

ABSTRACT
Hutchinson-Gilford progeria syndrome (HGPS) is a sporadic, autosomal dominant disorder characterized by premature and accelerated aging symptoms leading to death at the mean age of 14.6 years usually due to cardiovascular complications. HGPS is caused by a de novo point mutation in the LMNA gene encoding the intermediate filament proteins lamins A and C which are structural components of the nuclear lamina. This mutation leads to the production of a truncated toxic form of lamin A, issued from aberrant splicing and called progerin. Progerin accumulates in HGPS cells’ nuclei and is a hallmark of the disease. Small amounts of progerin are also produced during normal aging. HGPS cells and animal preclinical models have provided insights into the molecular and cellular pathways that underlie the disease and have also highlighted possible mechanisms involved in normal aging. This review reports recent medical advances and treatment approaches for patients affected with HGPS.
KEYWORDS: HGPS, Progerin, FTI, ZOPRA, AON, Rapamycin, Metformin, MG132
Introduction
Hutchinson-Gilford progeria syndrome (HGPS; OMIM #176670) is a rare genetic disorder which affects 1 in 4–8 million children with symptoms resembling physiological aging that include growth impairment, very thin skin, loss of subcutaneous fat, alopecia, osteoporosis and heart disease leading to shortened life span and death at about 14.6 years [1,2]. HGPS was first described at the end of the XIX century by Jonathan Hutchinson and Hastings Gilford [3,4]. It was only more than 100 years later, in 2003, that the heterozygous, de novo mutation c.1824C>T, p.G608G (NM_170707.3) responsible for this accelerated-aging disease was found to be located within exon 11 of the LMNA gene that encodes lamins A and C [5,6]. Lamin A is a nuclear protein belonging to type V intermediate filaments. It is synthetized as a precursor called prelamin A. Prelamin A undergoes a multistep post-translational processing, including cysteine farnesylation by farnesyl transferase (FTase) on its C-terminal CaaX motif, then cleavage of the remaining 3 amino acids by the metallopeptidase ZMPSTE24. The C-terminal cysteine is then carboxymethylated by the isoprenylcysteine carboxylmethyltransferase (ICMT). Finally, the last 15 amino acids are cleaved again by ZMPSTE24 to produce the unfarnesylated, mature lamin A. HGPS mutation activates a cryptic donor splice site in exon 11 of the LMNA gene that leads to deletion of 50 amino acids near the C terminus, abrogating the second ZMPSTE24 cleavage site and resulting in accumulation of a truncated and permanently farnesylated prelamin A called progerin that incorporates abnormally into the nuclear lamina and exerts multiple toxic effects [7]. At the cellular level, HGPS is characterized by dramatic defects in nuclear envelope structure and function [8,9]. Primary fibroblasts from HGPS patients exhibit reduced proliferation as well as premature senescence [10], impaired DNA repair mechanisms [2,11–13], increased reactive oxygen species production [14], mitochondrial dysfunction [15], loss of peripheral heterochromatin [16–18] and telomere attrition [18]. Through these alterations, progerin accumulation results in loss of peripheral heterochromatin caused by the decrease in the repressive histone marks H3K9me3, H3K27me3 and H3K27 methyltransferase EZH2, increase in H4K20me3 [9,16,19], reduced levels of heterochromatin protein 1 (HP1) [20] and Lap2α [9]. Progerin also impairs the formation of DNA repair foci due to recruitment deficiency of the DNA double-strand break (DSB) repair factors p53-binding protein 1 (53BP1), Rad50 and Rad51 at DNA damage sites [21,22]. On the other hand, altered signaling pathways have been described in HGPS cells [23]. Among them, altered extracellular matrix synthesis caused by disturbed Wnt/β-catenin signaling [24], affected Notch signaling [20], hyperactivation of NF-Κb in response to inflammation [25] and impaired NRF2 (Nuclear factor erythroid-2-Related Factor 2) transcriptional activity resulting in increased chronic oxidative stress [26].
A wide spectrum of treatment strategies, targeting several processes with different specificities, has been proposed to correct the defects in HGPS: (i) to directly “repair” the disease -causing mutation; (ii) to inhibit pre-mRNA aberrant splicing leading to progerin mRNA production; (iii) to decrease the toxicity of isoprenylated and methylated progerin; (iv) to induce progerin clearance; (v) to decrease the noxious downstream effects linked to progerin accumulation [27] (Figure 1). These approaches will be presented below and grouped following their individual action.
Figure 1.
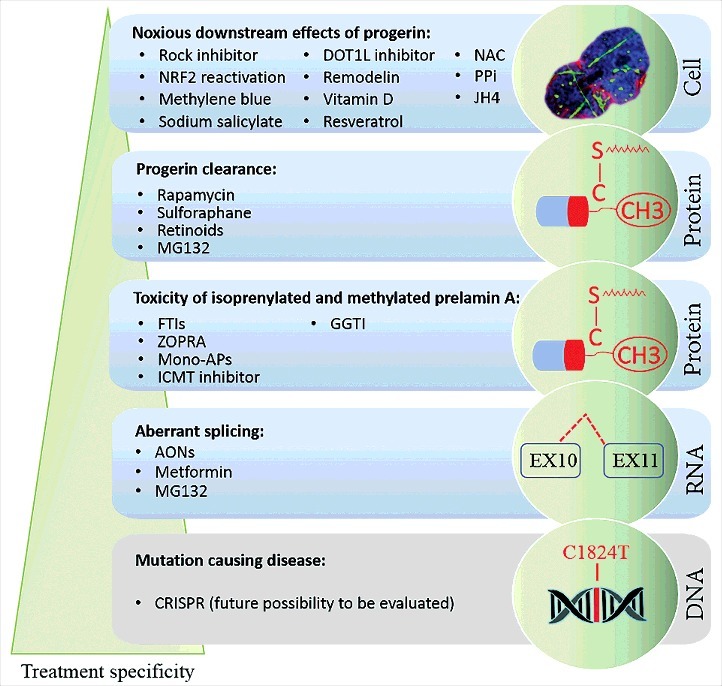
Specificity of a wide spectrum of treatment strategies for progeria. Several therapeutic strategies for progeria are shown. In decreasing order for their target specificity, treatment strategies can be mentioned as follows: the mutation-causing disease repair by genome editing (CRISPR as a future possibility to be evaluated), the inhibition of pre-mRNA aberrant splicing (Antisense OligoNucleotides: AONs, Metformin, MG132), the reduction of isoprenylated and methylated progerin levels (Farnesyl transferase inhibitors: FTIs, ZOledronate-PRAvastatin: ZOPRA, Monoaminopyrimidines: Mono-APs, Isoprenylcysteine CarboxyMethyl Transferase (ICMT) inhibitor, GeranylGeranylTransferase Inhibitor: GGTI-2147), the induction of progerin clearance (Rapamycin, Sulforaphane, Retinoids, MG132) and the reduction of the noxious downstream effects due to progerin accumulation (Rock inhibitor: Y-27632, Nuclear factor erythroid-2-Related Factor 2: NRF2 reactivation, Methylene blue, sodium salicylate, DOT1L inhibitor: epz-4777, Remodelin, Vitamin D, Resveratrol, N-Acetyl-Cysteine: NAC, inorganic pyrophosphate: PPi and JH4).
Prelamin A Isoprenylation and methylation inhibitors: Lonafarnib, Zoledronate/Pravastatin, Monoaminopyrimidines and isoprenylcysteine carboxyl methyltransferase inhibitor
The aberrant splice event that gives rise to progerin leads to the deletion of the ZMPSTE24 cleavage site normally used to remove the farnesylated carboxy terminus from prelamin A during posttranslational processing. Consequently, permanently farnesylated progerin remains anchored to the inner nuclear membrane resulting in dominant-negative disruption of the nuclear scaffold upon progerin dimerization with wild-type lamins [28]. Knowledge of these steps predicted that blocking farnesylation using farnesyltransferase inhibitor (FTI) drugs would decrease progerin production and toxicity. FTI are small molecules which reversibly bind to the farnesyltransferase CAAX binding site [29]. Blocking farnesylation of progerin with FTIs restored normal nuclear architecture and resulted in significant reductions in nuclear blebbing both in transiently transfected HeLa, HEK 293, NIH 3T3 cells and human HGPS fibroblasts [8,30–33]. In transgenic HGPS murine models treated with FTIs, bone mineralization, and weight are improved, lifespan is extended [34,35] and cardiovascular defects are prevented [36]. In 2007, the above studies led to the initiation of a prospective single-arm clinical trial (ClinicalTrials.gov, NCT00425607), using an FTI called lonafarnib, which was originally developed for the treatment of cancer. A cohort of 25 HGPS patients between 3 and 16 years of age were included in this trial and received lonafarnib for a minimum of 2 years. In 2012, researchers reported that some children with HGPS receiving lonafarnib showed a modest improvement in weight gain: nine patients’ rates of weight gain increased by ≥50%, six patients’ rates of weight gain decreased by ≥50%, and 10 remained stable with respect to the rate of weight gain. Other results from this trial showed improvements in vascular stiffness including decreases in arterial pulse wave velocity by a median of 35% in 18 subjects, in addition to increases in skeletal rigidity (median percent increases by 40–50% in axial rigidity, 170–228% in flexural rigidity, and 167–229% in torsional rigidity) as well as sensorineural hearing and bone mineral density (≥3% increase at one or more sites in 76% of children compared with 40% of the participants who exhibited decreases at one or more sites) [37]. Although FTI treatment increased mean survival by 1.6 years [38] it was reported that progerin may become alternatively prenylated by geranylgeranyltransferase I when farnesyltransferases are inhibited; indeed, the simultaneous presence of both farnesyltransferase inhibitor (FTI-277) and GeranylGeranylTransferase I inhibitor (GGTI-2147) led to a substantial amount of prelamin A accumulation compared with the effect of each inhibitor alone [39], thereby explaining the limited beneficial effects of FTI monotherapy. Accordingly, authors hypothesized that blocking both protein farnesylation and geranylgeranylation could minimize the possibility of alternative prenylation events that confer resistance to FTIs. Acting on the synthetic pathway of farnesyl pyrophosphate, a co-substrate of farnesyltransferase and a precursor of geranylgeranyl pyrophosphate, we and our close collaborator Carlos Lopez-Otin (Spain) demonstrated the synergistic effect of a combination of ZOledronate (N-BisPhosphonate) and PRAvastatin (statin) (ZOPRA) and their effectiveness to reduce prenylation and rescue HGPS cells defects and the progeroid phenotypes of Zmpste24−/− mice, including improvement of growth retardation, loss of weight, lipodystrophy, hair loss and bone defects. Likewise, the longevity of these mice was substantially extended [39]. This preclinical work allowed our clinical team to design and conduct a phase II, monocentric, open-label, single arm clinical trial (ClinicalTrials.gov, NCT00731016) using ZOPRA to evaluate its safety and efficacy in 12 HGPS patients included for 3.5 years each, with partially positive results including improvement of weight gain and bone density defects, without severe adverse effects (De Sandre‐Giovannoli, Sigaudy et al, submitted). Although trials using FTIs (Lonafarnib) and ZOPRA showed some efficacy for given parameters, these drugs could not be considered as a cure, and further research was needed to implement more effective therapeutic approaches for patients. A single-arm triple therapy trial (ClinicalTrials.gov, NCT00879034) was designed to administer pravastatin, zoledronate and lonafarnib and sought to further improve disease parameters by additionally inhibiting progerin prenylation, given that each enzyme functions along the protein prenylation pathway. The results of this trial on 37 participants with HGPS which began in 2009 in Boston revealed additional bone mineral density benefit but no major benefit beyond that seen with lonafarnib alone [40].
However, caution should be taken when considering the long-term treatment effects of FTIs, as FTIs have been shown to cause lethal cardiomyopathy in mouse models of progeria because of non-farnesylated prelamin A accumulation [41]. On the other hand, among 21.608 small molecules tested in induced pluripotent stem cell (iPSC) lines derived from HGPS patients, Nissan and colleagues identified several compounds, called monoaminopyrimidines (mono-APs), as new modulators of farnesylation, resulting, in vitro, in improved phenotypes associated with HGPS [42]. This class of protein farnesylation inhibitors, targeting both farnesyl pyrophosphate synthase and farnesyl transferase, acts like a combination of FTIs and bisphosphonates.
In addition to isoprenylation, another consequence of defective posttranslational modification to prelamin A in progeria was reported implicating the carboxy methylation that is mediated by isoprenylcysteine carboxyl methyltransferase (ICMT). In this context, Ibrahim and colleagues revealed that, in vitro treatment with ICMT inhibitor, increased proliferation and delayed senescence in human HGPS fibroblasts. Furthermore, ICMT inhibition by lentiviral short hairpin RNAs (shICMT) increased body weight, normalized grip strength, as well as prevented bone fractures and death in Zmpste24-deficient mice [43].
Autophagy-activating drugs: Rapamycin, Sulforaphane, Retinoids and MG132
Rapamycin, an immunosuppressive agent that is used to prevent the rejection of transplanted organs, promoted progerin clearance through autophagy, improved the abnormal nuclear shape, delayed the onset of cellular senescence of HGPS fibroblasts [44,45] and rescued the chromatin phenotype of cultured fibroblasts, including histone methylation status and BAF and LAP2alpha distribution patterns [46]. Otherwise, studies on Lmna−/− mouse model for dilated cardiomyopathy and muscular dystrophy, showed that pharmacologic reversal of elevated mTORC1 signaling by rapamycin improves cardiac and skeletal muscle function and enhances survival [47]. Using the same mouse model, Liao et al. showed that rapamycin treatment results in lifespan extension associated with increased body weight and fat content [48]. Although these results show beneficial effects on lmna KO mice, this model does not mimic the genetics and pathophysiology of HGPS in patients. A phase I/II monocentric trial (NCT02579044) of Everolimus, an agent derived from rapamycin, in combination with Lonafarnib, is currently conducted at the Clinical and Translational Study Unit (CTSU) of Boston Children's Hospital. Rapamycin inhibits the activity of mTOR (Mammalian Target Of Rapamycin), known to regulate a large panel of cellular functions including protein synthesis, cell growth, cytoskeleton rearrangements, transcription, immune responses or autophagy. Therefore, much caution should be used while performing the direct translation of the in vitro results to children affected with progeria. Furthermore, since rapamycin is also known to inhibit adipogenesis [49,50], precautions should be taken when using this compound in HGPS patients who are affected with generalized lipoatrophy and lipodystrophy.
Sulforaphane, an antioxidant derived from cruciferous vegetables, has been also described to enhance progerin clearance by autophagy and to reverse the cellular hallmarks of HGPS in vitro [51]. Intermittent treatment with Sulforaphane and Lonafarnib separately rescued the HGPS cellular phenotype [52]. Furthermore, since the LMNA promoter contains a retinoic acid responsive element, two recent studies suggested that either retinoids alone [53] or in a combination with rapamycin [54] reduce the amount of progerin and reverse aging defects in HGPS patient skin fibroblasts. These drugs may be beneficial in progeria treatment, but in vivo extensive studies should be performed before translating them into clinical trials.
We recently showed that progerin is sequestered into abnormally shaped Promyelocytic-Nuclear Bodies (PML-NB), identified as novel biomarkers in progeria. We identified MG132 as being effective on progerin degradation. Indeed, MG132 induces progerin nucleocytoplasmic translocation after a transition through the nucleolus, and progerin clearance through macroautophagy in HGPS patient fibroblasts as well as in HGPS patient iPSC-derived mesenchymal stem cells (MSCs) and Vascular Smooth Muscle Cells (VSMCs). MG132 treatment improves cellular HGPS phenotypes, reduces cellular senescence and enhances viability and proliferation in HGPS fibroblasts. In vivo, through MG132 treatment, progerin expression decreases in skeletal muscle from LmnaG609G/G609G mice. Altogether, we demonstrate progerin reduction based on MG132 action and shed light on a promising class of molecules towards a potential therapy for children with HGPS [55]. The observed specific effect of MG132 on progerin downregulation, sparing normal A-type lamins, further supports the idea that besides typical Progeria, other prelamin A-associated diseases might benefit from the same treatment.
Downregulation of Prelamin A aberrant splicing: Antisense oligonucleotides, Metformin and MG132
The efficiency of an antisense therapeutic approach using morpholino antisense oligonucleotides (AON) in sterically blocking the aberrant LMNA splicing site leading to progerin production has been previously proven in vitro on human HGPS patients’ cells and in vivo on a knock-in LmnaG609G/G609G mouse model [56]. Indeed, Osorio et al. tested the combined administration of two AONs: “MmEx11” targeting the exon 11 aberrant splice site activated by the progeria mutation in order to hamper its use, and “MmEx10”, targeting the physiological exon 10 splice site, in order to reinforce the action of the first AON, by shifting splicing events towards lamin C production. Similar results were obtained by another group on the same LmnaG609G/G609G mouse model and in HGPS fibroblasts, but using another antisense oligonucleotide (ASO) that reduces the binding of the splicing factor SRSF-2 to exon 11 LMNA pre-mRNA, confirming the efficacy of this approach [57].
Some patients carry other LMNA mutations affecting exon 11 splicing and are named “HGPS-like” patients [58]. They also produce Progerin and/or other truncated Prelamin A isoforms (Δ35 and Δ90). Recently, we showed that downregulation of progerin and other truncated or wild type Prelamin A isoforms can be achieved via the antisense therapeutic approach either in HGPS-like and Mandibuloacral Dysplasia type B (MAD-B) patients’ cells [59], establishing a preclinical proof of principle for the use of antisense morpholino oligonucleotides in HGPS, HGPS-like and MAD-B syndromes. Nonetheless, the administration to children of the same molecules administered to mice, i.e. vivo-morpholinos, could not be envisaged due to their known toxicity. Therefore, the choice of AON chemistry and route of administration will be particularly important for future therapeutic approaches involving splicing-modulation.
In human HGPS primary fibroblasts and mouse LmnaG609G/G609G 56, the HGPS mutation induces the use of an internal 5’ cryptic splice site within exon 11 of the LMNA pre-mRNA, leading to an aberrant alternative splicing and production of a truncated form of prelamin A (progerin). In 2011, the RNA-binding protein SRSF-1 (for Serine/arginine-Rich Splicing Factor 1) was shown to favor this aberrant alternative splicing [60]. On the other hand, it has been shown that SRSF-1 expression is transcriptionally regulated by the antidiabetic drug Metformin [61]. Based on these findings, Egesipe et al. demonstrated that Metformin decreases SRSF-1 and progerin expression in mesenchymal stem cells derived from HGPS induced pluripotent stem cells (HGPS MSCs) and in several other in vitro models of HGPS, i.e., human primary HGPS fibroblasts and LmnaG609G/G609G mouse fibroblasts, resulting in improved nuclear shape abnormalities and premature osteoblastic differentiation of HGPS MSCs [62,63].
In HGPS fibroblasts and in vivo via intramuscular injections of MG132 in LmnaG609G/G609G mice, we recently demonstrated that, in addition to activating macroautophagy leading to progerin degradation, MG132 strongly reduces progerin production through downregulation of SRSF-1, controlling prelamin A mRNA aberrant splicing [55]. Further in vivo studies, particularly on the LmnaG609G/G609G mouse model, will be needed to evaluate the effects of Metformin and MG132 on HGPS phenotype improvement.
Reduction of progerin downstream toxic effects
Few approaches have been described to counteract the altered downstream pathways caused by progerin accumulation, including nuclear shape abnormalities, ROS generation, accumulation of oxidized proteins, mitochondrial dysfunction [14,64], cell senescence and NF-kB activation, leading to the secretion of high levels of the proinflammatory cytokines IL-6, CXCL1, and TNF-α [25,65]. Treatment of progeroid fibroblasts with the ROS scavenger N-acetyl cysteine (NAC) reduced the levels of un-repairable DSB and improved their growth rates in culture [64]. Similarly, it has also been shown that rho-associated protein kinase (ROCK) regulates mitochondrial ROS generation by modulating the interaction between Rac1b and cytochrome c [66]. Accordingly, in vitro treatment of HGPS fibroblasts with ROCK inhibitor (Y-27632) resulted in decreased ROS levels and induced recovery of mitochondrial function along with a reduction in the frequency of abnormal nuclear morphology and DNA double-strand breaks [66]. Elevated levels of ROS and oxidative stress were also lowered by reactivation of NRF2, whose transcriptional activity is impaired in HGPS cells, resulting in improvements of cellular HGPS defects [26]. Interestingly, MG132 seems to be a potentially effective drug to be used in the prevention of oxidative damage in HGPS cells through the activation of the NRF2 signaling pathway [67–69]. Mitochondrial dysfunction was reported in HGPS fibroblasts as well as in HGPS mouse models [70,71]. To rescue mitochondrial defects, Xiong et al. showed that treatment of HGPS cells with Methylene Blue, an antioxidant compound known to stimulate mitochondrial function, improves not only the mitochondrial morphology and function but also rescues the premature aging phenotypes in HGPS cells including nuclear morphology, perinuclear heterochromatin loss and corrects misregulated gene expression [72]. In LmnaG609G/+ mice, mitochondrial dysfunction in vascular smooth muscle cells (VSMCs) results in increased tissue-nonspecific alkaline phosphatase activity and diminished ATP synthesis. Accordingly, VSMCs have an impaired capacity to synthesize extracellular pyrophosphate, a major inhibitor of vascular calcification [71]. Using LmnaG609G/G609G, Villa-Bellosta et al. showed that aortic vascular calcification due to defective pyrophosphate production is counterbalanced systemically by inorganic pyrophosphate (PPi) treatment [71].
In order to explore the therapeutic potential of NF-kB inhibition on HGPS disease parameters, Osorio et al. showed that crossing Zmpste24−/− mice with transgenic mice displaying reduced NF-κB signaling extends longevity and prevents the development of progeroid features. In the same study, and to the same end, the authors also showed that sodium salicylate treatment efficiently prevents NF-kB activation and associated disease phenotypes in Zmpste24-deficient mice, while also extending longevity in the LmnaG609G/G609G model [25]. Accordingly, NF-κB activation impairs somatic cell reprogramming in aging by eliciting the reprogramming repressor DOT1L. The identification of this molecular mechanism has allowed to translate this information into a therapeutic approach, indeed, DOT1L inhibition by epz-4777 extended longevity and prevented aging-associated alterations in progeroid mice [73]. In the same way, MG132 is also known to inhibit the secretion of proinflammatory cytokines and attenuation of IκB degradation, resulting in the abolition of NF-κB activation [74-76]. These findings, together with the fact that other models of normal and HGPS accelerated aging show arteriosclerotic lesions with calcification and inflammation [77], support the idea that inflammation is a major regulator of the aging process also in progeria and could be improved by several inflammation-reducing treatments, including MG132. Importantly, MG132 was reported to have a significant preventive and therapeutic effect on cardiovascular and renal injury [67,68,78], oxidative damage [69,79] and on accelerated atherosclerosis in rabbits [80]. These features are exhibited by HGPS patients who might benefit from the same therapeutic effects. On the other hand, compounds screening showed that Remodelin, by inhibiting the lamina interacting SUN1-associated acetyl-transferase protein NAT10, improves nuclear shape and fitness of both progeric and lamin A/C–depleted cells, as observed by decreased levels of the DNA double-strand break markers γH2AX and autophosphorylated ATM (ataxia telangiectasia mutated), decreased DNA damage signaling and improved chromatin and nucleolar organization [81].
Among the proteins affected by progerin accumulation is the vitamin D receptor (VDR) whose levels are reduced in HGPS cells. In addition, VDR knockout mice develop a premature aging phenotype similar to HGPS patients [82,83]. Interestingly, Kreienkamp et al. recently showed that reconstituting VDR signaling via 1α,25-dihydroxyvitamin D3 (1,25D), the active hormonal form of vitamin D, improves HGPS phenotypes, including nuclear morphological abnormalities, DNA repair defects, and premature senescence [84]. Another important target of progerin is lamin A/C. Indeed, progerin shows strong binding affinity for lamin A/C exerting a negative dominant effect. On this basis, Lee et al. identified new chemicals (JH4) that can block the interaction between progerin and lamin A/C through direct interaction with progerin. Treatment with JH4 alleviated nuclear deformation and reversed senescence and growth arrest markers. Furthermore, administration of JH4 to LmnaG609G/G609G resulted in a marked improvement of several progeria phenotypes including grip strength, body weight, organ size and cell density, as well as lifespan extension [85].
Resveratrol, a SIRT1 activator that interacts with lamin A [86], has been proposed as an alternative treatment for progeria. Indeed, in the premature aging mouse model Zmpste24−/− [87], due to the negative dominant effect of progerin, SIRT-1 is weakly associated with the nuclear matrix: as a result deacetylase activity is decreased, leading to a rapid depletion of adult stem cells. By increasing SIRT1 interaction with lamin A, Resveratrol rescues adult stem cell decline and alleviates progeroid features in Zmpste24−/− mice [86]. Although this model reproduces the progeroid syndromes phenotype, the underlying molecular mechanism is different from that of progeria. Further studies, using a model that produces progerin by the same abnormal splicing mechanism observed in humans, as the LmnaG609G/G609G model, will be needed to evaluate the impact of Resveratrol on the disease phenotypes reversion. Another study revealed that resveratrol treatment of an osteoblasts-specific progerin-expressing mouse model [88] does not show overall beneficial effects [89]. Resveratrol efficacy has been suboptimal due to poor bioavailability of this compound and variable dose-linked effects, limiting the full potential of SIRT1 activation treatment. A novel formulation of micronized resveratrol, SRT501, has been developed with improved bioavailability, tolerance and significant pharmacologic effect [90]. SRT501 open a new interesting research path on this class of molecules towards the development of new therapeutic approaches for progeria.
Recently, an in vivo study showed that partial reprogramming by short-term cyclic expression of Oct4, Sox2, Klf4, and c-Myc (OSKM) ameliorates cellular and physiological hallmarks of aging and prolongs lifespan in LmnaG609G/G609G mice [91,92].
Conclusion
Progress in progeria research led to a growing number of promising therapeutic candidates (Figure 2). However, most of these approaches lack sufficient in vitro and in vivo preclinical data to be transposed to patients. Mostly, evidence of efficacy in an adapted progeria animal model is required to exclude toxicity and perform tentative determination of optimal doses, dose frequencies, administration routes, and evaluation endpoints. A specific drug formulation requires advanced preclinical toxicity testing to avoid serious side effects that would prohibit long‐term use. It is worth to note that, in HGPS preclinical studies, the ability of a drug to preferentially target the cardiovascular system should be evaluated, in that it represents the major pathophysiological target in the disorder, leading to premature death. Likewise, among treatment efficacy readouts, cardiovascular measures would be preferred. Increasing our understanding of the disease biology will also be important to further elucidate which impaired pathways are most relevant to the disease, in order to identify both other treatment pathophysiological targets and other outcome measures. A limiting factor in advancing the development of therapeutic approaches for progeria is that studies are performed on primary cultures of patient's cells or animal models due to the limited number of autopsy specimens from HGPS patients. To reproduce key features of HGPS, iPSCs [93,94] and in vitro 3D tissue model using human iPSC-derived cells [95] thus provide relevant tools for drug screening.
Figure 2.
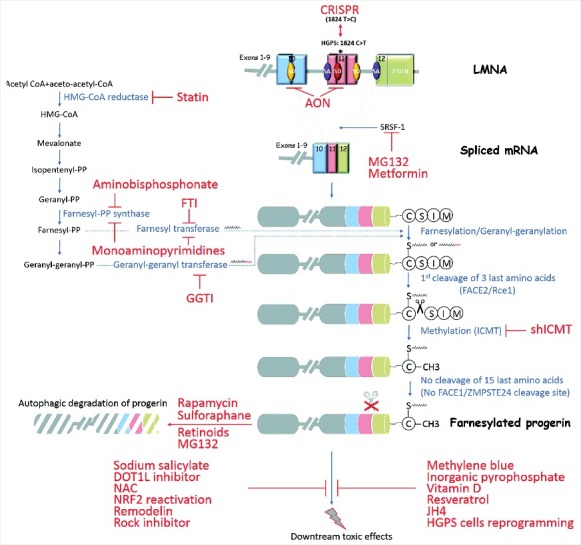
Treatment strategies for progeria and their targets. Envisaged treatments for progeria target several mechanisms triggering the disease. By acting on the genetic cause, recent advances in genome editing using the CRISPR technique could be beneficial to repair the mutation causing the disease. Other molecules target the aberrant splicing process leading to the production of a truncated protein, by blocking the splice site with AONs or by inhibiting the splicing factor SRSF-1 (Metformin, MG132). During prelamin A maturation, treatments inhibiting isoprenylation (farnesylation or geranylation) or the methylation have shown their effectiveness in reducing progerin toxicity (Statins, aminobisphonates, FTIs, GGTI-2147, monoaminopyrimidines, shICMT). To counter the accumulation of progerin, several molecules have been described as activators of autophagy leading to progerin degradation (Rapamycin, Sulphoraphane, Retinoids and MG132). Finally, by acting on the reduction of the noxious downstream effects of progerin, some approaches have shown their effectiveness against inflammation (sodium salicylate, DOT1L inhibition by epz-4777), oxidative stress (NAC, NRF2 reactivation), nuclear shape abnormalities (Remodelin), mitochondrial abnormalities (Rock inhibitor: Y-27632, Methylene blue), impaired pyrophosphate metabolism (inorganic pyrophosphate: PPi), vitamin D receptor signaling (vitamin D), decreased deacetylase activity (Resveratrol), Progerin/Lamin AC interaction (JH4) or aging hallmarks by reprogramming HGPS cells to pluripotency in vitro and in vivo.
To date, the amelioration of disease phenotypes in HGPS mouse models by the different tested therapies is limited, and this is even more true in HGPS patients. One could speculate that the age of the HGPS participant patients in the clinical trials could determine the outcomes of the treatments, but according to the clinical trial using lonafarnib, the authors stated that no associations between age at time of treatment and outcome measures that were improved at end of study were identified. At the same time, it should be noted that given the limited number of participants, it is difficult to correlate age with outcomes of the treatment.
As research progresses, it becomes clear that targeting the pathophysiology of progeria to a single level will likely be insufficient to significantly alleviate the signs and change the normal course of such a multi-organ devastating disease. Altogether, there is a major rationale in targeting progerin at different levels. Addressing therapies in HGPS associated to progerin accumulation may thus rely on multi-approaches combination, including its decreased production, increased degradation, or downstream noxious cascades.
As a future therapeutic approach that still remains to be evaluated for HGPS is the Clustered Regularly Interspaced Short Palindromic Repeats/Cas protein (CRISPR/Cas) for in vivo gene editing [96–98] and repair of the disease-causing mutation. On the other hand, Adeno-Associated Virus (AAVs) have gained popularity, in the last years, as safe gene delivery vectors, due to their ability to mediate long-term expression in both non-dividing and dividing cells, with specific tissue tropism [99–101]. In this regard, AAVs constitute interesting CRISPR/Cas9 delivery candidates to specifically repair the progeria-causing mutation. Preferentially, AAV serotype should target VSMCs, since they are involved in heart attack and stroke that represent the main cause of death in progeria patients. To overcome the limited packaging capacity of AAV (∼ 4.8 kb), small Cas9 orthologues, including Staphylococcus aureus-derived Cas9 (SaCas9, 3.16 kb) [102], has been used to package the Cas9 and its gRNA into a single AAV delivery vehicle for in vivo genome editing. CRISPR libraries should be designed to ideally select those able to target the mutation both in mouse and human genes with high on-target activity and low off-target activity, so that the effectiveness of the same gRNA could be studied first on human HGPS fibroblasts and then in vivo on the Lmna G609G/G609G mice.
Recent studies have shown the involvement of microRNAs and in particular, mir-9 which prevents progerin accumulation in HGPS neurons. Therefore, another possibility for developing new therapeutic approaches for progeria would be the modulation of microRNAs, taking into consideration off-targets that, again, must be evaluated in vitro and in vivo before transposition to humans [103].
Funding Statement
This work was supported by grants from Institut National de la Santé et de la Recherche Médicale (INSERM), Aix-Marseille University, A*Midex Foundation (VinTAGE Program) and the Association Française contre les Myopathies (AFM grant MNH-Decrypt 2011–2015 and TRIM-RD 2016–2020 to NL). This study is part of the FHU A*MIDEX project MARCHE n.ANR-11-IDEX-001-02 funded by the “Investissement d'avenir” French governmental program, managed by the French National Research Agency (ANR).
Abbreviations
- AON
Antisense Oligonucleotides;
- ATM
Ataxia Telangiectasia Mutated;
- BAF
Barrier-to-Autointegration Factor;
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats;
- DOT1L
Disruptor Of Telomeric silencing 1-Like;
- DSB
Double-Strand Break;
- EZH2
Enhancer of Zeste Homolog 2 polycomb repressive complex 2 subunit;
- FTase
Farnesyltransferase;
- FTI
Farnesyl Transferase Inhibitor;
- GGTI
GeranylGeranylTransferase Inhibitor;
- HDR
Homology-Directed Repair;
- HGPS
Hutchinson-Gilford Progeria Syndrome;
- HP1
Heterochromatin Protein 1;
- ICMT
Isoprenylcysteine CarboxylMethylTransferase;
- KLF4
Krüppel-Like Factor 4.
- Lap2α
Lamina-associated Polypeptide 2α;
- MAD-B
Mandibuloacral Dysplasia type B;
- Mtor
Mammalian Target Of Rapamycin;
- MSC
Mesenchymal Stem Cells;
- NAC
N-Acetyl Cysteine;
- NAT10
N-acetyltransferase 10;
- NFκB
Nuclear Factor kappa B;
- NHEJ
Non-Homologous End Joining;
- NRF2
Nuclear Factor-E2-related factor 2;
- OCT4
Octamer-binding transcription factor 4;
- PML-NB
ProMyelocytic Leukemia-Nuclear Body;
- ROS
Reactive oxidative species;
- SIRT-1
Sirtuin-1;
- SOX2
Sex determining region Y-box 2;
- SRSF-1
Serine/arginine-Rich Splicing Factor 1;
- TNF
Tumor Necrosis Factor;
- VSMC
Vascular Smooth Muscle Cells;
- ZOPRA
ZOledronate-PRAvastatin;
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603–24. [DOI] [PubMed] [Google Scholar]
- [2].Merideth MA, Gordon LB, Clauss S, et al.. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hutchinson J. Congenital absence of hair and mammary glands with atrophic condition of the skin and its appendages, in a boy whose mother had been almost wholly bald from Alopecia Areata from the age of six. Med Chir Trans. 1886;69:473–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gilford H. On a condition of mixed premature and immature development. Med Chir Trans. 1897;80:17-46:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].De Sandre-Giovannoli A, Bernard R, Cau P, et al.. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. [DOI] [PubMed] [Google Scholar]
- [6].Eriksson M, Brown WT, Gordon LB, et al.. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Goldman RD, Shumaker DK, Erdos MR, et al.. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Capell BC, Erdos MR, Madigan JP, et al.. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huang S, Chen L, Libina N, et al.. Correction of cellular phenotypes of Hutchinson-Gilford Progeria cells by RNA interference. Hum Genet. 2005;118:444–50. [DOI] [PubMed] [Google Scholar]
- [11].Burtner CR, Kennedy BK. Progeria syndromes and ageing: What is the connection? Nat Rev Mol Cell Biol. 2010;11:567–78. [DOI] [PubMed] [Google Scholar]
- [12].Liu B, Wang J, Chan KM, et al.. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11:780–5. [DOI] [PubMed] [Google Scholar]
- [13].Hilton BA, Liu J, Cartwright BM, et al.. Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. FASEB J. 2017;31:3882–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Viteri G, Chung YW, Stadtman ER. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech Ageing Dev. 2010;131:2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rivera-Torres J, Acin-Perez R, Cabezas-Sanchez P, et al.. Identification of mitochondrial dysfunction in Hutchinson-Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J Proteomics. 2013;91:466–77. [DOI] [PubMed] [Google Scholar]
- [16].Columbaro M, Capanni C, Mattioli E, et al.. Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell Mol Life Sci. 2005;62:2669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kubben N, Adriaens M, Meuleman W, et al.. Mapping of lamin A- and progerin-interacting genome regions. Chromosoma. 2012;121:447–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].McCord RP, Nazario-Toole A, Zhang H, et al.. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23:260–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shumaker DK, Dechat T, Kohlmaier A, et al.. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103:8703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10:452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu Y, Wang Y, Rusinol AE, et al.. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. Faseb J. 2008;22:603–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Manju K, Muralikrishna B, Parnaik VK. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J Cell Sci. 2006;119:2704–14. [DOI] [PubMed] [Google Scholar]
- [23].Prokocimer M, Barkan R, Gruenbaum Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell. 2013;12:533–43. [DOI] [PubMed] [Google Scholar]
- [24].Hernandez L, Roux KJ, Wong ESM, et al.. Functional coupling between the extracellular matrix and nuclear Lamina by wnt signaling in Progeria. Dev Cell. 2010;19:413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Osorio FG, Barcena C, Soria-Valles C, et al.. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012;26:2311–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kubben N, Zhang W, Wang L, et al.. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell. 2016;165:1361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cau P, Navarro C, Harhouri K, et al.. Nuclear matrix, nuclear envelope and premature aging syndromes in a translational research perspective. Seminars Cell Dev Biol. 2014;29:125–47. [DOI] [PubMed] [Google Scholar]
- [28].Delbarre E, Tramier M, Coppey-Moisan M, et al.. The truncated prelamin A in Hutchinson-Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum Mol Genetics. 2006;15:1113–22. [DOI] [PubMed] [Google Scholar]
- [29].Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res. 2006;47:15–31. [DOI] [PubMed] [Google Scholar]
- [30].Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genetics. 2005;14:2959–69. [DOI] [PubMed] [Google Scholar]
- [31].Toth JI, Yang SH, Qiao X, et al.. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102:12873–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang SH, Bergo MO, Toth JI, et al.. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A. 2005;102:10291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mallampalli MP, Huyer G, Bendale P, et al.. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:14416–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yang SH, Meta M, Qiao X, et al.. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006;116:2115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yang SH, Andres DA, Spielmann HP, et al.. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J Clin Invest. 2008;118:3291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Capell BC, Olive M, Erdos MR, et al.. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc Natl Acad Sci U S A. 2008;105:15902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gordon LB, Kleinman ME, Miller DT, et al.. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109:16666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gordon LB, Massaro J, D'Agostino RB, et al.. Impact of Farnesylation inhibitors on survival in Hutchinson-Gilford Progeria syndrome. Circulation. 2014;130:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Varela I, Pereira S, Ugalde AP, et al.. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14:767–72. [DOI] [PubMed] [Google Scholar]
- [40].Gordon LB, Kleinman ME, Massaro J, et al.. Clinical Trial of the Protein Farnesylation inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson-Gilford Progeria Syndrome. Circulation. 2016;134:114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Davies BS, Barnes RH, 2nd Tu Y, et al.. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genetics. 2010;19:2682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Blondel S, Egesipe AL, Picardi P, et al.. Drug screening on Hutchinson Gilford progeria pluripotent stem cells reveals aminopyrimidines as new modulators of farnesylation. Cell Death Dis. 2016;7:e2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ibrahim MX, Sayin VI, Akula MK, et al.. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science. 2013;340:1330–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cao K, Graziotto JJ, Blair CD, et al.. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011;3:89ra58. [DOI] [PubMed] [Google Scholar]
- [45].Graziotto JJ, Cao K, Collins FS, et al.. Rapamycin activates autophagy in Hutchinson-Gilford progeria syndrome: implications for normal aging and age-dependent neurodegenerative disorders. Autophagy. 2012;8:147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cenni V, Capanni C, Columbaro M, et al.. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria (vol 55, e36, 2011). Eur J Histochem. 2013;57:283–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ramos FJ, Chen SC, Garelick MG, et al.. Rapamycin reverses elevated mTORC1 signaling in Lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Translational Med. 2012;4:144ra103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liao CY, Anderson SS, Chicoine NH, et al.. Rapamycin reverses metabolic deficits in Lamin A/C-Deficient mice. Cell Rep. 2016;17:2542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cho HJ, Park J, Lee HW, et al.. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem Bioph Res Co. 2004;321:942–8. [DOI] [PubMed] [Google Scholar]
- [50].Yeh WC, Bierer BE, Mcknight SL. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3t3-L1 cells. Proc Natl Acad Sci U S A. 1995;92:11086–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gabriel D, Roedl D, Gordon LB, et al.. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell. 2015;14:78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gabriel D, Shafry DD, Gordon LB, et al.. Intermittent treatment with farnesyltransferase inhibitor and sulforaphane improves cellular homeostasis in Hutchinson-Gilford progeria fibroblasts. Oncotarget. 2017;8:64809–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kubben N, Brimacombe KR, Donegan M, et al.. A high-content imaging-based screening pipeline for the systematic identification of anti-progeroid compounds. Methods. 2016;96:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pellegrini C, Columbaro M, Capanni C, et al.. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget. 2015;6:29914–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Harhouri K, Navarro C, Depetris D, et al.. MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol Med. 2017;9:1294–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Osorio FG, Navarro CL, Cadinanos J, et al.. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3:106ra7. [DOI] [PubMed] [Google Scholar]
- [57].Lee JM, Nobumori C, Tu Y, et al.. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J Clin Invest. 2016;126:1592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Barthelemy F, Navarro C, Fayek R, et al.. Truncated prelamin A expression in HGPS-like patients: a transcriptional study. Eur J Hum Genet. 2015;23:1051–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Harhouri K, Navarro C, Baquerre C, et al.. Antisense-based Progerin downregulation in HGPS-Like Patients' cells. Cells. 2016;5. pii: E31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lopez-Mejia IC, Vautrot V, De Toledo M, Behm-Ansmant I, et al.. A conserved splicing mechanism of the LMNA gene controls premature aging. Hum Mol Genetics. 2011;20:4540–55. [DOI] [PubMed] [Google Scholar]
- [61].Larsson O, Morita M, Topisirovic I, et al.. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc Natl Acad Sci U S A. 2012;109:8977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Egesipe AL, Blondel S, Cicero AL, et al.. Metformin decreases progerin expression and alleviates pathological defects of Hutchinson-Gilford progeria syndrome cells. NPJ Aging Mechan Dis. 2016;2:16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Park SK, Shin OS. Metformin alleviates ageing cellular phenotypes in Hutchinson-Gilford progeria syndrome dermal fibroblasts. Exp Dermatol. 2017;26:889–95. [DOI] [PubMed] [Google Scholar]
- [64].Richards SA, Muter J, Ritchie P, et al.. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum Mol Genetics. 2011;20:3997–4004. [DOI] [PubMed] [Google Scholar]
- [65].Tilstra JS, Clauson CL, Niedernhofer LJ, et al.. NF-kappaB in aging and disease. Aging Dis. 2011;2:449–65. [PMC free article] [PubMed] [Google Scholar]
- [66].Kang HT, Park JT, Choi K, et al.. Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Aging Cell. 2017;16:541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Cui W, Bai Y, Luo P, et al.. Preventive and therapeutic effects of MG132 by activating Nrf2-ARE signaling pathway on oxidative stress-induced cardiovascular and renal injury. Oxidative Med Cell Longevity. 2013;2013:306073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang Y, Sun W, Du B, et al.. Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: roles of Nrf2 and NF-kappaB. Am J Physiol Heart Circulatory Physiol. 2013;304:H567–78. [DOI] [PubMed] [Google Scholar]
- [69].Dreger H, Westphal K, Wilck N, et al.. Protection of vascular cells from oxidative stress by proteasome inhibition depends on Nrf2. Cardiovascular Res. 2010;85:395–403. [DOI] [PubMed] [Google Scholar]
- [70].Rivera-Torres J, Acin-Perez R, Cabezas-Sanchez P, et al.. Identification of mitochondrial dysfunction in Hutchinson-Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J Proteomics. 2013;91:466–77. [DOI] [PubMed] [Google Scholar]
- [71].Villa-Bellosta R, Rivera-Torres J, Osorio FG, et al.. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford Progeria Syndrome that is Ameliorated on Pyrophosphate treatment. Circulation. 2013;127:2442–51. [DOI] [PubMed] [Google Scholar]
- [72].Xiong ZM, Choi JY, Wang K, et al.. Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Aging Cell. 2016;15:279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Soria-Valles C, Osorio FG, Gutierrez-Fernandez A, et al.. NF-kappaB activation impairs somatic cell reprogramming in ageing. Nat Cell Biol. 2015;17:1004–13. [DOI] [PubMed] [Google Scholar]
- [74].Ortiz-Lazareno PC, Hernandez-Flores G, Dominguez-Rodriguez JR, et al.. MG132 proteasome inhibitor modulates proinflammatory cytokines production and expression of their receptors in U937 cells: involvement of nuclear factor-kappaB and activator protein-1. Immunology. 2008;124:534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hozhabri NST, Kim H, Varanasi V. NF-kappa B inhibitor MG132 enhances differentiation and collagen expression of dental pulp stem cells. Faseb J. 2014;28 [Google Scholar]
- [76].Ahmed AS, Ahmed M, Li J, et al.. Proteasome inhibitor MG132 modulates inflammatory pain by central mechanisms in adjuvant arthritis. Int J Rheum Dis. 2017;20:25–32. [DOI] [PubMed] [Google Scholar]
- [77].Olive M, Harten I, Mitchell R, et al.. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30:2301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Chatterjee PK, Yeboah MM, Dowling O, et al.. Nicotinic acetylcholine receptor agonists attenuate septic acute kidney injury in mice by suppressing inflammation and proteasome activity. PloS One. 2012;7:e35361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Miao X, Cui W, Sun W, et al.. Therapeutic effect of MG132 on the aortic oxidative damage and inflammatory response in OVE26 type 1 diabetic mice. Oxidative Med Cell Longevity. 2013;2013:879516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Feng B, Zhang Y, Mu J, et al.. Preventive effect of a proteasome inhibitor on the formation of accelerated atherosclerosis in rabbits with uremia. J Cardiovascular Pharmacol. 2010;55:129–38. [DOI] [PubMed] [Google Scholar]
- [81].Larrieu D, Britton S, Demir M, et al.. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014;344:527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bouillon R, Carmeliet G, Verlinden L, et al.. Vitamin D and human health: Lessons from Vitamin D receptor null mice. Endocr Rev. 2008;29:726–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Plum LA, DeLuca HF. Vitamin D, disease and therapeutic opportunities. Nat Rev Drug Discovery. 2010;9:941–55. [DOI] [PubMed] [Google Scholar]
- [84].Kreienkamp R, Croke M, Neumann MA, et al.. Vitamin D receptor signaling improves Hutchinson-Gilford progeria syndrome cellular phenotypes. Oncotarget. 2016;7:30018–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lee SJ, Jung YS, Yoon MH, et al.. Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J Clin Invest. 2016;126:3879–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Liu BH, Ghosh S, Yang X, et al.. Resveratrol Rescues SIRT1-Dependent Adult Stem Cell Decline and Alleviates Progeroid Features in Laminopathy-Based Progeria. Cell Metab. 2012;16:738–50. [DOI] [PubMed] [Google Scholar]
- [87].Varela I, Pereira S, Ugalde AP, et al.. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14:767–72. [DOI] [PubMed] [Google Scholar]
- [88].Schmidt E, Nilsson O, Koskela A, et al.. Expression of the Hutchinson-Gilford progeria mutation during osteoblast development results in loss of osteocytes, irregular mineralization, and poor biomechanical properties. J Biol Chem. 2012;287:33512–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Strandgren C, Nasser HA, McKenna T, et al.. Transgene silencing of the Hutchinson-Gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J. 2015;29:3193–205. [DOI] [PubMed] [Google Scholar]
- [90].Howells LM, Berry DP, Elliott PJ, et al.. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases–safety, pharmacokinetics, and pharmacodynamics. Cancer Prevention Res. 2011;4:1419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ocampo A, Reddy P, Martinez-Redondo P, et al.. In Vivo Amelioration of age-associated Hallmarks by partial reprogramming. Cell. 2016;167:1719–33 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Ocampo A, Reddy P, Izpisua Belmonte JC. Anti-aging strategies based on cellular reprogramming. Trends Mol Med. 2016;22:725–38. [DOI] [PubMed] [Google Scholar]
- [93].Liu GH, Barkho BZ, Ruiz S, et al.. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zhang JQ, Lian QZ, Zhu GL, et al.. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and Mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45. [DOI] [PubMed] [Google Scholar]
- [95].Atchison L, Zhang HY, Cao K, et al.. A tissue engineered blood vessel model of Hutchinson-Gilford Progeria Syndrome using human iPSC-derived smooth muscle cells. Sci Rep-Uk. 2017;7:8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. [DOI] [PubMed] [Google Scholar]
- [98].Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genetics. 2014;15:445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discovery. 2017;16:387–99. [DOI] [PubMed] [Google Scholar]
- [101].Suzuki K, Tsunekawa Y, Hernandez-Benitez R, et al.. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ran FA, Cong L, Yan WX, et al.. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–U98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Frankel D, Delecourt V, Harhouri K, et al. MicroRNAs in hereditary and sporadic premature aging syndromes and other laminopathies. Aging Cell. 2018;e12766. [DOI] [PMC free article] [PubMed] [Google Scholar]