

Abstract
Mutations in LMNA, encoding A-type lamins, are responsible for laminopathies including muscular dystrophies, lipodystrophies, and premature ageing syndromes. LMNA mutations have been shown to alter nuclear structure and stiffness, binding to partners at the nuclear envelope or within the nucleoplasm, gene expression and/or prelamin A maturation. LMNA-associated lipodystrophic features, combining generalized or partial fat atrophy and metabolic alterations associated with insulin resistance, could result from altered adipocyte differentiation or from altered fat structure.
Recent studies shed some light on how pathogenic A-type lamin variants could trigger lipodystrophy, metabolic complications, and precocious cardiovascular events. Alterations in adipose tissue extracellular matrix and TGF-beta signaling could initiate metabolic inflexibility. Premature senescence of vascular cells could contribute to cardiovascular complications. In affected families, metabolic alterations occur at an earlier age across generations, which could result from epigenetic deregulation induced by LMNA mutations. Novel cellular models recapitulating adipogenic developmental pathways provide scalable tools for disease modeling and therapeutic screening.
KEYWORDS: Lamin A/C, lipodystrophy, adipose tissue, differentiation, senescence, extracellular matrix, anticipation, epigenetics, induced pluripotent stem cells, metreleptin
Introduction
Lipodystrophic syndromes are rare diseases characterized by generalized or partial fat atrophy (lipoatrophy) and metabolic alterations resulting from insulin resistance (glucose tolerance abnormalities, dyslipidemia, non-alcoholic fatty liver disease). Obesity and lipodystrophy share similar metabolic defects thus illustrating the complex relationships between deregulation of adipose tissue and systemic metabolism. Several authors postulate that a personal threshold controls the individual capacity to store nutrient excess as triglycerides in the lipid droplet of white subcutaneous adipocytes [1–3] When the nutrient intake exceeds adipose tissue storage capacity, this results in ectopic lipid deposition in non-adipose tissues and in metabolic inflexibility. These alterations characterize both metabolically-unhealthy obesity and lipodystrophic diseases. Accordingly, a recent genomic study, performed in the general population, has revealed that limited peripheral adipose storage capacity is a major determinant of insulin resistance [4].
Important questions remain unanswered regarding factors that modulate the personal threshold of adipocyte expandability. These factors depend on multiple mechanisms, notably formation of new adipocytes from precursors (adipogenesis) and biogenesis, maintenance and regulation of the adipocyte lipid droplet, but also interactions between adipocytes and other cellular and extracellular components of adipose tissue, and cross-talks between the different body fat depots and other organs.
Although lipodystrophic syndromes are rare diseases, deciphering their pathogenic mechanisms would provide valuable insights into adipose tissue physiology, notably regarding its capacity to adequately expand and maintain metabolic homeostasis. Major advances have been achieved since 1999 thanks to the identification of several monogenic causes of lipodystrophic syndromes [5–18]. Their description is far beyond the scope of this review, but it is worth mentioning that pathophysiological molecular mechanisms of most of them involve defects in adipocyte differentiation, in biogenesis and/or structural properties of the adipocyte lipid droplet, in triglyceride synthesis and/or in lipolysis [19]. This further stresses the important role played by the lack of lipid storage in the occurrence of post-receptor insulin resistance (Figure 1) [20]. LMNA mutations are responsible for the most frequent genetic form of lipodystrophy. However, the mechanisms by which these molecular variants alter adipocyte function remain largely unknown. Strikingly, the different LMNA mutations, located all along the gene, give rise to very diverse clinical phenotypes of laminopathies, comprising not only lipodystrophic syndromes but also dystrophic myopathies, neuropathies, premature ageing syndromes and rare overlapping syndromes. This very large clinical spectrum associated with LMNA defects, illustrates the pathophysiological complexity of laminopathies [21].
Figure 1.
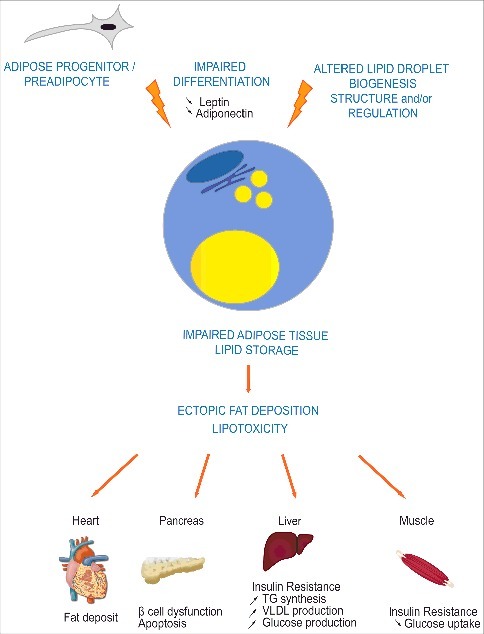
Impaired adipose tissue lipid storage induces metabolic complications in lipodystrophic syndromes. Most genes involved in lipodystrophic syndromes have been shown to regulate adipocyte differentiation, triglycerides synthesis, lipolysis, and/or lipid droplet structure or biogenesis. Impaired storage of excess energy as triglycerides in adipocytes leads to ectopic fat deposition and lipotoxicity in several tissues such as muscle, heart, liver and pancreas, resulting in post-receptor insulin resistance, dyslipidemia and liver steatosis.
After a brief overview of the pathogenic mechanisms that have been discussed since the discovery of the first laminopathies in 1999 [22], we will propose an update of some recent studies on LMNA-related lipodystrophies, dealing with some genetic, preclinical and fundamental aspects, which allow to refine or readdress the pathophysiological hypotheses.
Several pathogenic mechanisms contribute to laminopathies
A- and B-type lamins are nuclear proteins belonging to the intermediate filaments family. Lamins have been shown to play an essential role in nuclear function.
Lamins A and C, both encoded by LMNA, are developmentally regulated in most lineage precursors and are expressed in differentiated cells. They represent A-type lamins main isoforms and interact with the ubiquitous B-type lamins, encoded by LMNB1 and LMNB2. The lamin functional domains are organized into a short N-terminal head domain, a central alpha helical rod domain, driving lamin dimerization and polymerization, and a large C-terminal tail, containing the nuclear localization signal and an immunoglobulin-like fold domain with multiple binding properties [23]. A CaaX motif, at the C-terminal end, allows post-translational farnesylation of both prelamin A isoform produced by LMNA and B-type lamins. While B-type lamins retain the farnesyl moiety, thus increasing their affinity for the inner nuclear membrane, prelamin A undergoes further post-translational modifications. Farnesylated prelamin A is finally cleaved by the ZMPSTE24/FACE-1 metalloproteinase, removing its farnesylated C-terminal end, and producing a mature, non-farnesylated lamin A [24].
Lamin filaments form the lamina meshwork at the nucleoplasmic side of the inner nuclear membrane, which provides a structural support for the nucleus [25,26], and controls the functional organization of interphase chromatin [27]. At the inner nuclear periphery, lamins interact with several inner nuclear membrane proteins. Among them, the SUN-domain proteins span the inner nuclear membrane and bind to the KASH domain of proteins embedded in the outer nuclear membrane, which, in turn, bind to cytoskeletal proteins. All these proteins together form a complex that links the nucleoskeleton to the cytoskeleton [28–30]. Lamin-associated nuclear envelope proteins can impact on chromatin, and influence the spatial positioning of developmental genes in a tissue-specific manner [31,32]. Through these multistep interactions, lamins control nuclear stiffness and mechano-sensitivity, which are strongly modified during stem cell differentiation [33–35]. In addition, A- type lamin filaments, although mainly localized at the nuclear periphery, are also found in the nucleoplasm, where they interact with lamina-associated protein 2alpha (LAP2alpha), a modulator of cell-cycle progression and apoptosis [36], and where they regulate several other signaling proteins and transcription factors [37]. Lamins also bind DNA and histones, ensuring the formation of multiprotein complexes associated with chromatin, able to regulate the expression of genes such as retinoblastoma protein (Rb) and barrier-to-integration factor (BAF) [37]. Importantly, lamins organize chromatin at the nuclear periphery through lamin-associated domains (LAD) [38], and regulate interactions with epigenetic factors such as the Polycomb group of proteins [39]. Thus, there is increasing evidence that A-type lamins epigenetically influence stem cell differentiation and tissue-specific developmental programs [40–42]. As many structural and regulatory roles of A-type lamins are impaired by LMNA mutations, the pathophysiological mechanisms of the different laminopathies could involve distinct pathways.
Defects in adipocyte differentiation in LMNA-associated lipodystrophies
The clinical features of LMNA-associated lipodystrophic syndromes are reviewed by David Araujo-Vilar in this journal issue. The typical familial partial lipodystrophy of the Dunnigan type (FPLD2, OMIM #151660) is mainly due to heterozygous amino acid substitutions at the 482nd position in the C-terminal domain of A-type lamins, the most frequent being the p.Arg482Trp variant. Closely related lipodystrophic phenotypes are due to other point mutations in the immunoglobulin-fold domain [6,7,43]. In contrast to LMNA mutations involved in muscular dystrophies or cardiomyopathies, lipodystrophy-causing mutations do not disrupt the tridimensional structure of A-type lamins but modify a positively charged amino acid at the surface of their C-terminal domain [44,45].
In accordance, several studies have confirmed that LMNA mutations specific for lipodystrophies result in modified interactions of the protein C-terminal domain with distinctive partners in vitro. Thus, in vitro studies have revealed that two FPLD-causing mutations, LMNA p.Gly465Asp and p.Lys486Asn, alter the lamin A C-terminal tail SUMOylation, a posttranslational modification known to regulate the localization, interactions and functions of proteins [46]. The LMNA p.Arg482Leu mutation down-regulates Notch signaling in mesenchymal stem cells, decreasing their adipogenic potential [47]. SREBP1c, an important transcription factor driving adipogenesis, binds differently wild-type and lipodystrophy-causing lamin A variants [48,49]. In addition, the LMNA p.Arg482Trp and p.Arg482Gln mutations impair the interaction between lamin A and DNA in vitro [50]. It has been shown that lamin A, SREBP1 and its DNA responsive elements form ternary complexes in vitro, and that Arg482Trp lamin A deregulates SREBP1 activity in patients’ cells [51], suggesting that it could disrupt adipocyte differentiation. The expression of Arg482Trp or Arg482Gln lamin A, but also overexpression of wild-type lamin A, inhibit adipocyte differentiation of 3T3-L1 cells [52]. Recently, the Fragile X related protein (FXR1P), a mRNA binding protein, was identified as a lamin A partner at the nuclear envelope. Its expression and localization inside the nucleus are modified in the presence of lamin A bearing lipodystrophy-causing mutations [53]. The expression of Arg482Trp lamin A in human adipose stem cells increases FXR1P protein expression and impairs adipocyte differentiation through a process involving epigenetic and conformational changes in chromatin organization [42,53].
At the clinical level, FPLD2 illustrates how the different body fat depots, which are characterized by distinct developmental origins [54], respond in a very different manner to the same constitutional LMNA mutation. Indeed, while patients’ subcutaneous fat mass at the limbs and buttocks level is severely decreased, the mass of cervical, facial, perineal and visceral depots is increased. In addition, the lipodystrophic phenotype becomes apparent generally after puberty, and is more pronounced in women [43,55,56]. In agreement with the hypothesis of impaired adipogenesis induced by LMNA mutations, we and others reported that expression of adipogenic genes was altered in adipose tissue from patients with FPLD2, both at thigh [57,58] and cervical levels [59], with a decreased expression of the master adipogenic factor PPAR-gamma. Dystrophic features characterized not only lipoatrophic adipose tissue, but also lipomatous areas, and accumulated cervical fat, from patients with FPLD2 [57–59].
In addition to FPLD2, due to hotspot mutations in the C-terminal region, lipodystrophic features are also observed in uncommon forms of complex laminopathies due to mutations affecting different protein domains of A type-lamins. These mixed forms associate lipodystrophy and muscular and/or cardiac symptoms [60–62], and also often signs of premature aging [63–69]. Mandibulo-acral dysplasia, due to mutations in LMNA or ZMPSTE24, was identified as the first laminopathy associating premature ageing and lipodystrophic features [63,64]. This phenotypic combination was further observed with other LMNA mutations in typical Hutchinson-Gilford progeria [66,67] or in atypical progeroid syndromes [65,68,69].
In that setting, similar cellular defects have been observed in different laminopathies. Nuclear abnormal morphology and nuclear envelope disorganization appear as hallmarks of human cultured laminopathic cells, independently of the associated clinical presentation [66,67,70–75]. The typical LMNA mutation responsible for Hutchinson-Gilford progeria results in the expression of a constitutively farnesylated prelamin A pathogenic variant, called progerin. Although other lipodystrophy-causing mutations in LMNA do not directly modify the proteolytic maturation site of the protein, they could secondarily alter its maturation and result in prelamin A accumulation. Accordingly, we and others observed an accumulation of prelamin A in cells and/or tissues from patients with FPLD2 or other LMNA-related lipodystrophies associated or not with progeroid signs [57,76,77]. Although this prelamin A accumulation is controversial [78], some HIV protease inhibitors, used as antiretroviral drugs, and involved in the development of a lipodystrophic syndrome with premature cellular senescence [75], were also shown to increase the cellular amount of farnesylated prelamin A through inhibition of ZMPSTE24 [77,79]. Prelamin A accumulation, by sequestrating SREBP1c at the nuclear periphery, may alter adipogenesis [76,80,81]. Accumulated prelamin A could sequestrate the transcription factor Sp1, resulting in altered extracellular matrix gene expression and adipose lineage differentiation of human mesenchymal stem cells [82]. In addition, prelamin A and progerin were shown to induce the recruitment of the chromatin remodeling factor BAF inside the nucleus, which could result in altered gene expression [83,84].
However, accumulation of farnesylated prelamin A is not mandatory for the development of lipodystrophic diseases upon expression of lipodystrophy-causing lamin A variant. We have described a pathogenic homozygous frameshift mutation in LMNA, leading to the synthesis of a prelamin A variant lacking the consensus CaaX farnesylation site. This variant results in the expression of a non-farnesylated form of prelamin A, without any production of mature lamin A, and is responsible for a severe lipodystrophic syndrome [85]. Other studies also showed that observation of prelamin A accumulation may depend on the antibodies used, and is not a prerequisite for lipodystrophy diseases [78,86].
Taken as a whole these studies show that lipodystrophy-causing LMNA mutations could result in several defects leading to defective adipocyte differentiation. Several recent studies extend and refine these hypotheses, further linking pathogenic molecular mechanisms to clinical features.
Early extracellular matrix alterations in lipodystrophic laminopathies
Altered adipose tissue extracellular matrix (ECM), acknowledged as a major contributor to metabolic alterations associated with obesity [87], has also been observed in patients with lipodystrophic syndromes of different etiologies [15,17,59,88].
Le Dour et al generated transgenic mice overexpressing the human p.Arg482Gln pathogenic lamin A variant specifically in adipose tissue, and also studied a transgenic mice expressing higher levels of p.Arg482Gln lamin A [89]. The severity of the lipodystrophic phenotype in mice probably depends on the level of expression of p.Arg482Gln lamin A, since only the latter mouse model displayed a decreased capacity to accumulate body fat, associated with decreased insulin sensitivity and liver steatosis [90]. However, ECM alterations were observed in adipose tissue from mice overexpressing p.Arg482Gln lamin A only in fat tissue, similar to those reported in adipose tissue from patients with FPLD2, even though these mice did not show overt lipoatrophy. This suggests that these tissular abnormalities may be early defects in the pathogenesis of the disease [59,90]. Indeed, human and mice subcutaneous adipose tissue expressing p.Arg482Gln lamin A displayed increased fibrosis and decreased mean adipocyte area. In addition, the level of gene expression of fibronectin, which binds type 1 collagen and is involved in the maintenance of adipocyte shape, was increased. Conversely, the level of gene expression of elastin, a major component of elastic fibers providing strength and flexibility to connective tissue, and of decorin, which also binds to type 1 collagen and participates to matrix assembly, was decreased. Similar ECM abnormalities were also observed in cultured fibroblasts from patients with FPLD2 or other LMNA-associated lipodystrophic syndromes. These abnormalities were linked to activation of TGF-beta signaling, a driver of matrix deposition, and were associated with increased expression and activity of matrix metalloproteinase 9, an endopeptidase that degrades ECM proteins.
These results suggest that an early detrimental remodeling of fat ECM develops upon expression of lipodystrophy-causing lamin A variants in adipose tissue. This could hamper adipocyte differentiation and contribute to the limited capacity of fat storage, previously shown to induce adipose tissue dysfunction, fatty acid spillover to non-adipose organs, lipotoxicity and associated metabolic defects in humans (Figure 2).
Figure 2.

An early detrimental remodeling of adipose tissue extracellular matrix could contribute to the pathophysiology of LMNA-associated lipodystrophies. Extracellular matrix alterations induced by lipodystrophy-causing LMNA mutations could hamper adipocyte differentiation and limit the expandability of adipose tissue, triggering adipocyte dysfunction and metabolic defects.
ECM alterations with fibrosis, altered metalloproteinase activity and/or increased TGF-beta signaling have been previously described in LMNA-linked cardiomyopathy [91,92], mandibulo-acral dysplasia [93–95], and Hutchinson-Gilford progeria (Figure 3) [96]. This suggests that ECM alterations, triggered by several LMNA mutations in different tissues, could globally contribute to the pathophysiology of laminopathies, and that antagonists of TGF-beta may have potential therapeutic benefit in these diseases.
Figure 3.

Several laminopathies are characterized by tissular fibrosis, increased TGF-beta signaling and/or metalloproteinase expression/activity. Extracellular matrix alterations at the level of adipose tissue, vascular wall or heart have been described in several laminopathies and participate to the clinical phenotype and to the complications of laminopathies. 59,90-96
Premature senescence and osteoblast-like differentiation of smooth vascular cells in lipodystrophic laminopathies
Cardiovascular events are frequent and precocious in patients with FPLD2, inasmuch as they are frequently exposed to dyslipidemia, insulin resistance and/or diabetes [97,98]. However, in addition to metabolic risk factors, FPLD2-associated LMNA mutations could have a direct impact on the vascular wall cells.
In Hutchinson-Gilford progeria, severe premature atherosclerosis leads to myocardial infarction and strokes, the major causes of patients’ death at a mean age of 14.6 years. This has been linked to accumulation of progerin, a farnesylated mutated form of prelamin A expressed in patient's cells [99–101]. We observed, in FPLD2, that p.Arg482Trp prelamin A accumulated abnormally at the nuclear envelope and induced endothelial cell dysfunction with increased oxidative stress and cellular senescence [98]. Additionally, we recently showed that several LMNA mutations, either leading to a lipodystrophy typical of the FPLD2 type, or associated with signs of premature ageing, also triggered vascular smooth muscle cell senescence with osteoblastic transdifferentiation and calcification [102]. This could lead to early vascular calcifications, as observed in patients [102]. All together, these studies suggest that LMNA mutations responsible for lipodystrophies may directly affect the arterial wall, resulting in early atherosclerosis and vascular calcification, in addition to atherosclerotic lesions resulting from associated metabolic risk factors (Figure 4). In human induced pluripotent stem cells, p.R482W lamin A was recently shown to deregulate the network of genes involved in early vascular differentiation, which is also in favor of a cell-autonomous origin of endothelial cell dysfunction in FPLD2 [103]. This should encourage researchers to develop therapeutic strategies aiming at minimizing the cellular amount and toxicity of pathogenic A-type lamin variants, not only in Hutchinson-Gilford progeria but also in LMNA-linked lipodystrophic diseases.
Figure 4.

Vascular effects of LMNA mutations causing lipodystrophy. A. Prelamin-A physiologically undergoes a complex post-translational maturation process affecting its C-terminal CaaX motif. After farnesylation of the carboxy-terminal cysteine, aaX amino acids are removed, farnesyl-cysteine is carboxymethylated, then the 15 C-terminal amino acids are cleaved by the metalloprotease ZMPSTE24 to produce mature lamin A. B. Lipodystrophy-causing mutations in LMNA lead to accumulation of farnesylated prelamin A and nuclear envelope disorganization. C. Accumulation of prelamin A pathogenic variants at the nuclear envelope induces oxidative stress, inflammation and cellular senescence. These cellular alterations contribute to endothelial cell dysfunction and to osteoblastic transdifferentiation of vascular smooth muscle cell, promoting atherosclerosis and vascular calcification.
Anticipation of metabolic complications in lipodystrophic laminopathies
The study of our cohort, that represents the largest cohort of familial forms of LMNA-associated lipodystrophies reported to date (85 patients from 24 families), revealed that diabetes and hypertriglyceridemia occurred at an earlier age over successive generations (Figure 5) [104]. This happened independently of the potential earlier screening of metabolic alterations over time. In contrast, lipodystrophy, which is the earliest clinical feature, appeared at similar age in all patients. Notably, body mass index and total fat mass were similar in patients from different generations, showing that these factors cannot account by themselves for this observation. This major decrease in the age at onset of metabolic complications provides one of the very rare examples of anticipation in a Mendelian disorder that does not fit with the well-known model of trinucleotide repeat disorders [105]. Recognition of this phenomenon is important for proper management of the disease. In this regard, we propose to perform presymptomatic genetic screening during childhood in affected families, with reinforcement of metabolic monitoring and adoption of preventive lifestyle measures in young affected patients.
Figure 5.
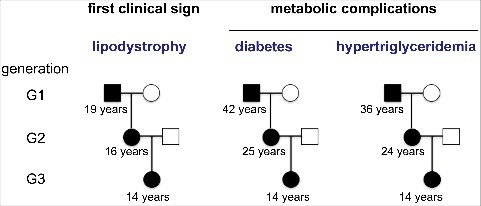
Anticipation of metabolic complications in lipodystrophic laminopathies. The comparison of the natural history and disease severity in familial forms of lipodystrophic syndromes due to LMNA pathogenic variants reveals similar characteristics and age at onset for lipodystrophy, but an anticipation of metabolic complications over generations. The mean age at onset of each clinical sign is indicated below symbols representing patients from different generations.
This anticipation phenomenon has to be considered in the light of recent studies, showing that LMNA mutations could modify, by epigenetic mechanisms, the chromatin architecture regulated by lamins during development [38,106–108]. Complications of lipodystrophic laminopathies might thus be sensitive to environmental agents triggering chromatin rearrangements. As an example, several environmental stressors, including prenatal exposure to parental disease, have been shown to drive type 2 diabetes inheritance at the epigenetic level [109]. Disruptions in gene expression could alter either adipocyte [42,110,111] or myogenic [41] cell differentiation, depending on the type of LMNA mutation. These studies, reviewed by Briand and Collas in this issue, suggest that lamin A mutations could differently alter the cell fate of different cell lineages. They provide major keys to understand how different pathogenic mutations in the same LMNA gene can lead to tissue-specific phenotypes in humans.
Recombinant leptin therapy in the management of metabolic alterations in patients with LMNA-associated lipodystrophies
International guidelines for the management of lipodystrophy syndromes were recently published [112]. Patients with LMNA-associated lipodystrophies are mainly treated with therapies recommended for classical diabetes and dyslipidemia. Among them, diet and exercise are of major importance to reduce insulin resistance and metabolic complications. Metformin is a first-line therapy for insulin resistance and diabetes. Hypoglycemic agents, including insulin, can be useful, although their efficiency has not been specifically studied in these rare diseases. Lipids should also be managed in accordance with guidelines for the general population, although stricter targets for LDL-cholesterol may be discussed in the presence of several metabolic and cardiovascular risk factors. There is no current treatment that can reconstitute adipose tissue, but plastic surgery can be helpful when lipodystrophy causes psychological distress and/or physical discomfort [112].
Leptin deficiency, which correlates with the decreased amount of body fat, was shown to contribute to metabolic complications of lipodystrophies, whatever their underlying molecular mechanisms, both in mice [113,114] and humans [115,116]. Recombinant leptin (metreleptin) therapy is approved in the US and Japan for the treatment of lipodystrophic syndromes, and is available through named compassionate programs in several European countries. Treatment with metreleptin decreases insulin resistance, hyperglycemia, dyslipidemia and liver steatosis in hypoleptinemic lipodystrophic patients, in part independently of an improved control of deregulated eating behavior [115–118]. However, this treatment is less efficient in partial forms of lipodystrophies than in generalized ones [119]. We have recently shown that metreleptin improves not only insulin sensitivity, but also insulin secretion in patients with lipodystrophies, which could result from decreased lipotoxicity in pancreatic islets [120]. Insulin secretion also improved under metreleptin therapy in the subgroup of patients with lipodystrophic syndromes due to LMNA mutations, either of the FPLD2 type or associated with mixed laminopathic phenotypes. We also confirmed that the effect of metreleptin on glucose control was related to the severity of baseline hyperglycemia. In addition, we observed, in these patients, that one-year metreleptin therapy significantly decreased the plasma concentrations of proprotein convertase subtilisin/kexin type 9 (PCSK9) [121]. PCSK9 is an endogenous inhibitor of LDL receptor that increases LDL-cholesterol circulating levels. In accordance, metreleptin-mediated decrease in PCSK9 was associated with a reduction in the level of the proatherogenic apolipoprotein B [121].
These results further stress that metreleptin improves the metabolic consequences of LMNA-associated lipodystrophies and should be integrated in the therapeutic strategy [112].
New cellular tools to study adipocyte development in vitro
As stated above, it is likely that developmental defects affecting the adipocyte lineage underlie, at least in part, important pathophysiological mechanisms leading to lipodystrophic laminopathies.
In that setting, reprogramming of patients’ primary cells into human induced pluripotent stem cells (hiPSCs) provides a relevant cellular model for pathophysiological studies. One limitation of this strategy is the incomplete knowledge of developmental pathways leading towards the distinct human adipose depots. Indeed, several types of adipocytes co-exist in the human body. Whereas adipocytes store excess energy as triglycerides, brown adipocytes are able to dissipate energy through mitochondrial thermogenesis [122]. A third type of adipocytes, called beige adipocytes, displays thermogenic properties upon activation, and could therefore be a relevant target to treat metabolic complications of diabetes [123].
Most of the current protocols for in vitro adipocyte differentiation of hiPSCs are based on generation of embryoid bodies and/or derivation of mesenchymal stem cells prior to adipose differentiation [124]. A strategy involving overexpression of adipogenic transcription factors has also been proposed [125], but this results in a bypass of developmental pathways and therefore hampers pathophysiological studies.
Guénantin, Briand et al have recently set up an efficient protocol allowing the differentiation of hiPSCs into adipose progenitors with a dual white and beige differentiation potential [126]. This protocol recapitulates adipocyte developmental processes in vitro through mesodermal then adipose stem cells stages. Engraftment of hiPSC-derived progenitor cells allowed the generation of a well-organized human adipose tissue in vivo.
This new iPS cell-based tool may be particular relevant to study adipose differentiation in lipodystrophic laminopathies. In addition, this unlimited source of adipocytes could provide a valuable material for drug screening and further development of targeted therapeutic approaches (Figure 6).
Figure 6.
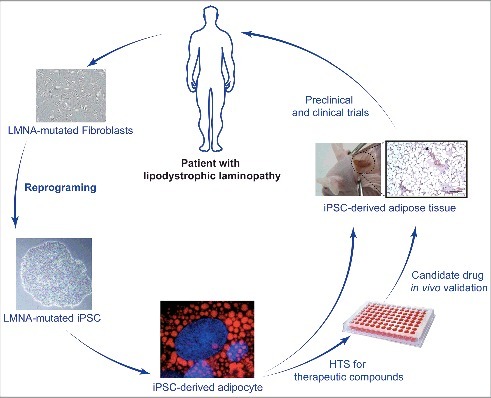
A new cellular tool for studying adipocyte development in vitro. Patients’ primary cells reprogramming into induced Pluripotent Stem Cells (iPSCs) allow to study several relevant cells types that would be otherwise inaccessible. This also avoids using non-physiological lamin overexpression strategies. Differentiation of iPS cells originating from patients with lipodystrophic laminopathies into adipocytes through a developmentally relevant protocol also provides an unlimited source of cells for high throughput screening (HTS) of therapeutic compounds, opening perspectives for the treatment of these rare diseases.
Acknowledgments
The authors thank Sorbonne Université, former Université Pierre and Marie Curie (UPMC), Institut National de la Santé et de la Recherche Médicale (INSERM), Institute of CardioMetabolism (ICAN), ANRS (France REcherche Nord&Sud Sida-hiv Hépatites), Région Ile-de-France STEM-Pole, Association Française d'Etude et de Recherche sur l'Obésité (AFERO), Fondation de France, Fondation pour la Recherche Médicale, Société Francophone du Diabète (SFD), for the financial support to our research team at Saint Antoine Research Centre, Paris, France.
References
- [1].Cuthbertson DJ, Steele T, Wilding JP, Halford JC, Harrold JA, Hamer M, Karpe F. What have human experimental overfeeding studies taught us about adipose tissue expansion and susceptibility to obesity and metabolic complications? Int J Obes (Lond). 2017;41(6):853–865. [DOI] [PubMed] [Google Scholar]
- [2].Stefan N, Schick F, Haring HU. Causes, Characteristics, and Consequences of Metabolically Unhealthy Normal Weight in Humans. Cell Metab. 2017;26(2):292–300. [DOI] [PubMed] [Google Scholar]
- [3].Pellegrinelli V, Carobbio S, Vidal-Puig A. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia. 2016;59(6):1075–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan J, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402(6764):880–3. [DOI] [PubMed] [Google Scholar]
- [6].Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9(1):109–12. [DOI] [PubMed] [Google Scholar]
- [7].Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, Schmidt H, Brabant G, Kumar S, Durrington PN, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24(2):153–6. [DOI] [PubMed] [Google Scholar]
- [8].Magre J, Delepine M, Khallouf E, Gedde-Dahl T, Jr., Van Maldergem L Sobel E, Papp J, Meier M, Megarbane A, Bachy A, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28(4):365–70. [DOI] [PubMed] [Google Scholar]
- [9].Agarwal AK, Arioglu E, De Almeida S Akkoc N, Taylor SI, Bowcock AM, Barnes RI, Garg A. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet. 2002;31(1):21–3. [DOI] [PubMed] [Google Scholar]
- [10].George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1129–34. [DOI] [PubMed] [Google Scholar]
- [12].Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, Park YE, Nonaka I, Hino-Fukuyo N, Haginoya K, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119(9):2623–33. doi: 10.1172/JCI38660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rubio-Cabezas O, Puri V, Murano I, Saudek V, Semple RK, Dash S, Hyden CS, Bottomley W, Vigouroux C, Magre J, et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1(5):280–7. doi: 10.1002/emmm.200900037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, Martinez de Villarreal L, dos Santos HG, Garg A. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87(6):866–72. doi: 10.1016/j.ajhg.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gandotra S, Le Dour C Bottomley W, Cervera P, Giral P, Reznik Y, Charpentier G, Auclair M, Delepine M, Barroso I, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364(8):740–8. doi: 10.1056/NEJMoa1007487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, Le Merrer M Le Luyer B, Heron D, Mathieu-Dramard M, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93(1):141–9. doi: 10.1016/j.ajhg.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Weedon MN, Ellard S, Prindle MJ, Caswell R, Lango Allen H, Oram R, Godbole K, Yajnik CS, Sbraccia P, Novelli G, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45(8):947–50. doi: 10.1038/ng.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Payne F, Lim K, Girousse A, Brown RJ, Kory N, Robbins A, Xue Y, Sleigh A, Cochran E, Adams C, et al. Mutations disrupting the Kennedy phosphatidylcholine pathway in humans with congenital lipodystrophy and fatty liver disease. Proc Natl Acad Sci U S A. 2014;111(24):8901–6. doi: 10.1073/pnas.1408523111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vigouroux C, Caron-Debarle M, Le Dour C Magre J, Capeau J. Molecular mechanisms of human lipodystrophies: from adipocyte lipid droplet to oxidative stress and lipotoxicity. Int J Biochem Cell Biol. 2011;43(6):862–76. [DOI] [PubMed] [Google Scholar]
- [20].Semple RK. EJE PRIZE 2015: How does insulin resistance arise, and how does it cause disease? Human genetic lessons. Eur J Endocrinol. 2016;174(5):R209–23. [DOI] [PubMed] [Google Scholar]
- [21].Worman HJ. Nuclear lamins and laminopathies. J Pathol. 2012;226(2):316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21(3):285–8. [DOI] [PubMed] [Google Scholar]
- [23].Stuurman N, Heins S, Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol. 1998;122(1-2):42–66. [DOI] [PubMed] [Google Scholar]
- [24].Barrowman J, Hamblet C, George CM, Michaelis S. Analysis of prelamin A biogenesis reveals the nucleus to be a CaaX processing compartment. Mol Biol Cell. 2008;19(12):5398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Aebi U, Cohn J, Buhle L, Gerace L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature. 1986;323(6088):560–4. [DOI] [PubMed] [Google Scholar]
- [26].Turgay Y, Eibauer M, Goldman AE, Shimi T, Khayat M, Ben-Harush K, Dubrovsky-Gaupp A, Sapra KT, Goldman RD, Medalia O. The molecular architecture of lamins in somatic cells. Nature. 2017;543(7644):261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Benavente R, Krohne G. Involvement of nuclear lamins in postmitotic reorganization of chromatin as demonstrated by microinjection of lamin antibodies. J Cell Biol. 1986;103(5):1847–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zastrow MS, Vlcek S, Wilson KL. Proteins that bind A-type lamins: integrating isolated clues. J Cell Sci. 2004;117(Pt 7):979–87. [DOI] [PubMed] [Google Scholar]
- [29].Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006;172(1):41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Barateau A, Buendia B. In Situ Detection of Interactions Between Nuclear Envelope Proteins and Partners. Methods Mol Biol. 2016;1411:147–58. [DOI] [PubMed] [Google Scholar]
- [31].Worman HJ, Schirmer EC. Nuclear membrane diversity: underlying tissue-specific pathologies in disease? Curr Opin Cell Biol. 2015;34:101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Birendra K, May DG, Benson BV, Kim DI, Shivega WG, Ali MH, Faustino RS, Campos AR, Roux KJ. VRK2A is an A-type lamin-dependent nuclear envelope kinase that phosphorylates BAF. Mol Biol Cell. 2017;28(17):2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341(6149):1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Osmanagic-Myers S, Dechat T, Foisner R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015;29(3):225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Heo SJ, Driscoll TP, Thorpe SD, Nerurkar NL, Baker BM, Yang MT, Chen CS, Lee DA, Mauck RL. Differentiation alters stem cell nuclear architecture, mechanics, and mechano-sensitivity. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dechat T, Korbei B, Vaughan OA, Vlcek S, Hutchison CJ, Foisner R. Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J Cell Sci. 2000;113 Pt 19:3473–84. [DOI] [PubMed] [Google Scholar]
- [37].Naetar N, Ferraioli S, Foisner R. Lamins in the nuclear interior – life outside the lamina. J Cell Sci. 2017;130(13):2087–2096. [DOI] [PubMed] [Google Scholar]
- [38].Lund E, Oldenburg AR, Delbarre E, Freberg CT, Duband-Goulet I, Eskeland R, Buendia B, Collas P. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013;23(10):1580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cesarini E, Mozzetta C, Marullo F, Gregoretti F, Gargiulo A, Columbaro M, Cortesi A, Antonelli L, Di Pelino S, Squarzoni S, et al. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J Cell Biol. 2015;211(3):533–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Naetar N, Foisner R. Lamin complexes in the nuclear interior control progenitor cell proliferation and tissue homeostasis. Cell Cycle. 2009;8(10):1488–93. [DOI] [PubMed] [Google Scholar]
- [41].Perovanovic J, Dell'Orso S, Gnochi VF, Jaiswal JK, Sartorelli V, Vigouroux C, Mamchaoui K, Mouly V, Bonne G, Hoffman EP. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci Transl Med. 2016;8(335):335ra58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Oldenburg A, Briand N, Sorensen AL, Cahyani I, Shah A, Moskaug JO, Collas P. A lipodystrophy-causing lamin A mutant alters conformation and epigenetic regulation of the anti-adipogenic MIR335 locus. J Cell Biol. 2017;216(9):2731–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Vigouroux C, Magre J, Vantyghem MC, Bourut C, Lascols O, Shackleton S, Lloyd DJ, Guerci B, Padova G, Valensi P, et al. Lamin A/C gene: sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes. 2000;49(11):1958–62. [DOI] [PubMed] [Google Scholar]
- [44].Dhe-Paganon S, Werner ED, Chi YI, Shoelson SE. Structure of the globular tail of nuclear lamin. J Biol Chem. 2002;277(20):17381–4. [DOI] [PubMed] [Google Scholar]
- [45].Krimm I, Ostlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, Bonne G, Courvalin JC, Worman HJ, Zinn-Justin S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure. 2002;10(6):811–23. [DOI] [PubMed] [Google Scholar]
- [46].Simon DN, Domaradzki T, Hofmann WA, Wilson KL. Lamin A tail modification by SUMO1 is disrupted by familial partial lipodystrophy-causing mutations. Mol Biol Cell. 2013;24(3):342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Perepelina K, Dmitrieva RI, Ignatieva E, Borodkina A, Kostareva A, Malashicheva A. Lamin A/C mutation associated with lipodystrophy influences adipogenic differentiation of stem cells through interaction with Notch signaling. Biochem Cell Biol. 2017. [DOI] [PubMed] [Google Scholar]
- [48].Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet. 2002;11(7):769–77. [DOI] [PubMed] [Google Scholar]
- [49].Duband-Goulet I, Woerner S, Gasparini S, Attanda W, Konde E, Tellier-Lebegue C, Craescu CT, Gombault A, Roussel P, Vadrot N, et al. Subcellular localization of SREBP1 depends on its interaction with the C-terminal region of wild-type and disease related A-type lamins. Exp Cell Res. 2011; 317( 20): 2800–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Stierle V, Couprie J, Ostlund C, Krimm I, Zinn-Justin S, Hossenlopp P, Worman HJ, Courvalin JC, Duband-Goulet I. The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry. 2003;42(17):4819–28. [DOI] [PubMed] [Google Scholar]
- [51].Vadrot N, Duband-Goulet I, Cabet E, Attanda W, Barateau A, Vicart P, Gerbal F, Briand N, Vigouroux C, Oldenburg AR, et al. The p.R482W substitution in A-type lamins deregulates SREBP1 activity in Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2015;24(7):2096–109. [DOI] [PubMed] [Google Scholar]
- [52].Boguslavsky RL, Stewart CL, Worman HJ. Nuclear lamin A inhibits adipocyte differentiation: implications for Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2006;15(4):653–63. [DOI] [PubMed] [Google Scholar]
- [53].Oldenburg AR, Delbarre E, Thiede B, Vigouroux C, Collas P. Deregulation of Fragile X-related protein 1 by the lipodystrophic lamin A p.R482W mutation elicits a myogenic gene expression program in preadipocytes. Hum Mol Genet. 2014;23(5):1151–62. [DOI] [PubMed] [Google Scholar]
- [54].Billon N, Dani C. Developmental origins of the adipocyte lineage: new insights from genetics and genomics studies. Stem Cell Rev. 2012;8(1):55–66. [DOI] [PubMed] [Google Scholar]
- [55].Garg A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab. 2000;85(5):1776–82. [DOI] [PubMed] [Google Scholar]
- [56].Araujo-Vilar D, Loidi L, Dominguez F, Cabezas-Cerrato J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Horm Metab Res. 2003;35(1):29–35. [DOI] [PubMed] [Google Scholar]
- [57].Araujo-Vilar D, Lattanzi G, Gonzalez-Mendez B, Costa-Freitas AT, Prieto D, Columbaro M, Mattioli E, Victoria B, Martinez-Sanchez N, Ramazanova A, et al. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J Med Genet. 2009;46(1):40–8. [DOI] [PubMed] [Google Scholar]
- [58].Araujo-Vilar D, Victoria B, Gonzalez-Mendez B, Barreiro F, Fernandez-Rodriguez B, Cereijo R, Gallego-Escuredo JM, Villarroya F, Paneda-Menendez A. Histological and molecular features of lipomatous and nonlipomatous adipose tissue in familial partial lipodystrophy caused by LMNA mutations. Clin Endocrinol (Oxf). 2012;76(6):816–24. [DOI] [PubMed] [Google Scholar]
- [59].Bereziat V, Cervera P, Le Dour C Verpont MC, Dumont S, Vantyghem MC, Capeau J, Vigouroux C, Lipodystrophy Study G. LMNA mutations induce a non-inflammatory fibrosis and a brown fat-like dystrophy of enlarged cervical adipose tissue. Am J Pathol. 2011;179(5):2443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med. 2002;112(7):549–55. [DOI] [PubMed] [Google Scholar]
- [61].van der Kooi AJ, Bonne G, Eymard B, Duboc D, Talim B, Van der Valk M, Reiss P, Richard P, Demay L, Merlini L, et al. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology. 2002;59(4):620–3. [DOI] [PubMed] [Google Scholar]
- [62].Decaudain A, Vantyghem MC, Guerci B, Hecart AC, Auclair M, Reznik Y, Narbonne H, Ducluzeau PH, Donadille B, Lebbe C, et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J Clin Endocrinol Metab. 2007;92(12):4835–44. [DOI] [PubMed] [Google Scholar]
- [63].Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D'Apice MR, Massart C, Capon F, Sbraccia P, Federici M, Lauro R, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71(2):426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12(16):1995–2001. [DOI] [PubMed] [Google Scholar]
- [65].Caux F, Dubosclard E, Lascols O, Buendia B, Chazouilleres O, Cohen A, Courvalin JC, Laroche L, Capeau J, Vigouroux C, et al. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J Clin Endocrinol Metab. 2003;88(3):1006–13. [DOI] [PubMed] [Google Scholar]
- [66].De Sandre-Giovannoli A Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300(5628):2055. [DOI] [PubMed] [Google Scholar]
- [67].Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Garg A, Subramanyam L, Agarwal AK, Simha V, Levine B, D'Apice MR, Novelli G, Crow Y. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab. 2009;94(12):4971–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Soria-Valles C, Carrero D, Gabau E, Velasco G, Quesada V, Barcena C, Moens M, Fieggen K, Mohrcken S, Owens M, et al. Novel LMNA mutations cause an aggressive atypical neonatal progeria without progerin accumulation. J Med Genet. 2016. [DOI] [PubMed] [Google Scholar]
- [70].Vigouroux C, Auclair M, Dubosclard E, Pouchelet M, Capeau J, Courvalin JC, Buendia B. Nuclear envelope disorganization in fibroblasts from lipodystrophic patients with heterozygous R482Q/W mutations in the lamin A/C gene. J Cell Sci. 2001;114(Pt 24):4459–68. [DOI] [PubMed] [Google Scholar]
- [71].Favreau C, Dubosclard E, Ostlund C, Vigouroux C, Capeau J, Wehnert M, Higuet D, Worman HJ, Courvalin JC, Buendia B. Expression of lamin A mutated in the carboxyl-terminal tail generates an aberrant nuclear phenotype similar to that observed in cells from patients with Dunnigan-type partial lipodystrophy and Emery-Dreifuss muscular dystrophy. Exp Cell Res. 2003;282(1):14–23. [DOI] [PubMed] [Google Scholar]
- [72].Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101(24):8963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Muchir A, Medioni J, Laluc M, Massart C, Arimura T, van der Kooi AJ, Desguerre I, Mayer M, Ferrer X, Briault S, et al. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve. 2004;30(4):444–50. [DOI] [PubMed] [Google Scholar]
- [74].Lombardi F, Gullotta F, Columbaro M, Filareto A, D'Adamo M, Vielle A, Guglielmi V, Nardone AM, Azzolini V, Grosso E, et al. Compound heterozygosity for mutations in LMNA in a patient with a myopathic and lipodystrophic mandibuloacral dysplasia type A phenotype. J Clin Endocrinol Metab. 2007;92(11):4467–71. [DOI] [PubMed] [Google Scholar]
- [75].Barateau A, Vadrot N, Vicart P, Ferreiro A, Mayer M, Heron D, Vigouroux C, Buendia B. A Novel Lamin A Mutant Responsible for Congenital Muscular Dystrophy Causes Distinct Abnormalities of the Cell Nucleus. PLoS One. 2017;12(1):e0169189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Capanni C, Mattioli E, Columbaro M, Lucarelli E, Parnaik VK, Novelli G, Wehnert M, Cenni V, Maraldi NM, Squarzoni S, et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum Mol Genet. 2005;14(11):1489–502. [DOI] [PubMed] [Google Scholar]
- [77].Caron M, Auclair M, Donadille B, Bereziat V, Guerci B, Laville M, Narbonne H, Bodemer C, Lascols O, Capeau J, et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007;14(10):1759–67. [DOI] [PubMed] [Google Scholar]
- [78].Tu Y, Sanchez-Iglesias S, Araujo-Vilar D, Fong LG, Young SG. LMNA missense mutations causing familial partial lipodystrophy do not lead to an accumulation of prelamin A. Nucleus. 2016;7(5):512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Coffinier C, Hudon SE, Farber EA, Chang SY, Hrycyna CA, Young SG, Fong LG. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc Natl Acad Sci U S A. 2007;104(33):13432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Maraldi NM, Capanni C, Mattioli E, Columbaro M, Squarzoni S, Parnaik WK, Wehnert M, Lattanzi G. A pathogenic mechanism leading to partial lipodistrophy and prospects for pharmacological treatment of insulin resistance syndrome. Acta Biomed. 2007;78 Suppl 1:207–15. [PubMed] [Google Scholar]
- [81].Maraldi NM, Capanni C, Lattanzi G, Camozzi D, Facchini A, Manzoli FA. SREBP1 interaction with prelamin A forms: a pathogenic mechanism for lipodystrophic laminopathies. Adv Enzyme Regul. 2008;48:209–23. [DOI] [PubMed] [Google Scholar]
- [82].Ruiz de Eguino G, Infante A, Schlangen K, Aransay AM, Fullaondo A, Soriano M, Garcia-Verdugo JM, Martin AG, Rodriguez CI. Sp1 transcription factor interaction with accumulated prelamin a impairs adipose lineage differentiation in human mesenchymal stem cells: essential role of sp1 in the integrity of lipid vesicles. Stem Cells Transl Med. 2012;1(4):309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Capanni C, Cenni V, Haraguchi T, Squarzoni S, Schuchner S, Ogris E, Novelli G, Maraldi N, Lattanzi G. Lamin A precursor induces barrier-to-autointegration factor nuclear localization. Cell Cycle. 2010;9(13):2600–10. [DOI] [PubMed] [Google Scholar]
- [84].Capanni C, Squarzoni S, Cenni V, D'Apice MR, Gambineri A, Novelli G, Wehnert M, Pasquali R, Maraldi NM, Lattanzi G. Familial partial lipodystrophy, mandibuloacral dysplasia and restrictive dermopathy feature barrier-to-autointegration factor (BAF) nuclear redistribution. Cell Cycle. 2012;11(19):3568–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Le Dour C Schneebeli S, Bakiri F, Darcel F, Jacquemont ML, Maubert MA, Auclair M, Jeziorowska D, Reznik Y, Bereziat V, et al. A homozygous mutation of prelamin-A preventing its farnesylation and maturation leads to a severe lipodystrophic phenotype: new insights into the pathogenicity of nonfarnesylated prelamin-A. J Clin Endocrinol Metab. 2011;96(5):E856–62. [DOI] [PubMed] [Google Scholar]
- [86].Verstraeten VL, Caputo S, van Steensel MA, Duband-Goulet I, Zinn-Justin S, Kamps M, Kuijpers HJ, Ostlund C, Worman HJ, Briede JJ, et al. The R439C mutation in LMNA causes lamin oligomerization and susceptibility to oxidative stress. J Cell Mol Med. 2009;13(5):959–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Sun K, Tordjman J, Clement K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18(4):470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Jan V, Cervera P, Maachi M, Baudrimont M, Kim M, Vidal H, Girard PM, Levan P, Rozenbaum W, Lombes A, et al. Altered fat differentiation and adipocytokine expression are inter-related and linked to morphological changes and insulin resistance in HIV-1-infected lipodystrophic patients. Antivir Ther. 2004;9(4):555–64. [PubMed] [Google Scholar]
- [89].Wojtanik KM, Edgemon K, Viswanadha S, Lindsey B, Haluzik M, Chen W, Poy G, Reitman M, Londos C. The role of LMNA in adipose: a novel mouse model of lipodystrophy based on the Dunnigan-type familial partial lipodystrophy mutation. J Lipid Res. 2009;50(6):1068–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Le Dour C Wu W, Bereziat V, Capeau J, Vigouroux C, Worman HJ. Extracellular matrix remodeling and transforming growth factor-beta signaling abnormalities induced by lamin A/C variants that cause lipodystrophy. J Lipid Res. 2017;58(1):151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Holmstrom M, Kivisto S, Helio T, Jurkko R, Kaartinen M, Antila M, Reissell E, Kuusisto J, Karkkainen S, Peuhkurinen K, et al. Late gadolinium enhanced cardiovascular magnetic resonance of lamin A/C gene mutation related dilated cardiomyopathy. J Cardiovasc Magn Reson. 2011;13:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Chatzifrangkeskou M, Le Dour C Wu W, Morrow JP, Joseph LC, Beuvin M, Sera F, Homma S, Vignier N, Mougenot N, et al. ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum Mol Genet. 2016;25(11):2220–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Avnet S, Pallotta R, Perut F, Baldini N, Pittis MG, Saponari A, Lucarelli E, Dozza B, Greggi T, Maraldi NM, et al. Osteoblasts from a mandibuloacral dysplasia patient induce human blood precursors to differentiate into active osteoclasts. Biochim Biophys Acta. 2011;1812(7):711–8. [DOI] [PubMed] [Google Scholar]
- [94].Lombardi F, Fasciglione GF, D'Apice MR, Vielle A, D'Adamo M, Sbraccia P, Marini S, Borgiani P, Coletta M, Novelli G. Increased release and activity of matrix metalloproteinase-9 in patients with mandibuloacral dysplasia type A, a rare premature ageing syndrome. Clin Genet. 2008;74(4):374–83. [DOI] [PubMed] [Google Scholar]
- [95].Evangelisti C, Bernasconi P, Cavalcante P, Cappelletti C, D'Apice MR, Sbraccia P, Novelli G, Prencipe S, Lemma S, Baldini N, et al. Modulation of TGFbeta 2 levels by lamin A in U2-OS osteoblast-like cells: understanding the osteolytic process triggered by altered lamins. Oncotarget. 2015;6(10):7424–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30(11):2301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hegele RA. Premature atherosclerosis associated with monogenic insulin resistance. Circulation. 2001;103(18):2225–9. [DOI] [PubMed] [Google Scholar]
- [98].Bidault G, Garcia M, Vantyghem MC, Ducluzeau PH, Morichon R, Thiyagarajah K, Moritz S, Capeau J, Vigouroux C, Bereziat V. Lipodystrophy-linked LMNA p.R482W mutation induces clinical early atherosclerosis and in vitro endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2013;33(9):2162–71. [DOI] [PubMed] [Google Scholar]
- [99].McClintock D, Gordon LB, Djabali K. Hutchinson-Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc Natl Acad Sci U S A. 2006;103(7):2154–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Capell BC, Collins FS, Nabel EG. Mechanisms of cardiovascular disease in accelerated aging syndromes. Circ Res. 2007;101(1):13–26. [DOI] [PubMed] [Google Scholar]
- [101].Gordon LB, Kleinman ME, Massaro J, D'Agostino RB, Sr., Shappell H, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland RH, Nazarian A, et al. Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson-Gilford Progeria Syndrome. Circulation. 2016;134(2):114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Afonso P, Auclair M, Boccara F, Vantyghem MC, Katlama C, Capeau J, Vigouroux C, Caron-Debarle M. LMNA mutations resulting in lipodystrophy and HIV protease inhibitors trigger vascular smooth muscle cell senescence and calcification: Role of ZMPSTE24 downregulation. Atherosclerosis. 2016;245:200–11. [DOI] [PubMed] [Google Scholar]
- [103].Briand N, Guenantin AC, Jeziorowska D, Shah A, Mantecon M, Capel E, Garcia M, Oldenburg A, Paulsen J, Hulot JS, et al. The lipodystrophic hotspot lamin A p.R482W mutation deregulates the mesodermal inducer T/Brachyury and early vascular differentiation gene networks. Hum Mol Genet. 2018. [DOI] [PubMed] [Google Scholar]
- [104].Jeru I, Vatier C, Vantyghem MC, Lascols O, Vigouroux C. LMNA-associated partial lipodystrophy: anticipation of metabolic complications. J Med Genet. 2017;54(6):413–416. [DOI] [PubMed] [Google Scholar]
- [105].McInnis MG. Anticipation: an old idea in new genes. Am J Hum Genet. 1996;59(5):973–9. [PMC free article] [PubMed] [Google Scholar]
- [106].Ronningen T, Shah A, Oldenburg AR, Vekterud K, Delbarre E, Moskaug JO, Collas P. Prepatterning of differentiation-driven nuclear lamin A/C-associated chromatin domains by GlcNAcylated histone H2B. Genome Res. 2015;25(12):1825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Czapiewski R, Robson MI, Schirmer EC. Anchoring a Leviathan: How the Nuclear Membrane Tethers the Genome. Front Genet. 2016;7:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Poleshko A, Shah PP, Gupta M, Babu A, Morley MP, Manderfield LJ, Ifkovits JL, Calderon D, Aghajanian H, Sierra-Pagan JE, et al. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell. 2017;171(3):573-587 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Barres R, Zierath JR. The role of diet and exercise in the transgenerational epigenetic landscape of T2DM. Nat Rev Endocrinol. 2016;12(8):441–51. [DOI] [PubMed] [Google Scholar]
- [110].Paulsen J, Sekelja M, Oldenburg AR, Barateau A, Briand N, Delbarre E, Shah A, Sorensen AL, Vigouroux C, Buendia B, et al. Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol. 2017;18(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Elzeneini E, Wickstrom SA. Lipodystrophic laminopathy: Lamin A mutation relaxes chromatin architecture to impair adipogenesis. J Cell Biol. 2017;216(9):2607–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, Mungai L, Oral EA, Patni N, Rother KI, et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab. 2016;101(12):4500–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401(6748):73–6. [DOI] [PubMed] [Google Scholar]
- [114].Colombo C, Cutson JJ, Yamauchi T, Vinson C, Kadowaki T, Gavrilova O, Reitman ML. Transplantation of adipose tissue lacking leptin is unable to reverse the metabolic abnormalities associated with lipoatrophy. Diabetes. 2002;51(9):2727–33. [DOI] [PubMed] [Google Scholar]
- [115].Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570–8. [DOI] [PubMed] [Google Scholar]
- [116].Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia. 2010;53(1):27–35. [DOI] [PubMed] [Google Scholar]
- [117].Javor ED, Cochran EK, Musso C, Young JR, Depaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes. 2005;54(7):1994–2002. [DOI] [PubMed] [Google Scholar]
- [118].Schlogl H, Muller K, Horstmann A, Miehle K, Puschel J, Villringer A, Pleger B, Stumvoll M, Fasshauer M. Leptin Substitution in Patients With Lipodystrophy: Neural Correlates for Long-term Success in the Normalization of Eating Behavior. Diabetes. 2016;65(8):2179–86. [DOI] [PubMed] [Google Scholar]
- [119].Diker-Cohen T, Cochran E, Gorden P, Brown RJ. Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metab. 2015;100(5):1802–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Vatier C, Fetita S, Boudou P, Tchankou C, Deville L, Riveline J, Young J, Mathivon L, Travert F, Morin D, et al. One-year metreleptin improves insulin secretion in patients with diabetes linked to genetic lipodystrophic syndromes. Diabetes Obes Metab. 2016;18(7):693–7. [DOI] [PubMed] [Google Scholar]
- [121].Vatier C, Arnaud L, Prieur X, Guyomarch B, Le May C Bigot E, Pichelin M, Daguenel A, Vantyghem MC, Gautier JF, et al. One-year metreleptin therapy decreases PCSK9 serum levels in diabetic patients with monogenic lipodystrophy syndromes. Diabetes Metab. 2017;43(3):275–279. [DOI] [PubMed] [Google Scholar]
- [122].Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerback S, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360(15):1518–25. [DOI] [PubMed] [Google Scholar]
- [123].Chondronikola M, Volpi E, Borsheim E, Porter C, Annamalai P, Enerback S, Lidell ME, Saraf MK, Labbe SM, Hurren NM, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. 2014;63(12):4089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Hafner AL, Dani C. Human induced pluripotent stem cells: A new source for brown and white adipocytes. World J Stem Cells. 2014;6(4):467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Ahfeldt T, Schinzel RT, Lee YK, Hendrickson D, Kaplan A, Lum DH, Camahort R, Xia F, Shay J, Rhee EP, et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat Cell Biol. 2012;14(2):209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Guenantin AC, Briand N, Capel E, Dumont F, Morichon R, Provost C, Stillitano F, Jeziorowska D, Siffroi JP, Hajjar RJ, et al. Functional Human Beige Adipocytes From Induced Pluripotent Stem Cells. Diabetes. 2017;66(6):1470–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]