: 10.1093/toxsci/kfr304. [DOI] [PubMed] [Google Scholar]
- 26.Salinas PC. 2012. Wnt signaling in the vertebrate central nervous system: from axon guidance to synaptic function. Cold Spring Harb Perpect Biol 4:a008003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purro SA, Galli S, Salinas PC. 2014. Dysfunction of Wnt signaling and synaptic disassembly in neurodegenerative diseases. J Mol Cell Biol 6:75–80. doi: 10.1093/jmcb/mjt049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhardwaji D, Nager M, Camats J, David M, Benguira A, Adopazo Canti C, Herreros J. 2013. Chemokines induce axon outgrowth downstream of hepatocyte growth factor and TCF/beta-catenin signaling. Front Cell Neurosci 7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murase S, Mosser E, Schuman EM. 2002. Depolarization drives beta-catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 35:91–105. doi: 10.1016/S0896-6273(02)00764-X. [DOI] [PubMed] [Google Scholar]
- 30.Bamji SX, Rico B, Kimes N, Reichardt LF. 2006. BDNF mobilizes synaptic vesicles and enhances synaptic vesicles and enhances synapse formation by disrupting cadherin-beta-catenin interactions. J Cell Biol 174:289–299. doi: 10.1083/jcb.200601087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T, Liu X, Wang H, Moon RT, Malbon CC. 1999. Activation of a rat frizzled-1 promotes Wnt signaling and differentiation of mouse F9 teratocarcinoma cells via pathways that require Galpha (q) and Galpha (o) function. J Biol Chem 274:33539–33544. doi: 10.1074/jbc.274.47.33539. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Robbins JS, Kimmel AR. 2005. Rapid, Wnt-induced changes in GSK3beta association that regulate beta-catenin stabilization are mediated by Galpha proteins. Curr Biol 15:1989–1997. doi: 10.1016/j.cub.2005.10.050. [DOI] [PubMed] [Google Scholar]
- 33.Gori F, Lerner U, Ohlsson C, Baron R. 2015. A new WNT on the bone: WNT16, cortical bone thickness, porosity, and fractures. Bonekey Rep 4:669. doi: 10.1038/bonekey.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazieres J, You L, He B, Xu Z, Lee AY, Mikami I, McCormick F, Jablons DM. 2005. Inhibition of Wnt 16 in human acute lymphoblastoid leukemia cells containing the t(1;19) translocation induces apoptosis. Oncogene 11:5396–5400. doi: 10.1038/sj.onc.1208568. [DOI] [PubMed] [Google Scholar]
- 35.Silverio K, Davidson KC, James RG, Adams AM, Foster BL, Nociti FH Jr, Somermam MJ, Moon RT. 2012. Wnt/beta-catenin pathway regulates BMP2-mediated differentiation of dental follicle cells. J Periodontal Res 47:309–319. doi: 10.1111/j.1600-0765.2011.01433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ille F, Atanasoski S, Falk S, Ittner LM, Marki D, Buchmann-Moller S, Wurdak H, Suter U, Taketo MM, Sumner L. 2007. Wnt/BMP signal integration regulates the balance between proliferation and differentiation of neuroepithelial cells in the dorsal spinal cord. Dev Biol 304:394–408. doi: 10.1016/j.ydbio.2006.12.045. [DOI] [PubMed] [Google Scholar]
- 37.Guha U, Gomes W, Samantra J, Guptam M, Rice F, Kessler J. 2004. Target-derived BMP signaling limits sensory neuron number and the extent of peripheral innervation in vivo. Development 131:1175–1186. doi: 10.1242/dev.01013. [DOI] [PubMed] [Google Scholar]
- 38.Hodge L, Klassen M, Han B, Yiu G, Hurrel J, Howell A, Rosseau G, Lemaigre F, Tressier-Lavigne M, Wang F. 2007. Retrograde BMP signaling regulates trigeminal sensory neuron identitities and the formation of precise face maps. Neuron 55:572–586. doi: 10.1016/j.neuron.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Farkas L, Jasai J, Unsicker K, Krieglstein K. 1999. Characterization of bone morphogenetic protein family members as neurotropic factors for cultured sensory neurons. Neuroscience 92:227–235. doi: 10.1016/S0306-4522(98)00735-0. [DOI] [PubMed] [Google Scholar]
- 40.Osorio C, Chacon PJ, Kisiswa L, White M, Wyatt S, Rodriguez-Tebar A, Davies AM. 2013. Growth differentiation factor 5 is a key physiological regulator of dendrite growth during development. Development 140:4751–4762. doi: 10.1242/dev.101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dihlmann S, Kloor M, Fallsehr C, von Knebler Doeberitz M. 2005. Regulation of AKT1 expression by beta-catenin/Tcf/Lef signaling in colorectal cancer cells. Carninogenesis 26:1503–1512. doi: 10.1093/carcin/bgi120. [DOI] [PubMed] [Google Scholar]
- 42.Fang D, Hawke D, Zhang Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. 2007. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282:11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fukumoto S, Hsieh C-M, Maemura K, Layne MD, Yet S-F, Lee K-H, Matsui T, Rosenzweig A, Taylor WG, Rubin JS, Perrella MA, Lee M-E. 2001. Akt participation in the Wnt signaling pathway through dishevelled. J Biol Chem 276:17479–17483. doi: 10.1074/jbc.C000880200. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Shemezis JR, McQuinn ER, Wang J, Sverdlov M, Chenn A. 2013. AKT activation by N-cadherin regulates beta-catenin signaling and neuronal differentiation during cortical development. Neural Dev 8:7. doi: 10.1186/1749-8104-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaler P, Godasi BN, Augenlicht L, Klampfer L. 2009. The NF-kB/Akt-dependent induction of Wnt signaling in colon cancer cells by macrophages and IL-1b. Cancer Microenviron 2:69–80. doi: 10.1007/s12307-009-0030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langhammer T-S, Roolf C, Krohn S, Kretzchmar C, Huebner R, Rolfs A, Freund M, Junghass C. 2013. PI3/K/Akt signalling interacts with Wnt/Beta-catenin signaling but does not induce an accumulation of beta-catenin in the nucleus of acute lymphoblastic leukemia cell lines. Blood 122:4886. [Google Scholar]
- 47.Kim S-E, Lee W-J, Choi K-Y. 2007. The PI3 kinase-Akt pathway mediates Wnt3a-induced proliferation. Cell Signal 19:511–518. doi: 10.1016/j.cellsig.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Franke TF, Kaplan DR, Cantley LC. 1997. PI3K: downstream AKTion blocks apoptosis. Cell 88:435–437. doi: 10.1016/S0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 49.Manning B, Toker A. 2017. AKT/PKB signaling: navigating the network. Cell 169:381–405. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanekura K, Hashimoto Y, Kita Y, Sasabe J, Aiso S, Nishimoto I, Matsuoka M. 2005. A Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggerred by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J Biol Chem 280:4532–4543. doi: 10.1074/jbc.M410508200. [DOI] [PubMed] [Google Scholar]
- 51.Diez H, Garrido JJ, Wandosell F. 2012. Specific roles of Akt iso forms in apoptosis and axon growth regulation in neurons. PLoS One 74:e32715. doi: 10.1371/journal.pone.0032715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie R, Cheng M, Li M, Xiong X, Daadi M, Sapolsky RM, Zhao H. 2013. Akt isoforms differentially protect against stroke-induced neuronal injury by regulating mTOR activities. J Cereb Blood Flow Metab 33:1875–1885. doi: 10.1038/jcbfm.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carpenter D, Hsiang C, Jiang X, Osorio N, BenMohamed L, Jones C, Wechsler SL. 2015. The herpes simplex virus type 1 (HSV-1) latency-associated transcript (LAT) protects cells against cold shock induced apoptosis by maintaining phosphorylation of protein kinase B (AKT). J Neurovirol 21:568–575. doi: 10.1007/s13365-015-0361-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li S, Carpenter D, Hsiang C, Wechsler SL, Jones C. 2010. The herpes simplex virus type 1 latency-associated transcript (LAT) locus inhibits apoptosis and promotes neurite sprouting in neuroblastoma cells following serum starvation by maintaining active AKT (protein kinase B). J Gen Virol 91:858–866. doi: 10.1099/vir.0.015719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of HSV-1 in neurons. PLoS Pathog 8:e1002540. doi: 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Camarena V, Kobayashi M, Kim JK, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schenck A, Goto-Silva L, Collinet C, Rhin M, Giner A, Haberman B, Brand M, Zerial M. 2008. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 133:486–497. doi: 10.1016/j.cell.2008.02.044. [DOI] [PubMed] [Google Scholar]
- 58.Majumdar D, Nebhan CA, Hu L, Anderson B, Webb DJ. 2011. An APPL1/Akt signaling complex regualtes dendritic spine and synapse formation in hippocampal neurons. Mol Cell Neurosci 46:633–644. doi: 10.1016/j.mcn.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tortelote G, Hernandez-Hernandez J, Quaresma AJC, Nickerson JA, Imbalzano AN, Rivera-Perez JA. 2013. Wnt3 function in the epiblast is required for the maintenance but not the initiation of gastrulation in mice. Dev Biol 374:164–173. doi: 10.1016/j.ydbio.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simonetti M, Agarwal N, Strosser S, Bali KK, Karaulanov E, Kamble R, Pospisilova B, Kurejova M, Birchmeier W, Niehrs C, Heppenstall P, Kunier R. 2014. Wnt-Fzd signaling sensitizes peripheral sensory neurons via distinct noncanonical pathways. Neuron 83:104–121. doi: 10.1016/j.neuron.2014.05.037. [DOI] [PubMed] [Google Scholar]
- 61.Skronska-Wasek W, Mutze K, Baarsma HA, Bracke KR, Alsafadi HN, Lehmann M, Costa R, Stornaiuolo M, Novellino E, Bruselle GG, Wagner DE, Yildrim AO, Konigshoff M. 2017. Reduced Frizzled receptor 4 expression prevents Wnt/beta-catenin driven alveolar lung repair in chronic obstructive pulmonary disease. Am J Resp Crit Care Med 196:172–185. doi: 10.1164/rccm.201605-0904OC. [DOI] [PubMed] [Google Scholar]
- 62.Fernandez A, Huggins IJ, Perna L, Brafman D, Lu D, Yao S, Gaasterland T, Carson DA, Willert K. 2014. The WNT receptor FZD7 is required for maintenance of the pluripotent state in human embryonic stem cells. Proc Natl Acad Sci U S A 111:1409–1414. doi: 10.1073/pnas.1323697111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin T. 2016. Current understanding on role of the Wnt Signaling pathway effector TCFL7 in glucose homeostasis. Endocrine Rev 37:254–277. doi: 10.1210/er.2015-1146. [DOI] [PubMed] [Google Scholar]
- 64.Kendziorra E, Ahlborn K, Spitzner M, Rave-Frank M, Emons G, Gaedke J, Kramer F, Wolff HA, Becker H, Beissbarth T, Ebner R, Ghadimi BM, Purop T, Ried T, Grade M. 2011. Silencing of the Wnt transcription factor TCF4 sensitizes colorectal cancer cells to (chemo-)radiotherapy. Carcinogenesis 32:1824–1831. doi: 10.1093/carcin/bgr222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharma N, Jadhav SW, Bapat SA. 2010. CREBBP re-arrangements affect protein function and lead to aberrant neuronal differentiation. Differentiation 79:218–231. doi: 10.1016/j.diff.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 66.Hsieh J, Kodjabachian L, Rebbert ML, Rattner A, Smallwood PM, Samos CH, Nusse R, Dawid IB, Nathans J. 1999. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature 398:431–436. doi: 10.1038/18899. [DOI] [PubMed] [Google Scholar]
- 67.Kawanon Y, Kypta R. 2003. Secreted antagonists of the Wnt signaling pathway. J Cell Sci 116:2677–2634. doi: 10.1242/jcs.00470. [DOI] [PubMed] [Google Scholar]
- 68.Warrier S, Balu SK, Kumar AP, Willward M, Dharmarajan A. 2013. Wnt antagonist, secreted frizzled-related protein 4 (sFRP4), increases chemotherapeutic response of glioma stem-like cells. Oncol Res 21:93–102. doi: 10.3727/096504013X13786659070154. [DOI] [PubMed] [Google Scholar]
- 69.Li Y, Rankin SA, Sinner D, Kenny AP, Kenny PA, Zorn AM. 2008. SFRP5 coordinates foregut specification and morphogenesis by antagonizing both canonical and noncanonical Wnt11 signaling. Genes Dev 22:3050–3063. doi: 10.1101/gad.1687308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kormish J, Sinner D, Zorn AM. 2010. Interactions between SOX factors and Wnt/beta-catenin signaling in development and disease. Dev Dyn 239:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cappuccio I, Calderone A, Busceti CL, Biagioni F, Pontarelli F, Bruno V, Storto M, Terstappen GT, Gaviraghi G, Fornai F, Battaglia G, Melchiorri D, Zukin S, Nicoletti F, Caricasole A. 2005. Induction of dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J Neurosci 25:2647–2657. doi: 10.1523/JNEUROSCI.5230-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, Storto M, Gaviraghi G, Terstappen GC, Nicoletti F. 2004. Induction of dickkopf 1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer's brain. J Neurosci 24:6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Niehrs C. 2006. Function and biological roles of the dickkopf family of Wnt modulators. Oncogene 25:7469–7481. doi: 10.1038/sj.onc.1210054. [DOI] [PubMed] [Google Scholar]
- 74.Cruciat CM, Niehrs C. 2013. Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harb Perpect Biol 5:a015081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nusse R, Clever H. 2017. Wnt/beta-catenin signaling, diseases, and emerging therapeutic modalities. Cell 169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 76.Zorn AM. 2001. Wnt signaling: antagonistic Dicckopf. Curr Biol 11:R592–R595. doi: 10.1016/S0960-9822(01)00360-8. [DOI] [PubMed] [Google Scholar]
- 77.Jiang Y, Inman M, Zhang Y, Posadas NA, Jones C. 2004. A mutation in the latency related gene of bovine herpesvirus 1 (BHV-1) inhibits protein expression of a protein from open reading frame 2 (ORF-2) and an adjacent reading frame during productive infection. J Virol 78:3184–3189. doi: 10.1128/JVI.78.6.3184-3189.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang Y, Hossain A, Winkler MT, Holt T, Doster A, Jones C. 1998. A protein encoded by the latency-related gene of bovine herpesvirus 1 is expressed in trigeminal ganglionic neurons of latently infected cattle and interacts with cyclin-dependent kinase 2 during productive infection. J Virol 72:8133–8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pearce LR, Komander D, Alessi DR. 2010. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 80.Tremblay R, Sikorska M, Sandhu JK, Lanthier P, Ribecco-Lutkiewicz M, Bani-Yaghoub M. 2010. Differentiation of mouse Neuro-2A cells into dopamine neurons. J Neuroscience Methods 186:60–67. doi: 10.1016/j.jneumeth.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 81.Manning BD, Cantley LC. 2007. AKT/PKB: navigating down stream. Cell 129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Scheid MP, Woodgett JR. 2003. Unraveling the activation of protein kinase B/Akt. FEBS Lett 546:108–112. doi: 10.1016/S0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 83.Martelli A, Tabellini G, Bressanin D, Ognibene A, Goto K, Coco L, Evangelisti C. 2012. The emerging multiple roles of nuclear Akt. Biochim Biophys Acta 1823:2168–2178. doi: 10.1016/j.bbamcr.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 84.Gao G, Li J-T, Kong L, Tao L, Wei L. 2009. Human herpesviruses miRNAs statistically preferentially target host genes involved in cell signaling and adhesion/junction pathways. Cell Res 19:665–667. doi: 10.1038/cr.2009.45. [DOI] [PubMed] [Google Scholar]
- 85.Winkler MT, Doster A, Sur JH, Jones C. 2002. Analysis of bovine trigeminal ganglia following infection with bovine herpesvirus 1. Vet Microbiol 86:139–155. doi: 10.1016/S0378-1135(01)00498-9. [DOI] [PubMed] [Google Scholar]
- 86.Lambert C, Cisternas P, Inestrosa NC. 2016. Role of Wnt signaling in central nervous system injury. Mol Neurobiol 53:2297–2311. [DOI] [PubMed] [Google Scholar]
- 87.Rosso SB, Inestrosa NC. 2013. Wnt signaling in neuronal maturation and synaptogenesis. Cell Neurosci 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lemmon MA, Schlessinger J. 2010. Cell signaling by receptor tyrosine kinases. Cell 141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tian Q, Feetham MC, Tao WA, He XC, Li L, Aebersold R, Hood L. 2004. Proteomic analysis identifies that 14-3-3zeta interacts with beta-catenin and facilitates its activation by Akt. Proc Natl Acad Sci U S A 101:15370–15375. doi: 10.1073/pnas.0406499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee G, Goretsky T, Managlia E, Dirisina R, Singh AP, Brown JB, May R, Yang G-Y, Ragheb JW, Evers BM, Weber CR, Turner JR, He XC, Katzman RB, Li L, Barrett TB. 2010. Phosphoinositide 3-kinase signaling mediates beta-catenin activation in intestinal epithelial stem and progenitor cells in colitis. Gastroenterology 139:869–881. doi: 10.1053/j.gastro.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao S, Fu J, Liu X, Wang T, Zhang J, Zhao Y. 2012. Activation of Akt/GSK-3beta/beta-catenin signaling pathway is involved in survival of neurons after traumatic brain injury in rats. Neurol Res 34:400–407. doi: 10.1179/1743132812Y.0000000025. [DOI] [PubMed] [Google Scholar]
- 92.Ahn J, Rong R, Liu XS, Ye KQ. 2004. PIKE/nuclear PI 3-kinase signaling mediates the antiapoptotic functions of NGF in the nucleus. EMBO J 23:3995–4006. doi: 10.1038/sj.emboj.7600392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ackermann M, Peterhans E, Wyler R. 1982. DNA of bovine herpesvirus type 1 in the trigeminal ganglia of latently infected calves. Am J Vet Res 43:36–40. [PubMed] [Google Scholar]
- 94.Brown GA, Field HJ. 1990. Experimental reactivation of bovine herpesvirus 1 (BHV-1) by means of corticosteroids in an intranasal rabbit model. Arch Virol 112:81–101. doi: 10.1007/BF01348987. [DOI] [PubMed] [Google Scholar]
- 95.Hage JJ, Glas RD, Westra HH, Maris-Veldhuis MA, Van Oirschot JT, Rijsewijk FA. 1998. Reactivation of latent bovine herpesvirus 1 in cattle seronegative to glycoproteins gB and gE. Vet Microbiol 60:87–98. doi: 10.1016/S0378-1135(97)00152-1. [DOI] [PubMed] [Google Scholar]
- 96.Homan EJ, Easterday BC. 1983. Experimental latent and recrudescent bovine herpesvirus-1 infections in calves. Am J Vet Res 44:309–313. [PubMed] [Google Scholar]
- 97.Jones C, Newby TJ, Holt T, Doster A, Stone M, Ciacci-Zanella J, Webster CJ, Jackwood JM. 2000. Analysis of latency in cattle after inoculation with a temperature sensitive mutant of bovine herpesvirus 1 (RLB106). Vaccine 18:3185–3195. doi: 10.1016/S0264-410X(00)00106-7. [DOI] [PubMed] [Google Scholar]
- 98.Rock D, Lokensgard J, Lewis T, Kutish G. 1992. Characterization of dexamethasone-induced reactivation of latent bovine herpesvirus 1. J Virol 66:2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sheffy BE, Davies DH. 1972. Reactivation of a bovine herpesvirus after corticosteroid treatment. Proc Soc Exp Biol Med 140:974–976. doi: 10.3181/00379727-140-36592. [DOI] [PubMed] [Google Scholar]
- 100.Workman A, Perez S, Doster A, Jones C. 2009. Dexamethasone treatment of calves latently infected with bovine herpesvirus 1 (BHV-1) leads to activation of the bICP0 early promoter, in part by the cellular transcription factor C/EBP-alpha. J Virol 83:8800–8809. doi: 10.1128/JVI.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mastroiacovo F, Busceti CL, Biagioni F, Moyanova SG, Meisler MH, Battaglia G, Caricasole A, Bruno V, Nicoletti F. 2009. Induction of the Wnt antagonist, Dickopf-1, contributes to the development of neuronal death in models of brain focal ischemia. J Cereb Blood Flow Metab 29:264–276. doi: 10.1038/jcbfm.2008.111. [DOI] [PubMed] [Google Scholar]
- 102.Rossol-Voth R, Rossol S, Schutt KH, Corridori S, de Cian W, Falke D. 1991. In vivo protective effect of tumour necrosis factor alpha against experimental infection with herpes simplex virus type 1. J Gen Virol 72:143–147. doi: 10.1099/0022-1317-72-1-143. [DOI] [PubMed] [Google Scholar]
- 103.Berwick DC, Harvey K. 2012. The importance of Wnt signaling for neurodegeneration in Parkinson's disease. Biochem Soc Trans 40:1123–1128. doi: 10.1042/BST20120122. [DOI] [PubMed] [Google Scholar]
- 104.Ali-Harthi. 2012. Wnt/beta-catenin and its diverse physiological cell signaling pathways in neurodegenerative and neuro-psychiatric disorders. J Neuroimmune Pharm 7:725–730. doi: 10.1007/s11481-012-9412-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Magana R, Giles N, Adcroft K, Keeney D, Wood F, Fear M, Dharmarajan A. 2008. Secreted frizzled related protein-4 (sFRP4) promotes epidermal differentiation and apoptosis. Biochem Biophys Res Commun 377:606–611. doi: 10.1016/j.bbrc.2008.10.050. [DOI] [PubMed] [Google Scholar]
- 106.Bhuvanalakshmi G, Arfuso F, Millward M, Dharmarajan A, Warrier S. 2015. Secreted frizzled-related protein 4 inhibits glioma stem-like cells by reversing epithelial to mesenchymal transition, inducing apoptosis and decreasing cancer stem cell properties. PLoS One 10:e0127517. doi: 10.1371/journal.pone.0127517. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 107.Lacher M, Siegenthaler A, Jager R, Yan X, Hett S, Xuan L, Saurer S, Lareu RR, Dharmarajan AM, Friis R. 2003. Role of DDC-4/sFRP-4, a secreted frizzled-related protein, as the onset of apoptosis in mammary involution. Cell Death Differ 10:528–538. doi: 10.1038/sj.cdd.4401197. [DOI] [PubMed] [Google Scholar]
- 108.Prasad A, Remick J, Zeichner SL. 2013. Activation of human herpesvirus replication by apoptosis. J Virol 87:10641–10650. doi: 10.1128/JVI.01178-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Du T, Zhou G, Roizman B. 2012. Induction of apoptosis accelerates reactivation from latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc Natl Acad Sci U S A 109:14616–14621. doi: 10.1073/pnas.1212661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kook I, Henley C, Meyer F, Hoffmann F, Jones C. 2015. Bovine herpesvirus 1 productive infection and the immediate early transcription unit 1 are stimulated by the synthetic corticosteroid dexamethasone. Virology 484:377–385. doi: 10.1016/j.virol.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 111.El-Mayet FS, Sawant L, Thungunutla P, Jones C. 2017. Combinatorial effects of the glucocorticoid receptor and Krüppel-like transcription factor 15 on bovine herpesvirus 1 transcription and productive infection. J Virol 91:e00904-17. doi: 10.1128/JVI.00904-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oakley RH, Cidlowski JA. 2013. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rhen T, Cidlowski JA. 2005. Antiinflammatory action of glucocorticoids–new mechanisms of old drugs. N Engl J Med 353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 114.Smoak KL, Cidlowski JA. 2004. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Aging Dev 125:697–706. doi: 10.1016/j.mad.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 115.Kook I, Doster A, Jones C. 2015. Bovine herpesvirus 1 regulatory proteins are detected in trigeminal ganglionic neurons during the early stages of stress-induced escape from latency. J Neurovirol 21:585–591. doi: 10.1007/s13365-015-0339-x. [DOI] [PubMed] [Google Scholar]
- 116.Lovato L, Inman M, Henderson G, Doster A, Jones C. 2003. Infection of cattle with a bovine herpesvirus 1 (BHV-1) strain that contains a mutation in the latency related gene leads to increased apoptosis in trigeminal ganglia during the transition from acute infection to latency. J Virol 77:4848–4857. doi: 10.1128/JVI.77.8.4848-4857.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Perez S, Inman M, Doster A, Jones C. 2005. Latency-related gene encoded by bovine herpesvirus 1 promotes virus growth and reactivation from latency in tonsils of infected calves. J Clin Microbiol 43:393–401. doi: 10.1128/JCM.43.1.393-401.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Perez S, Lovato L, Zhou J, Doster A, Jones C. 2006. Comparison of inflammatory infiltrates in trigeminal ganglia of cattle infected with wild type BHV-1 versus a virus strain containing a mutation in the LR (latency-related) gene. J Neurovirol 12:392–397. doi: 10.1080/13550280600936459. [DOI] [PubMed] [Google Scholar]
- 119.Perez S, Meyer F, Saira K, Doster A, Jones C. 2008. Premature expression of the latency-related RNA encoded by bovine herpesvirus 1 correlates with higher levels of beta interferon RNA expression in productively infected cells. J Gen Virol 89:1338–1345. doi: 10.1099/vir.0.83481-0. [DOI] [PubMed] [Google Scholar]
- 120.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300. [Google Scholar]
- 124.Frizzo da Silva L, Kook I, Doster A, Jones C. 2013. Bovine herpesvirus 1 regulatory proteins, bICP0 and VP16, are readily detected in trigeminal ganglionic neurons expressing the glucocorticoid receptor during the early stages of reactivation from latency. J Virol 87:11214–11222. doi: 10.1128/JVI.01737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]