

Abstract
To overcome the challenges in the analysis of protein glycosylation, we have developed a comprehensive and universal tool through permethylation of glycopeptides and their tandem mass spectrometric analysis. This method has the potential to simplify glycoprotein analysis by integrating glycan sequencing and glycopeptide analysis in a single experiment. Moreover, glycans with unique glycosidic linkages, particularly from prokaryotes, which are resistant to enzymatic or chemical release, could also be detected and analyzed by this methodology. Here we present a strategy for the permethylation of intact glycopeptides, obtained via controlled protease digest, and their characterization by using advanced mass spectrometry. We used bovine RNase B, human transferrin, and bovine fetuin as models to demonstrate the feasibility of the method. Remarkably, the glycan patterns, glycosylation site, and their occupancy by N-glycans are all detected and identified in a single experimental procedure. Acquisition on a high resolution tandem-MSn system with fragmentation methodologies such as high-energy collision dissociation (HCD) and collision induced dissociation (CID), provided the complete sequence of the glycan structures attached to the peptides. The behavior of 20 natural amino acids under the basic permethylation conditions was probed by permethylating a library of short synthetic peptides. Our studies indicate that the permethylation imparts simple, limited, and predictable chemical transformations on peptides and do not interfere with the interpretation of MS/MS data. In addition to this, permethylated O-glycans in unreduced form (released by β elimination) were also detected, allowing us to profile O-linked glycan structures simultaneously.
Graphical abstract
The focus of numerous studies in the elucidation of complex biological processes and diseases has recently converged on the structural analysis of glycoproteins. Protein glycosylation plays important roles in distinct functions in all organisms, ranging from microbes to highly organized species in both animal and plant kingdoms. Glycosylation is ubiquitously important in host–pathogen interactions, cellular communication, cell adhesion, molecular trafficking and clearance, immunosurveillance, receptor functions at the membrane level, and other intriguing processes.1–3 Unusual glycosylation has been deduced as the etiology behind various human pathological conditions such as hereditary disorders,4,5 cardiovascular and liver disorders,6–9 neurological diseases,2 muscular dystrophies,10 and different forms of cancer.11,12 Both qualitative (aberrant glycan structures) and quantitative perturbations in the highly heterogeneous and complex glycans of both N-linked and/or O-linked glycoconjugate types have been associated with these abnormal conditions.4,12
The development of highly sensitive, quantitatively and qualitatively accurate and reliable analytical methods and instrumentation is warranted by the complex relationships of glycan structures and their functions. Advancements in mass spectrometry, microarray technologies, chromatographic systems, and nuclear magnetic resonance (NMR) as well as improved sample preparation techniques have yielded great progress in the field of glycobiology. These advances have been driven by the very low sample amounts obtainable from clinical sources, the extremely heterogeneous nature of glycan occupancies and structures, the need for high-throughput reliable analysis methods, and recent discoveries of more unusual glycoconjugates.13–16
Recently, mass spectrometry has emerged as one of the most powerful tools for the characterization of glycoproteins.13 Glycoprotein profiling comprises the structural characterization of both the glycan and peptide moieties as well as pinpointing the site of attachment of the glycans to the peptides. Development of advanced high-resolution tandem MS/MS techniques has enabled simultaneous analysis of intact glycopeptides and the site of glycan-protein attachment to a significant extent.14,15 However, characterization of isomeric glycan species, including sequence and glycosidic linkage positions, is very difficult due to potential coelution and frequently identical fragmentation patterns. Even though high-performance liquid chromatography and tandem mass spectrometry (MS/MS) of proteolytic glycopeptides provide information on the composition and linear sequences of glycans in addition to protein sequence and site of glycan attachment, this method does not give detailed information about glycan branching and linkages. Detection of multiple glycoforms of glycopeptides, which tend to be poorly ionized and are often present as minor fractions relative to peptides, is currently a challenging task in the glycoproteomic profiling of glycoproteins.17,18 Moreover, glycopeptides of low charge state are poor candidates for the electron transfer dissociation (ETD) fragmentation methodology, which is presently state-of the art technology for accurate site mapping of post-translational modification on peptides.19 Multifaceted work-flows for the enrichment of glycopeptides from among the peptides are often associated with profiling of glycoprotein from a complex proteome.
Because of the limitations of unmodified glycopeptide analysis, detailed structural analysis of glycans, including the determination of monosaccharide sequence and linkage positions, is mostly performed on glycans released from the proteins by either enzymatic (peptide-N-glycosidase F; PNGase F) or chemical methods (β-elimination or hydrazinolysis). Released glycans are often derivatized to improve sensitivity, and permethylation is the method of choice for comprehensive characterization of glycans.17 Glycan permethylation replaces every free hydroxyl group (−OH) with a methoxy group (−OCH3) and leads to more useful product ion spectra for the characterization of glycan isomers. Sequential fragmentation (MSn) of glycans by CID enables the characterization of branching and monosaccharide attachment patterns in the glycans. Groups involved in glycosidic linkages are not methylated and thus provide information on signature differences by imparting a 14 Da mass deficiency to glycosidic and cross ring fragments.17,20,21 The permethylation process simplifies sample purification, improves the sensitivity of detection, stabilizes carbohydrates against in-source fragmentation, and allows simultaneous analysis of basic, acidic, and neutral residues.22–25 Nonetheless, this classical approach forfeits the potential site of glycan attachment information and thus necessitates parallel processing of released peptides for the determination of glycosylation sites. Moreover, several challenges have been reported in the effective release of glycans from the peptides.26 While alternate endoglycosidase enzymes (peptide-N-glycosidase A; PNGase A) are available to deglycosylate plant and insect N-glycopeptides, there are no known enzymes that are able to release glycans from bacterial glycoproteins, which frequently feature unusual sugar residues in the glycan-protein linkage, e.g., the bacillosamine derivative in C. jejuni.26 Furthermore, there are other unique classes of glycan attachments, including extensive O-glycosylation of 4(R)-hydroxy-L-proline (Hyp) residues on the polyproline chains of plant arabinogalactan proteins, and these glycans are not susceptible to any of the available glycosidases or conventional chemical release methods.27
Permethylation, which was commonly used for mass spectrometric peptide characterization decades ago became less popular due to some unwanted side reactions, concerns over the stability of peptides during permethylation, and development of several alternative mass spectrometric technologies.28,29 The 20 natural amino acids that constitute biological proteins feature a great variety of functional groups, many of which can reasonably be expected to be sensitive to the relatively strong basic conditions of the permethylation reaction. However, there are few reports that describe the behavior of amino acids under such conditions.28
The work presented here combines the analytical power of glycan permethylation with glycopeptide analysis, preserving site-specific information for comprehensive glycoprotein analysis. Thus, we envisaged the possibility of retaining the site of glycan attachment of N-linked glycopeptides by direct permethylation of glycopeptides and their analysis by tandem mass spectrometry. Permethylation of glycan-amino acid conjugates obtained by exhaustive digestion of the glycoprotein to amino acids by nonspecific protease was reported previously.20,26 However, the complete digestion of the protein abolished all sequence information, and therefore site mapping of the glycopeptide was not addressed in these reports. We have instead focused our studies on the permethylation of glycopeptides that are large enough to retain sequence-identifying information. Here, we report the effective permethylation of peptides and glycopeptides and show the tandem mass spectrometric analysis of the resulting permethylated glycopeptides, yielding attachment sites, isomeric glycan distributions, and glycan sequence and linkage determination. The methodology presented here combines two or more lengthy sample preparation workflows into a one-pot, three-step procedure, greatly simplifying the whole process of glycoprotein analysis. Moreover, the simpler workflow for sample processing before mass spectrometric analysis can enhance the applicability of the methodology for the characterization of large sample sets of glycoproteins with minimal skill level involvement. This tool has the potential to enable high-throughput comprehensive characterization of glycosylation where analysis of site of glycan attachment is often important.
EXPERIMENTAL PROCEDURES
General Materials
Bovine RNase B, bovine fetuin, dithiothreitol (DTT), iodoacetamide (IAA), and methyl iodide (MeI) were purchased from Sigma-Aldrich. Human transferrin and Pronase were purchased from Roche Diagnostics. Sequencing-grade modified trypsin was purchased from Promega. For the peptide synthesis, amino acid derivatives and Wang resin were purchased from Merck Millipore. All other chemical reagents were purchased from AnaSpec and Sigma-Aldrich, unless otherwise mentioned. Synthetic peptides were either purchased (American Peptide Company, CA; LifeTein, NJ) or in-house synthesized on a CEM Liberty peptide synthesizer 908505 equipped with a UV detector. Mass spectrometric data acquisition was performed in the positive ion mode on a Thermo Scientific LTQ Orbitrap Fusion Tribrid mass spectrometer and on AB SCIEX MALDI TOF/TOF 5800 (Applied Biosystem MDS Analytical Technologies) mass spectrometer.
Synthesis of Peptides
The peptides were synthesized on the 0.1 mmol scale using Wang resin as a solid support on microwave-assisted automated peptide synthesizer using N-α-Fmoc-protected amino acids and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU)/1-hydroxy benzotriazole (HOBt) as the activating reagents and N, N-diisopropylethylamine (DIPEA) as the base. The coupling of the first amino acid to Wang resin was performed manually in CH2Cl2/N, N-dimethylformamide (DMF) (9:1) using HOBt, 4-dimethylaminopyridine (DMAP), and N, N′-diisopropylcarbodiimide (DIC). Further, amino acids were coupled on CEM Liberty peptide synthesizer using standard protocol. Cleavage of peptide from resin support and removal of side chain protecting groups were performed at room temperature by treatment with 50% trifluoroacetic acid (TFA) in CH2Cl2 (v/v) for 4–6 h. The resin was then filtered off and peptides were precipitated in ice cold diethyl ether and recovered by centrifugation.
Permethylation of Synthetic Peptides
We have performed permethylation based on a procedure developed for glycans,24,25 with some modifications. To the lyophilized synthetic peptides (25 μg), dissolved in 200 μL of anhydrous dimethyl sulfoxide (DMSO), 300 μL of sodium hydroxide (NaOH) base in anhydrous DMSO (prepared separately by mixing NaOH and DMSO) was added and vortexed. A volume of 100 μL of MeI was added to the sample, and the reaction mixture was vortexed vigorously for 10 min. The permethylation reaction was quenched by the addition of 2 mL of ddH2O, bubbled off excess MeI by a stream of nitrogen gas, and purified by a C18 solid phase extraction (SPE) cartridge. Cysteine containing peptides were treated with DTT and alkylated by IAA before permethylation.
Protease Digestion
A volume of 25 μL of digestion buffer (50 mM aqueous ammonium bicarbonate (NH4HCO3)) was added to 25 μL of glycoprotein (100 μg) solution. To the glycoprotein sample, 25 μL of 25 mM DTT was added, and the mixture was incubated at 45 °C for 45 min. The sample was treated with 25 μL of 90 mM IAA and incubated at room temperature for 45 min (in the dark). The protein sample was digested after dialysis by adding 5 μL of sequencing-grade trypsin (1 μg/μL) or Pronase (0.6 μg/μL) in order to obtain a protein to enzyme ratio of 1:20 or 1:30, respectively, and incubated at 37 °C for 3–24 h. The digests were heated at 100 °C for 5 min to stop the protease activity, evaporated to dryness by lyophilization, and stored at −30 °C until permethylation reaction.
Permethylation of Glycopeptides
To the lyophilized protease digest (25–100 μg), dissolved in 200 μL of anhydrous DMSO, 300 μL of NaOH base in anhydrous DMSO was added and vortexed. A volume of 100 μL of MeI was added to the sample, and the reaction mixture was vortexed vigorously for 10 min. The permethylation reaction was quenched by the addition of 2 mL of ddH2O. Excess MeI was bubbled off by a stream of nitrogen gas, and subsequently 2 mL of CH2Cl2 was added to extract permethylated glycopeptides, the mixture was vortexed thoroughly for 10 min, and the CH2Cl2 layer was washed five times with 2 mL of ddH2O. The CH2Cl2 layer was removed carefully and transferred to a clean glass tube and subsequently dried under a stream of nitrogen gas. The efficiency of the extraction was checked by MALDI-MS and gas chromatography (GC) of both layers (Supp. Figure S3).
MALDI-TOF MS Analysis
The entire amount of glycopeptides or synthetic peptides after permethylation was dissolved in 20 μL of methanol. A 2 μL portion of the resulting sample solution was mixed with 2 μL of 2,5-dihydroxy benzoic acid (DHB) matrix (20 μg in 1:1 methanol/ddH2O) and spotted on a MALDI plate. The samples were analyzed on a MALDI-TOF MS instrument in reflector positive ion mode.
ESI-MS Analysis by Direct Infusion
A 2 μL aliquot of the permethylated glycopeptides or synthetic peptides dissolved in 20 μL of methanol was mixed with 98 μL of 50% v/v 1 mM NaOH solution in methanol. Permethylated glycopeptides and synthetic peptides were infused on an Orbitrap Fusion Tribrid mass spectrometer through an NSI probe. The MS1 and MS2 spectra (CID, HCD) were acquired at 120 000 and 15 000 resolution by a simple precursor scan and total ion mapping (TIM) program, respectively. Selected individual peaks of glycan fragments at MS2 were further fragmented by CID at the MS3, MS4, or MS5 level and detected on an Orbitrap/ion trap.
Structure Interpretation
Assignment of permethylated peptides and glycopeptides structures were performed manually based on the high-resolution MS and MSn fragmentation patterns with the aid of software such as ChemBioDraw Ultra 14.0, GlycoWorkbench 1.1, Data Explorer V4.5, and Xcalibur 3.0 software.
RESULTS AND DISCUSSION
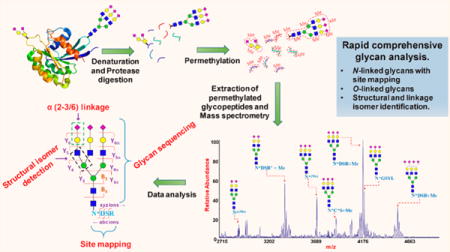
The goal of our present analysis procedure is the development of a tool for the rapid site mapping of glycan attachment and comprehensive glycan structure analysis, by exploring the structural leverage inherent in the process of glycopeptide permethylation, followed by tandem MS (Figure 1).
Figure 1.
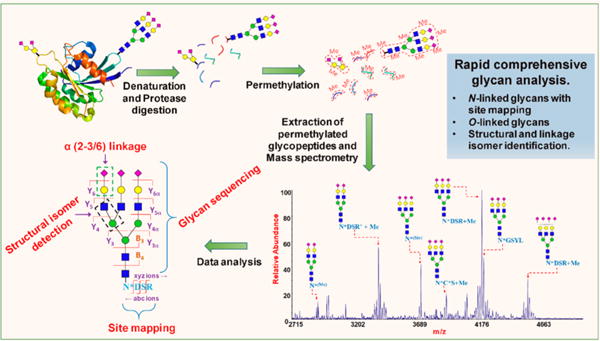
Strategy for the analysis of glycoprotein by integrating glycomics and glycoprotemics in a single experiment through the permethylation of glycopeptides and their tandem mass spectrometry analysis.
Behavior of Amino Acids under Permethylation Conditions
Although derivatization by permethylation has been a routine method for the detailed structural characterization of released glycans, there are only a few recent reports mentioning the permethylation of peptides.20,23,24 Before the advent of high-resolution tandem mass spectrometry, permethylation was commonly used for mass spectrometric peptide characterization. However, some side reactions that were observed in those early studies, concerns over the stability of peptides during permethylation, and development of several alternative mass spectrometric technologies caused peptide permethylation to fall out of favor and to be used only rarely.28,29 Nevertheless, the mass spectrometric characterization of glycosylated peptides, which has gained tremendous importance in recent years, poses challenges for which permethylation offers attractive advantages in obtaining detailed structural information, including linkage and branching data.20–24 We have now found that, although side reactions do occur during the permethylation of peptides, they are limited to a few amino acids, are henceforth predictable, and do not prevent the interpretation of mass spectra and the characterization of peptide sequence and glycan structures. The procedure causes amino acids with free amino or sulfhydryl groups to be converted into onium ions, receiving a positive charge, and these onium ions undergo β-elimination if an acidic proton is present at the β-position. Accordingly, though the side chain amino group of lysine and sulfhydryl group of methionine both become onium ions, only methionine experiences β-elimination, because, in lysine, the protons in the β-position relative to the ammonium group are not acidic (Figure 2). In some instances, methanol was β-eliminated from methylated serine and threonine (Figure 3, entries PerMe – S# and PerMe – T# and Supp. Figure S25).
Figure 2.
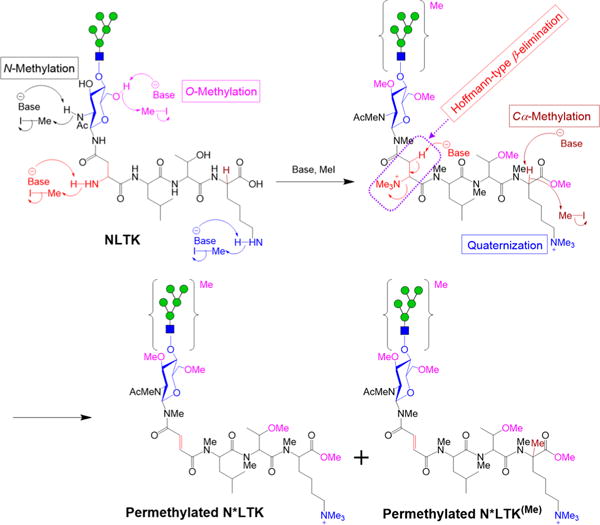
Permethylation of glycopeptides by treating with MeI in the presence of DMSO/NaOH base. N-methylation, O-methylation, and Cα-methylation were observed on the glycopeptide along with quarternization of the side chain ε-amino group of lysine and the N-terminal amino group of asparagine. Hoffmann-type β elimination of the quarternized N-terminal amino group of asparagine was observed, where acidic β proton is available. *, loss of NMe3.
Figure 3.
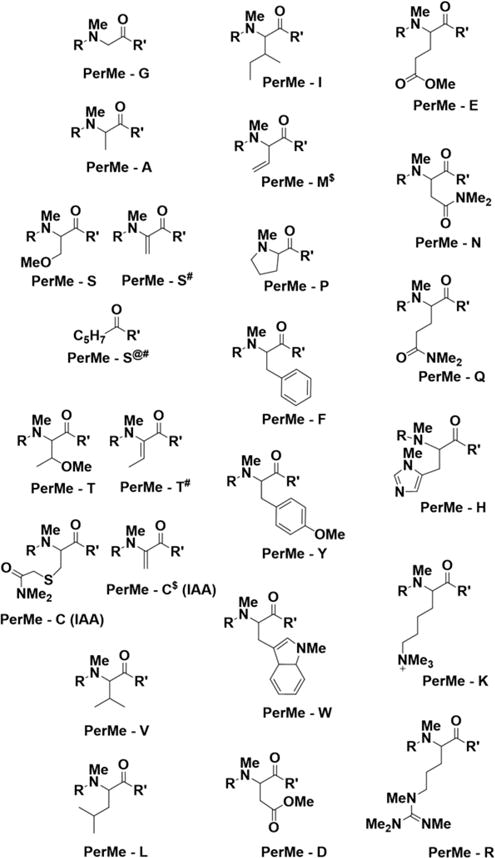
Modifications and elimination of substituents observed on the amino acids upon permethylation of peptides and glycopeptides. Standard notations were used for labeling amino acids. #loss of OMe, @loss of NMe3, and $loss of SMe.
An obvious approach to prevent the various side reactions would be to block the amino and sulfhydryl groups using classical protecting group chemistry. Our preliminary experiments included N-acetylation of free amino groups to prevent quaternization and β-elimination during the permethylation of peptides. However, this treatment, using addition of acetic anhydride in sodium bicarbonate buffer, was sometimes associated with peptide bond cleavage, partial acetylation and added a further step to the procedure (Supp. Figure S1).28,30 For these reasons, we wondered if the permethylation of peptides and glycopeptides could be accomplished directly on unprotected starting material. We observed in initial experiments on a limited number of glycopeptides that direct permethylation of underivatized glycopeptides avoided peptide bond cleavage, simplified the overall procedure, and yielded products whose generation could be rationalized rather easily. To investigate whether these findings could be generalized to peptides comprising any of the 20 natural amino acids and to understand how each amino acid behaves under the alkaline conditions of permethylation, we performed permethylation on a peptide library, which collectively comprises all the 20 amino acids in varying combinations, and characterized the products by MSn (Figure 3, Supp. Figures S8–S25, and Supp. Tables S1–S17). The results indicate that all amino, amido, hydroxyl, and sulfhydryl groups are methylated in the procedure along with minor methylation reaction on the α-carbon of some amino acids (C-methylation).28,31 Methylation of amino groups, including those found in the side chains of lysine, histidine, and arginine, as well as the N-terminal α-amino group, proceeded to the quaternary ammonium salt.28 If the N-terminal residue had a β-proton, β-elimination of the quaternized N-terminal amino group occurred consistently. Furthermore, we found elimination of the sulfhydryl groups of unprotected cysteine and thioether of methionine. However, protection of cysteine by the customary protecting group iodoacetamide prevented elimination of the sulfhydryl group (Supp. Figures S13 and S14 and Supp. Tables S7 and S8). We have observed unusual behavior of serine when it is located at the N-terminus of a peptide. Loss of both the quaternized N-terminal amino group as well as the side chain methoxy group and further, as yet unknown, modification was consistently found in the permethylation of N-terminal serine. The final product of this reaction contained a group with a mass consistent with the composition C5H7 (Figure 3, entry PerMe – S@# and Supp. Figures S17 and S20, Supp. Tables S11 and S14). Experiments to determine the structure and the mechanism of formation of this product as well as experiments with N-terminal threonine are underway in our laboratory. We have found this transformation in every peptide with serine at the N-terminus, leading us to conclude that the present lack of knowledge of its precise structure does not hinder the identification of glycopeptides.
It is to be noted that all studied glycopeptides, covering a wide range of hydrophilicity and including glycopeptides with quaternizable groups, consistently partitioned to the organic layer upon permethylation (Supp. Figures S2 and S3). We observed almost complete recovery of permethylated glycopeptide in the organic layer upon liquid–liquid extraction as the peaks corresponding to permethylated glycopeptides were not detected in the water layer (Supp. Figure S3A,B). In addition to this, we have performed monosaccharide composition analysis by GC-MS on both the organic and water layers and observed monosaccharides only in the organic layer (Supp. Figure S3C,D). Results similar to that of organic extraction were obtained when C18 SPE was performed for the cleanup of glycopeptides after permethylation (Supp. Figure S4). In contrast, unglycosylated peptides with quaternizable groups tended to partition into the aqueous layer and had to be purified by C18 SPE. This additional step is unnecessary in our method, which presents an added advantage of permethylation.
Permethylated peptides were characterized by precursor ion spectra at high-resolution and MS2 fragmentation by CID. On the basis of MS2 fragmentation of the peptide library, modifications and elimination of substituents on the peptides during the permethylation reaction were deduced (Figure 3, Supp. Tables S1–17).
Optimization of Protease Digestion of Glycoproteins
Although confident glycosylation site identification requires a minimum peptide length of 3 amino acids, peptides should be rather small (<6 amino acids) in order to simplify the data interpretation. This defines a preliminary working range of usable peptide length for the purposes of this approach as 3–6 amino acids. Protein digestion using specific proteases such as trypsin is useful for rapid sequencing of the generated glycopeptides and peptides since the sequences of generated peptides can be easily predicted. Such specific proteases can be used to prepare glycopeptides for permethylation if the glycan-bearing peptide happens to fall into the 3–6 amino acid range, and we have employed trypsin for the digestion of RNase B, which results in a 4-amino acid glycopeptide. However, since the number of amino acid residues on the digested peptides depends on the distribution of digestion sites, specific proteases are not generally applicable to form suitable glycopeptides. To address this problem, we employed nonspecific protease Pronase for the digestion of glycoproteins, where the number of amino acid residues on the peptides ranging from one to several amino acids could be generated.32 Glycosylation provides some protection from proteolysis to the peptide bonds in the vicinity of the glycosylation site, making it possible to obtain small glycopeptides of a tunable size range.33,34 The process used here includes denaturation by disulfide reduction and carbamidomethylation before Pronase digestion. The digestion conditions of Pronase, such as the ratio of protease to glycoprotein, digestion buffer, and duration of digestion, were optimized (Supp. Figure S2). We obtained glycopeptides with 3–6 amino acid residues using a 3 h digestion with Pronase at the ratio of 1/30 with respect to glycoprotein concentration (37 °C in 50 mM NH4HCO3) (Supp. Figures S2). This condition was found as an optimum condition for all the glycoproteins tested, namely, bovine RNase B, human transferrin, and bovine fetuin (Figures 4–6).
Figure 4.
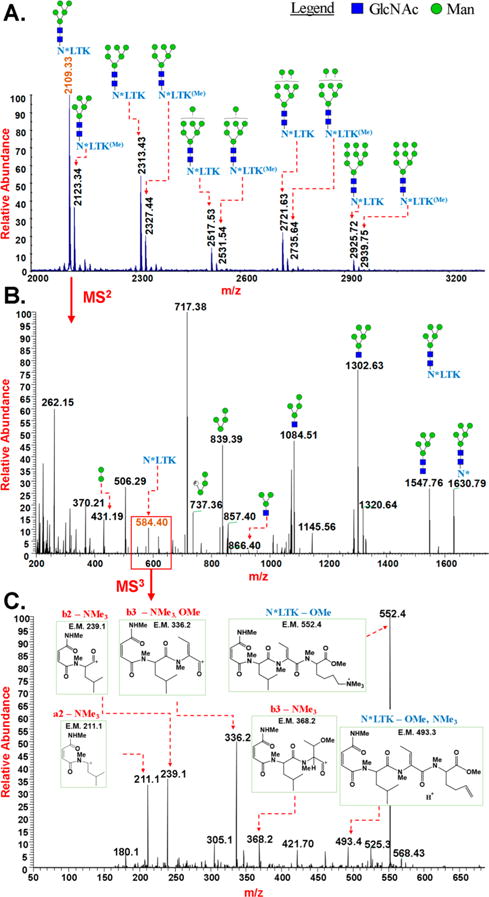
Analysis of tryptic digest of RNase B by permethylation of glycopeptides and mass spectrometry: (A) MALDI-MS spectrum of multiple glycoforms of permethylated glycopeptide NLTK of RNase B, (B) CID MS2 of permethylated glycopeptide NLTK of RNase B with high mannose structures, (C) CID MS3 of permethylated peptide fragment NLTK. *loss of NMe3; (Me)Cα-methylation.
Figure 6.
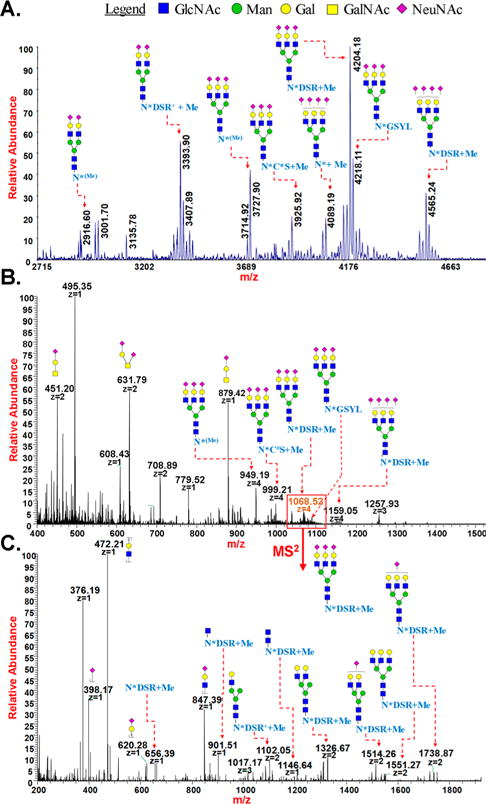
Analysis of Pronase digest of fetuin by permethylation of glycopeptides and mass spectrometry: (A) MALDI-MS spectrum of multiple glycoforms of permethylated glycopeptides “NCS”, “NDSR”, and “NGSYL” of fetuin; (B) ESI-MS spectra of O-glycans (released during permethylation) and multiple glycoforms of permethylated N-linked glycopeptides “NCS”, “NDSR”, and “NGSYL” of fetuin; and (C) HCD MS2 of permethylated glycopeptide “NDSR” of fetuin with triantennary sialylated structures. *loss of NMe3; (Me)Cα-methylation.
Permethylation of Glycopeptides Obtained from Protease Digest
Direct permethylation of the protease digest was performed using the standard NaOH/DMSO-methyl iodide (MeI) system,24,25 after the complete evaporation of aqueous digestion buffer. The permethylation reaction was quenched by the addition of ddH2O, and the permethylated glycopeptides were extracted with CH2Cl2. As mentioned in the previous section, we have found efficient partitioning of permethylated, including quaternized, glycopeptides into the organic layer (Supp. Figure S3).
During mass spectrometry analysis, the higher mass of permethylated glycopeptides relative to permethylated peptides enabled direct analysis of permethylated glycopeptides by MALDI-MS and ESI-MS without interferences from permethylated peptides (Figure 1 and Supp. Figure S3). One of the major challenges in the conventional underivatized glycopeptide analysis is the appearance of peptides and glycopeptides in the same mass range, affecting the detection of glycopeptides from among all the species present. This is further exacerbated by the ion-suppressing influence that peptides exert on glycopeptides. Characteristic signature ions have been used to select glycopeptides from among nonglycosylated peptides,35,36 but this step is not necessary in our protocol since permethylation increases the mass difference between peptides and glycopeptides significantly, and thus they are well separated in the parent spectra. The Pronase digestion produces small peptides, and the permethylation increases the glycan mass, resulting in simple mass separation of peptides and glycopeptides, so that one can isolate by simply selecting the appropriate mass range in MALDI-MS or ESI-MS (Supp. Figure S3). In addition, the permethylation also prevents ion suppression and enhances the detection sensitivity for glycopeptides. Even though the sample preparation includes five ddH2O washes of the CH2Cl2 layer, the mass spectra did not indicate any bias toward the uncharged permethylated glycopeptides, in fact, the opposite is the case, as evidenced by stronger signals in mass spectrometry. It appears that the permanent charge of the onium species enhances the ionization efficiency of quaternized glycopeptides (Figures 4–6).
Tandem MS/MS Analysis of Permethylated Glycopeptides and Glycosylation Site Mapping
RNase B was digested with both trypsin and Pronase for the direct permethylation and mass spectrometric characterization. With both digestions, glycoforms with the same sets of high mannose structures were detected (Figure 4 and Supp. Figure S2). Trypsin digestion produced glycopeptide NLTK, comprising N60, and MS analysis revealed that elimination of the N-terminal amino group of asparagine had occurred upon permethylation (Figure 4). Moreover, the side chain amino group of lysine was trimethylated, imparting a positive charge to the glycopeptide. Nevertheless, the positive charge did not prevent partitioning into the organic CH2Cl2 layer during the extraction of the aqueous permethylation mixture. While complete permethylation of both the glycan (hydroxyl and amido groups) and peptide (hydroxyl, amido, and amino groups) portions was achieved, we also observed an additional methyl group on the glycopeptide. MS3 spectra of the NTLK peptide fragment of the permethylated glycopeptide indicated an extra methyl group on the α carbon of lysine (Figures 2 and 4, Supp. Figures S10 and S11, and Supp. Tables S4 and S5). Permethylation of a synthetically prepared peptide, NLTK, also showed methylation on the α carbon of lysine, consistent with earlier observation (Figure 2, Supp. Figures S15 and S16, and Supp. Tables S9 and S10). The identification of the NLTK glycopeptide located the site of glycosylation, based on knowledge of the amino acid sequence of the protein (Figure 4).
In order to demonstrate the applicability of glycopeptide permethylation for the characterization of biantennary sialylated N-linked glycosylation, human transferrin was subjected to controlled Pronase digestion and the resulting products were permethylated directly. Analysis of the permethylated glycopeptides by MALDI-MS detected both known sites of N-glycosylation. Two glycopeptides involving site N432, dipeptide NK and tripeptide NKS, bearing biantennary disialylated complex N-glycans, were detected. The expected loss of the N-terminal amino group of asparagine was observed on both peptides, along with quaternization of the lysine side chain amino group. Loss of the terminal amino group was also observed at the asparagine of the peptide NVT, at the site N630 of transferrin. Peaks associated with peptide α-carbon methylation were also observed in both cases. MSn experiments of the permethylated glycopeptides provided the glycan structures, together with the sequence of peptides, on both sites of N-linked glycosylation of human transferrin (Figure 5 and Supp. Figure S5). Site-specific heterogeneity in the glycosylation of human transferrin has been suggested to be involved in some biological events and aberrant glycosylation conditions.37 Identification of abnormal glycosylation at each of the two N-linked glycosylation sites of transferrin has been correlated with various pathological conditions such as congenital disorders of glycosylation (CDG) and several liver dysfunctions.38 Development of methodology for high-throughput screening of clinical samples for the detection of abnormality in the glycosylation at the N-glycosylation sites of transferrin would facilitate pathological screening tests and study of unexplained etiology of diseases related to aberrant glycosylation.38,39 Using mass spectrometric analysis of the permethylated Pronase digest of transferrin by a high-resolution precursor scan and MSn fragmentation, we detected N-linked glycans and their glycoforms at both glycosylation sites of human transferrin simultaneously. We also demonstrated the detection of structural and linkage isomers by the MSn analysis of permethylated glycopeptides of transferrin (Supp. Figure S26.)
Figure 5.
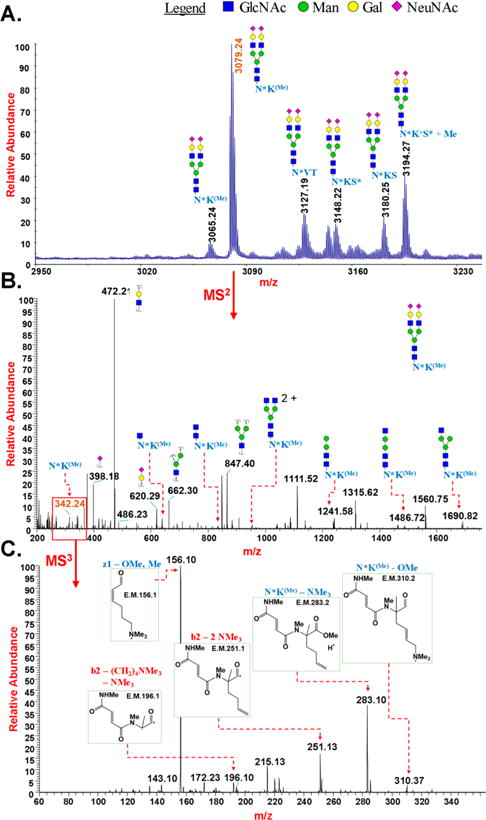
Analysis of Pronase digest of transferrin by permethylation of glycopeptides and mass spectrometry: (A) MALDI-MS spectrum of multiple glycoforms of permethylated glycopeptide “NK”, “NKS”, and “NVT” of transferrin; (B) HCD MS2 of permethylated glycopeptide “NK” of transferrin with biantennary sialylated structures; and (C) CID MS3 of permethylated peptide fragment “NK”. *loss of NMe3; (Me)Cα-methylation.
Bovine fetuin has three sites of N-glycosylation and several sites of O-glycosylation.40 By direct permethylation of the Pronase digest of bovine fetuin and MALDI-MS analysis, N-linked glycosylation at all the three sites was identified (Figure 6A). We observed bi- and triantennary sialylated complex N-glycans. Elimination of the N-terminal amino group and the cysteine thiol group as well as Cα-methylation were observed on the peptide NCS at the site N99. Similarly, elimination of the N-terminal amino group, trimethylation on the guanidine group of arginine, and Cα-methylation occurred on the peptide NDSR at the site N156 (Figure 6B,C). For peptide NGSYL at site N176, elimination of the N-terminal amino group was the only detected modification. More experiments are being conducted to identify the position and pattern of Cα-methylation on these peptides. In addition to this, a glycopeptide with a single N-linked asparagine was also detected from the Pronase digest of fetuin (Figure 6 and Supp. Figure 6).
Tandem MS/MS Analysis of Permethylated O-Glycans
In general, the glycan-protein linkage of O-glycans is not stable under the permethylation conditions.41 This means that the attachment site of O-glycans cannot be determined directly by permethylation of glycopeptides without additional labeling. However, the presence of the permethylated O-glycans allows their structural characterization. Accordingly, the permethy-lated, released O-glycans were detected in their unreduced form, and their structures were determined by MS2 analysis, the outcome of which was in agreement with previous reports (Figure 6B and Supp. Figure S7).40
Structural Isomer Determination by the MSn Analysis of Permethylated Glycopeptides
For the purpose of this methodology, we conducted experiments to distinguish topological and linkage isomerism. We speak of topological isomerism when the difference between isomers lies in the branching pattern and of linkage isomerism when the isomers have the same branching pattern but different linkage positions on the same residue.42 We chose the high mannose glycan of RNase B to demonstrate how our method can be used to identify glycan structural isomers at a single glycosylation site because this isomerism has been well characterized previously.42 To this end, we selected the Man5GlcNAc2 glycoform of permethylated glycopeptide NLTK (m/z 2109) for targeted MSn fragmentation (Figure 7A), using the characteristic mass differences imparted by permethylation between the different glycan branching patterns.
Figure 7.
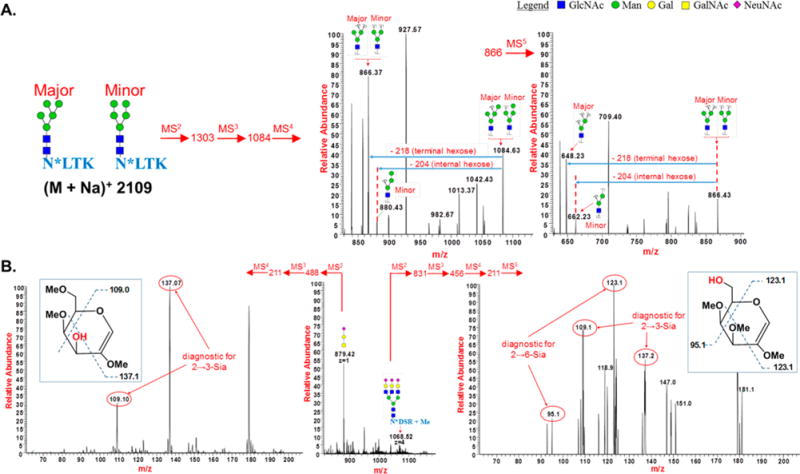
Identification of structural and linkage isomers by the MSn of permethylated glycopeptides: (A) detection of structural isomers among the high mannose glycoforms of RNase B by MSn of permethylated glycopeptides-selected regions of MSn spectra were shown; (B) detection of linkage isomers in the sialylated N-glycans (both α 2,3 and α 2,6) of fetuin; and O-glycan (only α 2,3) of fetuin by MSn of permethylated glycopeptides. MSn spectra of selected peaks were shown.
Loss of a permethylated terminal hexose residue, for example, leads to a 218-u mass difference, while loss of an internal hexose residue is associated with a 204-u mass difference, and a branching hexose residue gives a 190-u mass difference. As the glycan is successively deconstructed by CID fragmentation, the presence or absence of these characteristic mass differences reveals the identity of the different isomeric glycan structures. Three different topologies of the glycan Man5GlcNAc2 have been identified previously.42 We intended here only to demonstrate that the isomeric glycan portions of permethylated glycopeptides can be structurally characterized in the same way as that of released permethylated glycans. Therefore, we have limited the characterization to only two of the topological isomers of the glycan Man5GlcNAc2, of which one was predominant (major isomer) and one was minor. MS2 fragmentation of the permethylated glycopeptide MS peak at m/z 2109 generated the fragment ion m/z 1302 corresponding to the loss of the peptide portion and one GlcNAc from the reducing end of the glycan. Loss of terminal Man (m/z 218) upon MS3 fragmentation led to generation of the fragment m/z 1084. Subsequent MS4 fragmentation of ion m/z 1084 produced fragment ions m/z 866 and m/z 880, which corresponds to loss of terminal (m/z 218) and internal (m/z 204) Man, respectively. While fragment ion m/z 866 could be formed from both major and minor isomers, ion m/z 880 could only be formed from the minor isomer. MS5 fragmentation of ion m/z 866 generated ions m/z 648 and m/z 662, corresponding to loss of terminal (m/z 218) and internal (m/z 204) Man, respectively. Fragment ion m/z 648 could only be formed from the major isomer, whereas ion m/z 662 could only be formed from the minor isomer (Figure 7A).
Beside the topological isomers that can be encountered, linkage isomers are also common in glycans. We chose the two most common ways that sialic acid is linked to galactose in glycans, either through O-3 (2 → 3-linked) or O-6 (2 → 6-linked) to demonstrate the usefulness of our method for differentiating linkage isomers (Figure 7B). We subjected N-linked permethylated glycopeptide NDSR, bearing a trianten-nary, trisialylated glycan and a permethylated sialylated O-glycan from bovine fetuin to sequential MSn. Differentiation of 2 → 3- and 2 → 6-linked sialic acids (NeuNAc) can be achieved through a series of MSn fragmentation steps of the galactose residue to which NeuNAc was linked previously.43 The galactose fragment is then further broken down by cross-ring cleavages, revealing the position of nonmethylated oxygen, which was involved in the linkage to NeuNAc. ESI-MS2 of permethylated triantennary glycopeptides of fetuin, as their lithium adducts, generated the trisaccharide NeuNAc-Gal-GlcNAc (m/z 831). Neutral loss of NeuNAc during the MS3 fragmentation of this trisaccharide lead to generation of disaccharide Gal-GlcNAc (m/z 456). Similarly, MS4 of this disaccharide liberated free, partially methylated Gal mono-saccharide (m/z 211) with free hydroxyl groups at the linkage sites. MS5 of this ion (m/z 211) induced cross ring cleavages leading to the generation of fragments characteristic of the linkage of Gal to NeuNAc, as per previous reports.43 Fragments with m/z 109 and 137 are diagnostic for the NeuNAc- 2 → 3-Gal linkage, and fragments with m/z 95 and 123 are diagnostic for the NeuNAc 2 → 6-Gal linkage. Thus, we observed the presence of both 2 → 3 and 2 → 6 NeuNAc-Gal linkages in the N-glycopeptide NDSR of fetuin. Conversely, sequential ESI-MSn fragmentation of the permethylated released O-glycan NeuNAc-Gal-GalNAc (m/z 863) from the Pronase digest of fetuin, through the intermediate at m/z 488, led to the partially methylated Gal (m/z 211) with free hydroxyl group at the linkage sites. MS4 of partially methylated Gal (m/z 211) induced cross ring cleavages leading to the generation of fragments characteristic to the linkage of NeuNAc to Gal (Figure 7B). Here, treatment of the sialylated O-glycan released during the permethylation of fetuin glycopeptides revealed the presence only the 2 → 3- NeuNAc-Gal linkage, in agreement with previous studies.43,44
Increased sensitivity, mass accuracy and ability to perform tandem MSn experiments along with better ion isolation capability of modern mass spectrometers enables broad structural determination of glycopeptides. Permethylation has been employed for decades for released glycan characterization and offers several advantages over conventional LC–MS/MS characterization of glycopeptides without derivatization. The “scar” (lack of methyl group on oxygens previously involved in glycosidic linkages) that is generated during the fragmentation of a permethylated glycan enables the accurate characterization isomers of the glycans. Structural information on O-glycans, N-glycans with the site of attachments and linkages of terminal sialic acids at both N-linked and O-glycans along with glycoforms isomer detection were all obtained by the mass spectrometry of permethylated glycopeptides. Thus, we have demonstrated that MS of directly permethylated glycopeptides is able to provide detailed structural information on glycans in the glycopeptide context.
Although glycopeptide permethylation is currently focused on the analysis of isolated glycoproteins, the advantages can be further exploited by combining MSn detection with prior LC separation, potentially enabling comprehensive analysis of protein mixtures. Studies to optimize the conditions for the LC–MS/MS experiments are underway in our laboratory. In this regard, significant efforts in the development of software for automatic data interpretation of permethylated glycopeptide spectra are required for the characterization of a large sample set.44–48 We are in the process of building a plugin for the Grits software toolbox (http://www.grits-toolbox.org/) with the goal of automated annotation of permethylated glycopeptide spectra. Development of the automated sample handling technique for the protease digestion and permethylated in a 96-well plate format is also underway.
CONCLUSIONS
The methodology demonstrated here drastically reduces the degree of difficulty associated with the comprehensive characterization of glycoprotein primary structure by combining several analytical workflows into a single, one-pot experiment based on the permethylation of intact glycopeptides. Although permethylation entails some chemical modifications in the peptide portion of glycopeptides, these modifications have been extensively studied here and shown to be predictable. Even though a variety of glycan release (enzymatic and chemical) strategies are available, comprehensive release of N-glycans by enzymes are limited by the steric constraints of protein and/or lack of enzymes for certain unusual glycan attachments. Controlled proteolysis of protein to shorter glycopeptides and direct permethylation enables the identification of both N-linked and O-linked glycans along with the site of attachment of N-linked glycans. We also have shown that, in contrast to released glycan sequencing and direct glycopeptide analysis, this tool facilitates identification of site of attachment of N-glycans along with characterization of both N-linked and O-linked glycans simultaneously with isomeric information.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (NIH) funded research, Grant R21GM122633; resource for biomedical glycomics, Grant P41GM103490; instrument grant, Grant S10OD018530; and U.S. Department of Energy grant, Grant DE-SC0015662, to the Complex Carbohydrate Research Center. We thank Dr. Geert-Jan Boons for providing the facility to synthesize peptides and Ian Black for performing the GC/MS experiments. We would also like to thank Dr. Russell W. Carlson for critical reading of the manuscript.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.7b01730.
Supporting figures (S1–S26) and supporting tables (S1–S17) with MS analysis of permethylated glycopeptides, synthetic peptides, and MSn spectra (PDF)
ORCID
Asif Shajahan: 0000-0002-4804-1130
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Lowe JB, Marth JD. Annu Rev Biochem. 2003;72:643–691. doi: 10.1146/annurev.biochem.72.121801.161809. [DOI] [PubMed] [Google Scholar]
- 2.Ohtsubo K, Marth JD. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 3.Varki A. Cell. 2006;126:841–845. doi: 10.1016/j.cell.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 4.Freeze HH, Aebi M. Curr Opin Struct Biol. 2005;15:490–498. doi: 10.1016/j.sbi.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Marquardt T, Denecke J. Eur J Pediatr. 2003;162:359–379. doi: 10.1007/s00431-002-1136-0. [DOI] [PubMed] [Google Scholar]
- 6.Shao B, Yago T, Setiadi H, Wang Y, Mehta-D’souza P, Fu J, Crocker PR, Rodgers W, Xia L, McEver RP. Proc Natl Acad Sci U S A. 2015;112:8661–8666. doi: 10.1073/pnas.1507712112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callewaert N, Van Vlierberghe H, Van Hecke A, Laroy W, Delanghe J, Contreras R. Nat Med. 2004;10:429–434. doi: 10.1038/nm1006. [DOI] [PubMed] [Google Scholar]
- 8.Zhong M, Zhang H, Reilly JP, Chrisitie JD, Ishihara M, Kumagai T, Azadi P, Reilly MP. Arterioscler, Thromb, Vasc Biol. 2015;35:1570–1578. doi: 10.1161/ATVBAHA.115.305337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komarowska I, Coe D, Wang G, Haas R, Mauro C, Kishore M, Cooper D, Nadkarni S, Fu H, Steinbruchel DA, Pitzalis C, Anderson G, Bucy P, Lombardi G, Breckenridge R, Marelli-Berg FM. Immunity. 2015;42:1087–1099. doi: 10.1016/j.immuni.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muntoni F, Torelli S, Brockington M. Neurotherapeutics. 2008;5:627–632. doi: 10.1016/j.nurt.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vasconcelos-Dos-Santos A, Oliveira IA, Lucena MC, Mantuano NR, Whelan SA, Dias WB, Todeschini AR. Front Oncol. 2015;5:138. doi: 10.3389/fonc.2015.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lakshminarayanan V, Supekar NT, Wei J, McCurry DB, Dueck AC, Kosiorek HE, Trivedi PP, Bradley JM, Madsen CS, Pathangey LB, Hoelzinger DB, Wolfert MA, Boons GJ, Cohen PA, Gendler SJ. PLoS One. 2016;11:e0145920. doi: 10.1371/journal.pone.0145920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan S, Chen R, Aebersold R, Brentnall TA. Mol Cell Proteomics. 2011;10:R110.003251. doi: 10.1074/mcp.R110.003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Wong CH, Chin A, Taguchi A, Taylor A, Hanash S, Sekiya S, Takahashi H, Murase M, Kajihara S, Iwamoto S, Tanaka K. Nat Protoc. 2011;6:253–269. doi: 10.1038/nprot.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah P, Wang X, Yang W, Eshghi ST, Sun S, Hoti N, Chen L, Yang S, Pasay J, Rubin A, Zhang H. Mol Cell Proteomics. 2015;14:2753–2763. doi: 10.1074/mcp.M115.047928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhuo Y, Yang JY, Moremen KW, Prestegard JH. J Biol Chem. 2016;291:20085–20095. doi: 10.1074/jbc.M116.740050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shajahan A, Heiss C, Ishihara M, Azadi P. Anal Bioanal Chem. 2017;409:4483–4505. doi: 10.1007/s00216-017-0406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao L, Qu Y, Zhang Z, Wang Z, Prytkova I, Wu S. Expert Rev Proteomics. 2016;13:513–522. doi: 10.1586/14789450.2016.1172965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mechref Y. Current Protocols in Protein Science. Vol. 68. Wiley; New York: 2012. Use of CID/ETD Mass Spectrometry to Analyze Glycopeptides; pp. 12.11.1–12.11.11. Chapter 12, Unit 12.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schiel JE, Smith NJ, Phinney KW. J Mass Spectrom. 2013;48:533–538. doi: 10.1002/jms.3196. [DOI] [PubMed] [Google Scholar]
- 21.Ashline D, Singh S, Hanneman A, Reinhold V. Anal Chem. 2005;77:6250–6262. doi: 10.1021/ac050724z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morelle W, Michalski JC. Nat Protoc. 2007;2:1585–1602. doi: 10.1038/nprot.2007.227. [DOI] [PubMed] [Google Scholar]
- 23.Hakomori S. J Biochem. 1964;55:205–208. [PubMed] [Google Scholar]
- 24.Ciucanu I, Kerek F. Carbohydr Res. 1984;131:209–217. [Google Scholar]
- 25.Anumula KR, Taylor PB. Anal Biochem. 1992;203:101–108. doi: 10.1016/0003-2697(92)90048-c. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, McNally DJ, Nothaft H, Szymanski CM, Brisson JR, Li J. Anal Chem. 2006;78:6081–6087. doi: 10.1021/ac060516m. [DOI] [PubMed] [Google Scholar]
- 27.Owens NW, Stetefeld J, Lattova E, Schweizer F. J Am Chem Soc. 2010;132:5036–5042. doi: 10.1021/ja905724d. [DOI] [PubMed] [Google Scholar]
- 28.Morris HR, Dickinson RJ, Williams DH. Biochem Biophys Res Commun. 1973;51:247–255. doi: 10.1016/0006-291x(73)90535-4. [DOI] [PubMed] [Google Scholar]
- 29.Dikler S, Kelly JW, Russell DH. J Mass Spectrom. 1997;32:1337–1349. doi: 10.1002/(SICI)1096-9888(199712)32:12<1337::AID-JMS599>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 30.Morris HR, Panico M, Barber M, Bordoli RS, Sedgwick RD, Tyler A. Biochem Biophys Res Commun. 1981;101:623–631. doi: 10.1016/0006-291x(81)91304-8. [DOI] [PubMed] [Google Scholar]
- 31.Priddle JD, Rose K, Offord RE. Biochem J. 1976;157:777–780. doi: 10.1042/bj1570777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Anal Chem. 2003;75:5628–5637. doi: 10.1021/ac034414x. [DOI] [PubMed] [Google Scholar]
- 33.Seipert RR, Dodds ED, Lebrilla CB. J Proteome Res. 2009;8:493–501. doi: 10.1021/pr8007072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zauner G, Koeleman CAM, Deelder AM, Wuhrer M. J Sep Sci. 2010;33:903–910. doi: 10.1002/jssc.200900850. [DOI] [PubMed] [Google Scholar]
- 35.Saba J, Dutta S, Hemenway E, Viner R. Int J Proteomics. 2012;2012:560391. doi: 10.1155/2012/560391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh C, Zampronio CG, Creese AJ, Cooper HJ. J Proteome Res. 2012;11:4517–4525. doi: 10.1021/pr300257c. [DOI] [PubMed] [Google Scholar]
- 37.Satomi Y, Shimonishi Y, Hase T, Takao T. Rapid Commun Mass Spectrom. 2004;18:2983–2988. doi: 10.1002/rcm.1718. [DOI] [PubMed] [Google Scholar]
- 38.Freeze HH, Chong JX, Bamshad MJ, Ng BG. Am J Hum Genet. 2014;94:161–175. doi: 10.1016/j.ajhg.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guillard M, Wada Y, Hansikova H, Yuasa I, Vesela K, Ondruskova N, Kadoya M, Janssen A, Van den Heuvel LP, Morava E, Zeman J, Wevers RA, Lefeber DJ. J Inherited Metab Dis. 2011;34:901–906. doi: 10.1007/s10545-011-9311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang LJ, Lin JH, Tsai JH, Chu YY, Chen YW, Chen SL, Chen SH. J Chromatogr A. 2014;1371:136–145. doi: 10.1016/j.chroma.2014.10.046. [DOI] [PubMed] [Google Scholar]
- 41.Goetz JA, Novotny MV, Mechref Y. Anal Chem. 2009;81:9546–9552. doi: 10.1021/ac901363h. [DOI] [PubMed] [Google Scholar]
- 42.Prien JM, Ashline DJ, Lapadula AJ, Zhang H, Reinhold VN. J Am Soc Mass Spectrom. 2009;20:539–556. doi: 10.1016/j.jasms.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Science. 2008;320:373–376. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morimoto K, Nishikaze T, Yoshizawa AC, Kajihara S, Aoshima K, Oda Y, Tanaka K. Bioinformatics. 2015;31:2217–2219. doi: 10.1093/bioinformatics/btv110. [DOI] [PubMed] [Google Scholar]
- 45.Hu H, Khatri K, Klein J, Leymarie N, Zaia J. Glycoconjugate J. 2016;33:285–296. doi: 10.1007/s10719-015-9633-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aiyetan P, Zhang B, Zhang Z, Zhang H. MOJ Proteom Bioinform. 2014;1:4. doi: 10.15406/mojpb.2014.01.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jansen BC, Reiding KR, Bondt A, Hipgrave Ederveen AL, Palmblad M, Falck D, Wuhrer M. J Proteome Res. 2015;14:5088–5098. doi: 10.1021/acs.jproteome.5b00658. [DOI] [PubMed] [Google Scholar]
- 48.Lynn KS, Chen CC, Lih TM, Cheng CW, Su WC, Chang CH, Cheng CY, Hsu WL, Chen YJ, Sung TY. Anal Chem. 2015;87:2466–2473. doi: 10.1021/ac5044829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.