

Abstract
Huntington’s Disease (HD) is caused by a CAG repeat expansion that encodes a polyglutamine (polyQ) expansion in the HD disease protein, huntingtin (HTT). PolyQ expansion promotes misfolding and aggregation of mutant HTT (mHTT) within neurons. The cellular pathways, including ubiquitin-dependent processes, by which mHTT is regulated remain incompletely understood. Ube2W is the only ubiquitin conjugating enzyme (E2) known to ubiquitinate substrates at their amino (N)-termini, likely favoring substrates with disordered N-termini. By virtue of its N-terminal polyQ domain, HTT has an intrinsically disordered amino terminus. In studies employing immortalized cells, primary neurons and a knock-in (KI) mouse model of HD, we tested the effect of Ube2W deficiency on mHTT levels, aggregation and neurotoxicity. In cultured cells, deficiency of Ube2W activity markedly decreases mHTT aggregate formation and increases the level of soluble monomers, while reducing mHTT-induced cytotoxicity. Consistent with this result, the absence of Ube2W in HdhQ200 KI mice significantly increases levels of soluble monomeric mHTT while reducing insoluble oligomeric species. This study sheds light on the potential function of the non-canonical ubiquitin-conjugating enzyme, Ube2W, in this polyQ neurodegenerative disease.
Keywords: Huntington’s disease, Huntingtin, ubiquitination, ubiquitin-conjugating enzyme, Ube2W, protein misfolding, neurodegeneration
Introduction
The accumulation of ubiquitinated proteins is a pathological hallmark shared by many neurodegenerative diseases including Huntington’s disease (HD). HD is the most common among nine polyglutamine (polyQ) diseases, caused by CAG repeat expansions that encode a polyQ stretch in the disease proteins. In HD, when the polyQ length exceeds a threshold close to 40 glutamines, the HD protein huntingtin (HTT) becomes pathogenic (1). HD patients typically develop involuntary movements such as chorea and dystonia, neuropsychiatric symptoms and cognitive deficits. Pathologically, HD displays preferential degeneration of the striatum, with the medium spiny neurons expressing dopamine D2 receptors, DARPP-32 and encephalin being particularly vulnerable(2–5).
In HTT, the N-terminal 17 amino acids and adjacent polyQ stretch are largely unstructured and disordered, contributing to the relative “disorderedness” of this domain of the protein(6). Mutant HTT (mHTT) becomes insoluble and forms aggregates known as inclusion bodies in neuronal nuclei, perinuclear regions and neurites(7–9). These inclusion bodies contain N-terminal fragments of HTT, ubiquitin, proteasomal components and numerous other proteins(8). The presence of ubiquitin and proteasomal subunits in HD inclusions supports the importance of ubiquitin-dependent pathways in HD. However, the precise roles of ubiquitination pathways in HD are not fully understood. Studies have shown that numerous components of ubiquitin-dependent systems can contribute to and alter HTT degradation, aggregation and cytotoxicity(10–12). K11, K48 and K63-linked polyubiquitin-modified proteins have been identified in HD inclusions, suggesting a regulatory role for the proteasomal and/or autophagy systems in inclusion formation(13, 14). Furthermore, ubiquitin-like molecules such as SUMO have been implicated as regulators of HTT aggregation(15, 16).
Ubiquitin (Ub) conjugation requires the sequential action of enzymes to target ubiquitin to substrates: Ub activating enzyme (E1), Ub conjugating enzyme (E2) and Ub ligase (E3). Given the diversity in Ub-chain lengths, linkages and substrate attachment sites, dramatically different kinds of ubiquitination can occur. The diversity of ubiquitination patterns is primarily achieved by different combinations of E2/E3 pairs. Recently, Ube2W was identified as the only E2 that initiates ubiquitination at the α-amino group of the N-termini of proteins, preferably recognizing substrates with disordered N-termini (17–19). Ube2W can function with various ubiquitin ligases including the C terminus of Hsc-70-interacting protein (CHIP) and the BRCA1/BARD1 complex to mono-ubiquitinate select substrates at their amino-termini(17, 18, 20–22). Ube2W null mice show an incompletely penetrant multi-organ defect and post-natal lethality, suggesting an important function of N-terminal ubiquitination by Ube2W(23).
The relatively disordered nature of the N-terminal domain of HTT predicts it to be a potential candidate target for Ube2W. In this study, we employ a range of model systems to study the effect of Ube2W on HTT protein levels, aggregation and neurotoxicity.
Materials and Methods
Animals
Ube2W germline KO mice were generated as described in our previous publication(23). Ube2W neuronal KO mice were generated by crossing Ube2W Flox/Flox mice with Nestin promoter driven Cre transgenic mice from Jackson Laboratory (B6.Cg-Tg(Nescre)1kln/J), in which Cre is expressed in central nervous systems. All mice in this study were maintained on a pure C57BL/6 genetic background, housed in cages with a maximum number of five animals and maintained in a standard 12-hour light/dark cycle with food and water ad libitum. Genotyping was performed using DNA isolated from tail biopsy at the time of weaning, otherwise indicated. Genotype was determined by PCR amplification of a fragment of gene-of-interest. For crosses to a mouse model of HD, we utilized male HD-KI Q200 (HdhQ200) mouse model expressing murine Htt with ~200 CAG repeats (CAG sizing is verified by Laragen) (24, 25). Mice were euthanized at the specified ages, anesthetized with ketamine/xylazine, and perfused transcardially with phosphate-buffered saline.
PCR primers
For Ube2W KO genotyping: 5′AAAGGAAGAGCCCAGTATGGACCCT3’ and 5’AGAGTCCCTGCAGCTATTAC3’; Cre genotyping: 5’GTCCAATTTACTGACCGTACACC3’, 5’GTTATTCGGATCATCAGCTACACC3’, 5’CTAGGCCACAGAATTGAAAGATCT3’ and 5’GTAGGTGGAAATTCTAGCATCATCC; Flox genotyping: 5’AAAGGAAGAGCCCAGTATGGACCCT3’ and 5’TGTGTTTTGTTTTAATCTTTCTGGCC3’; Hdh genotyping: 5’CCCATTCATTGCCTTGCTG3’ and 5’GCGGCTGAGGGGGTTGA3’; Hdh qRT-PCR: 5’TTGTGTTAGATGGTGCCGAT3’ and 5’GTTGAAGGGCCAGAGAAGAG3’.
Transfection and immunofluorescence imaging
Human embryonic kidney 293 (HEK293) cells were cultured in DMEM, supplemented with 10% FBS, 100U/ml penicillin/streptomycin. Transfections were carried out with lipofectamine 2000 (Invitrogen) as described previously(25). GFP fluorescence was visualized with an Olympus IX-71 fluorescence microscope.
Plasmids
pcDNA3.1-Httex1Q103-GFP and pGW1-mApple have been reported previously(25–27); pCMV6-Ube2W plasmid was obtained from Origene (RC204985), C91A and W144E mutations were introduced using Quikchange Lightning Site-Directed Mutagenesis (#210518, Agilent Technologies) and sequence-verified; pGW1–Httex1-(Q17 or Q72)-EGFP plasmids were kindly provided by Dr. Steven Finkbeiner(28).
Western blotting
Protein lysates from HEK293 cells were generated by lysis in radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors (Complete-mini; Roche Diagnostics, Indianapolis, IN), followed by sonication and centrifugation at 16,000 rpm for 5 mins at 4 °C. The supernatants were collected for analysis. For transfection experiments, transfected cells were lysed with 0.5% Triton X-100 lysis buffer containing 150 mM NaCl, 20 mM Tris/HCl, pH 8.0, 5 mM EDTA and complete Mini Protease Inhibitor tablets. Insoluble pellet from centrifugation was further lysed with 2X Laemmli buffer. Total protein concentration was determined using the BCA method (Pierce, Rockford, IL) and samples were stored at −80 °C. Proteins were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and corresponding polyvinylidene difluoride membranes were incubated overnight at 4 °C with the following primary antibodies: rabbit anti-Ube2W (1:1,000; 15920-1-AP; Protein Tech Group), mouse anti-GAPDH (1:10,000; MAB374; EMD Millipore), rabbit anti- α tubulin (1:10,000; #2144; Cell Signaling Technology), rabbit anti-GFP (1:1,000; sc-8334; Santa Cruz Biotechnology), mouse anti-polyglutamine (1:2,000; MAB1574; EMD Millipore), mouse anti-Huntingtin (1:250, MAB2166, EMD Millipore), rabbit anti-Hsp40 (1:1,000; #4868, Cell Signaling Technology), rabbit anti-Hsp60 (1:1,000; ab46798, abcam), mouse anti-Hsp70 (1:1,000; SPA-810, Enzo Life Sciences) mouse anti-Hsp90 (1:1,000; SAP-830, Enzo Life Sciences), anti-ubiquitin (1:2,000; z0458, Dako), and rabbit anti-p62 (1:1000; 5114S, Cell Signaling Technology). Bound primary antibodies were visualized by incubation with a peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody (1:10,000; Jackson Immuno Research Laboratories, West Grove, PA) followed by treatment with the ECL-plus reagent (Western Lighting; PerkinElmer, Waltham, MA) or Pico/Dura Western Blotting Detection System (Pierce) and exposure to films for images.
35S-Methionine labeling and immunoprecipitation
24h after transfection, cells were cultured in depletion media for 30 mins and labeled with 100 μCi of 35S-Met (Perkin Elmer) in Cys/Met-free DMEM. After 20 min of labeling, cells were rapidly rinsed with ice-cold PBS then lysed in RIPA buffer (pH 7.4) containing protease inhibitors (Roche). Lysates were immunoprecipitated overnight with anti-GFP, boiled in SDS sample buffer with 0.1 M DTT, and analyzed by 10% Tris-Acetate SDS-PAGE followed by autoradiography.
Histological analysis
All tissues were fixed for 96hrs at 4 °C in 4% Formaldehyde solution in PBS, transferred to 30% sucrose in PBS, processed, and sectioned. For immunohistochemical staining, tissue underwent antigen retrieval in boiling citrate-based buffer (0.01 mol/L citric acid, pH 6.8) for 20 mins at 80 °C. Endogenous peroxidases were quenched with 3% H2O2, followed by blocking in 5% goat serum and incubation with primary antibodies (anti-Huntingtin 1:250, sc8767, Santa Cruz Biotechnology). Bound antibodies were detected with the Vector M.O.M. peroxidase (Vector Laboratories, Burlington, CA), using SigmaFast diaminobenzidene as a peroxidase substrate (Jackson ImmunoResearch, West Grove, PA).
Primary neuron culture and transfection
Cortical neurons were dissected from P0 WT or Ube2W KO pups and cultured at 0.6 × 106 cells per mL for 4 d in vitro before transfection, as described previously(29). Transfection of primary neurons was performed using Lipofectamine 2000 (Invitrogen). A total of 0.2 μg DNA (total) and 0.5 μL Lipofectamine 2000 were used per well in 96-well plates. Cells were incubated with Lipofectamine/DNA complexes for 20 min at 37 °C before rinsing and changing to normal culture media. Rest of the transfection was performed according to manufacture recommendation.
Automated fluorescence microscopy
Cultured primary mouse neurons were imaged by automated fluorescent microscopy (26–28, 30). The system consists of an inverted microscope (Nikon Eclipse Ti) equipped with the Perfect Focus System (Nikon), a high-numerical aperture 20× objective lens, a digital camera, a Lambda XL lamp (Sutter) and an ASI MS2000 stage to automatically control the platform. Neurons were imaged inside a thermo chamber, in which temperature and CO2 concentration was maintained at 37°C and 5% respectively.
Image analysis
25 raw images were taken from each well of primary neurons. Raw images were stitched together to represent larger areas. According to the sequence and alignment, stitched images were stacked together to represent time-elapse change of fluorescence. Neuronal survival was calculated by a software algorithm developed in ImageJ (26). The time of death for each neuron was recorded as the last time a neuron was confirmed to be alive. Intensities of transfected proteins were determined automatically by segmentation of neuronal cell bodies in ImageJ and measurement of mean pixel intensity within each region of interest. Statistical analyses and cumulative hazard plots were generated using the survival package within R (26).
RNA extraction and qRT-PCR
RNA was extracted from forebrain of mice using TRIzol® (Life Technologies) and further purified using the RNeasy kit with on-column DNase I digestion (Qiagen). For qPCR, 1 μg of RNA was reverse-transcribed using iScript™. 0.5 μl was used with the SYBR® GreenMasterMix and each reaction was performed in triplicate. qRT-PCR was performed on the Bio-Rad iCycler with MyIQ single color real-time PCR detection system module with the following parameters: 95°C at 3 min, (95°C 10 s, 55°C 30 s) Å~ 40, 95°C 1 min, 55°C 1 min. The fold change in transcript level was calculated using the ΔΔCt method(31). Gapdh was used as control. The primers used for qRT-PCR are listed above. For figure 6A and 6B, qRT-PCR was performed with TaqMan® Gene Expression Master Mix (#4369016, Life technologies). qRT-PCR was performed on the Applied Biosystems 7500 real-time PCR system and the remainder of the transfection protocol was per the manufacturer’s suggestions. Primers used for TaqMan qRT-PCR are from Life technologies (Drd2: Mm00438545_m1, Ppp1r1b: Mm00454892_m1 and Actb: Mm00607939_s1).
FIGURE 6.
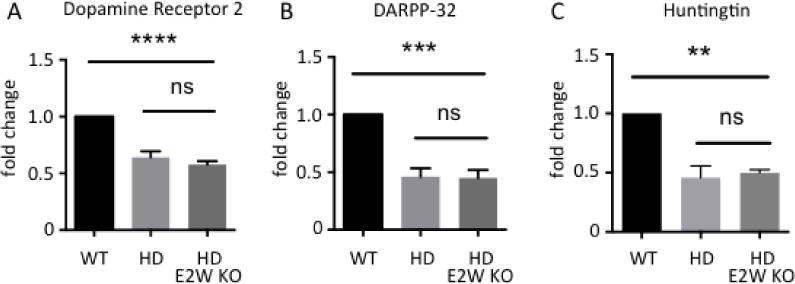
Ube2W deficiency does not alter transcript levels of striatal neuronal markers in HdhQ200 KI mice. Dopamine receptor 2 (Drd2) (A), DARPP-32 (Ppp1r1b) (B) and Huntingtin (Hdh) (C) transcript levels are measured by qRT-PCR amplification. RNA was extracted from WT, HdhQ200 KI, and HdhQ200 het KI/Ube2W KO mice forebrain. Results are normalized to β-actin transcript levels. Results were quantified and plotted as graphs. Graphs show means +/− SEM; ns, not significant; **, p<0.01; ***, p<0.001; ****, p<0.0001. n=4.
Results
Ube2W increases HTT inclusion formation in cultured cells
Httex1Q103-GFP overexpression in HEK293 cells results in nuclear inclusion formation (Fig 1A). Using fluorescence microscopy, we visualized the formation of GFP-positive HTT inclusions when Ube2W or functional mutants of Ube2W were expressed with Httex1Q103-GFP. Mutating Ube2W’s enzymatic active site cysteine, C91, to alanine eliminates the ability of Ube2W to transfer Ub to substrates while still allowing Ube2W to bind substrates(17, 20). In contrast, amino acid W144 near the C-terminus of Ube2W is critically important for substrate binding, and mutating this residue to glutamic acid eliminates substrate binding(17). Both the C91 and the W144 mutants are predicted to disrupt Ube2W-mediated ubiquitination. When WT Ube2W is coexpressed with Httex1Q103, inclusion number and size significantly increases, whereas coexpression of either Ube2W mutant markedly decreases Httex1Q103 inclusion number and average size (Fig 1B-D). Cell number were quantified, revealing that neither WT Ube2W nor either mutant alters cell viability.
FIGURE 1.
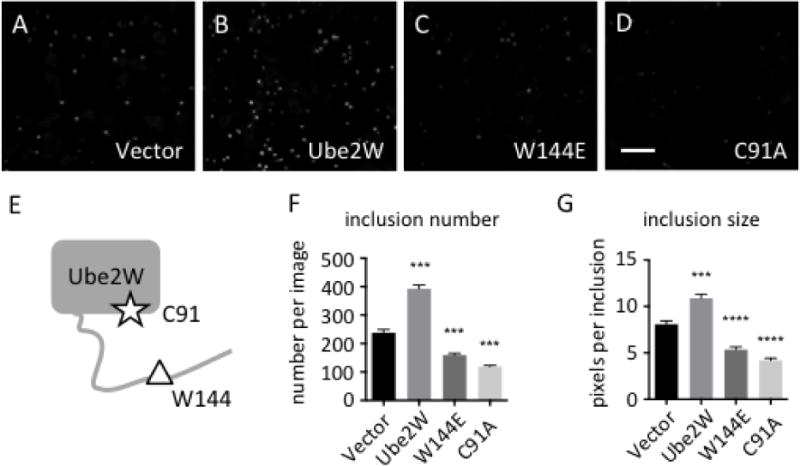
Ube2W alters Httex1Q103-GFP inclusion formation in HEK293 cells
A-D. In HEK293 cells, Httex1Q103 inclusions were visualized by GFP fluorescence 48 hrs after transfection.
Httex1Q103-GFP was co-expressed with vector alone (A), Ube2W (B), Ube2W-W144E (C) or Ube2W-C91A (D). Scale bar=30 μm.
E. Schematic of Ube2W structure illustrates relative positions of C91 (the active site cysteine) and W144 (needed for substrate binding).
F,G. Httex1Q103 inclusion number (F) and size (G) from all four groups were plotted. Graphs show means +/− SEM; ***, p<0.001; ****, p<0.0001. n=8 images(F), n>34 inclusions (G).
Biochemically, mHTT exists in at least three distinct states in cells: soluble monomers, soluble oligomers and insoluble aggregates(32, 33). Accordingly, lysates from transfected cells were separated into soluble and insoluble fractions based on solubility in the nonionic detergent Triton-X100. Coexpression of Ube2W-W144E significantly increases soluble monomeric mHTT whereas WT Ube2W and Ube2W-C91A do not lead to a statistically significant change in soluble mHTT monomers (Fig 2A and B). In the insoluble fraction mHTT partitions as two species discernible by gel electrophoresis: high molecular weight (HMW) mHTT in the stacking gel and monomeric mHTT in the resolving gel. Both Ube2W mutants significantly reduced HMW mHTT in the insoluble fraction whereas WT-Ube2W did not (Fig 2A and C). The same trend can be observed for monomeric mHTT in the insoluble fraction, possibly due to the solubilization of HMW HTT species.
FIGURE 2.
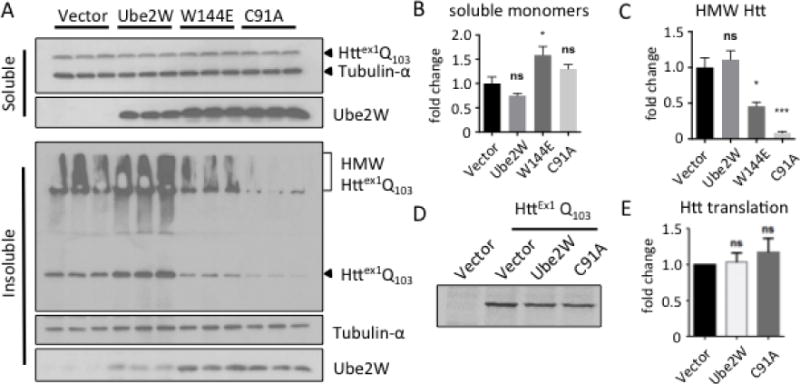
Ube2W alters solubility of Httex1Q103
A. Western Blot of lysates from HEK293 cells transfected with Httex1Q103 and Ube2W or its mutants, Ube2W-W144E or Ube2W-C91A. Upper panel is the TritonX-100 soluble fraction and lower panel is the TritonX-100 insoluble fraction. Immunoblots were performed with anti-GFP (visualizing Httex1Q103), anti-tubulin-α, and anti-Ube2W. (15 μg total protein/lane.) Arrowheads indicate monomeric Httex1Q103, Ube2W or tubulin, and bracket indicates high molecular weight (HMW) Httex1Q103 in stacking gel.
B. Quantification of soluble HTT monomers in upper panel of Fig 2A. Graphs show means +/− SEM; ns, not significant; *, p<0.05. n=3.
C. Quantification of HMW HTT species in lower panel of Fig 2A. Graphs show means +/− SEM; ns, not significant; *, p<0.05; ***, p<0.001. n=3.
D. Autoradiograph of a representative 35S-methionine pulse-chase experiment followed by GFP-IP and gel electrophoresis.
E. Quantification of autoradiograph in Fig 2D. Graphs show means +/− SEM; ns, not significant. n=3.
The effect of Ube2W on HTT most likely occurs post-translationally. But as an E2 that ubiquitinates N-termini, Ube2W could act cotranslationally by interacting with the nascent N-terminal polypeptide as it exits the ribosome and thus alter the rate of HTT protein synthesis itself. To study whether Ube2W affects HTT synthesis, we measured HTT translation using 35S-methionine pulse-labeling. HEK293 cells co-expressing Httex1Q103 with WT Ube2W or Ube2W-C91A were pulse-labeled 20 min with 35S-methionine to label all newly synthesized proteins, then immediately lysed. Anti-GFP immunoprecipitation (IP) captured the soluble Httex1Q103-GFP present in cell lysates and newly synthesized radiolabeled HTT was visualized by autoradiography. The rate of Httex1Q103 translation appeared similar regardless of which form of Ube2W was co-expressed (Fig 2D and E). Efforts to determine HTT half-life using pulse-chase labeling were not successful, most likely due to HTT’s tendency to aggregate and become inaccessible to gel autoradiography over the long chase times needed.
Ube2W deficiency results in decreased mHTT inclusion formation and reduced neurotoxicity
To investigate the consequences of Ube2W deficiency in vivo, we generated primary neuronal cultures from WT or conditional Ube2W KO mice in which Nestin-Cre expression was used to delete Ube2W in the nervous system. To assess the potential relationship between Ube2W and Httex1Q72-mediated neurotoxicity, we measured single-cell fluorescence and cell survival by automated fluorescence microscopy (AFM) (26–28, 30, 33). Using longitudinal AFM, we plotted the risk of death for neurons expressing EGFP alone (as a control), normal (nonpathogenic) repeat Httex1Q17-EGFP, or expanded (mutant) repeat Httex1Q72-EGFP, and assessed for differences in neuronal survival among conditions with Cox proportional hazards analysis. Qualitative measures used to estimate cell death in these assays (loss of fluorescence or disruption of cell integrity) are equally sensitive as conventional measures such as staining for apoptotic markers (33)(Fig 3A).
FIGURE 3.
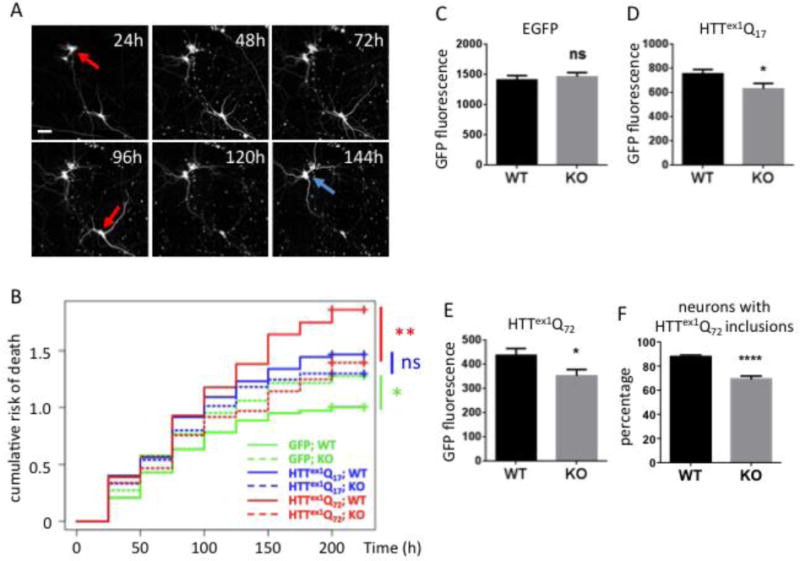
Ube2W deficiency results in decreased Httex1Q72 inclusion formation and increased neuronal survival
A. Representative images from automated fluorescence microscopy. Primary cortical neurons from WT or Ube2W KO mice were transfected with mApple and Httex1Q72-EGFP, and survival was determined by repeated imaging at regular intervals. The last time at which the cell was noted to be alive (red arrows) was used as the time of death. Cells that survive the entire length of the experiment (blue arrow) were censored (analyzed as living cell at the end of experiment). Scale bar=25 μm.
B. Cumulative risk of death over time for WT and Ube2W KO neurons transfected with EGFP, Httex1Q17-EGFP and Httex1Q72-EGFP. Results were pooled from 16 wells per condition, with experiments performed in duplicate. ns, not significant; *, p<0.05; **, p<0.01. n>250.
C-F. Quantification of EGFP signals (C-E) or inclusion formation (F) from experiments as in panel A, 48 hours after transfection. Graphs show means +/− SEM; ns, not significant; *, p<0.05; ****, p<0.0001. n>232 for all analyses.
GFP-expressing neurons lacking Ube2W showed significantly increased rates of death compared to GFP-expressing WT neurons, suggesting that Ube2W plays a role in neuronal health, at least in the cell culture environment employed here. The expression of Httex1-EGFP, whether normal or expanded repeat, increased neuronal cell death. As expected, Httex1Q-EGFP was more toxic than Httex1Q17-EGFP in WT neurons, revealing a glutamine repeat length-dependent toxic effect.
Despite the apparent role of Ube2W in cultured neuronal health, the toxicity of mutant Httex1Q72-EGFP was significantly reduced by the absence of Ube2W (comparison of solid versus dashed red lines in figure 3B). In contrast, the presence or absence of Ube2W did not have a significant effect on the survival of neurons expressing nonpathogenic Httex1Q17-EGFP (comparison of solid versus dashed blue lines in figure 3B).
To assess the effect of Ube2W on Httex1 expression levels, we measured the fluorescence signal separately in each of hundreds of EGFP- or Httex1Qn-EGFP-expressing neurons 24 hours after transfection, a time when Httex1Q72-EGFP inclusion formation is minimal. In neurons expressing EGFP alone, fluorescence did not differ between WT and Ube2W KO neurons (Fig 3C). In contrast, Ube2W KO significantly decreased the fluorescence intensity of both Httex1Q-EGFP and Httex1Q72-EGFP (Fig 3D, E). In comparison to Ube2W WT neurons, Ube2W KO neurons displayed ~20% fewer Httex1Q72-EGFP inclusions (Fig 3F). The increased population of inclusion-negative neurons in the absence of Ube2W continued to show diffuse mHTT signal to the endpoint of the experiment (216 hr).
Absence of Ube2W increases soluble, monomeric mutant Htt in a knock-in mouse model of HD
The above results in neurons transiently expressing an expanded Htt fragment support the view that Ube2W regulates mutant Htt levels, aggregation and toxicity. To further study Ube2W’s effect on full-length mutant Htt in vivo, we crossed Ube2W germline KO mice to heterozygous HdhQ200 KI mice(34, 35). This HD mouse model displays cytoplasmic aggregation foci by 9 weeks of age, followed by neuronal intranuclear inclusion by 20 weeks of age (34, 35). At 32 weeks of age, frontal brain lysates including the cortex and striatum (known vulnerable regions in HD) were homogenized in RIPA buffer, separated by gel electrophoresis and visualized by anti-Htt immunoblot. Strikingly, soluble mutant HttQ200 monomers were increased approximately 5-fold in the absence of Ube2W (Fig 4A and B). A similar increase in soluble HttQ200 was also observed in 46 week-old in Ube2W KO/HdhQ200 KI mice (data not shown). Moreover, in HdhQ200 KI mice the absence of Ube2W also resulted in an approximately 3-fold increase in WT (i.e. nonexpanded) Htt monomers (Fig 4A and B). By contrast, in Ube2W KO mice, WT Htt levels were not significantly altered, suggesting that the observed increase in WT Htt in HdhQ200 mice lacking Ube2W occurs only in the presence of mutant HttQ200.
FIGURE 4.
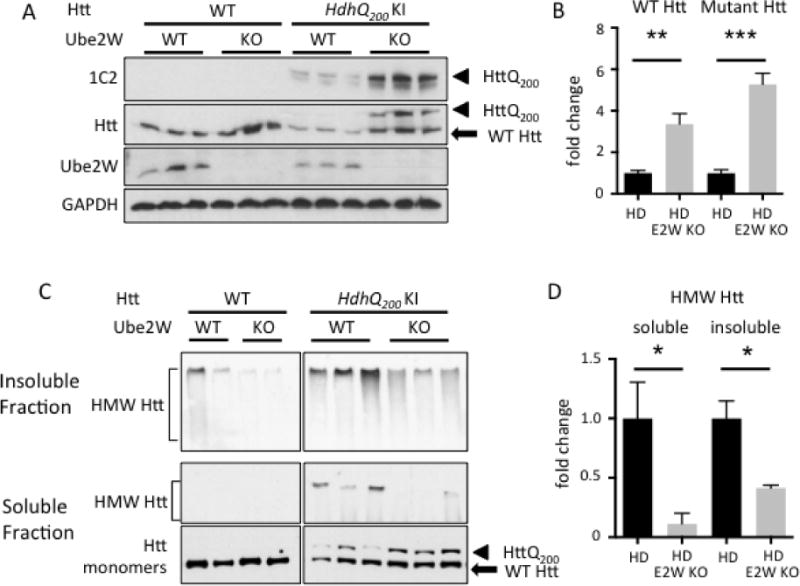
Ube2W deficiency increases soluble Htt levels in HdhQ200 KI mice
A. Western Blot of brain lysates from WT, Ube2W KO, HdhQ200 KI, or HdhQ200 KI/Ube2W KO mice. Immunoblot were performed with anti-polyQ (1C2), anti-Htt, anti-Ube2W and anti-GAPDH antibodies. (40 μg total protein/lane.)
B. Quantification of Blot against Htt in panel A.
C. Western Blot of soluble and insoluble fraction of mouse brain lysates from WT, Ube2W KO, HdhQ200 heterozygous (het) KI, HdhQ200 het KI with Ube2W KO mice. Blotted with anti-Htt antibody.
D. Quantification of HMW Htt blotted in panel C.
Arrowheads represent mutant HttQ200, arrow represents WT Htt, and bracket indicates high molecular weight (HMW) Htt in stacking gel. Graphs show means +/− SEM; *, p<0.05; **, p<0.01; ***, p<0.001. n=3.
In these experiments, in which RIPA buffer was the lysis buffer and samples were analyzed by SDS-PAGE, no Htt signal was observed in the stacking gel corresponding to detergent-resistant HMW Htt species. To better visualize non-monomeric Htt species, we used a milder detergent to fractionate brain lysate into soluble and insoluble fractions (15, 36) (see method section). Absence of Ube2W significantly decreased the level of HMW Htt signal, presumably representing aggregated Htt, in both detergent soluble and insoluble fractions (Fig 4C and D).
HttQ200 inclusion bodies in striatum are not altered by Ube2W deficiency
Htt-containing inclusions within neurons are a pathological hallmark of HD. Due to its hyperexpansion, HttQ200 forms widespread neuronal inclusions in neurons of the striatum, cortex, hippocampus and cerebellum in HdhQ200 KI mice, beginning by ~20 weeks of age (35). To determine whether the marked increase in HttQ200 monomers correlated with a change in size or number of inclusion bodies, we performed immunostaining with an N-terminal Htt antibody on brain sections from 32 and 46 week old mice (Fig 5A and B). The striatum of HdhQ200 KI mice showed an equally robust presence of inclusions in the presence or absence of Ube2W, with no significant difference noted in inclusion number or size at 32 or 46 weeks of age (Fig 5C and D). Both in the presence and absence of Ube2W, Htt inclusions immunostained positively for ubiquitin (data not shown). These results suggest that Ube2W preferentially affects levels of soluble and intermediate species of HttQ200 that are detectable by gel electrophoresis and western blotting, without altering the level of insoluble, aggregated disease protein that accumulates over time within inclusions.
FIGURE 5.
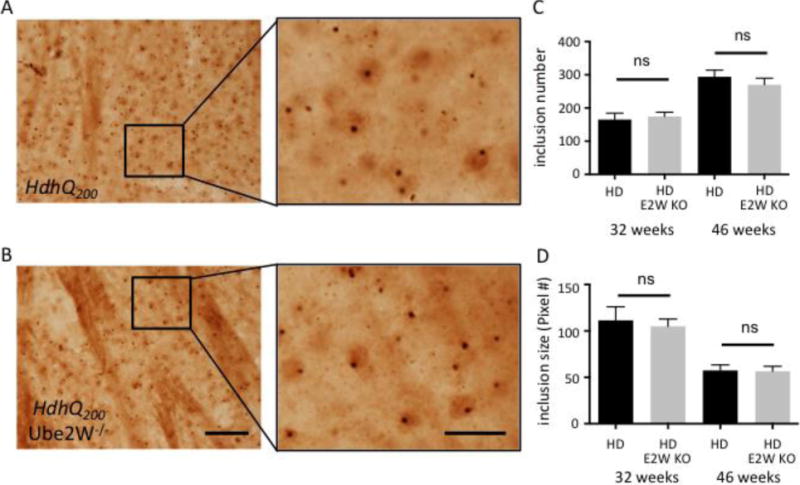
Absence of Ube2W does not alter HttQ200 inclusion levels in HdhQ200 KI mice
A and B. Htt immunostaining of striatum showing HttQ200 intranuclear inclusions in 46 week old HdhQ200 KI (A) and HdhQ200 KI/Ube2W KO mice (B). Magnified regions from insets are shown in the right panels. Scale bar, left panels=200 μm; Scale bar, right panels=50 μm.
C and D. HttQ200 inclusion number (C) and size (D) from 32 and 46 week old mice were plotted. Graphs show means +/− SEM; ns, not significant. n=10 images (C), n>10 images (D).
Ube2W deficiency does not alter transcript levels of Htt or striatal markers in HdhQ200 mice
Medium spiny neurons of the striatum are selectively vulnerable in HD and are susceptible in HdhQ200 mice. Expression levels of two striatal neuronal markers, Dopamine D2 receptors (Drd2) and DARPP-32 (Ppp1r1b), decline dramatically over the course of disease in human HD patients and animal models (24, 35, 37–39). Transcript levels of Drd2 and Ppp1r1b are sensitive measurements of HD pathology in mice (5). Indeed, at 46 weeks of age transcript levels for both markers are markedly decreased in HttQ200 het KI mice (Fig 6A and B). Ube2W deficiency had little effect upon Drd2 and Ppp1r1b transcript levels in HdhQ200 mice (Fig 6A and B). Thus, the presence or absence of Ube2W does not result in detectable changes to HttQ200-mediated striatal neuron dysfunction, assessed by these striatal neuronal markers.
Transcriptional analysis also allowed us to establish that the observed changes in soluble levels of mutant Htt in the absence of Ube2W are not simply due to an effect on Htt expression. By qRT-PCR, we investigated whether Ube2W affects WT or mutant Hdh transcript levels. Ube2W deficiency did not alter levels of the Hdh transcript (Fig 6C), arguing that Ube2W instead affects Htt levels co-translationally or, more likely, post-translationally.
Levels of key components of protein quality control are unchanged in Ube2W KO mice
Given Ube2w’s role in ubiquitin pathways, we investigated whether global ubiquitination in HttQ200 het KI mice is altered by loss of Ube2W. Levels of ubiquitin-positive HMW species in mouse brain lysate did not significantly differ in the presence or absence of Ube2W (Figure 7A). Molecular chaperones and autophagy are important regulators of mutant polyQ protein processing generally, and of HTT processing, accumulation and HD pathology specifically. Accordingly, we sought to determine whether the Ube2W-mediated increase in HttQ200 soluble monomers reflects a change in key components of the neuronal protein quality control machinery. The heat shock protein (hsp) family of molecular chaperones has been implicated in maintenance of protein homeostasis. We measured expression levels of four key hsp family members (hsp40, hsp60, hsp70 and hsp90) as well as the autophagy marker P62. Of these, only hsp70 showed a decrease in Ube2W−/−, HdhQ200 mice (Fig 7).
FIGURE 7.
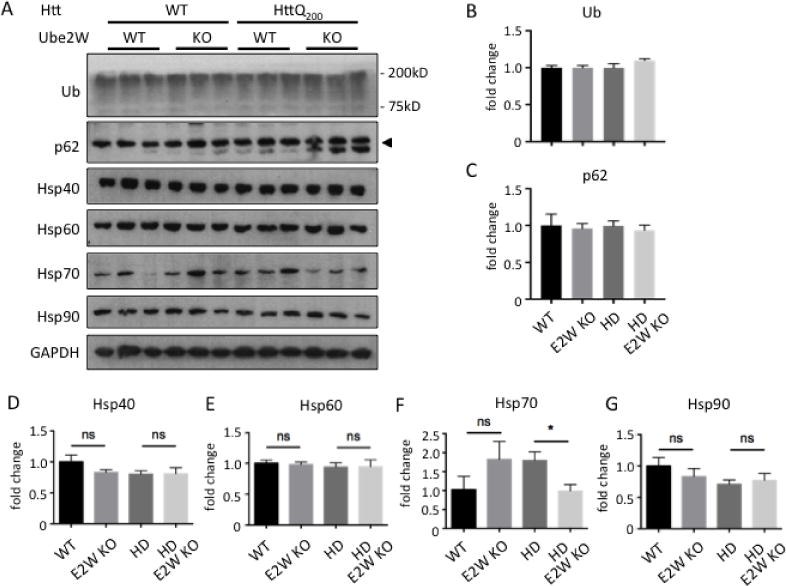
Levels of key components of protein quality control are unchanged in Ube2W KO mice.
A. Western Blot of mouse forebrain lysates from WT, Ube2W KO, HdhQ200 het KI, HdhQ200 het KI with Ube2W KO mice. Blotted with anti-ubiquitin, anti-p62, anti-Hsp40, anti-Hsp60, anti-Hsp70 and anti-Hsp90 antibodies. (40 μg total protein loaded per lane.)
B–G. Quantification of the indicated proteins from Western blots in panel A. Graphs show means +/− SEM; ns, not significant; *, p<0.05; n=3.
A decrease in hsp70 has been reported in various HD mouse models (24, 40) but whether it correlates with HD progression is not known. Collectively, these results suggest that Ube2W deficiency does not lead to the observed increase in soluble HttQ200 monomers simply by changing levels of key components of protein quality control.
Discussion
We undertook this study because the disordered nature of the mHTT N-terminus suggested it as a target for Ube2W, an E2 that uniquely ubiquitinates N-termini and may preferentially act on disordered N-termini. Ubiquitin pathways actively regulate mHTT levels, IB formation and disease progression in model systems (13, 15, 16, 41–45), but the role of Ube2W in HD has not been addressed. Here, we utilized a range of model systems to show that Ube2W deficiency increases soluble mHTT levels while reducing aggregation and neurotoxicity in primary cortical neurons. Our results further support the importance of ubiquitin pathways in HD and shed light on a possible function of Ube2W in modulating HD pathogenesis.
We observed a significant increase in soluble mHTT monomers when Ube2W is functionally inactive or eliminated, accompanied by increased neuronal survival in cultured cells (Fig 1, 2 and 3). mHTT can exist in at least three distinct states in cells: soluble monomers, soluble oligomers and insoluble aggregates(32, 33, 46). mHTT oligomers that precede large scale aggregate formation are considered to be more toxic than monomers or aggregates(32, 47–50). For example, using FRET confocal microscopy, researchers showed that neuronal cells containing oligomers die faster than those with either monomers or inclusions(32). And in drosophila, polyphenol epigallocatechin-gallate reduces oligomeric species by promoting inclusion formation with a corresponding protective effect towards mHTT neurotoxicity(48). In contrast to toxic oligomers, mHTT insoluble aggregates may be neuroprotective, possibly in part by recruiting and neutralizing toxic oligomers(29, 32, 33, 51, 52). In the current study, the increase in mHTT monomers and reduced neurotoxicity in the absence of Ube2W could result from reduced mHTT oligomerization or increased levels of readily soluble mHTT complexes that may lack the toxicity of more insoluble oligomers and would electrophorese as monomers on denaturing gels.
What is the mechanism by which Ube2W alters solubility of mHTT? We offer several hypotheses: 1. N-terminal ubiquitination of HTT stabilizes the protein, ultimately leading to increased HTT aggregation; 2. N-terminal ubiquitination of HTT alters the probability of other post-translational modification(s) to the protein, thus indirectly promoting HTT aggregation; 3. Ube2W acts indirectly on HTT by ubiquitinating one or more proteins that regulate HTT, such as SUMO-2, a documented Ube2W substrate that is known to increase mHTT aggregation(15, 36); and, least likely, 4. Ube2W acts directly or indirectly on HTT through a mechanism unlinked to its ubiquitinating function (53, 54). Further studies are required to determine whether indeed Ube2W directly ubiquitinates HTT or acts in a more indirect manner to alter behavior of the disease protein.
The biological role of Ube2W-mediated N-terminal ubiquitination remains poorly understood. Researchers have identified at least 13 proteins that can be N-terminally ubiquitinated(18, 55–63). All but two known substrates of Ube2W (ataxin-3 and LMP2A) are targeted for proteasomal degradation by N-terminal ubiquitination via K48-linked polyubiquitination. We previously showed an accumulation of relatively disordered proteins in Ube2W KO mouse testis, an organ in which Ube2W is highly expressed, suggesting that Ube2W-mediated N-terminal ubiquitination functions as a degradation signal(23). While these lines of evidence favor the view that Ube2W-mediated N-terminal ubiquitination can target proteins for degradation, Ube2W strictly monoubiquitinates substrates and does not itself specify subsequent Ub chain linkage(17, 18, 20, 21, 64). And, of course, cellular functions of ubiquitination are not limited to degradation pathways. For example, working together with Ube2N/Ube2V2, Ube2W is able to facilitate the formation of K63-linked ubiquitin chains on TRIM5α and TRIM21, which regulates their reverse transcription activity rather than their degradation (21, 22). The potential functions of N-terminal ubiquitination in non-degradative pathways cannot be ruled out as yet.
Besides direct N-terminal ubiquitination by Ube2W, alternative mechanisms of action, such as an effect on SUMOylation, could underlie the alteration in HTT monomers we observed. SUMO-2 itself can be N-terminally monoubiquitinated by Ube2W in vitro(19), though the biological function of this signal is still unknown. Conceivably, Ube2W could ubiquitinate SUMO-2 that is already conjugated to HTT. SUMO-2 modification of Httex1 decreases mHTT solubility, favoring mHTT accumulation and decreasing monomer levels(15); conceivably, Ube2W-mediated ubiquitination of SUMO attached to mHTT underlies this decreased solubility of mHTT. Similar to our finding here with Ube2W mutants, knockdown of an E3 SUMO ligase that enhances SUMO modification of HTT significantly decreases insoluble mHTT species(15). Further mechanistic studies will need to be carried out to study the biologic function of Ube2W on SUMOylation, ubiquitination and mHTT processing and its relationship to mHTT solubility and aggregation.
Our analysis of key protein quality control components in the chaperone and autophagy pathways revealed a reduction only in Hsp70 levels in HD KI mice lacking Ube2W. We would not expect this reduction in Hsp70 to lead to increased levels of soluble, mutant Htt as observed in HD KI mice lacking Ube2W. Thus, despite the fact that Hsp70 has been implicated in Htt handling and toxicity in numerous studies (see, for examples, 65, 66), the isolated reduction of Hsp70 suggests that the effects of Ube2W on mutant Htt are not principally an indirect effect on protein quality control pathways.
Our observation that eliminating Ube2W increased the solubility of full length mutant Htt in HD KI mice complemented our results in transfected cells and cultured neurons expressing an N-terminal mutant Htt fragment. Collectively, these results suggest Ube2W regulates mutant Htt levels and aggregation. Unexpectedly, however, we did not observe an effect of Ube2W on Htt inclusions in the brains of HD KI mice; inclusions appear to be equally abundant in HD KI mice whether or not Ube2W is present. This discrepancy may reflect the substantial differences between cells transiently overexpressing an Htt fragment and neurons continually expressing physiological concentrations of the full length disease protein. It also implies that the slow development of Htt inclusion formation in KI mice and human HD is complex, and that Ube2W is only one of many factors that influence the process.
Our results in cultured neurons suggest Ube2W also modulates Htt toxicity. An important limitation of our study, however, is that we cannot translate this finding of an effect on toxicity to the mouse model. The relationship between disease protein aggregation and toxicity remains an unsettled question in many neurodegenerative diseases, including HD. Our focus here was on the biochemical relationship of Ube2W to Htt, not on the broader question of whether modulating Htt aggregation or solubility affects toxicity in vivo. Our studies were not powered to carry out behavioral phenotypic measurements, and such measurements were not performed on mice in this study. Our findings provide a framework for a future, larger study that can determine the role of Ube2W in modulating Htt toxicity in vivo.
Acknowledgments
This work was supported by the National Institutes of Health Grants R01 AG034228 (to H.L.P), R01 NS090390 (to L.M.T), K99 NS073936 (to K.M.S) and NSF GRFP 2013137284 (to J.O). The authors also received supported from the University of Michigan Protein Folding Diseases Initiative.
Footnotes
Conflicts of Interest: The authors declare no competing financial interests.
References
- 1.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–47. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 2.Augood SJ, Faull RL, Love DR, Emson PC. Reduction in enkephalin and substance P messenger RNA in the striatum of early grade Huntington’s disease: a detailed cellular in situ hybridization study. Neuroscience. 1996;72:1023–36. doi: 10.1016/0306-4522(95)00595-1. [DOI] [PubMed] [Google Scholar]
- 3.Ginovart N, Lundin A, Farde L, Halldin C, Bäckman L, Swahn CG, Pauli S, Sedvall G. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain. 1997;120(Pt 3):503–14. doi: 10.1093/brain/120.3.503. [DOI] [PubMed] [Google Scholar]
- 4.Sapp E, Ge P, Aizawa H, Bird E, Penney J, Young AB, Vonsattel JP, DiFiglia M. Evidence for a preferential loss of enkephalin immunoreactivity in the external globus pallidus in low grade Huntington’s disease using high resolution image analysis. Neuroscience. 1995;64:397–404. doi: 10.1016/0306-4522(94)00427-7. [DOI] [PubMed] [Google Scholar]
- 5.Crook ZR, Housman DE. Dysregulation of dopamine receptor D2 as a sensitive measure for Huntington disease pathology in model mice. Proc Natl Acad Sci U S A. 2012;109:7487–92. doi: 10.1073/pnas.1204542109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baias M, Smith PES, Shen K, Joachimiak LA, Żerko S, Koźmiński W, Frydman J, Frydman L. Structure and Dynamics of the Huntingtin Exon-1 N-Terminus: A Solution NMR Perspective. J Am Chem Soc. 2017;139:1168–1176. doi: 10.1021/jacs.6b10893. [DOI] [PubMed] [Google Scholar]
- 7.DiFiglia M. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science (80-) 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 8.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–48. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 9.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19:2522–34. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Todi SV, Paulson HL. Balancing act: deubiquitinating enzymes in the nervous system. Trends Neurosci. 2011;34:370–382. doi: 10.1016/j.tins.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20:1242–1253. doi: 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- 12.Schipper-Krom S, Juenemann K, Reits EAJ. The ubiquitin-proteasome system in huntington’s disease: Are proteasomes impaired, initiators of disease, or coming to the rescue? Biochem Res Int. 2012 doi: 10.1155/2012/837015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim KL, Lim GGY. K63-linked ubiquitination and neurodegeneration. Neurobiol Dis. 2011;43:9–16. doi: 10.1016/j.nbd.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Atkin G, Paulson H. Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci. 2014;7:63. doi: 10.3389/fnmol.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Rourke JG, Gareau JR, Ochaba J, Song W, Raskó T, Reverter D, Lee J, Monteys AM, Pallos J, Mee L, Vashishtha M, Apostol BL, Nicholson TP, Illes K, Zhu YZ, Dasso M, Bates GP, Difiglia M, Davidson B, Wanker EE, Marsh JL, Lima CD, Steffan JS, Thompson LM. SUMO-2 and PIAS1 modulate insoluble mutant huntingtin protein accumulation. Cell Rep. 2013;4:362–75. doi: 10.1016/j.celrep.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–4. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 17.Vittal V, Shi L, Wenzel DM, Scaglione KM, Duncan ED, Basrur V, Elenitoba-Johnson KSJ, Baker D, Paulson HL, Brzovic PS, Klevit RE. Intrinsic disorder drives N-terminal ubiquitination by Ube2w. Nat Chem Biol. 2014 doi: 10.1038/nchembio.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scaglione KM, Basrur V, Ashraf NS, Konen JR, Elenitoba-Johnson KSJ, Todi SV, Paulson HL. The ubiquitin-conjugating enzyme (E2) ube2w ubiquitinates the N terminus of substrates. J Biol Chem. 2013;288:18784–18788. doi: 10.1074/jbc.C113.477596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatham MH, Plechanovová A, Jaffray EG, Salmen H, Hay RT. Ube2W conjugates ubiquitin to α-amino groups of protein N-termini. Biochem J. 2013;453:137–45. doi: 10.1042/BJ20130244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scaglione KM, Zavodszky E, Todi SV, Patury S, Xu P, Rodríguez-Lebrón E, Fischer S, Konen J, Djarmati A, Peng J, Gestwicki JE, Paulson HL. Ube2w and Ataxin-3 Coordinately Regulate the Ubiquitin Ligase CHIP. Mol Cell. 2011;43:599–612. doi: 10.1016/j.molcel.2011.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fletcher AJ, Christensen DE, Nelson C, Tan CP, Schaller T, Lehner PJ, Sundquist WI, Towers GJ. TRIM5α requires Ube2W to anchor Lys63-linked ubiquitin chains and restrict reverse transcription. EMBO J. 2015 doi: 10.15252/embj.201490361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fletcher AJ, Mallery DL, Watkinson RE, Dickson CF, James LC. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1507534112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang B, Merillat SA, Vincent M, Huber AK, Basrur V, Mangelberger D, Zeng L, Elenitoba-Johnson K, Miller RA, Irani DN, Dlugosz AA, Schnell S, Scaglione KM, Paulson HL. Loss of the Ubiquitin-conjugating Enzyme UBE2W Results in Susceptibility to Early Postnatal Lethality and Defects in Skin, Immune, and Male Reproductive Systems. J Biol Chem. 2016;291:3030–42. doi: 10.1074/jbc.M115.676601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng L, Tallaksen-Greene SJ, Wang B, Albin RL, Paulson HL. The de-ubiquitinating enzyme ataxin-3 does not modulate disease progression in a knock-in mouse model of Huntington disease. J Huntingtons Dis. 2013;2:201–15. doi: 10.3233/JHD-130058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng L, Wang B, Merillat SA, N Minakawa E, Perkins MD, Ramani B, Tallaksen-Greene SJ, Costa M do C, Albin RL, Paulson HL. Differential recruitment of UBQLN2 to nuclear inclusions in the polyglutamine diseases HD and SCA3. Neurobiol Dis. 2015;82:281–288. doi: 10.1016/j.nbd.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barmada SJ, Ju S, Arjun A, Batarse A, Archbold HC, Peisach D, Li X, Zhang Y, Tank EMH, Qiu H, Huang EJ, Ringe D, Petsko GA, Finkbeiner S. Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc Natl Acad Sci U S A. 2015;112:7821–6. doi: 10.1073/pnas.1509744112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, Tsvetkov A, Pleiss M, Li X, Peisach D, Shaw C, Chandran S, Finkbeiner S. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10:677–85. doi: 10.1038/nchembio.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller J, Arrasate M, Shaby BA, Mitra S, Masliah E, Finkbeiner S. Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into huntington’s disease molecular pathogenesis. J Neurosci. 2010;30:10541–50. doi: 10.1523/JNEUROSCI.0146-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 30.Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30:639–49. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet. 2008;17:345–56. doi: 10.1093/hmg/ddm311. [DOI] [PubMed] [Google Scholar]
- 33.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 34.Heng MY, Detloff PJ, Paulson HL, Albin RL. Early alterations of autophagy in Huntington disease-like mice. Autophagy. 2010;6:1206–8. doi: 10.4161/auto.6.8.13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heng MY, Duong DK, Albin RL, Tallaksen-Greene SJ, Hunter JM, Lesort MJ, Osmand A, Paulson HL, Detloff PJ. Early autophagic response in a novel knock-in model of Huntington disease. Hum Mol Genet. 2010;19:3702–20. doi: 10.1093/hmg/ddq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ochaba J, Monteys AM, O’Rourke JG, Reidling JC, Steffan JS, Davidson BL, Thompson LM. PIAS1 Regulates Mutant Huntingtin Accumulation and Huntington’s Disease-Associated Phenotypes In Vivo. Neuron. 2016;90:507–520. doi: 10.1016/j.neuron.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heng MY, Tallaksen-Greene SJ, Detloff PJ, Albin RL. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington’s disease. J Neurosci. 2007;27:8989–98. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, Greengard P. Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc Natl Acad Sci. 2000;97:6809–6814. doi: 10.1073/pnas.120166397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Dellen A, Welch J, Dixon RM, Cordery P, York D, Styles P, Blakemore C, Hannan AJ. N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington’s disease mice. Neuroreport. 2000;11:3751–7. doi: 10.1097/00001756-200011270-00032. [DOI] [PubMed] [Google Scholar]
- 40.Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R, Mahal A, Smith DL, Woodman B, Bates GP. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389–405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- 41.Maynard CJ, Böttcher C, Ortega Z, Smith R, Florea BI, Díaz-Hernández M, Brundin P, Overkleeft HS, Li JY, Lucas JJ, Dantuma NP. Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment. Proc Natl Acad Sci U S A. 2009;106:13986–91. doi: 10.1073/pnas.0906463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ortega Z, Díaz-Hernández M, Maynard CJ, Hernández F, Dantuma NP, Lucas JJ. Acute polyglutamine expression in inducible mouse model unravels ubiquitin/proteasome system impairment and permanent recovery attributable to aggregate formation. J Neurosci. 2010;30:3675–88. doi: 10.1523/JNEUROSCI.5673-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhat KP, Yan S, Wang CE, Li S, Li XJ. Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc Natl Acad Sci U S A. 2014;111:5706–11. doi: 10.1073/pnas.1402215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Wang CE, Orr A, Tydlacka S, Li SH, Li XJ. Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J Cell Biol. 2008;180:1177–89. doi: 10.1083/jcb.200709080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, Davidson BL, Rebagliati MR, Paulson HL. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci. 2005;25:9152–61. doi: 10.1523/JNEUROSCI.3001-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weiss A, Klein C, Woodman B, Sathasivam K, Bibel M, Régulier E, Bates GP, Paganetti P. Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington’s disease. J Neurochem. 2008;104:846–58. doi: 10.1111/j.1471-4159.2007.05032.x. [DOI] [PubMed] [Google Scholar]
- 47.Leitman J, Ulrich Hartl F, Lederkremer GZ. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat Commun. 2013;4:2753. doi: 10.1038/ncomms3753. [DOI] [PubMed] [Google Scholar]
- 48.Ehrnhoefer DE, Duennwald M, Markovic P, Wacker JL, Engemann S, Roark M, Legleiter J, Marsh JL, Thompson LM, Lindquist S, Muchowski PJ, Wanker EE. Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum Mol Genet. 2006;15:2743–51. doi: 10.1093/hmg/ddl210. [DOI] [PubMed] [Google Scholar]
- 49.Nucifora LG, Burke KA, Feng X, Arbez N, Zhu S, Miller J, Yang G, Ratovitski T, Delannoy M, Muchowski PJ, Finkbeiner S, Legleiter J, Ross CA, Poirier MA. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J Biol Chem. 2012;287:16017–28. doi: 10.1074/jbc.M111.252577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, Leavitt BR, Hayden MR. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A. 2005;102:11402–7. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, Kim TW, Williams M, Reddy PH, Tagle D, Boyce FM, Won L, Heller A, Aronin N, DiFiglia M. Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci. 1999;19:964–73. doi: 10.1523/JNEUROSCI.19-03-00964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–57. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- 53.Gafni J, Hermel E, Young JE, Wellington CL, Hayden MR, Ellerby LM. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem. 2004;279:20211–20. doi: 10.1074/jbc.M401267200. [DOI] [PubMed] [Google Scholar]
- 54.Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the Caspase-6 Site Is Required for Neuronal Dysfunction and Degeneration Due to Mutant Huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 55.Trausch-Azar JS, Lingbeck J, Ciechanover A, Schwartz AL. Ubiquitin-Proteasome-mediated degradation of Id1 is modulated by MyoD. J Biol Chem. 2004;279:32614–9. doi: 10.1074/jbc.M403794200. [DOI] [PubMed] [Google Scholar]
- 56.Breitschopf K, Bengal E, Ziv T, Admon A, Ciechanover A. A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 1998;17:5964–73. doi: 10.1093/emboj/17.20.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reinstein E, Scheffner M, Oren M, Ciechanover A, Schwartz A. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: targeting via ubiquitination of the N-terminal residue. Oncogene. 2000;19:5944–50. doi: 10.1038/sj.onc.1203989. [DOI] [PubMed] [Google Scholar]
- 58.Aviel S, Winberg G, Massucci M, Ciechanover A. Degradation of the epstein-barr virus latent membrane protein 1 (LMP1) by the ubiquitin-proteasome pathway. Targeting via ubiquitination of the N-terminal residue. J Biol Chem. 2000;275:23491–9. doi: 10.1074/jbc.M002052200. [DOI] [PubMed] [Google Scholar]
- 59.Ikeda M, Ikeda A, Longnecker R. Lysine-independent ubiquitination of Epstein-Barr virus LMP2A. Virology. 2002;300:153–9. doi: 10.1006/viro.2002.1562. [DOI] [PubMed] [Google Scholar]
- 60.Ben-Saadon R, Fajerman I, Ziv T, Hellman U, Schwartz AL, Ciechanover A. The tumor suppressor protein p16(INK4a) and the human papillomavirus oncoprotein-58 E7 are naturally occurring lysine-less proteins that are degraded by the ubiquitin system. Direct evidence for ubiquitination at the N-terminal residue. J Biol Chem. 2004;279:41414–21. doi: 10.1074/jbc.M407201200. [DOI] [PubMed] [Google Scholar]
- 61.Coulombe P, Rodier G, Bonneil E, Thibault P, Meloche S. N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol Cell Biol. 2004;24:6140–50. doi: 10.1128/MCB.24.14.6140-6150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuo ML, den Besten W, Bertwistle D, Roussel MF, Sherr CJ. N-terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev. 2004;18:1862–74. doi: 10.1101/gad.1213904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trausch-Azar J, Leone TC, Kelly DP, Schwartz AL. Ubiquitin proteasome-dependent degradation of the transcriptional coactivator PGC-1{alpha} via the N-terminal pathway. J Biol Chem. 2010;285:40192–200. doi: 10.1074/jbc.M110.131615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol. 2007;14:941–948. doi: 10.1038/nsmb1295. [DOI] [PubMed] [Google Scholar]
- 65.Wacker JL, Huang SY, Steele AD, Aron R, Lotz GP, Nguyen Q, Giorgini F, Roberson ED, Lindquist S, Masliah E, Muchowski PJ. Loss of Hsp70 Exacerbates Pathogenesis But Not Levels of Fibrillar Aggregates in a Mouse Model of Huntington’s Disease. J Neurosci. 2009;29:9104–9114. doi: 10.1523/JNEUROSCI.2250-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guzhova IV, Lazarev VF, Kaznacheeva AV, Ippolitova MV, Muronetz VI, Kinev AV, Margulis BA. Novel mechanism of Hsp70 chaperone-mediated prevention of polyglutamine aggregates in a cellular model of huntington disease. Hum Mol Genet. 2011;20:3953–3963. doi: 10.1093/hmg/ddr314. [DOI] [PubMed] [Google Scholar]