

ABSTRACT
Interphase phosphorylation of lamin-A,C depends dynamically on a cell's microenvironment, including the stiffness of extracellular matrix. However, phosphorylation dynamics is poorly understood for diseased forms such as progerin, a permanently farnesylated mutant of LMNA that accelerates aging of stiff and mechanically stressed tissues. Here, fine-excision alignment mass spectrometry (FEA-MS) is developed to quantify progerin and its phosphorylation levels in patient iPS cells differentiated to mesenchymal stem cells (MSCs). The stoichiometry of total A-type lamins (including progerin) versus B-type lamins measured for Progeria iPS-MSCs prove similar to that of normal MSCs, with total A-type lamins more abundant than B-type lamins. However, progerin behaves more like farnesylated B-type lamins in mechanically-induced segregation from nuclear blebs. Phosphorylation of progerin at multiple sites in iPS-MSCs cultured on rigid plastic is also lower than that of normal lamin-A and C. Reduction of nuclear tension upon i) cell rounding/detachment from plastic, ii) culture on soft gels, and iii) inhibition of actomyosin stress increases phosphorylation and degradation of lamin-C > lamin-A > progerin. Such mechano-sensitivity diminishes, however, with passage as progerin and DNA damage accumulate. Lastly, transcription-regulating retinoids exert equal effects on both diseased and normal A-type lamins, suggesting a differential mechano-responsiveness might best explain the stiff tissue defects in Progeria.
KEYWORDS: matrix, stiffness, lamin-A, progeria, HGPS, mechanobiology
Introduction
Hutchinson-Gilford Progeria Syndrome (‘HGPS’ or ‘Progeria’) is a premature aging disease that resembles normal aging in many key respects [1–3]. Shared defects include atherosclerosis, stiffening of skin, muscle attrition, weakening of bones, and fibrosis across many solid tissues [3]. However, tissues most severely affected by Progeria are those which are mechanically stressed and stiff, whereas soft tissues including brain, bone marrow, and blood appear unaffected [4] (Fig. 1A). This stiff versus soft dichotomy seems independent of lineage and developmental origin, and raises the question of whether a defective mechanical response contributes to the pathology of Progeria.
Figure 1.
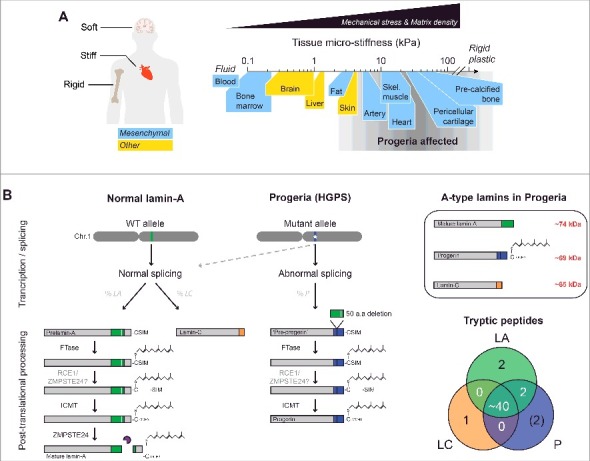
HGPS aging defects are most pronounced in mechanically stressed tissues. (A) HGPS disproportionately affects stiff and mechanically stressed tissues (e.g. skeletal/cardiac muscle) while soft tissues (e.g. brain, marrow) appear normal, regardless of lineage or developmental origin. (B) Post-translational processing of normal lamin-A/C and progerin. The truncated mutant, progerin, retains a C-terminus farnesyl group. Upper right box: A-type lamin isoforms differ in molecular weight (MW) by ∼5 kDa. Lower right: Venn diagram of unique and common A-type lamin peptides. The majority of the peptides are shared by all three isoforms, with the exception of isoform-specific peptides at the C-terminus. ‘FTase’ = Farnesyl transferase; ‘RCE1’ = Ras converting CAAX endopeptidase 1; ‘ZMPSTE24’ = Zinc metallopeptidase STE24; ‘ICMT’ = Isoprenylcysteine carboxyl methyltransferase.
Progeria is typically caused by a point mutation in one allele of LMNA that activates a cryptic splice site to produce ‘progerin’, a C-terminal mutant that lacks 50 amino acids [5,6] and thereby retains a farnesyl group that is cleaved off in normal lamin-A [7] (Fig. 1B). Farnesylation favors binding to the inner lipid leaflet of the nucleus [8] and, consistent with membrane viscosity impeding diffusion [9], the permanently farnesylated B-type lamins show very low molecular mobility (as GFP-fusions) [10,11] similar to prelamin-A and progerin. In contrast, mature lamin-A and its truncated spliceform, lamin-C, are both mobile and exchange dynamically between the lamina and the nucleoplasm (in ‘3D’) [10]. Movement along or within the lamina (in ‘∼2D’) is relatively hindered; however, interphase phosphorylation of lamin-A/C at multiple residues clearly enhances mobility in either direction/mode by promoting rapid disassembly of filaments and solubilization into the nucleoplasm [12]. In particular, phosphorylation at serines 22, 390, and 392 near the head and tail domains has been shown to exert dominant effects on nucleoplasmic localization. While the precise functions of phosphorylated, nucleoplasmic lamin-A/C during interphase are still unclear [13,14], phospho-solubilization promotes lamin-A/C interaction with several key regulatory factors (e.g. LAP2α [15] and significantly alters the mechanical properties of the nucleus [16]. Given the many structural and protective functions of the lamins at the nuclear periphery [17–21], regulation of mobility and assembly dynamics by such post-translational modifications (PTMs) suggests some mechanical relation to the stiff tissue defects seen in Progeria.
Contributions to disease from cell-extrinsic factors such as ‘tissue stiffness’ is consistent with surprising conclusions from mosaic mouse models22: mice with 50% of cells expressing farnesylated lamin-A in all tissues maintain a normal lifespan, while mice with 100% of cells expressing farnesylated lamin-A die within weeks of birth. Conventional cultures of these cells on rigid tissue culture plastic leads to premature senescence and/or apoptosis, as is also observed with related progeroid cells having low amounts of normal lamin-A/C23, but the in vitro phenotype is rescued by cultures on almost any type of extracellular matrix (ECM) [23,24], which is typically softer than plastic by many orders of magnitude. Furthermore, with cells depleted of lamin-A/C, migration through small rigid pores has shown that nuclear stress induces apoptosis [25]. Failure to dynamically remodel the nuclear envelope and protect the nucleus from mechanical stress might thus provide some explanation for why defects in HGPS patients are limited to stiff tissues.
Soft tissues (e.g. marrow) as well as stiff tissues (e.g. muscle) almost always have within a perivascular niche a population of mesenchymal stem cells (MSCs), which are key contributors to fibrosis [26]. Fibrosis is in turn a mechanosensitive process that affects MSC nuclei [27,28], and is a major hallmark of both normal and premature aging of solid tissues. Understanding MSC responses to microenvironmental properties can therefore provide fundamental insight into processes of relevance to many tissues and organs affected in disease or not. In standard cultures, MSCs (and closely related vascular smooth muscle cells [29]) that are differentiated from HGPS patient-derived iPS cells (HGPS iPS-MSCs) exhibit the highest levels of progerin, nuclear abnormalities, and DNA damage [30]. However, any effect of matrix stiffness or mechanical stress remains unknown.
Cytoskeletal tension on the nucleus suppresses interphase phosphorylation of normal A-type lamins [16,31], which otherwise promotes their solubilization into the nucleoplasm and subsequent degradation [16,32–34]. In particular, lamin-A/C phosphorylation is low in cells on rigid surfaces that lead to stress fibers (such as tissue culture plastic), but increases rapidly (<1 hr) upon enzymatic detachment which disrupts the cytoskeleton and leads to cell and nuclear rounding (as seen during mitosis) [35]. Soft ECM similarly causes cell/nuclear rounding and increases phosphorylation of lamin-A and C. Whether the presence of a C-terminal farnesyl group can affect mechanosensitive phospho-solubilization of A-type lamins is unclear, but de-farnesylation is reportedly required for phosphorylation at serine 2236. Here, we develop a new mass spectrometry (MS)-based method for quantitation of intact lamins and their phosphorylation states in HGPS iPS-MSCs that are exposed to different mechanical environments.
Results
Stoichiometries of lamins in HGPS-derived iPS-MSCs quantified by FEA-MS
Progerin is the product of one of two alleles and might naively be expected to compose half of all A-type lamin protein, but past immunoblots of human progeria cells or tissues show disproportionately less progerin compared to normal lamin-A/C [37,38]. Quantitative immunoblotting for protein levels is of course extremely powerful, but precise measurements of protein stoichiometry can be a particular challenge unless an antibody binds with equal affinity to each protein band. We therefore developed a label-free mass spectrometry (MS)-based method for simultaneous quantitation of progerin, the normal lamins, and their phosphorylation states in HGPS patient-derived iPS-mesenchymal stem cells (HGPS iPS-MSCs). Briefly, fine-excision alignment mass spectrometry (FEA-MS) exploits molecular weight differences (ΔMW) between A-type lamins (Fig. 1B, upper right box) by sectioning SDS-PAGE gels into narrow slices (<1 mm3) along the electrophoresis direction (Fig. 2A). A custom device with equally spaced blades allows for precise sectioning of the gel into seven or more slices per lane spanning the lamin MW range (60∼80 kDa) (Fig. 2A). Known amounts of synthetic phospho-peptides (each containing a phosphorylated serine residue: ‘pSer22’ and ‘pSer390’) and their non-phosphorylated forms (‘Ser22’ and ‘Ser390’) are injected into samples from adjacent replicates (Fig. 2A, upper right), which adds precision to MS peak alignment and calibrated quantitation (see Materials and Methods). FEA-MS sample lanes are flanked by replicate lanes for immunoblotting.
Figure 2.
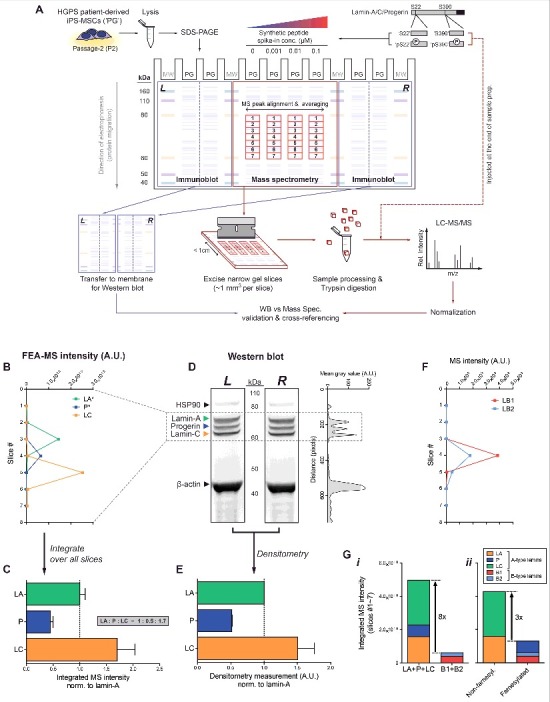
Quantification of lamin isoform stoichiometries by fine-excision alignment mass spectrometry (FEA-MS). (A) FEA-MS pipeline. HGPS iPS-MSC lysates (‘PG’) are injected into 4 middle SDS-PAGE lanes. Known concentrations of synthetic phospho-peptide standards (each containing a well-known serine residue: ‘pSer22’ and ‘pSer390’) and their non-phosphorylated counterparts (‘Ser22’ and ‘Ser390’) are injected into adjacent lanes to allow for (i) better detection by MS peak alignment, (ii) averaging over replicates, and (iii) calibrated quantitation of peptide/protein concentrations. An extended (>1h) SDS-PAGE run achieves sufficient separation of A-type lamin isoforms, allowing for excision of the gel into multiple narrow slices along the electrophoresis direction with a custom device of equally spaced parallel blades. FEA-MS sample lanes are flanked by 2 additional lanes on either side (left and right), which are used for parallel immunoblot analyses. ‘MW’ = molecular weight standards. (B) Intensity profiles of lamin-A (‘LA’), progerin (‘P’), and lamin-C (‘LC’) determined by FEA-MS and plotted vs slice #. (C) Summation of FEA-MS signal over all gel slices (#1-7) quantifies A-type lamin stoichiometries, LA : P : LC ∼ 1 : 0.5 : 1.7. (D) Aligned left & right (‘L’ & ‘R’) flank Western blots for anti-lamin-A/C show three distinct bands for LA, P, and LC. Right inset: line profile plot of immunoblot densitometry signal. (E) Quantification of LA : P : LC by densitometry is consistent with that by FEA-MS. (F) Line profile plot of normalized MS intensities of B-type lamins (‘B1’ & ‘B2’). (G) Stacked bar graph illustrating summed intensities of each isoform across all gel slices. (i) Total A-type lamins are >8-fold more abundant than B-type lamins, but (ii) the ratio of non-farnesylated : farnesylated lamins decreases to ∼3 with progerin expression.
Isoform-specific peptides (Fig. 1B, lower right Venn diagram) were used to generate separate intensity plots for lamin-A (‘LA’), progerin (‘P’), and lamin-C (‘LC’) in slices 2–6 (Fig. 2B & S1A). Signal-to-noise ratios calculated for isoform-specific peptides proved highest at the respective peaks (Fig. S1B). Summed MS intensities gave A-type lamin isoform stoichiometries of (LA : P : LC) = (1 : 0.5 : 1.7) in HGPS iPS-MSCs (Fig. 2C; Fig. S1C,D), in agreement with densitometry of Western blots (Fig. 2D,E). We showed previously that the iPS-MSCs studied here have a normal karyotype [39] with one allele expressing normal (LA + LC) and one expressing progerin (P), and so normal-to-diseased protein ratios of (2.7 : 0.5) likely suggest differences in protein or mRNA stability.
The total MS intensity of A-type lamins was also 8-fold greater than that of B-type lamins (lamin-B1, ‘LB1’, plus lamin-B2, ‘LB2’) (Fig. 2F & G-i, Fig. S1E), such that lamin-A:B ∼ 8:1. Normal primary bone-marrow derived human MSCs (hMSCs) have a similar lamin-A:B ∼ 10:131, which is consistent with iPS differentiation toward MSCs. On the other hand, expression of progerin in these HGPS iPS-MSCs effectively decreased the (non-farnseylated : farnesylated) lamin ratio to ∼3:1 (Fig. 2G-ii). Since lamin-A:B is a key determinant of nuclear response to mechanical stress [25,31], the latter stoichiometry raised questions of how progerin responds to various mechanobiological perturbations.
Progerin and lamin-B's are both depleted from mechanically-induced nuclear blebs
To assess whether C-terminal farnesylation causes progerin to respond differently to mechanical stress, HGPS iPS-MSCs were seeded onto transwell membranes for migration through 3 μm pores (Fig. 3A), which are typical of pores in stiff tissues [25]. Despite the well-documented reduction in motility and disrupted nucleus-cytoskeleton connections in Progeria cells [40–42], the patient-derived iPS-MSCs were fully capable of migrating through narrow constrictions. The cells that migrated to the bottom of the transwells (∼40% of cells in 48h) further exhibited nuclear blebs that are typical of mechanically-induced nuclear envelope rupture [25,39] (Fig. 3B). With these cells, however, progerin could not be easily distinguished from normal LA and LC due to antibody cross-reactivity. Human A549 lung carcinoma cells with a low LMNA background (via shLMNA) were therefore transfected with GFP-LA or GFP-progerin and seeded onto the same transwell membranes to assess any differences in their migration-induced response. LMNA knockdown in A549s resulted in a higher number of cells that migrated compared to wild type (‘WT’) (Fig. S2A), consistent with past studies demonstrating that a soft nucleus facilitates migration through narrow pores [25]. Migrating cells as (fraction of cells migrating to bottom) was rescued to baseline upon transfection with WT GFP-LA, but was further reduced with expression of GFP-progerin (Fig. S2A).
Figure 3.
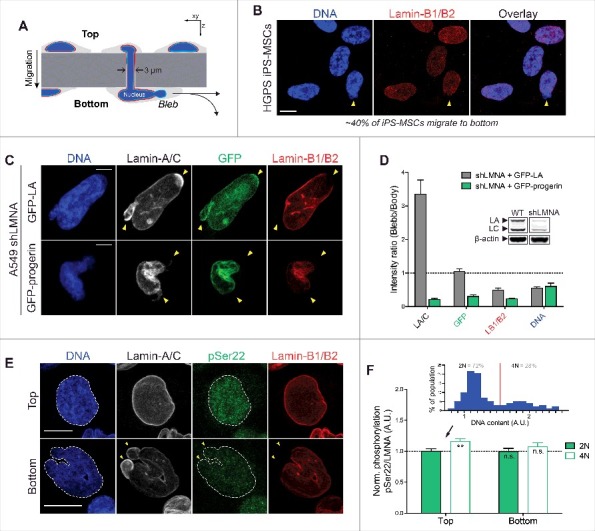
Farnesylated progerin, as with lamin-B1/B2, is depleted from nuclear blebs following constricted migration through narrow pores. (A) Cartoon illustrating transwell migration of cells. Pore diameter = 3 μm; Polystyrene membrane thickness ∼10 μm. (B) Confocal images of HGPS iPS-MSCs that migrate to the bottom of narrow 3 μm pores (∼40% of seeded cells migrate to bottom in 48h) show typical nuclear blebs with lamin-B1/2 depletion. Scale bar = 10 μm. (C) Representative images of nuclei exhibiting characteristic blebs following constricted migration. Lamin-A/C is enriched in sites of nuclear blebs (as seen by immunofluorescence and GFP signal), but GFP-progerin is depleted from blebs, as are the farnesylated lamins-B1 and B2. Scale bar = 5μm. (D) Quantitation of nuclear bleb/body fluorescence intensity ratio. Inset: immunoblot of WT and shLMNA cells showing residual LA and LC. (E) Confocal images of WT A549 cells at the top & bottom of the transwell membrane. Phosphorylated lamin-A/C (‘pSer22’) is seen in the nucleoplasm of interphase nuclei. Cells that migrate to the bottom show nuclear blebs with lamin-A/C enrichment and lamin-B1/B2 depletion (yellow arrowheads). Scale bar = 10 μm. (F) While lamin-A/C is phosphorylated ∼10-15% higher in ‘4N’ vs ‘2N’ cells (inset), normalized phosphorylation (as ‘pSer22/LMNA’) does not change significantly before and after migration.
As with the patient-derived iPS-MSCs, the GFP-LA expressing A549s that managed to migrate to the bottom of the transwells exhibited mechanically-induced nuclear blebs (Fig. 3C). Blebs were again depleted of B-type lamins but were enriched in lamin-A/C immunofluorescence signal, which accounts for endogenous LA & C as well as transfected GFP-LA (Fig. 3D). No such enrichment was seen however with GFP fluorescence (accounting for LA only), suggesting that the ∼3-fold enrichment in anti-lamin-A/C might be a result of preferential accumulation of endogenous LC (Fig. 3D, immunoblot inset). Nuclear blebs in the shLMNA + GFP-progerin cells showed progerin depletion as well as a dominant negative effect on LA and LC (Fig. 3C,D). Progerin's absence from blebs suggested that it responds more similarly to lamin-B's than LA or LC upon nuclear constriction and/or rupture. Permanent C-terminus farnesylation is one possible explanation, but the mobility and localization of A-type lamins can also be regulated dynamically by interphase phosphorylation, which promotes solubilization of filaments from the lamina into the nucleoplasm [12]. Confocal immunofluorescence with an anti-phospho-LA/C-Ser22 antibody (‘pSer22’) indeed revealed significant nucleoplasmic signal, but the localization and overall levels of phosphorylated A-type lamins were unaffected by constricted migration (Fig. 3E,F) regardless of DNA content (‘2N’ vs ‘4N’, Fig. 3F upper inset). Migration resulted in a decrease in % 4N cells (Fig. S2B), consistent with recent reports showing pore migration suppresses late cell cycle (G2) [43], and the same cells showed slightly higher pSer22 as expected [44]. Due to the cross-reactivity of the pSer22 antibody, however, any differences in phosphorylation responses of progerin relative to normal LA/C remained unclear and were best assessed by FEA-MS and immunoblots.
Basal phosphorylation of progerin and lamin-A is 2-fold lower than that of lamin-C
To clarify any isoform-specific differences in interphase phosphorylation of A-type lamins including endogenous progerin, we examined HGPS iPS-MSCs on rigid culture plastic. The iPS-MSCs were first analyzed by immunofluorescence, which again revealed characteristic nuclear blebs that were enriched in A-type lamins and depleted of B-type lamins (Fig. 4A), consistent with images after pore migration of both iPS Progeria cells and normal A549 cells (Fig. 3). Phosphorylated A-type lamins were again found mostly in the nucleoplasm (Fig. 4A-i) and appeared capable of diffusing into lamin-B-depleted nuclear blebs (Fig. 4A-ii). Nucleoplasmic signal in the vast majority of the population was clearly attributable to interphase phosphorylation, as very few cells (<1%) were mitotic with the expected cytoplasmic distributions of lamin-A/C and pSer22 (Fig. 4B).
Figure 4.
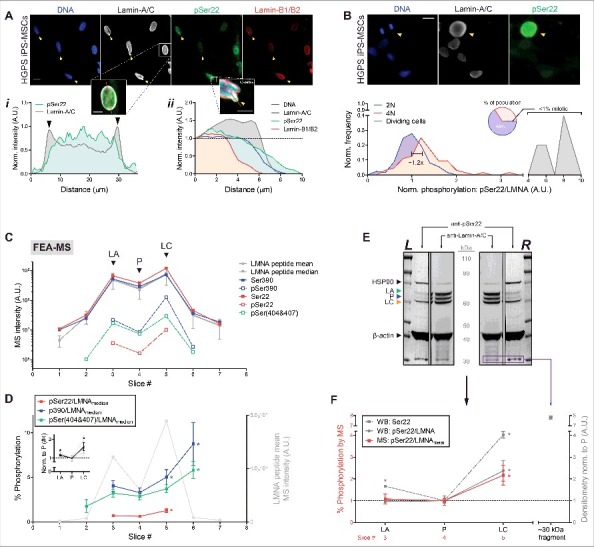
Progerin and lamin-A phosphorylation in HGPS iPS-MSCs is 2-fold lower than that of lamin-C. (A) Immunofluorescence images of HGPS iPS-MSCs on rigid culture plastic showing lamin-A/C-enriched, lamin-B-depleted nuclear blebs (yellow arrowheads). Scale bar = 10 μm. (i) Intensity profile (along red line) of pSer22 primarily in the nucleoplasm and lamin-A/C at the nuclear periphery. (ii) Intensity profile (along red line) of a nuclear bleb enriched in lamin-A/C and depleted of lamin-B. (B) Immunofluorescence images of a dividing cell next to non-dividing cells. Bottom histogram illustrates normalized frequency distribution of pSer22/LMNA, with respect to DNA content (‘2N’ vs ‘4N’). Pie chart: mitotic cells with hyper-phosphorylated lamin-A/C are extremely rare (<1%). Scale bar = 10 μm. (C) Semi-log profile plot of A-type lamin peptide MS intensities. Intensities of non-phosphorylated (endogenous) peptides ‘Ser22’ and ‘Ser390’ are close to the average and median intensities of all lamin-A/C peptides (gray). (D) Normalized phosphorylation stoichiometries (signal of phosphorylated peptide divided by the median signal of all lamin-A/C peptides; e.g. ‘pSer22/LMNAmedian’) quantified for Ser22, Ser390, and the doubly phosphorylated pSer(404&407). Normalized phosphorylation of lamin-C (slices 5–7) is ∼2-fold higher than that of LA or progerin (slices 1–4). Inset: average %-phosphorylation normalized to slice #4, which corresponds to the progerin peak. *p < 0.05. (E) Left and right SDS-PAGE ‘flank’ lanes cut and analyzed in parallel by Western blot, using anti-lamin-A/C and anti-pSer22 separately. (F) Quantitation of normalized phosphorylation at Ser22 (densitometry signal of pSer22 divided by that of total lamin-A/C; ‘pSer22/LMNA’) is consistent with that by FEA-MS (‘pSer22/LMNAmean’). A low MW (∼30 kDa) degradation fragment is visible in both blots (purple box, C) and is highly phosphorylated at Ser22.
For more rigorous quantitation of phosphorylation of the three A-type lamin isoforms, we then calibrated our FEA-MS analysis with synthetic versions of tryptic peptides that have well-documented phospho-serines at Ser22 and Ser390, which are near the head and tail domains of lamin-A/C, respectively [14]. Injections of known amounts of both phosphorylated (‘pSer22’ and ‘pSer390’) and non-phosphorylated (‘Ser22’ and ‘Ser390’) peptides into adjacent replicate lanes (Fig. 2A) allowed for MS peak alignment and transfer of identifications from the ‘spike-in’ lanes to the ‘endogenous’ sample lane containing no synthetic peptide. Intensities of injected synthetic peptides exhibited robust linearity versus spike-in amounts over several orders of magnitude (all R2 > 0.97 in slices #3-5; Fig. S3A), which provides confidence in MS quantitation.
Analysis by FEA-MS revealed that progerin (P) phosphorylation is slightly lower than that of intact LA, and is ∼2-fold lower than that of intact LC in HGPS iPS-MSCs (Fig. 4C). All three A-type lamin isoforms exhibited phosphorylation stoichiometries (‘% phosphorylation’) of 0.5∼10% (Fig. 4D). This was determined by three different normalization methods: the ratio of phosphorylated peptide intensity divided by 1) the intensity of its non-phosphorylated counterpart (e.g. ‘pSer22/Ser22’), 2) the mean intensity of all lamin-A/C peptides (e.g. ‘pSer22/LMNAmean’), and 3) the median of all lamin-A/C peptides (e.g. ‘pSer22/LMNAmedian’) (Fig. S3B). All three normalization methods produced consistent trends showing phosphorylation of LC > LA ≥ P (Fig. 4B, inset). In addition to the Ser22 and Ser390 spike-in sites, FEA-MS also detected a doubly phosphorylated peptide with ‘pSer404&407’, which followed similar trends as Ser390, a neighboring tail-domain phospho-site (Fig. 4C,D; Fig. S3B-iii). LC having the highest phosphorylation is consistent with LC being the most mobile and mechanosensitive A-type isoform in vitro [45–47]. On the other hand, phosphorylation of LB1 and LB2 at analogous sites was not detected (Table S1) despite abundant signal from the non-phosphorylated control peptides. Undetectably low phosphorylation of the B-type lamins is consistent with the hypothesis that C-terminal farnesylation somehow suppresses phosphorylation and solubilization of lamins, as likewise suggested by progerin and LB1/B2's depletion from mechanically-induced nuclear blebs (Fig. 3B,C).
Western blots for anti-pSer22 validated the above FEA-MS trends, showing the highest normalized phosphorylation of LC (anti-pSer22 densitometry signal divided by that of total anti-lamin-A/C: ‘pSer22/LMNA’), followed by LA, then P (Fig. 4C,D). Low MW bands (30-40 kDa) that immuno-stained for lamin-A/C also stained intensely with anti-pSer22 (as plotted for the lowest band at ∼30 kDa, purple box; Fig. 4D); this is consistent with previous studies of interphase phosphorylation favoring degradation into smaller fragments [16,32–34]. Since phosphorylation of A-type lamins increases upon release of cytoskeletal tension on the nucleus [16,31], these measurements raised questions of whether responses to mechanical perturbations are also isoform-dependent.
Phosphorylation of A-type lamins increases with low tension but mechanosensitivity is lost with passage of iPS-MSCs
To assess whether the difference in baseline phosphorylation levels across A-type lamin isoforms influence their responses to mechanical stress, well-spread HGPS iPS-MSCs on rigid plastic were treated with low concentrations of trypsin for tens of minutes to induce cell rounding and detachment from the substrate (Fig. 5A). In culture, lamin-A/C levels in adult cells including primary hMSCs decrease rapidly (<1h) upon cell rounding, with increased phosphorylation and turnover of lamin-A/C dependent on actomyosin contractility [16]. Lower mechanical tension on the nucleus (e.g. cell/nuclear rounding) increases phosphorylation and solubilization of lamin-A/C into the nucleoplasm, which in turn favors its degradation [16,32,34]. Intact A-type lamins in early passage (P2) HGPS iPS-MSCs likewise decreased in level upon cell rounding and detachment by trypsinization (up to 45 min), with correspondingly higher phosphorylation at Ser22 (Fig. 5A-i). Low MW (∼40 kDa and lower) degradation fragment bands were again clearly visible and increased in intensity with cell/nuclear rounding (Fig. 5A-i, immunoblot), correlating with elevated pSer22 signal but anti-correlating with the decrease in intact lamins. Effects were most pronounced once again for LC > LA > P. However, the rapid response to cell rounding was lost at high passage (>P7) (Fig. 5A-ii).
Figure 5.

Low nuclear tension increases A-type lamin phosphorylation and degradation, but mechanosensitivity is lost with passage in HGPS iPS-MSCs. (A) (i) Full-length Western blots of cells rounded and detached from their substrate using low concentrations of trypsin, probed with anti-lamin-A/C (left) and anti-pSer22 (right). A-type lamins (LA, P, and LC) in the intact MW range (60-80 kDa) decrease with cell rounding, which anti-correlates with the increase in phosphorylation and the increase in low MW degradation fragment bands (≤ 40 kDa). Bottom plot: Normalized phosphorylation (‘pSer22/LMNA’) of intact LA, P, and LC vs rounding time. (ii) Sensitivity to low tension (cell/nuclear rounding) is blunted with cell passage (P7). (B) Immunoblot of P7 HGPS iPS-MSCs cultured on stiff vs soft gels. Lower bar graph: fold change in densitometry signal (soft/stiff) of LA, P, and LC. Gray bar (right) indicates fold change of total A-type lamins in early passage primary hMSCs. (C) Cartoon and Immunofluorescence images of HGPS iPS-MSCs on rigid culture plastic treated with blebbistatin (25 μM, 2h) or DMSO control. (D) Nucleus 2D projected area decreases with blebbistatin treatment. (E) (i) Fraction (%) of cells with nuclear blebs. (ii) Nucleus circularity measurements. Nuclei with circularity < ∼0.65 (red box) correspond to those with more than one bleb. (F) Immunofluorescence measurements of LA/P/LC (A-type lamin) abundance in DMSO vs blebbistatin treated iPS-MSCs. LB1/B2 (B-type lamins) remain unaffected by myosin-II inhibition. (G) (i) Normalized phosphorylation (as ‘pSer22/LMNA’) measured in DMSO vs blebbistatin treated iPS-MSCs. (ii) Fraction (%) of ‘2N’ vs ‘4N’ cells (by DNA content) remain unchanged with blebbistatin treatment. (H) Representative immunofluorescence images of LA/C, LB1/2, and γH2AX in HGPS iPS-MSCs (P1 and P4) and normal primary hMSCs. Scale bar = 10 μm. (I) Lamin-A:B ratio (total A : B-type lamin ratio) and cell/nuclear size increase with higher passage in iPS-MSCs. (J) Quantification of progerin intensity (using anti-progerin, top) and % cells with >1 γH2AX foci (bottom) determined by immunofluorescence.
Cell and nuclear rounding can also be achieved and sustained for days by culturing cells on soft gels as opposed to stiff gels which promote cell spreading per conventional cultures on glass or plastic. Quantitative immunoblotting of P7 iPS-MSCs cultured on soft or stiff collagen-coated gels showed decreases in LC > LA > P on soft gels relative to stiff (Fig. 5B & Fig. S4A), and a ∼40 kDa fragment band was evident only in cells on soft gels, consistent with degradation under these sustained low tension conditions. Immunofluor-escence showed normal primary hMSCs are more mechano-responsive, at least at low passage (Fig. 5B: gray bar, adapted from Dingal et al [27]) and likewise showed a clear mechano-response in lower passage iPS-MSCs (Fig. S4B).
To further clarify the effects of tension on lamin phosphorylation and degradation, low-passage (P2) iPS-MSCs on rigid culture plastic were treated for 2h with the myosin-II inhibitor, blebbistatin, and analyzed by immunofluorescence (Fig. 5C). Treatment with blebbistatin caused nuclear rounding (smaller projected area, Fig. 5D) and significantly reduced the fraction of nuclei with blebs (Fig. 5C and Fig. 5E-i,ii), consistent with inhibition of actomyosin tension. Reduced actomyosin stress also caused a rapid decrease in A-type lamin levels (but not B-type lamins) (Fig. 5F), concomitant with an increase in normalized phosphorylation (in both ‘2N’ and ‘4N’ cells) (Fig. 5G-i). Furthermore, the fraction of 2N vs 4N cells remained unaffected with blebbistatin treatment (Fig. 5G-ii), removing the possibility of any confounding effects of cell cycle shift. Once again, higher passage (P7) iPS-MSCs were unaffected (Fig. S4C).
Loss of sensitivity with passage coincided with (i) an increase in cell and nuclear area (Fig. 5H,I), (ii) accumulation of progerin (Fig. 5J, top), and (iii) a greater number of cells with γH2AX foci, which is a marker of DNA damage (Fig. 5H,J-bottom). The findings thus suggest that regulation of the intact A-type lamins (LC > LA > P) by tension-mediated phosphorylation and turnover diminishes with passage number and possibly with cell senescence, which has been shown in many contexts to be accelerated with accumulation of progerin [48–50]. The rapid and distinct mechano-responses at the level of protein for the normal and diseased A-type lamin isoforms raised questions of whether or not they also respond differently to transcriptional regulation.
Lamin-A, progerin, and lamin-C respond equally to transcriptional regulation by retinoids
Progerin protein levels measured by FEA-MS were clearly below the levels expected from allele ratios (Fig. 1B), and were also found to be less mechanosensitive than normal LA/C after both acute and sustained perturbations (Fig. 5A-G). It is conceivable that through some allele-specific mechanisms (such as positioning the mutated sequence more in heterochromatin) progerin expression occurs at an unperturbable, low level similar to lamin-B1/B2 in the MSCs. It is also conceivable that splicing mechanisms are differentially regulated (Fig. 1B). HGPS iPS-MSCs were therefore cultured on soft or stiff collagen-coated gels and treated with retinoid compounds that are known to regulate LMNA gene expression. All-trans retinoic acid (‘RA’) and CD1530 are retinoid agonists that repress LMNA promoter activity [31,51], while antagonist (‘AGN’) and CD2665 upregulate LMNA promoter activity. RA is a vitamin-A metabolite with potent effects in differentiation that is normally ∼10 nM in serum. Treatment of early passage (P2) HGPS iPS-MSCs with 1 μM CD1530 or CD2665 revealed significant changes in LMNA expression on stiff gel cultures but not on soft (Fig. 6A), consistent with recent reports demonstrating that a stiff matrix is required in order to sensitize cells to these compounds [51]. Quantitative densitometry of A-type lamins in RA/AGN treated cells cultured on rigid plastic further revealed that regulation by retinoids is not isoform-specific in these early passage (P2) HGPS iPS-MSCs (Fig. 6B and Fig. S4D): LA, P, and LC all responded equally, resulting in ∼30% repression with RA and up to ∼10% upregulation with AGN. These results agree with recent reports [52] and suggest that regulatory perturbations of the mutated LMNA gene and transcript are similar to the normal allele, with effects likely independent of phospho-degradation (Fig. S4E). Nonetheless, as seen with mechano-regulation (Fig. 5A-E), sensitivity to retinoids diminished with passage (Fig. 6C), indicating that expression responses to both soluble and insoluble microenvironmental cues are blunted over time.
Figure 6.
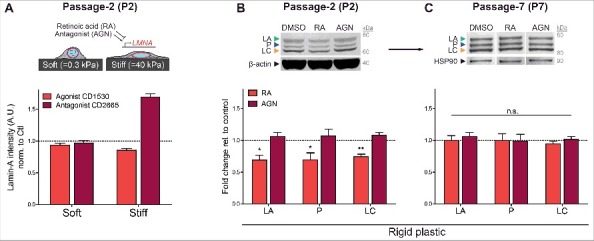
Transcriptional regulation of LA, P, and LC by retinoid compounds. (A) Top: cartoon illustrating HGPS iPS-MSCs on soft/stiff gels treated with retinoid compounds, all-trans retinoic acid (RA) and antagonist (AGN). Bottom: immunofluorescence quantitation of total A-type lamins after treatment with retinoids CD1530 (agonist) and CD2665 (antagonist) on soft or stiff gel cultures. (B) Western blot of early passage (P2) HGPS iPS-MSCs treated with 1 μM RA or AGN on rigid plastic. (C) Sensitivity to transcriptional regulation by retinoids decreases with passage.
Discussion
FEA-MS complements and extends antibody-based methods for quantifying stoichiometries of lamins and phosphorylation states in HGPS patient-derived cells that are differentiated to MSCs with abundant lamin-A. Additional steps in the workflow could improve quantitation (e.g. injection of synthetic peptides unique to each isoform), but our measurements of LA : P : LC (1: 0.5 : 1.7) and %-phosphorylation (0.5-10%) proved consistent with immunoblot trends (Fig. 2B-E,Fig. 3A-C) and with total lamin-A:B stoichiometry in primary MSCs (Fig. 2G). New phospho-specific antibodies should be useful for further assessments, including degradation involving pSer404 (Fig. S4E). The ratio of normal : mutant A-type lamins, (LA + LC) : P, was found to be far below 1:1 despite the iPS-MSCs having a normal karyotype [39], suggesting differences in mRNA stability [53] and/or inefficiencies in the activation of the cryptic progerin splice site [54]. Progerin protein also interacts more strongly with the proteasome [55] and is suggested to be a selective target of autophagy (as is prelamin-A) [56–58], which could favor a basal rate of degradation. However, intact progerin was found here to be least responsive in its dynamic mechanosensitive phosphorylation and degradation (Fig. 7). These molecular observations in a model cell type found in most organs begin to provide some insight into why Progeria [3] primarily afflicts tissues that we pointed out are normally stiff and more stressed mechanically (Fig. 1A), whereas soft tissues including brain, bone marrow, and blood are unaffected [4].
Figure 7.
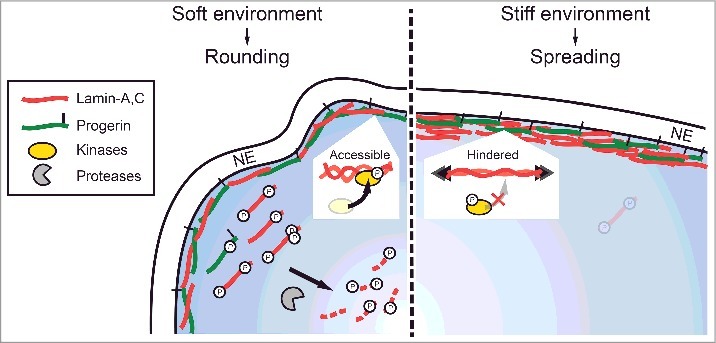
Progerin phospho-degradation is least responsive to mechanical perturbations. Cartoon illustrating phosphorylation and turnover of A-type lamins in soft (rounded, low nuclear stress) and stiff (well-spread, high nuclear stress) environments.
Mechanically induced nuclear blebs after constricted migration were depleted of progerin, which appeared to behave more like B-type lamins rather than LA or LC (Fig. 3C,D). This observation is consistent with nuclear blebs seen in HGPS cells in culture [59,60] (Fig. 4A,B). GFP-progerin also prevented the enrichment of endogenous LA and LC to nuclear blebs, suggesting a dominant negative effect on lamin mobility and remodeling dynamics. The findings are consistent with reports showing decreased nucleoplasmic localization of A-type lamins in HGPS fibroblasts [61]. Super-resolution imaging suggests A-type lamins form distinct filament networks [62,63], but the dominant negative effect seen here could reflect interactions between progerin and LA/C that are stronger than those between A-type and B-type lamins; in addition to ‘hetero-filaments’ of mixed LA-P or LC-P filaments, shared binding partners [64,65] might also suppress mobility under mechanical perturbations.
Consistent with low mobility of progerin, its steady state phosphorylation in iPS-MSCs on rigid culture plastic proved slightly lower than that of intact LA and far lower than that of LC. Turnover of progerin also exhibited lower sensitivity to mechanical perturbations than that of LA or LC: upon cell/nuclear rounding in early passage iPS-MSCs (Fig. 5A&G), all three A-type isoforms decreased in level with increased phosphorylation at Ser22. Of the three isoforms, intact LC was again most responsive to the reduction in nuclear stress, consistent with its highest baseline phosphorylation (Fig. 4C-F), followed by LA, then P. One appealing explanation for progerin's lower sensitivity to mechanical regulation is that farnesylation limits stress-induced conformational changes in lamin dimers that increase their affinity for modifying enzymes [16,31]. It is also clear from studies of Ser22Asp that such a phospho-mimetic is more nucleoplasmic and soluble than WT LMNA [12], and results in a significantly softer nucleus [16]. The low MW phospho-bands could therefore derive from intact LA/P/LC. Determining which A-type lamin yields such degradation peptides will be a challenge but is essential to clarifying the LC > LA > P mechanosensitive dynamics of the intact proteins.
Sensitivity of all A-type isoforms to matrix stiffness in 2D adhesion and 3D migration as well as to retinoid compounds (soluble transcriptional regulators) was blunted by passage, consistent with loss of mechano-sensitivity reported for primary MSCs [27]. A dampened response to extracellular inputs is consistent with an increasingly senescent phenotype that correlates with progerin accumulation, DNA damage, and enlargement of cells and nuclei in higher-passage MSCs (Fig. 5C-E). Given that progerin is known to reduce force propagation to the nuclear interior [66], interfere with mitosis [67], compromise stem cell differentiation potential [68], and induce senescence [48–50], these findings point to a potential positive feedback loop in which progerin drives a cell into premature senescence, which in turn favors further accumulation of progerin by limiting its sensitivity to upstream regulatory factors. The findings also imply that therapeutic efforts to modulate progerin levels in vivo (e.g. with farnesyl transferase inhibitors [69]) could become increasingly ineffective over time, which provides further motivation and rationale for early and accurate diagnosis and intervention.
Materials and methods
Differentiation and maintenance of patient-derived iPS-MSCs
Induced pluripotent stem cell (iPSC) lines derived from primary fibroblasts were obtained from ‘The Progeria Research Foundation Cell & Tissue Bank’, University of Ottawa. Differentiation of the iPSCs into mesenchymal stem cells (iPSC-MSCs) was achieved as described in Zou et al [70]. Briefly, iPS culture medium was replaced with MSC medium (low glucose Dulbecco's modification of Eagle's medium (DMEM, Invitrogen) with 10% fetal bovine serum (FBS, Sigma Aldrich) and 1% penicillin/streptomycin) three days after splitting. The MSC culture medium was refreshed every 2 days. After 14 days of culture, the cells were trypsinized (0.25% trypsin/1 mM EDTA, Difco-Sigma) and expanded in MSC medium on 0.1% gelatin coated dishes (BD). Upon confluency (typically 3–5 days), cells were trypsinized (0.025% trypsin-EDTA) and regularly passaged at ∼1:3 ratio. A morphologically homogeneous population of fibroblast-like cells became evident typically after the third passage, at which point the cells were assessed for MSC phenotypic characteristics and differentiation potential.
Fine-excision mass spectrometry (FEA-MS)
HGPS patient-derived iPS-MSC lysates (see Immunoblotting section) were injected into four middle lanes of an SDS-PAGE gel (15-well NuPAGE 4‐12% Bis‐Tris; Invitrogen) and four additional lanes (two on each side) spaced by molecular weight (MW) standards (Fig. 1B). Gel electrophoresis was run for 10 min at 100 V, then subsequently for >1 hr at 160 V to allow maximum possible separation of the lamin MW range (60 – 80 kDa). A custom device made of >8 equally spaced blades (1 mm apart) was used to excise the SDS-PAGE into 7 narrow slices per lane (∼1 mm3 in volume) vertically along the direction of electrophoresis. The excised gel slices were prepared for LC-MS/MS processing following the protocol outlined in Swift et al [31]. Briefly, the gel slices were washed in 50% 0.2 M ammonium bicarbonate (AB), 50% acetonitrile (ACN) solution for 30 min at RT. The washed slices were lyophilized, incubated with a reducing agent (20 mM TCEP in 25 mM AB solution), then alkylated (40 mM iodoacetamide (IAM) in 25 mM AB solution). The gel sections were lyophilized again before in-gel trypsinization (20 mg/mL sequencing grade modified trypsin, Promega) overnight at 37°C with gentle shaking. The resulting tryptic peptides were extracted by adding 50% digest dilution buffer (60 mM AB solution with 3% formic acid). Known concentrations of phosphorylated lamin-A peptide standards (GenScript) containing well-documented phospho-serine residues (‘pSer22’ and ‘pSer390’) and their non-phosphorylated counterparts (‘Ser22’ and ‘Ser390’) were spiked-into the final tryptic peptide solutions (0.001, 0.01, 0.1 μM into lanes #6, 7, 8, respectively), per Fig. 1B. Samples were injected (∼10 nL) into a high-pressure liquid chromatography (HPLC) system coupled to a hybrid LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) via a nano-electrospray ion source.
Raw data from each MS sample was processed separately using MaxQuant (version 1.5.3.8, Max Planck Institute of Biochemistry). MaxQuant's built-in Label-Free Quantification (LFQ) algorithm was employed with full tryptic digestion and up to 2 missed cleavage sites. Peptides were searched against a human FASTA database compiled from UniProt, plus contaminants and a reverse decoy database. The software's decoy search mode was set as ‘revert’ and a MS/MS tolerance limit of 20 ppm was used, along with a false discovery rate (FDR) of 1%. The minimum number of amino acid residues per tryptic peptide was set to 7, and MaxQuant's ‘match between runs’ feature was used for transfer of MS2 peak identifications across samples. All other parameters were run under default settings. The MaxQuant output tables were then fed into its custom bioinformatics suite, Perseus (version 1.5.2.4), for protein annotation and sorting.
The LFQ intensity for ‘Prelamin-A/C’ (normalized intensity value incorporating signal from all LMNA peptides) was plotted against gel slice # to generate an intensity line profile with two distinct peaks at slices 3 and 5, respectively. Isoform-specific peptides (those belonging to lamin-A only (‘LA’), lamin-A or progerin (‘LA/P’), and lamin-C only (‘LC’) in Fig. S1A; Venn diagram) were used to calculate pair-wise intensity ratios for each slice range (e.g. LA:LC ratio in slice #3). The pair-wise ratios were then used to generate isoform-specific intensity plots (Fig. S1A, middle plot) which, when summed, preserved the total A-type isoform abundance (‘LA / P / LC’ in gray). Final adjusted line profiles for each isoform (‘LA*’, ‘P*’, and ‘LC’, Fig. S1A, rightmost plot) were generated based on the symmetry of the LA and LC peaks: since LA (∼74 kDa) was close to zero at all slices except #3 and LC ∼ 0 at all slices but #5, progerin was also assumed to have one distinct peak at slice #4 with minimal contribution to ‘LA/P’ in slice #3. The ratio of LA signal at slice #2 / #3 (LA2/LA3) and that of LC signal at slice #4 / #5 (LC4/LC5) were thus averaged to estimate the fractional decrease of progerin signal from slice #4 to #3, to estimate progerin's signal in slice #3, P*3 (which was found to be ∼ 0). Isoform stoichiometries were computed by summing intensities over all slices (1-7), or alternatively, by integrating the best-fit Gaussian functions.
Immunoblotting
Cell pellets were rinsed with PBS and suspended in ice‐cold 1x NuPAGE LDS buffer (Invitrogen; diluted 1:4 in 1x RIPA buffer, plus 1% protease inhibitor cocktail (Sigma), 1% β‐mercaptoethanol (Sigma)), and lysed by sonication on ice (10 × 3s pulses using a probe sonicator, at intermediate power setting). Lysed samples were then heated to ∼80 °C for 10 min and centrifuged at maximum RPM for 30 min at 4°C. SDS‐PAGE gels (NuPAGE 4‐12% Bis‐Tris; Invitrogen) were loaded with 5 – 15 μL of lysate per lane. Each sample was loaded in duplicates or triplicates (with varying loading volumes) for quantitative analysis. Lysates were diluted with additional 1x NuPAGE LDS buffer if necessary. Gel electrophoresis was run for 10 min at 100 V and 1 hr at 160 V. Electrophoresis-separated samples were then transferred to a polyvinylidene fluoride membrane using an iBlot Gel Transfer Device (Invitrogen). The membrane was blocked with 5% non-fat dry milk in TTBS buffer (Tris‐buffered saline, BioRad; with 0.1% Tween‐20), washed x3 in TTBS, then incubated with primary antibodies against: LMNA (CST, #4777), HSP90 (Abcam, #ab13495), β-actin (Santa Cruz, #sc-47778), and/or LMNA pSer22 (CST, #2026), LMNA pSer404 (EMD Millipore, #ABT1387) diluted in TBS to final concentrations of ∼1 μg/ml and incubated at 4 °C overnight. After washing x3 with TTBS, the membrane was incubated with 1:2000 diluted secondary Ab: anti‐mouse/rabbit HRP‐conjugated IgG (GE Healthcare), at RT for 1.5 hrs. The membrane was washed x3 again with TTBS and developed using ChromoSensor (GenScript) for ∼3 min at RT. Immunoblot images were obtained using a HP Scanjet 4850. Quantitative densitometry analysis was performed using ImageJ (NIH).
Transwell migration
Cells were seeded at densities of ∼300,000 cells/cm2 onto the top side of transwell filter membranes (Corning Inc.) and left to migrate under normal culture conditions for 24 hrs. The number of migrated cells on the bottom of the membrane are proportional to the number of cells added on the top in a given set of experiments, which allows for comparisons across conditions by normalizing to a control sample.
Lamin-A level modulation
A549 cells (a human lung carcinoma cell line) was cultured in Ham's F12 nutrient mixture (Gibco), supplemented with 10% FBS and 1% penicillin/streptomycin (Sigma). Overexpression of lamin-A was achieved by transfection with Lipofectamine 2000 (Invitrogen) for 24-hr. GFP-lamin-A and GFP-progerin were gifts from David M. Gilbert (Florida State University) and Tom Misteli (Addgene plasmid # 17653), respectively. For shRNA knockdown of LMNA, A549 cells were infected with lentiviral supernatants targeting lamin-A (TRCN000061833, Sigma) at a multiplicity of infection (MOI) of 10 in the presence of 80 μg/mL Polybrene (Sigma) for 24 hours. Transduced cells were then selected by treatment with 2 μg/mL puromycin (Sigma) for 30 days. Efficiency of knockdown was determined by immunoblot.
Immunofluorescence and imaging
Cells were first rinsed with pre-warmed PBS, fixed with 4% paraformaldehyde (PFA, Fisher) for 15 min, washed x3 with PBS, and permeabilized with 0.5% Triton-X (Fisher) in PBS for 10 min. Fixed and permeabilized cells were then blocked with 5% BSA in PBS for a minimum of 1.5 hrs. Samples were then incubated overnight with primary antibody solution in 0.5% BSA solution with gentle shaking at 4 °C. The primary antibodies used were: LMNA (1:500, CST, #4777), LMNB (1:500, Santa Cruz, #sc-6217), and γH2AX (EMD Millipore, #05-636). Samples were then washed x3 in 0.1% BSA in PBS and incubated with the corresponding secondary antibodies at 1:500 dilution for 1.5 hrs at RT (Alexa Fluor 488, 546 and 647 nm; Invitrogen). Immunostained cells on gels or glass coverslips were mounted with mounting media (Invitrogen ProLong Gold Antifade Reagent). Epifluorescence imaging was performed using an Olympus IX71 with a digital EMCCD camera (Cascade 512B, Photometrics) and a 40 × /0.6 NA objective. Confocal imaging was done in Leica TCS SP8 system with either a 63 × /1.4 NA oil-immersion or 40 × /1.2 NA water-immersion objective. Image analysis was done with ImageJ.
Synthesis of soft and stiff polyacrylamide (PA) gels for cell culture
Circular glass coverslips (Fisher Scientific; 18 mm) were first cleaned in boiling ethanol then subsequently in RCA solution (H2O : H2O2 : NH4OH = 2:1:1 by vol.) for 10 min each. The cleaned coverslips were then functionalized in ATCS solution (chloroform with 0.1% allytrichlorosilane (Sigma) plus 0.1% trimethylamine (Sigma)) for 1 hr. Fresh gel precursor solution for soft-stiff PA gels were prepared as previously described [31]. 1% ammonium persulphate (APS, Sigma) and 0.1% N,N,N’,N’-tetramethylethylenediamine (TEMED, Sigma) and were added to the precursor solutions to initiate gel polymerization, and 20 µl of the resulting mixture were added to each cleaned coverslip. The solutions were then covered with larger coverslips (Fisher Scientific; 25 mm) and incubated to allow for polymerization at RT for ∼45 min. Polymerized gels were rinsed x3 with PBS and the large coverslips were gently removed. To coat the gel with collagen-I, Sulfo-SANPAH cross-linker (50 μg/ml in 50 mM HEPES, G-Biosciences) was applied over the whole gel surface and photoactivated under 365nm UV light for 10 min. Excess Sulfo-SANPAH was washed away with PBS and collagen-I solution (0.2 mg/ml in 50mM HEPES) was then added and incubated overnight at RT with gentle shaking.
Supplementary Material
Funding Statement
This work was supported by the National Cancer Institute, U54
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 2010;11:567–78. [DOI] [PubMed] [Google Scholar]
- [2].Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res Rev. 2017;33:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gordon LB, Brown WT, Collins FS. Hutchinson-Gilford Progeria Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., eds. GeneReviews((R)); Seattle: (WA), 1993. [Google Scholar]
- [4].Worman HJ. Nuclear lamins and laminopathies. J Pathol. 2012;226:316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Eriksson M, Brown WT, Gordon LB, et al.. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol. 2016;145:401–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Davies BS, Fong LG, Yang SH, et al.. The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet. 2009;10:153–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Capell BC, Erdos MR, Madigan JP, et al.. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Goodwin JS, Drake KR, Remmert CL, et al.. Ras diffusion is sensitive to plasma membrane viscosity. Biophys J. 2005;89:1398–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dahl KN, Scaffidi P, Islam MF, et al.. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103:10271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moir RD, Yoon M, Khuon S, et al.. Nuclear lamins A and B1: different pathways of assembly during nuclear envelope formation in living cells. J Cell Biol. 2000;151:1155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kochin V, Shimi T, Torvaldson E, et al.. Interphase phosphorylation of lamin A. J Cell Sci. 2014;127:2683–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Naetar N, Ferraioli S, Foisner R. Lamins in the nuclear interior – life outside the lamina. J Cell Sci. 2017;130:2087–96. [DOI] [PubMed] [Google Scholar]
- [14].Torvaldson E, Kochin V, Eriksson JE. Phosphorylation of lamins determine their structural properties and signaling functions. Nucleus. 2015;6:166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dechat T, Korbei B, Vaughan OA, et al.. Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J Cell Sci. 2000;113(Pt 19):3473–84. [DOI] [PubMed] [Google Scholar]
- [16].Buxboim A, Swift J, Irianto J, et al.. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Current biology : CB. 2014;24:1909–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Aebi U, Cohn J, Buhle L, et al.. The nuclear lamina is a meshwork of intermediate-type filaments. Nature. 1986;323:560–4. [DOI] [PubMed] [Google Scholar]
- [18].Cho S, Irianto J, Discher DE. Mechanosensing by the nucleus: From pathways to scaling relationships. J Cell Biol. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dahl KN, Ribeiro AJ, Lammerding J. Nuclear shape, mechanics, and mechanotransduction. Circ Res. 2008;102:1307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gruenbaum Y, Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem. 2015;84:131–64. [DOI] [PubMed] [Google Scholar]
- [21].Turgay Y, Eibauer M, Goldman AE, et al.. The molecular architecture of lamins in somatic cells. Nature. 2017;543:261–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].de la Rosa J, Freije JM, Cabanillas R, et al.. Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion. Nature communications. 2013;4:2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hernandez L, Roux KJ, Wong ES, et al.. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell. 2010;19:413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Csoka AB, English SB, Simkevich CP, et al.. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell. 2004;3:235–43. [DOI] [PubMed] [Google Scholar]
- [25].Harada T, Swift J, Irianto J, et al.. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J Cell Biol. 2014;204:669–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kramann R, Schneider RK, DiRocco DP, et al.. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dingal PC, Bradshaw AM, Cho S, et al.. Fractal heterogeneity in minimal matrix models of scars modulates stiff-niche stem-cell responses via nuclear exit of a mechanorepressor. Nat Mater. 2015;14:951–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li CX, Talele NP, Boo S, et al.. MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat Mater. 2017;16:379–89. [DOI] [PubMed] [Google Scholar]
- [29].Liu GH, Barkho BZ, Ruiz S, et al.. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang J, Lian Q, Zhu G, et al.. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45. [DOI] [PubMed] [Google Scholar]
- [31].Swift J, Ivanovska IL, Buxboim A, et al.. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bertacchini J, Beretti F, Cenni V, et al.. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27:2145–55. [DOI] [PubMed] [Google Scholar]
- [33].Dingal PC, Discher DE. Systems mechanobiology: tension-inhibited protein turnover is sufficient to physically control gene circuits. Biophys J. 2014;107:2734–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Naeem AS, Zhu Y, Di WL, et al.. AKT1-mediated Lamin A/C degradation is required for nuclear degradation and normal epidermal terminal differentiation. Cell Death Differ. 2015;22:2123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sen S, Kumar S. Cell-Matrix De-Adhesion Dynamics Reflect Contractile Mechanics. Cell Mol Bioeng. 2009;2:218–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Moiseeva O, Lopes-Paciencia S, Huot G, et al.. Permanent farnesylation of lamin A mutants linked to progeria impairs its phosphorylation at serine 22 during interphase. Aging. 2016;8:366–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].McClintock D, Ratner D, Lokuge M, et al.. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PloS One. 2007;2:e1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Irianto J, Xia Y, Pfeifer CR, et al.. DNA Damage Follows Repair Factor Depletion and Portends Genome Variation in Cancer Cells after Pore Migration. Curr Biol: CB. 2017;27:210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Booth-Gauthier EA, Du V, Ghibaudo M, et al.. Hutchinson-Gilford progeria syndrome alters nuclear shape and reduces cell motility in three dimensional model substrates. Integrative biology: quantitative biosciences from nano to macro. 2013;5:569–77. [DOI] [PubMed] [Google Scholar]
- [41].Haque F, Mazzeo D, Patel JT, et al.. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem. 2010;285:3487–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang L, Yang W, Ju W, et al.. A proteomic study of Hutchinson-Gilford progeria syndrome: Application of 2D-chromotography in a premature aging disease. Biochem Biophys Res Commun. 2012;417:1119–26. [DOI] [PubMed] [Google Scholar]
- [43].Pfeifer CR, Xia Y, Zhu K, et al.. Cell cycle repression and DNA repair defects follow constricted migration. bioRxiv. 2017; [Google Scholar]
- [44].Akopyan K, Silva Cascales H, Hukasova E, et al.. Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol Cell. 2014;53:843–53. [DOI] [PubMed] [Google Scholar]
- [45].Broers JL, Kuijpers HJ, Ostlund C, et al.. Both lamin A and lamin C mutations cause lamina instability as well as loss of internal nuclear lamin organization. Exp Cell Res. 2005;304:582–92. [DOI] [PubMed] [Google Scholar]
- [46].González-Cruz RD, Sadick JS, Fonseca VC, et al.. Nuclear Lamin Protein C Is Linked to Lineage-Specific, Whole-Cell Mechanical Properties. Cell Mol Bioeng. 2018;11:131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pugh GE, Coates PJ, Lane EB, et al.. Distinct nuclear assembly pathways for lamins A and C lead to their increase during quiescence in Swiss 3T3 cells. J Cell Sci. 1997;110(Pt 19):2483–93. [DOI] [PubMed] [Google Scholar]
- [48].Benson EK, Lee SW, Aaronson SA. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J Cell Sci. 2010;123:2605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cao K, Blair CD, Faddah DA, et al.. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest. 2011;121:2833–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wheaton K, Campuzano D, Ma W, et al.. Progerin-Induced Replication Stress Facilitates Premature Senescence in Hutchinson-Gilford Progeria Syndrome. Mol Cell Biol. 2017;37: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ivanovska IL, Swift J, Spinler K, et al.. Cross-linked matrix rigidity and soluble retinoids synergize in nuclear lamina regulation of stem cell differentiation. Mol Biol Cell. 2017;28:2010–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lo Cicero A, Jaskowiak AL, Egesipe AL, et al.. A High Throughput Phenotypic Screening reveals compounds that counteract premature osteogenic differentiation of HGPS iPS-derived mesenchymal stem cells. Sci Rep. 2016;6:34798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rodriguez S, Coppede F, Sagelius H, et al.. Increased expression of the Hutchinson-Gilford progeria syndrome truncated lamin A transcript during cell aging. Eur J Hum Genet: EJHG. 2009;17:928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Reddel CJ, Weiss AS. Lamin A expression levels are unperturbed at the normal and mutant alleles but display partial splice site selection in Hutchinson-Gilford progeria syndrome. J Med Genet. 2004;41:715–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kubben N, Voncken JW, Demmers J, et al.. Identification of differential protein interactors of lamin A and progerin. Nucleus. 2010;1:513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cenni V, Capanni C, Columbaro M, et al.. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur J Histochem: EJH. 2011;55:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dou Z, Xu C, Donahue G, et al.. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pellegrini C, Columbaro M, Capanni C, et al.. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget. 2015;6:29914–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Goldman RD, Shumaker DK, Erdos MR, et al.. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Taimen P, Pfleghaar K, Shimi T, et al.. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc Natl Acad Sci U S A. 2009;106:20788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Vidak S, Kubben N, Dechat T, et al.. Proliferation of progeria cells is enhanced by lamina-associated polypeptide 2alpha (LAP2alpha) through expression of extracellular matrix proteins. Genes Dev. 2015;29:2022–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shimi T, Kittisopikul M, Tran J, et al.. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol Biol Cell. 2015;26:4075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Xie W, Chojnowski A, Boudier T, et al.. A-type Lamins Form Distinct Filamentous Networks with Differential Nuclear Pore Complex Associations. Curr Biol: CB. 2016;26:2651–8. [DOI] [PubMed] [Google Scholar]
- [64].Haque F, Lloyd DJ, Smallwood DT, et al.. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol. 2006;26:3738–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wilson KL, Foisner R. Lamin-binding Proteins. Cold Spring Harbor Perspect Biol. 2010;2:a000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Booth EA, Spagnol ST, Alcoser TA, et al.. Nuclear stiffening and chromatin softening with progerin expression leads to an attenuated nuclear response to force. Soft Matter. 2015;11:6412–8. [DOI] [PubMed] [Google Scholar]
- [67].Cao K, Capell BC, Erdos MR, et al.. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A. 2007;104:4949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10:452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gordon LB, Kleinman ME, Miller DT, et al.. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109:16666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zou L, Luo Y, Chen M, et al.. A simple method for deriving functional MSCs and applied for osteogenesis in 3D scaffolds. Sci Rep. 2013;3:2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.