

SUMMARY
The SRPK family of kinases regulates pre-mRNA splicing by phosphorylating serine/arginine (SR)-rich splicing factors, signals splicing control in response to extracellular stimuli, and contributes to tumorigenesis, suggesting that these splicing kinases are potential therapeutic targets. Here, we report the development of the first irreversible SRPK inhibitor, SRPKIN-1, which is also the first kinase inhibitor that forms a covalent bond with a tyrosine phenol group in the ATP-binding pocket. Kinome-wide profiling demonstrates its selectivity for SRPK1/2, and SRPKIN-1 attenuates SR protein phosphorylation at submicromolar concentrations. Vascular endothelial growth factor (VEGF) is a known target for SRPK-regulated splicing and, relative to the first-generation SRPK inhibitor SRPIN340 or small interfering RNA-mediated SRPK knockdown, SRPKIN-1 is more potent in converting the pro-angiogenic VEGF-A165a to the anti-angiogenic VEGF-A165b isoform and in blocking laser-induced neovascularization in a murine retinal model. These findings encourage further development of SRPK inhibitors for treatment of age-related macular degeneration.
In Brief
Hatcher et al. report the first irreversible SRPK1/2 inhibitor SRPKIN-1, which inhibits phosphorylation of serine/arginine (SR)-rich splicing factors protein and induces a VEGF alternative splicing isoform switch, leading to anti-angiogenesis in a wet CNV mouse model.
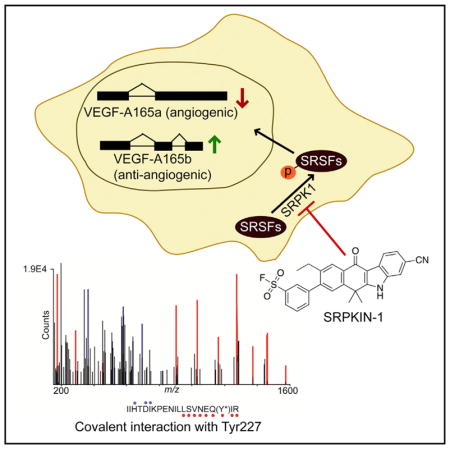
INTRODUCTION
Alternative pre-mRNA splicing in eukaryotic cells is a prevalent process for expanding the transcriptome complexity and proteome diversity, which is essential for maintaining both cellular and tissue homeostasis. This process is catalyzed by a complex cellular machine known as the spliceosome, which is composed of five small ribonucleoproteins and numerous protein co-factors (Wahl et al., 2009). Among them, the family of serine/arginine (SR)-rich splicing factors is involved in both constitutive and regulated splicing (Zhou and Fu, 2013), and their activities are regulated by several serine/threonine kinases. The first identified SR protein kinase is SRPK1 (Gui et al., 1994a, 1994b), which is conserved from yeast to humans (Siebel et al., 1999). The human genome encodes three SRPK genes, and SRPK1 has been detected in many human tissues, at varying levels of expression, while SRPK2 and SRPK3 exhibit tissue-specific expression in neurons and muscles, respectively (Wang et al., 1998; Nakagawa et al., 2005). In cells, most SRPK1 is localized in the cytoplasm where it catalyzes SR protein phosphorylation to facilitate their nuclear transport (Kataoka et al., 1999; Lai et al., 2001; Zhong et al., 2009), and this process is accelerated in response to extracellular stimuli (Nowak et al., 2010). Once in the nucleus, SRPK1 can synergize with additional SR protein kinases, such as the CLK family of kinases predominantly localized in the nucleus, to further phosphorylate SR proteins to promote spliceosome assembly (Aubol et al., 2016). During splicing, SR proteins become dephosphorylated by nuclear phosphatases, and like most phosphorylation-regulated proteins, SR proteins are regulated via this phosphorylation-dephosphorylation cycle in different cellular compartments (Misteli et al., 1998; Ngo et al., 2005; Huang and Steitz, 2001; Huang et al., 2003; Sanford et al., 2004).
This highly co-ordinated process is crucial for development and disease (Wang and Cooper, 2007; Cooper et al., 2009). Indeed, misregulation of SRPK1 expression induces a large number of aberrant alternative splicing events. In breast, colon, lung, prostate, and pancreatic cancer, for example, elevated SRPK1 levels are functionally linked to cell proliferation, migration, and trafficking, as well as angiogenesis and chemotherapy-induced resistance (Hayes et al., 2007; Gout et al., 2012; Mavrou et al., 2015). While cancer-associated splicing programs are likely regulated via a variety of mechanisms, some specific regulatory pathways have been well defined. For example, the enhanced production of the angiogenic isoform of vascular endothelial growth factor (VEGF) resulting from SRPK1 overexpression is a clear example of how splicing can impact disease progression (Amin et al., 2011; Gammons et al., 2014).
Angiogenesis, a biological process of new blood vessel formation, is critical for tumor growth, inflammatory disorders, and intraocular neovascular diseases. VEGF is a key regulator of angiogenesis through the activation of its cell surface receptor VEGF receptor (VEGFR), leading to endothelial cell proliferation. As an actively pursued therapeutic target, a plethora of small-molecule VEGFR inhibitors have been reported (Ivy et al., 2009). However, most Food and Drug Administration (FDA)-approved VEGFR inhibitors are pan-receptor tyrosine kinase (RTK) inhibitors, and developing a selective VEGFR inhibitor has been a challenge. Inhibition of VEGF signaling with a pan-VEGFR inhibitor has been shown to cause dose-dependent cellular toxicity (Richards, 2011; Duda et al., 2007). While targeting VEGF with small molecules has proven difficult, the use of VEGF-blocking antibodies such as Ranibizumab has been successfully used for treating age-related macular degeneration (AMD) (Rosenfeld et al., 2006; Gragoudas et al., 2004), an intraocular neovascularization disease caused by abnormal growth of blood vessels inside the eye (Seddon and Chen, 2004). However, antibody-based therapy is often associated with multiple risk factors, including infection, inflammation, and vitreous hemorrhage (Shima et al., 2008; Ventrice et al., 2013). Therefore, small-molecule inhibitors remain desirable for patients with AMD and cancer, either as a monotherapy or in combination with other anti-cancer agents.
Instead of blocking VEGFR, a different approach is to exploit its ligand VEGF by modulating alternative splicing. The VEGF gene is known to produce numerous isoforms, among which VEGF-A165a is the most predominant pro-angiogenic isoform in most cells and tissues. Alternative splicing can generate the anti-angiogenic VEGF-A165b isoform by using an alternative downstream 3′ splice site in exon 8 (Harper and Bates, 2008). Cancer cells tend to hijack VEGF-A165a while suppressing the expression of VEGF-A165b to accelerate cell growth and migration (Houck et al., 1991; Bates et al., 2002; Nowak et al., 2010). This raises the possibility to reverse this critical tumorigenic event by modulating VEGF splicing. Indeed, a previous study has demonstrated a critical role of SR proteins in the regulation of VEGF splicing and shown that inhibition of SRPK1 was able to switch VEGF-A165a to VEGF-A165b, thereby blocking angio-genesis in a colorectal cancer model, a kidney disease model (Denys-Drash syndrome), and in retinal angiogenesis induced by choroidal neovascularization (CNV) (Amin et al., 2011). Consistently, multiple studies have linked elevated SRPK1 expression to VEGF-A165a expression, increased likelihood of metastasis, and poor prognosis in cancer patients (Mavrou et al., 2015; Li et al., 2014; Gammons et al., 2014). These findings thus suggest a promising avenue for cancer intervention, which has led to the development of small-molecule inhibitor for SRPKs (Batson et al., 2017).
Realizing various drawbacks of reversible inhibitors developed to date, we aimed to derive an irreversible inhibitor against SRPKs based on our fortuitous discovery that the chemical scaffold for Alecitinib, a clinically approved anti-ALK inhibitor for treating non-small-cell lung cancer (Larkins et al., 2016), exhibits potent cross-reactivity against SRPK1. We have used structure-guided drug design to develop SRPKIN-1, which specifically and covalently binds SRPK1, blocks SR protein phosphorylation, induces VEGF-A165b at the expense of VEGF-A165a, and prevents angiogenesis in a mouse retina model.
RESULTS
A New Scaffold with Inhibitory Activity against SRPK1
Three chemical scaffolds have been described with inhibitory activities against SRPK1 (Batson et al., 2017; Morooka et al., 2015; Fukuhara et al., 2006; Siqueira et al., 2015; Székelyhidi et al., 2005; Gammons et al., 2013), and, among them, SPHINX31 was recently derived from SPHINX and reported to have much improved potency (Batson et al., 2017). Although high-throughput screening has been commonly used to generate lead compounds in kinase inhibitor projects, we chose to query existing kinase inhibitor libraries and known FDA-approved drugs for their potential abilities to target SRPK1, an approach that offers advantages in expediting structure-activity relationship (SAR) studies and optimizing pharmacological properties. By searching in-house profiling data on a panel of known kinase inhibitors, we encountered the FDA-approved ALK inhibitor Alectinib, which exhibited a significant capacity to target SRPK1 in a cell-based KiNativ profiling assay using HeLa cells (Figures 1A and 1B; Table S1; Patricelli et al., 2007). As expected, the compound potently inhibited the kinase activity of SRPK1 with an half maximal inhibitory concentration (IC50) of 11 nM by an in vitro kinase activity assay (Z-Lyte) (Figure 1C), which is significantly more potent than original SRPIN340 (IC50 = 367 nM; Fukuhara et al., 2006) and comparable with the recent published SRPK1 inhibitor SPHINX31 (IC50 = 6.0 nM; Batson et al., 2017). These observations raised an intriguing possibility that the anti-tumor effect of Alectinib might have benefited from its activities against SRPK1 and other kinases besides blocking ALK, which is currently under investigation in our laboratories.
Figure 1. Compound Structure and Biochemical Characterization of their Inhibitory Effects on SRPK1.

(A)The structures of all SRPK1 inhibitors and negative control compound used in this study.
(B)The potently bound kinases based upon KiNativ profiling of Alectinib. The HeLa cell was treated for 6 hr with 5 μM of Alectinib.
(C) In vitro kinase assays using the Z -LYTE Kinase assay technology (Invitrogen), which employs a fluorescence-based, coupled-enzyme format and is based on the differential sensitivity of phosphorylated and non-phosphorylated peptides to proteolytic cleavage.
See also Table S1.
As a part of our efforts to identify additional ALK inhibitors that may help overcome drug-resistant G1202R mutant (Hatcher et al., 2015), we generated a focused library based on the Alectinib scaffold. We took advantage of the new library to quickly probe the SAR of potential compounds against SRPK1. This led to the identification of JH-VII-139-1, in which a smaller pyrazole ring was installed to replace the 4-morpholinopiperidine moiety in Alectinib, resulting in a more active compound with an IC50 of 1.1 nM against SRPK1 (Figure 1C). In contrast, substitution with a dimethyl amino-piperidine ring (JH-VII-206-2) decreased the inhibitory activity against SRPK1 by ~1,000-fold (Figure 1C). Notably, JH-VII-139-1 and JH-VII-206-2 still share similar activities against other kinases, including ALK (data not shown), indicating that JH-VII-206-2 might be used as a specificity control for SRPK1 in some downstream functional analysis.
Structural Analysis of Inhibitor-Bound SRPK1
To understand how Alectinib binds to SRPK1, we determined the co-crystal structure of Alectinib bound to SRPK1ΔNS, an active version of SRPK1 in which the non-conserved N-terminal and spacer regions are truncated (Figure 2A). Comparison between the co-crystal structures of SRPK1ΔNS-Alectinib and ALK-Alectinib (PDB: 3AOX) revealed that Alectinib adopts a similar orientation and binding mode in the ATP-binding pockets of both kinases (Figure 2B). The nearly planar benzo[b]carbazole moiety of Alectinib lies close to the upper cleft of the ATP-binding pocket, making van der Waals contacts with side chains from both the small and large lobes (Arg84, Leu86, Val94, Ala107, Val167, Leu168, His170, and Leu220) (Figure 2C). The carbonyl group of the benzo[b]carbazole moiety directly hydrogen bonds with the backbone amide of Leu168 in the hinge region. This crucial hydrogen bond is conserved and mediated by the backbone of Met1199 in the ALK-Alectinib structure. Although the network of interactions formed between ALK and Alectinib via an ethylene glycol molecule is missing in the SRPK1-Alectinib structure, the water-mediated interaction between the cyano group of Alectinib with the invariant glutamate (Glu124 in SRPK1 and Glu1167 in ALK) of helix αC is conserved in both structures, indicating the importance of this water molecule for Alectinib binding to its target kinases (Figure 2D). This conserved water molecule also hydrogen bonds to the backbone amide of Leu498, forming a unique interaction between the cyano group and the DLG motif (canonically DFG motifs in other kinases) in the Mg2+ binding loop of SRPK1. In addition, Asp497 of the DLG motif also contacts the cyano group of Alectinib directly, which is absent in the ALK-Alectinib structure.
Figure 2. Crystal Structure of the SRPK1-Alectinib Complex.

(A) Overall structure of SRPK1 in complex with Alectinib. Helix αS1 of the non-conserved spacer region forms a continuous molecular surface adjacent to the ATP-binding pocket and is colored light orange.
(B) Comparison of the SRPK1-Alectinib and ALK-Alectinib complex structures. The two structures were superimposed using the carbons of their small lobes. SRPK1-Alectinib complex and ALK-Alectinib complex are colored purple and green, respectively. The unique helix αS1 of SRPK1 is colored light orange.
(C) Alectinib forms an extensive network of interactions with both the small and large lobes of SRPK1. Residues that interact with Alectinib are indicated.
(D) Alectinib hydrogen bonds with the backbone amide of Leu168 of SRPK1 (purple). A crucial solvent water molecule (blue) mediates a network of hydrogen bonds between Alectinib, the side chain of Glu124 and the backbone amide of Leu498 of SRPK1. All hydrogen bonds are denoted by black dotted lines. van der Waals interaction between Asp497 and Alectinib is denoted by pink dotted line. Extra contacts between the 4-morpholinopiperidine moiety of Alectinib and the side chains of Tyr227 and Leu231 of SRPK1 are observed. The structure of ALK-Alectinib complex is shown in green for comparison. The crucial water molecule in the ALK-Alectinib structure is colored red.
See also Table S2.
Catalytically active SRPK1 likely adopts a DFG-in conformation, thus favoring the formation of these SRPK1-specific interactions. SRPK1 contains a phenylalanine residue (Phe165), instead of a leucine in ALK, at the gatekeeper position, which may mediate π-π interaction with Alectinib. Although the 4-morpholinopiperidine moiety of Alectinib is positioned in a solvent-exposed region outside the ATP-binding pockets of both SRPK1 and ALK (Figure 2B), the extra helix αS1 from the spacer region of SRPK1 forms an extended molecular surface adjacent to the ATP-binding pocket to mediate extra interactions with the 4-morpholinopiperidine moiety via Tyr227 and Leu231 (Figure 2D).
This SRPK1-specific structural motif suggests that the smaller pyrazole ring in JH-VII-139-1 is more favored, whereas the dimethyl amino piperidine group in JH-VII-206-2 might have caused a steric clash with Tyr227, thus leading to the loss in potency observed with JH-VII-206-2. More importantly, our structural analyses also provided a framework for converting JH-VII-139-1 to an irreversible SRPK1 inhibitor via rational design by taking advantage of the unique Tyr227 that locates immediately adjacent to the ATP-binding pocket of SRPK1.
Development of SRPKIN-1 as an Irreversible Inhibitor for SRPK1
Following a successful strategy for improving the selectivity in the cases of Ibrutinib (Davids and Brown, 2014), AZD9291 (Finlay et al., 2014), and THZ1 (Kwiatkowski et al., 2014), we considered targeting Tyr227, which is located proximal to the morpholine group of Alectinib (Figure 2D) with the known tyrosine-reactive sulfonyl fluoride substituent (Hett et al., 2015). Reasoning that a benzenesulfonyl fluoride would maintain the required size in order to fit in the solvent-exposed region, which could also orient the warhead in the required position to covalently modify Tyr227, we replaced the pyrazole ring in JH-VII-139-1 with a 3-benzene-sulfonyl fluoride to generate a new compound, which we named SRPKIN-1. This substitution would potentially allow the formation of a covalent bond between the sulfonyl fluoride group of SRPKIN-1 and Tyr227 of SRPK1.
We next biochemically characterized SRPKIN-1 in a fixed time point assay, demonstrating that this new compound indeed maintained potent activity against SRPK1 (IC50 = 35.6 nM; Figure 1C). We also found that SRPKIN-1 also targeted the related SRPK2 (IC50 = 98 nM), while substantially reducing the activity on ALK (IC50 = 195 nM). To confirm the formation of a covalent bond between SRPKIN-1 and SRPK1, we performed capillary electrophoresis-mass spectrometry (CE-MS) analysis of recombinant protein incubated with DMSO or SRPKIN-1. Compound treatment produced a mass shift consistent with the covalent addition of a single molecule of SRPKIN-1 (-HF) to the protein (Figure 3A). Subsequent trypsin digest and CE-MS analysis of the resulting peptides demonstrated exclusive labeling of Tyr227 (Figure 3B).
Figure 3. The Mass Spectrum Labeling of SRPKIN-1 with SRPK1 Protein.

(A) Electropherograms (left), mass spectra (middle), and zero-charge mass spectra (right, overlayed) of SRPK1 protein treated with DMSO (top, red) or 10-fold molar excess of SRPKIN-1 (bottom, green) for 2 hr at room temperature. The observed mass shift of 452 Da is consistent with covalent labeling by SRPKIN-1 (with elimination of HF).
(B) Tandem MS spectrum of tryptic SRPK1 peptide (residues 209–229) with Y227 modified by SRPKIN-1. Ions of type b and y are indicated with blue and red glyphs, respectively. Y*, SRPKIN-1-modified tyrosine.
We next investigated whether this covalent modification resulted in enhanced selectivity toward SRPK1. To assess the selectivity kinome-wide, we again performed KiNativ profiling both before and after washout prior to the ATP-biotin labeling (Table S3; Patricelli et al., 2007). Following a 6-hr pre-treatment of HeLa cells with SRPKIN-1 at a fixed concentration of 5 μM, we observed striking selectivity for SRPK1 even before washout, and, in comparison, Alectinib showed broad interactions with numerous additional targets, including CHK2 and FER (Table S1). The selectivity of SRPKIN-1 for SRPK1 and SRPK2 became more striking after washout, while SRPKIN-1 binding to CaMK4 was clearly reversible, consistent with the formation of a covalent bond between SRPKIN-1 and SRPK1 (Figure 4A; Table S3). To know how selective of SRPKIN-1 in vitro condition, we performed a KinomeScan profiling with a large panel of more than 400 kinases. Consistent with its KiNativ profiling result, SRPKIN-1 demonstrated a weak binding (high score) with all the kinases in this panel (Table S4). Similar to Alectinib, which has been found to have a high score on SRPK1/2 with the KinomeScan profiling technology, SRPKIN-1 also displayed a high score on these kinases, indicating a technology limitation for SRPK1/2 may exist.
Figure 4. The Selectivity of SRPKIN-1.

(A) Comparison of the KiNativ profiling data for SRPKIN-1 on HeLa cell (in blue) with the data for Alectinib profiling at same condition (in red). The cutoff of inhibition difference between SRPKIN-1 and Alectinib is 30%.
(B) Cell-viability assays with SRPKIN-1, Alectinib, and JH-VII-206-2 on EML4-ALK Ba/F3 cells.
Finally, since HeLa cells express little to no ALK, we tested SRPKIN-1 along with Alectinib and JH-VII-206-2 against Ba/F3 cells transduced with EML4-ALK, which has been used extensively to test ALK inhibitors. Alectinib and JH-VII-206-2 showed potent inhibition of cell growth with a low nanomolar IC50, while SRPKIN-1 showed 50-fold less activity (Figure 4B), indicating that ALK is no longer inhibited by SRPKIN-1.
Attenuation of SR Protein Phosphorylation by SRPKIN-1
SRPK1 is known to phosphorylate the family of SR proteins characterized by the arginine/serine-rich domain (Gui et al., 1994a, 1994b), which takes place in a directional and processive manner (Ghosh and Adams, 2011). To investigate whether SRPKIN-1 affects the kinase activity of SRPK1 in a cellular context, we treated HeLa cells with SRPKIN-1 for 16 hr, lysed the cells, and performed a western blot with an antibody (mAb104) that recognizes phosphorylated epitopes in various SR proteins (Roth et al., 1990). For comparison, we similarly analyzed the effect of the first-rgeneration SRPK1 inhibitor SRPIN340. We first titrated the amounts of individual compounds in a broad range and then selected a specific concentration for direct comparison. Phosphorylation of multiple SR proteins was significantly reduced upon treatment with SRPKIN-1 at 200 nM, while SRPIN340 had a minimal effect even at a concentration of 10 μM under the same conditions (Figure S1).
We next performed the SR protein phosphorylation assay with or without washout. As shown in Figure 5A, SRPKIN-1 treatment at 200 nM significantly reduced SR protein phosphorylation at the steady state without washout, and continued to show the inhibitory effect after washout (note that the remaining levels of SR protein phosphorylation are likely due to the activity of other SR protein kinases, such as the CLK family of kinases). In comparison, the reversible compound JH-VII-139-1 was similar to SRPKIN-1 in reducing SR protein phosphorylation without washout, but the effect was lost after washout (Figure 5A). The negative control compound JH-VII-206-2 exhibited no inhibitory effect in this assay (Figure S1). These data demonstrated the potent effect of SRPKIN-1 in blocking phosphorylation of endogenous SR proteins.
Figure 5. SRPK1 Inhibitors Promote VEGF-A165b Expression by Inducing Alternative Splicing of the VEGF Pre-mRNA.

(A) Analysis of SR protein phosphorylation levels probed with mAb104 in HeLa cells following treatment with indicated compounds at 200 nM with or without washout. The ratio of remained phosphorylation SR protein upon the compound treatment versus actin was quantified. Standard deviation was calculated using the built-in stdev function in Excel.
(B) Effects of knock down of SRPK1 and/or SRPK2 on VEGF pre-mRNA splicing. VEGF-A165a and VEGF-A165b are indicated. No change was detected with SRPK1 overexpression. The VEGF-A165b/(VEGF-A165a + VEGF-A165b) ratios were quantified. Standard deviation was calculated using the built-in stdev function in Excel.
(C) Dosage-dependent effects of SRPKIN-1 on VEGF pre-mRNA splicing. The VEGF-A165b/(VEGF-A165a + VEGF-A165b) ratios were quantified. (D) Elevated expression of VEGF-A165b protein upon treatment with JH-VII-139-1 or SRPKIN-1.
See also Figure S1.
Switching VEGF Alternative Splicing toward Anti-angiogenic Isoform
Although inactivation of SR proteins or attenuation of SR protein phosphorylation is expected to induce a large number of alternative splicing events in the cell, as demonstrated earlier (Pandit et al., 2013; Zhou et al., 2012), we chose to characterize the newly developed SRPK1 inhibitor on a well-documented SR protein-dependent and SRPK1-regulated splicing event. As shown earlier, the VEGF pre-mRNA is predominantly spliced to generate the VEGF-A165a isoform in most cell types, and inhibition of SRPK1 is able to switch VEGF splicing to produce the VEGF-A165b isoform (Amin et al., 2011; Batson et al., 2017). This switch is of high biological importance because VEGF-A165a is a major ligand to bind and induce VEGFR2 phosphorylation, thereby promoting angiogenesis, whereas VEGF-A165b is able to bind VEGFR2 but fails to induce its phosphorylation (Bates et al., 2002; Woolard et al., 2004), therefore functioning as an antagonist for VEGF-A165a to block angiogenesis (Manetti et al., 2011; Rennel et al., 2008). We therefore focused on testing SRPKIN-1 on this critical alternative splicing event.
We first treated HeLa cells with the previously established small interfering RNAs against SRPK1 or SRPK2, or both (Zhou et al., 2012), to verify the role of SRPKs in switching VEGF splicing, which served as a reference for the biological effect in our hands. As expected, siSRPK1 and siSRPK2 both partially induced VEGF alternative splicing from VEGF-A165a to VEGF-A165b, and the combined treatment produced an additive effect, while SRPK1 overexpression showed no further effect on already dominant VEGF-A165a expression (Figure 5B). We next tested SRPKIN-1, observing a clear shift to VEGF-A165b in a dose-dependent manner, and a complete switch is achieved with 100 nM of SRPKIN-1 (Figure 5C). In comparison, we detected little effect with the first-rgeneration SRPK1 inhibitor SRPIN340, even at a concentration of 10 μM (Figure 5C), reminiscent of its weak effect on attenuating SR protein phosphorylation (Figure S1). We further showed that 200 nM of SRPKIN-1 significantly elevated the VEGF-A165b protein level and had a comparable effect with the reversible inhibitor JH-VII-139-1, but there was a much weaker effect with 10 μM of SRPIN340 (Figure 5D). These results demonstrated at least 50-fold increase in potency with SRPKIN-1 compared with SRPIN340 in a cellular context.
Potent Anti-angiogenesis Effect of New SRPK1 Inhibitors in an Animal Model
AMD is a slow progressing eye disease and is the major cause of age-related blindness among elderly people. One category of advanced AMD is wet CNV, which is characterized by extensive growth of blood vessels through the choroidal membrane to the retina (Zhang et al., 2012). CNV has been a well-established model for induced angiogenesis. As illustrated in Figure S2A, we first used a laser beam to induce burns in the Bruch’s membrane of the mouse eye and then injected a fluorescence-labeled dextran into one of the orbital plexus to visualize induced neovascularization in the retina/choroid layer a week after the injury (Du et al., 2013). Using this animal model, we examined the effect of our newly developed SRPK1 inhibitors in blocking laser-induced CNV.
Based on the observed cellular effects on SR protein phosphorylation and VEGF splicing we tested two doses at 50 and 300 nM for both the reversible compound JH-VII-139-1 and the irreversible compound SRPKIN-1, and, for comparison, we also examined SRPIN340 and the negative compound JH-VII-206-2 at concentrations of 20 μM and 300 nM, respectively. We found that neovascularization in SRPKIN-1-treated mice was significantly suppressed in a dose-dependent manner based upon measurement of the CNV area (Figures 6A and 6B). In comparison, the reversible JH-VII-139-1 compound exhibited a similar effect in this assay, while the reference compound SRPIN340 modestly suppressed angiogenesis, even at a concentration of 20 μM, and the negative compound JH-VII-206-2 showed no effect (Figures 6A and S2B). These data demonstrated the potent effect of the newly derived irreversible SRPK1 inhibitor in blocking angiogenesis in the AMD model.
Figure 6. SRPK1 Inhibitors Block Laser-Induced CNV in Mouse Eyes.

(A) Choroids were isolated and prepared as flat mounts. CNV spots are stained with fluorescein isothiocyanate-labeled isolectin and marked with dotted lines.
(B) CNV areas were measured and plotted. The p values were determined by Wilcox test.
See also Figure S2.
DISCUSSION
Rationally designed covalent kinase inhibitors have recently experienced a resurgence of interest (Singh et al., 2011), and some have been approved by the FDA, including osimertinib and Ibrutinib for targeting mutant EGFR and BTK, respectively. Covalent modification of a specific residue by covalent inhibitors can afford medicinal chemists the opportunity to gain greater selectivity, potency, and extended residence over reversible inhibitors (Singh et al., 2011; Bauer, 2015). However, due to a lack of targetable cysteines around the ATP pocket, covalent binding is not feasible for many kinases. Medicinal chemists have been making great efforts to expand the scope of both the nucleophile in target protein as well as an electrophilic warhead in the small-molecule inhibitor. A recent report on the discovery of a covalent inhibitor of mRNA-decapping scavenger enzyme DcpS highlighted the phenol group of tyrosine as a suitable nucleophile for the sulfonyl fluoride warhead (Hett et al., 2015).
In this study, we first solved the co-crystal structure of SRPK1 with the FDA-approved ALK inhibitor Alectinib, and demonstrated that this compound is also a potent reversible SRPK1 inhibitor. The kinase domain of SRPK1 is bifurcated by a non-conserved spacer domain in which an N-terminal spacer insert folds into a long helix (αS1) and interacts with the kinase small lobe. We observed that the side chains of Tyr227 and Leu231 of the helix lie at the mouth of the ATP-binding pocket and are 3.2 and 3.6 Å away from the morpholine ring of Alectinib, respectively. Inspired by these unique interactions and the accessibility of the OH group of Tyr227, we went on to introduce a sulfonyl fluoride warhead to convert the Alectinib scaffold into an irreversible binder of SRPK1. To our best knowledge, this is the first covalent kinase inhibitor that targets tyrosine, thus highlighting additional opportunities for developing novel covalent kinase inhibitors targeting tyrosine.
Alectinib was designed to be a more selective and potent ALK inhibitor than crizotinib, but, in addition to its activity on ALK, it also has activity against other kinases such as FER and CHK2 (Table S1). Covalent inhibitors usually achieve greater selectivity by eliminating off-targets that lack an appropriately positioned nucleophile. SRPKIN-1 shares a high degree of similarity with Alectinib, which is accommodated in the nucleotide-binding pocket. However, the hydrophilic morpholine ring in Alectinib is placed to the solvent-exposed region, leading to a significant increase on the overall binding affinity to ALK. The aryl sulfonyl fluoride in SRPKIN-1 is much more hydrophobic compared with morpholine, which results in a 10-fold loss of potency against ALK. This detrimental effect is also seen in a Ba/F3 cellular assay in which Alectinib inhibited the cell growth at single digital nanomolar concentration, whereas SRPKIN-1 only blocked ALK-dependent cell proliferation at micromolar concentrations. The inserted domain in SRPK1 severely interferes with the accessibility to the solvent-exposed region, but a small hydrophobic group with a sulfonyl fluoride is able to covalently modify SRPK1, resulting in enhanced potency and selectivity compared to the non-covalent compound. SRPK2, a paralog of SRPK1, was also inhibited by SRPKIN-1, although we have not yet tested its effect on SRPK3 due to the lack of its biochemical assay. Importantly, a Z-Lyte kinase biochemical assay indicated that SRPKIN-1 had little effect on CLK1 (IC50 > 10 μM).
The known direct cellular substrates of SRPKs are SR proteins. Dephosphorylation of SR proteins results in a VEGF splicing switch from VEGF-A165a to VEGF-A165b. In HeLa cells, both SRPKIN-1 and the less-selective reversible compound JH-VII-139-1 potently decreased the level of SR phosphorylation in a dose-dependent manner, leading to increased VEGF-A165b RNA as well as protein even at a dose of 200 nM. In comparison, the first-rgeneration SRPK1 inhibitor SRPIN340 barely showed such effects even at a dose of 10 μM. These data indicate that SRPKIN-1 is greater than 50-fold more potent than SRPIN340 in a cellular context. The superiority of SRPKIN-1 is also clear in a wet AMD mouse model where laser-induced neovascularization, which is highly dependent on VEGF-A165a expression, was monitored with the treatment of SRPKIN-1 in comparison with SRPN340.
AMD is a leading cause of vision loss among people aged 50 or older. More than ten million Americans suffer from the potentially disabling effects of macular degeneration. Most patients with macular degeneration have the dry form of the disease which is characterized by the presence of yellow deposits, called drusen, in the macula. Wet AMD is characterized by the growth of abnormal blood vessels from the choroid underneath the macula, which makes up the majority of patients who experience serious vision loss from the disease. No FDA-approved treatments exist yet for dry macular degeneration, although nutritional intervention may help prevent its progression to the wet form. Current treatments of wet AMD include injection of a monoclonal VEGF antibody, photodynamic therapy, and laser surgery (Abd et al., 2017). Given the complications associated with each of these treatments, much effort has been made in developing an effective small molecule directly targeting VEGFR; however, there are currently no FDA-approved small-molecule inhibitors of VEGFR for treatment of AMD. SRPKIN-1 shows a potential option for treating AMD based on its role in regulated splicing. It could be also useful for treating certain cancer types that show overexpression of SRPK1 and/or SR proteins because of their established contributions to tumorigenesis. Therefore, SRPKIN-1 could be an important stepping-stone for developing new therapeutics against AMD as well as cancers especially when the treatment is amenable with local drug delivery.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mAb104 | Xiangdong Fu lab in University of California San Diego | NA |
| Monoclonal Anti- VEGF-A165b 56/1 produced in mouse | EMD Millipore Corp | Cat#MABC595 |
| Monoclonal Anti-β-Actin produced in mouse | Sigma-Aldrich | Cat# A2228 RRID:AB_476697 |
| Goat anti mouse IgM-HRP | Santa Cruz Biotechnology | Cat# SC-2064 RRID:AB_631776 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 9-ethyl-8-iodo-6,6-dimethyl-11-oxo-6, 11-dihydro-5H-benzo[b]carbazole-3-carbonitrile (Compound 3) | Chemexpress | Cat#HY-76954 |
| SuperSignal West Pico chemiluminescent substrate | Thermo Fisher Scientific | Cat#34080 |
| SRPK1 protein | Jacky Ngo lab in Chinese University of Hong Kong | N/A |
| Trizol | Invitrogen | Cat#15596026 |
| xylazine | Vetone | Cat#510650 |
| tropicamide | Sandoz | Cat#PO-6567 |
| ketamine | Zoetis | Cat#000680 |
| isolectin | Thermo Fisher Scientific | Cat#I21413 |
| FITC-dextran | Sigma-Aldrich | Cat#FD2000S |
| isoflurane | Vetone | Cat#501017 |
| Critical Commercial Assays | ||
| SRPK1 Z′-LYTE Kinase assay | Life technology | N/A |
| SRPK2 Z′-LYTE Kinase assay | Life technology | N/A |
| KiNativ™ cell based kinase profiling assay | ActivX Bioscience | N/A |
| Deposited Data | ||
| SRPK1 in complex with Alectinib | This paper | 5XV7 https://www.rcsb.org/pdb/results/results.do?tabtoshow=Unreleased&qrid=759B5DEB |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | Cat# CCL-2 |
| Ba/F3 | Pasi Jänne lab in Dana-Farber Cancer Institute | N/A |
| MAb 104 Hybridoma cell line | ATCC | Cat#CRL-2067 |
| Experimental Models: Organisms/Strains | ||
| Wild type C57BL/6J mice | Jackson Laboratories | Stock#000664 |
| Oligonucleotides | ||
| VEGF165 forward | 5′-GGCAGCTTGAGTTAAACGAAC-3′ | N/A |
| VEGF165 reverse | 5′-ATGGATCCGTATCAGTCTTTCCTGG-3′ | N/A |
| Software and Algorithms | ||
| HKL2000 | HKL Research, Inc. | http://www.hkl-xray.com/download-instructions-hkl-2000 |
| CCP4i | Science & Technology Facilities Council Rutherford Appleton Laboratory | http://www.ccp4.ac.uk/ccp4i_main.php |
| PyMol | Schrodinger | https://pymol.org/2/ |
| Keyence BZ-II analyzer | Keyence | http://bz-ii-analyzer.software.informer.com/download/ |
| Others | ||
| Iridex OcuLight GL 532 nm laser photocoagulator | IRIDEX | NA |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nathanael Gray (nathanael_gray@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines Used in This Study
HeLa cells (female) were cultured in Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum and pen/strep. Ba/F3 cells (mouse cell line, the sex of cell line is not available) were maintained in RPMI-1640 containing L-glutamine, supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All cell cultures were incubated at 37°C in a humidified 5% CO2 atmosphere.
Animals Used in This Study
All animal experiments were approved by the UC San Diego Institutional Animal Care and Use Committee (IACUC), and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Wild type C57BL/6J mice (20 mice, 8 males and 12 females) were housed in a facility with well-ventilated racks, standard rodent chow and water. Laser-CNV experiments were performed when mice were 8 weeks old.
METHOD DETAILS
General Information for Chemical Synthesis
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on Bruker Avance (400 MHz) or Bruker A500 (500 MHz), and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Sunfire C18 column (19 × 50 mm, 5μM) using a gradient of 15–95% methanol in water containing 0.05% trifluoroacetic acid (TFA) over 22 min (28 min run time) at a flow rate of 20 mL/min. Purities of assayed compounds were in all cases greater than 95%, as determined by reverse-phase HPLC analysis.
Synthesis of SRPKIN-1
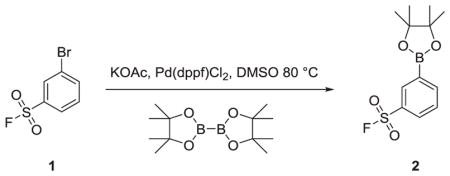
3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzenesulfonyl fluoride (2)
To a solution of 3-bromobenzenesulfonyl fluoride (500 mg, 2.09 mmol) in DMSO (10 mL) was added 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (584 mg, 2.30 mmol) and KOAc (616 mg, 6.27 mmol). The solution was degassed for 5 minutes in a sonicator, then Pd(dppf)Cl2 (46 mg, 0.062 mmol) was added and the mixture stirred at 80 °C for 1 hour at which point, TLC analysis indicated complete consumption of starting material. Water was added and the mixture extracted with EtOAc (3 X 50 mL), dried over MgSO4 and condensed to give a brown oil that was used without further purification (quantitative yield).

3-(3-cyano-9-ethyl-6,6-dimethyl-11-oxo-6,11-dihydro-5H-benzo[b]carbazol-8-yl)benzenesulfonyl fluoride (SRPKIN-1)
To a suspension of 9-ethyl-8-iodo-6,6-dimethyl-11-oxo-6,11-dihydro-5H-benzo[b]carbazole-3-carbonitrile (30 mg, 0.068 mmol) in 1-,4-dioxane (5 mL) was added 2M aqueous Na2CO3 (0.17 mL, 0.34 mmol) and 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) benzenesulfonyl fluoride (30 mg, 0.102 mmol) and the mixture degassed in a sonicator for 5 minutes. Pd(dppf)Cl2 (6 mg, 0.008 mmol) and tBuXPhos (5 mg, 0.012 mmol) were added and the mixture stirred for 1 hour at 90 °C at which time LCMS analysis indicated complete conversion of starting material to desired product. The mixture was purified directly by reverse phase HPLC using a gradient of 10–90% ACN in H2O to give the title compound as a beige solid (23 mg, 72% yield). 1H NMR (DMSO-d6, 500 MHz) δ 12.84 (s, 1H), 8.36 (d, J = 8 Hz, 1H), 8.23 (m, 2H), 8.12 (m, 1H), 8.04 (m, 2H), 7.93 (t, J = 8 Hz, 1H), 7.81 (s, 1H), 7.65 (m, 1H), 2.69 (q, J = 7 Hz, 2H), 1.79 (s, 6H), 1.17 (t, J = 7 Hz, 3H); 13C NMR 100 MHz (DMSO-d6) δ 179.46, 160.91, 146.25, 142.99, 142.82, 140.32, 137.72, 136.26, 132.37, 132.19, 131.64, 131.12, 128.75, 128.66, 128.09, 127.94, 126.17, 125.54, 122.19, 120.48, 116.97, 109.94, 105.36, 36.94, 25.67, 21.51, 15.43; MS m/z = 473.24 [M+1].
Kinase Z-Lyte Assay Protocol as Performed by Invitrogen
100 nL of the test compound (in 100% DMSO), 2.4 μL of Kinase buffer, 5 μL of 2× peptide/kinase mixture and 2.5 μL of 4X ATP are added to the well, resulting in a final 10 μL kinase reaction consisting of 3.74 – 37 ng SRPK1 (or SRPK2), 25 μM ATP and 2 μM Ser/Thr 18 in 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, 1 mM EGTA. After the 1 hour room temperature Kinase Reaction incubation, 5 μL of a 1:1024 dilution of Development Reagent A is added, mixed, and incubated at room temperature for 1 hour. The reaction is read on a fluorescent plate reader and the data analyzed.
KiNativ Profiling
Sample Preparation
HeLa cells were plated into 6–150 mm plates per treatment in growth medium (DMEM + 10%FBS + 1 % pen/strep). Cells were grown until 90% confluency. Cells were then treated with 5 μM Alectinib, 5 μM SRPKIN-1, or DMSO. After 3 hours, washout was performed on 3 of the 6 plates. Media were aspirated and cells were washed 3 times with DMEM. After washing, fresh DMEM was added to the plates. After 5 hours, growth medium was aspirated and cells were washed 3 times with cold PBS. Cells were harvested by scraping into 4 mL of cold PBS containing protease and phosphatase inhibitors. Cells were pelleted by centrifugation at 1350g at 4 °C for 5 minutes and immediately frozen in liquid N2. The frozen samples were shipped to ActivX for KiNativ analysis.
LC-MS/MS Analysis as provided by ActivX
The samples were then treated with ATP and ADP Acyl-nucleotide probes which were synthesized following a published procedure (Patricelli et al., 2007). Samples were analyzed by LC-MS/MS as described previously (Patricelli et al., 2011). Samples were analyzed on Thermo LTQ ion trap mass spectrometers coupled with Agilent 1100 series micro-HPLC systems with autosamplers, essentially as described, using a custom target list for the HeLa cell line comprising 282 unique kinase peptides that had been previously identified during the characterization of the HeLa cell line in data dependent mode (Patricelli et al., 2011; Nomanbhoy et al., 2016).
Data Analysis as Performed by ActivX
For signal extraction/quantitation, typically up to four ions were selected for based on their presence, intensity, and correlation to the reference MS/MS spectrum. The resulting chromatographic peaks from each run were then integrated and the integrated peak areas used to determine % inhibition values relative to control runs.
For each peptide quantitated, the MS signal for the adult myocyte samples relative to the MS signal for the neonatal myocyte samples was expressed as fold-change using the following equation:
All data points were visually verified, as were all datapoints showing variability outside of normal limits. Significance of datapoints changing more than 2-fold were determined according to the Student T-test (Excel 2010):
Array1: MS signals from control samples
Array2: MS signals from treated samples
Tails = 1 (one-tailed distribution)
Type = 2 (two-sample equal variance)
In cases where scores <0.04, the fold-change was considered significant.
In cases where the peptide signals in the adult myocyte samples were undetectable (noise), the fold-change value was preceded by “>”.
Crystallization and Data Collection
SRPK1ΔNS was expressed and purified as described previously (Ngo et al., 2007). Protein crystals were obtained by hanging drop vapor diffusion at 16°C in a reservoir condition containing 11% PEG 4000, 100mM sodium citrate (pH 5.6) and 200mM ammonium acetate. Different concentrations of Alectinib were diluted in soaking solution containing 13% PEG 4000, 100mM sodium citrate (pH 5.6) and 200mM ammonium acetate. Crystals of SRPKΔNS in complex with Alectinib were obtained by soaking the SRPK1ΔNS crystals with different concentrations of the inhibitor for 72 hours at 16 °C. Crystals were then cryo-protected in soaking solution supplemented with 20% ethylene glycol (v/v) and flash frozen in liquid nitrogen prior to data collection.
Crystals were screened and tested at the Center for Protein Science and Crystallography, the Chinese University of Hong Kong. The highest resolution dataset was collected using ADSC Quantum-315r CCD Area Detector at beamline BL13B1 of the National Synchrotron Radiation Research Center (NSRRC) at Taiwan. X-ray data were processed with the program HKL2000 (Otwinowski and Minor, 1997).
Structure Determination and Refinement
The structure of SRPK1ΔNS:Alectinib Complex was initially solved by molecular replacement using the program PHASER in the CCP4 program suite (McCoy et al., 2007). The structure of SRPKΔNS1 (PDB ID: 1WAK) was used as the search model. After restrained refinement using the program REFMAC, Fo-Fc density map revealed large positive peak and readily interpretable density for Alectinib at the ATP binding pocket. Model building was performed using Coot (Emsley et al., 2010) and the structure was refined with several cycles of TLS & restrained refinements using REFMAC (Murshudov et al., 1997) and manual refitting. Structure validation and conformational quality were assessed by PROCHECK (Laskowski et al., 1993). The model of the complex includes residues 67–238 and 477–655 residues of the kinase, 1 molecule of Alectinib, 4 molecules of ethylene glycol and 24 molecules of water.
Mass Spectrometry Analysis
Purified SRPK1 protein (5 μg) was treated with DMSO or SRPKIN-1 for 2 hours at room temperature with a 10-fold-molar excess of SRPKIN-1. Treated protein solutions were directly analyzed by CE-MS using a ZipChip CE system and autosampler (908 Devices, Boston, MA) interfaced to a QExactive HF mass spectrometer (ThermoFisher Scientific, San Jose, CA). Protein solutions were loaded for 10 seconds and separation performed at 500 V/cm for 6 minutes using an HR chip (22 cm separation channel) with a background electrolyte composed of 1% formic acid in 50% acetonitrile. Pressure assist was utilized and started at 1 minute. The mass spectrometer acquired profile spectra (15k resolution, 1E6 target, lock mass enabled) from m/z 300–2000. Mass spectra were deconvoluted using MagTran version 1.03 b2 (Zhang and Marshall, 1998). To identify the labeled tyrosine residue, treated protein was reduced (10 mM dithiothreitol), alkylated (22.5 mM iodoacetamide), and digested with trypsin overnight at 37 °C. Peptides were desalted using C18, dried by vacuum centrifugation, and reconstituted in 1% formic acid/50% acetonitrile with 100 mM ammonium acetate. Peptides were then analyzed by CE-MS as described above, except that peptide solution was loaded for 40 seconds, and the mass spectrometer was operated in data dependent mode and subjected the 5 most abundant ions in each MS scan (15k resolution, 1E6 target, lock mass enabled) to MS/MS (15k resolution, 2E5 target, 100 ms max inject time). Dynamic exclusion was enabled with a repeat count of 1 and an exclusion time of 6 seconds. MS/MS data was extracted to.mgf using mulitplierz scripts (Askenazi et al., 2009; Parikh et al., 2009) and searched against a forward-reverse human NCBI refseq database using Mascot version 2.2. Search parameters specified fixed carbamidomethylation of cysteine, and variable oxidation (methionine) and SRPKIN-1 modification (tyrosine). Precursor mass tolerance was set to 10 ppm and product ion tolerance was 25 mmu.
Preparation for mAb104 Antibody
mAb104 (ATCC CRL-2067™) hybridoma cells were thawed from liquid nitrogen tank and revived in Dulbecco’s modified Eagle’s Medium(DMEM) plus 10% fetal bovine serum. Cells were passaged at least once every two days. Healthy and round cells were pooled and grown in high densities in DMEM with 10% FBS for 2 days, and then in DMEM with 2% FBS for 5 days. Cell culture were spun at 5000 RPM for 15 min, and the supernatant was harvested and used directly for Western blotting.
BA/F3 CELL GROWTH ASSAY
Ba/F3 cells were seeded in 96-well plates and exposed to compounds at different concentrations in triplicate for 72 hours. Cell viability was evaluated using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) following the manufacturer’s instruction.
Inhibitor Treatment and Immunoblotting
HeLa Cells were grown in six well plates to 70% confluence and serum starved for overnight. Inhibitors or controls were added to serum free media and incubated for 16 hours. Following the incubation, cells were washed once with cold PBS buffer and immediately placed on ice. Cells were then harvested with lysis buffer (50 mM Tris-HCl, pH 7.5, 2% (w/v) SDS, 0.1% (w/v) bromophenol blue, 10% (v/v) glycerol, 100 mM DTT), and quickly moved to 95°C heating block for 10 min. Protein samples were loaded onto 12% SDS-PAGE gel (acrylamide:bisacrylamide = 29:1). Separated proteins were transferred to nitrocellulose membranes (Bio-rad), blocked with 5% BSA in TBS buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl), and incubated with primary antibody mAb104 or anti-VEGF-A165b overnight. Membranes were washed with TBST (TBS with 0.05% Tween 20) five times, 10 min each. Membranes were then blotted with secondary antibodies for 1 hour. For mAb104, goat anti mouse IgM-HRP diluted at 1:4000 (Santa Cruz Biotechnology) was used. For VEGF-A165b, rabbit anti mouse IgG-HRP dilutedat 1:10000 (Sigma-Aldrich) was used. Non-specific secondary antibodies were washed away in TBST for five times, 10 min each. Proteins were visualized using the enhanced chemiluminescent substrate (Thermo Scientific). Three independent biological replications were used in this treatment.
RT-PCR
Cells were washed with cold PBS once and then immediately lysed with Trizol (Invitrogen). 3 μg of total RNA was used for reverse transcription using SuperScript III Reverse Transcriptase (Invitrogen). 10% of the reverse transcription product was used for PCR with a pair of primers flanking exon 8a and 8b (Nowak et al., 2010). PCR products were resolved in 2.8% agarose gel and imaged with Bio-rad Gel Doc system. Three independent biological replications were used in this treatment.
Choroidal Neovascularization (CNV) Assay in Mice
Mice were anesthetized with an intraperitoneal injection of a mixture of ketamine and xylazine. Pupils were dilated with 1% topical tropicamide. A laser photocoagulation with slit-lamp delivery system was used to create four burns centering the optical disk with the following parameters: 100 mW power, 75 μm spot size, and 100ms duration. We only included burns that disrupted the Bruch’s membrane at the time of the laser treatment, which is indicated by the generation of a bubble. For chemical inhibition, mice were given an intravitreal injection of 1 μL of chemicals at five times the desired final concentrations (the vitriol body has a volume of ~5 μL) immediately after laser photocoagulation. An equal volume of DMSO was given in the same manner as the negative control. Five mice were used for each compound and DMSO treatment.
One week following laser photocoagulation, mice were anesthetized by isoflurane, and injected intravenously with 50 μL FITC-Dextran solution in PBS (50 mg/mL; 2 MDa). After 30 min, the mice were euthanized, their eyes were collected and fixed in paraformaldehyde (4% in PBS) for 1h. Choroids were then flat-mounted and stained with Alexa Fluor 594-conjugated isolectin GS-IB4. Slides were imaged at 10X magnification (Biorevo, Keyence), and images were quantified in Keyence software. The university of California San Diego (UCSD) institutional animal care and use committee (IACUC) approved animal use protocol.
QUANTIFICATION AND STATISTICAL ANALYSIS
For CNV assay in mice, slides were imaged at 10X magnification (Biorevo, Keyence), and images were quantified in Keyence software (copied from 965 to 966). Unpaired two-sample Wilcoxon test is used to generate the p-values (by calling the wilcox.test (Group A, Group B) function in R studio). Total Wild type C57BL/6J mice (20 mice, 8 males and 12 females) were used in this study.
DATA AND SOFTWARE AVAILABILITY
The coordinates of SRPK1 in complex with Alectinib has been deposited in the Protein Data Bank under the accession number 5XV7.
Supplementary Material
Highlights.
SRPKIN-1 is the first irreversible inhibitor for SRPK1/2 kinase
SRPKIN-1 is the first kinase inhibitor that targets a tyrosine residue covalently
SRPKIN-1 inhibits phosphorylation of serine/arginine (SR)-rich splicing factor proteins
SRPKIN-1 blocks angiogenesis in a CNV mouse model through VEGF alternative splicing
SIGNIFICANCE.
We demonstrated an evolution of a covalent SRPK1 inhibitor, SRPKIN-1 from the Alectinib scaffold with the guide of a complex crystallography of SRPK1 with Alectinib. Among the whole kinase family, SRPKIN-1 is also the first irreversible inhibitor that forms a covalent bond through a tyrosine instead of a commonly utilized cysteine residue. This provides a potential avenue for covalently targeting other kinases that possess an accessible tyrosine in their ATP pockets. Through covalent labeling, SRPKIN-1 displayed an exquisite selectivity and high potency for SRPK1, consistent with its efficient suppression of SR protein phosphorylation in cells. SRPKIN-1 treatment induced a critical switch in VEGF splicing to increase the expression of the anti-angiogenic isoform at the expense of the pro-angiogenic isoform, thus blocking angiogenesis in a wet CNV mouse model, indicating its potential therapeutic modality for both AMD and cancer. SRPKIN-1 will also serve as a useful tool to interrogate the function of SRPK1 in oncogenic processes.
Acknowledgments
We thank Walter Massefski in the core facility of NMR in Dana-Farber for the help on the compound NMR spectrum data collection and Nicholas Kwiatkowski and Calla M. Olson for critical reading of the manuscript, and to Jeffrey Kelly for advice regarding sulfonyl fluorides. C.Z. and J.C.K.N. were supported by a General Research Fund (14122615) from the Research Grants Council of Hong Kong SAR. G.W. was partially supported by a NCI training grant (CA067754). This work was supported by NIH grants GM052872 (to X.-D.F.), R01 CA136851-07 (to N.S.G.), and CA188881 (to J.A.M.).
Footnotes
Supplemental Information includes two figures and four tables and can be found with this article online at https://doi.org/10.1016/j.chembiol.2018.01.013.
AUTHOR CONTRIBUTIONS
Conceptualization, N.S.G., T.Z., and X.-D.F.; Compound Design, T.Z.; Compound Design and Synthesis, J.M.H.; Cellular Experiments, G.W., F.M., A.L.L., and C.E.P.; Crystallography, C.Z. and J.C.K.N.; Animal Studies, J.Z., G.W., W.W., S.P., and K.Z.; Mass Spectrum Labeling, S.B.F. and J.A.M.; Writing –Original Draft, T.Z.; Review & Editing, N.S.G., J.C.K.N., K.Z., and X.-D.F.
DECLARATION OF INTERESTS
Patent entitled as Inhibitors of Alk and Srpk And methods of use (WO 2017/053657 A1) has been filed.
References
- Abd AJ, Kanwar RK, Kanwar JR. Age-related macular degeneration: current therapeutics for management and promising new drug candidates. Drug Discov Today. 2017;22:1671–1679. doi: 10.1016/j.drudis.2017.07.010. [DOI] [PubMed] [Google Scholar]
- Amin EM, Oltean S, Hua J, Gammons MV, Hamdollah-Zadeh M, Welsh GI, Cheung MK, Ni L, Kase S, Rennel ES, et al. WT1mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell. 2011;20:768–780. doi: 10.1016/j.ccr.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askenazi M, Parikh JR, Marto JA. mzAPI: a new strategy for efficiently sharing mass spectrometry data. Nat Methods. 2009;6:240–241. doi: 10.1038/nmeth0409-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubol BE, Wu G, Keshwani MM, Movassat M, Fattet L, Hertel KJ, Fu XD, Adams JA. Release of SR proteins from CLK1 by SRPK1: a symbiotic kinase system for phosphorylation control of Pre-mRNA splicing. Mol Cell. 2016;63:218–228. doi: 10.1016/j.molcel.2016.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DO, Cui TG, Doughty JM, Winkler M, Sugiono M, Shields JD, Peat D, Gillatt D, Harper SJ. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002;62:4123–4131. [PubMed] [Google Scholar]
- Batson J, Toop HD, Redondo C, Babaei-Jadidi R, Chaikuad A, Wearmouth SF, Gibbons B, Allen C, Tallant C, Zhang J, et al. Development of potent, selective SRPK1 inhibitors as potential topical therapeutics for neovascular eye disease. ACS Chem Biol. 2017;12:825–832. doi: 10.1021/acschembio.6b01048. [DOI] [PubMed] [Google Scholar]
- Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today. 2015;20:1061–1073. doi: 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DG, Batchelor TT, Willett CG, Jain RK. VEGF-targeted cancer therapy strategies: current progress, hurdles and future prospects. Trends Mol Med. 2007;13:223–230. doi: 10.1016/j.molmed.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davids MS, Brown JR. Ibrutinib: a first in class covalent inhibitor of Bruton’s tyrosine kinase. Future Oncol. 2014;10:957–967. doi: 10.2217/fon.14.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Sun X, Guma M, Luo J, Ouyang H, Zhang X, Zeng J, Quach J, Nguyen DH, Shaw PX, et al. JNK inhibition reduces apoptosis and neovascularization in a murine model of age-related macular degeneration. Proc Natl Acad Sci USA. 2013;110:2377–2382. doi: 10.1073/pnas.1221729110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay MR, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, Bradbury RH, Brown SJ, Butterworth S, Campbell A, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57:8249–8267. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Fukuhara T, Hosoya T, Shimizu S, Sumi K, Oshiro T, Yoshinaka Y, Suzuki M, Yamamoto N, Herzenberg LA, Herzenberg LA, et al. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc Natl Acad Sci USA. 2006;103:11329–11333. doi: 10.1073/pnas.0604616103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammons MV, Lucas R, Dean R, Coupland SE, Oltean S, Bates DO. Targeting SRPK1 to control VEGF-mediated tumor angiogenesis in metastatic melanoma. Br J Cancer. 2014;111:477–485. doi: 10.1038/bjc.2014.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammons MV, Fedorov O, Ivison D, Du C, Clark T, Hopkins C, Hagiwara M, Dick AD, Cox R, Harper SJ, et al. Topical antiangiogenic SRPK1 inhibitors reduce choroidal neovascularization in rodent models of exudative AMD. Invest Ophthalmol Vis Sci. 2013;54:6052–6062. doi: 10.1167/iovs.13-12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh G, Adams JA. Phosphorylation mechanism and structure of serine arginine protein kinases. FEBS J. 2011;278:587–597. doi: 10.1111/j.1742-4658.2010.07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout S, Brambilla E, Boudria A, Drissi R, Lantuejoul S, Gazzeri S, Eymin B. Abnormal expression of the pre-mRNA splicing regulators SRSF1, SRSF2, SRPK1 and SRPK2 in non-small cell lung carcinoma. PLoS One. 2012;7:e46539. doi: 10.1371/journal.pone.0046539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR. VEGF inhibition study in ocular neovascularization clinical trial group, pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351:2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- Gui JF, Tronchere H, Chandler SD, Fu XD. Purification and characterization of a kinase specific for the serine- and arginine-rich pre-mRNA splicing factors. Proc Natl Acad Sci USA. 1994a;91:10824–10828. doi: 10.1073/pnas.91.23.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui JF, Lane WS, Fu XD. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994b;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer. 2008;8:880–887. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher JM, Bahcall M, Choi HG, Gao Y, Sim T, George R, Jänne PA, Gray NS. Discovery of inhibitors that overcome the g1202r anaplastic lymphoma kinase resistance mutation. J Med Chem. 2015;58:9296–9308. doi: 10.1021/acs.jmedchem.5b01136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes GM, Carrigan PE, Miller LJ. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res. 2007;67:2072–2080. doi: 10.1158/0008-5472.CAN-06-2969. [DOI] [PubMed] [Google Scholar]
- Hett EC, Xu H, Geoghegan KF, Gopalsamy A, Kyne RE, Jr, Menard CA, Narayanan A, Parikh MD, Liu S, Roberts L, et al. Rational targeting of active site tyrosine residues using sulfonyl fluoride probes. ACS Chem Biol. 2015;10:1094–1098. doi: 10.1021/cb5009475. [DOI] [PubMed] [Google Scholar]
- Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol. 1991;5:1806–1814. doi: 10.1210/mend-5-12-1806. [DOI] [PubMed] [Google Scholar]
- Huang Y, Steitz JA. Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol Cell. 2001;7:899–905. doi: 10.1016/s1097-2765(01)00233-7. [DOI] [PubMed] [Google Scholar]
- Huang Y, Gattoni R, Stevenin J, Steitz JA. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell. 2003;11:837–843. doi: 10.1016/s1097-2765(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nat Rev Clin Oncol. 2009;6:569–579. doi: 10.1038/nrclinonc.2009.130. [DOI] [PubMed] [Google Scholar]
- Kataoka N, Bachorik JL, Dreyfuss G. Transportin-SR, a nuclear import receptor for SR proteins. J Cell Biol. 1999;145:1145–1152. doi: 10.1083/jcb.145.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MC, Lin RI, Tarn WY. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc Natl Acad Sci USA. 2001;98:10154–10159. doi: 10.1073/pnas.181354098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkins E, Blumenthal GM, Chen H, He K, Agarwal R, Gieser G, Stephens O, Zahalka E, Ringgold K, Helms W, et al. FDA approval: Alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res. 2016;22:5171–5176. doi: 10.1158/1078-0432.CCR-16-1293. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Li XH, Song JW, Liu JL, Wu S, Wang LS, Gong LY, Lin X. Serine-arginine protein kinase 1 is associated with breast cancer progression and poor patient survival. Med Oncol. 2014;31:83. doi: 10.1007/s12032-014-0083-8. [DOI] [PubMed] [Google Scholar]
- Mavrou A, Brakspear K, Hamdollah-Zadeh M, Damodaran G, Babaei-Jadidi R, Oxley J, Gillatt DA, Ladomery MR, Harper SJ, Bates DO, et al. Serine arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene. 2015;34:4311–4319. doi: 10.1038/onc.2014.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti M, Guiducci S, Romano E, Ceccarelli C, Bellando-Randone S, Conforti ML, Ibba-Manneschi L, Matucci-Cerinic M. Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circ Res. 2011;109:e14–e26. doi: 10.1161/CIRCRESAHA.111.242057. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Caceres JF, Clement JQ, Krainer AR, Wilkinson MF, Spector DL. Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J Cell Biol. 1998;143:297–307. doi: 10.1083/jcb.143.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morooka S, Hoshina M, Kii I, Okabe T, Kojima H, Inoue N, Okuno Y, Denawa M, Yoshida S, Fukuhara J, et al. Identification of a dual inhibitor of SRPK1 and CK2 that attenuates pathological angiogenesis of macular degeneration in mice. Mol Pharmacol. 2015;88:316–325. doi: 10.1124/mol.114.097345. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nakagawa O, Arnold M, Nakagawa M, Hamada H, Shelton JM, Kusano H, Harris TM, Childs G, Campbell KP, Richardson JA, et al. Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by MEF2. Genes Dev. 2005;19:2066–2077. doi: 10.1101/gad.1338705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JC, Chakrabarti S, Ding JH, Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu XD, Ghosh G. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol Cell. 2005;20:77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Ngo JC, Gullingsrud J, Giang K, Yeh MJ, Fu XD, Adams JA, McCammon JA, Ghosh G. SR protein kinase 1 is resilient to inactivation. Structure. 2007;15:123–133. doi: 10.1016/j.str.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Nomanbhoy TK, Sharma G, Brown HE, Wu J, Aban A, Vogeti S, Alemayehu S, Sykes M, Rosenblum JS, Kozarich JW. Chemoproteomic evaluation of target engagement by the CDK4/CDK6 inhibitor palbociclib correlates with cancer cell response. Biochemistry. 2016;55:5434–5441. doi: 10.1021/acs.biochem.6b00629. [DOI] [PubMed] [Google Scholar]
- Nowak DG, Amin EM, Rennel ES, Hoareau-Aveilla C, Gammons M, Damodoran G, Hagiwara M, Harper SJ, Woolard J, Ladomery MR, et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J Biol Chem. 2010;285:5532–5540. doi: 10.1074/jbc.M109.074930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Sweet RM, Carter CW, editors. Methods in Enzymology. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, Huang J, Yeo GW, Ares M, Jr, Fu XD. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013;50:223–235. doi: 10.1016/j.molcel.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh JR, Askenazi M, Ficarro SB, Cashorali T, Webber JT, Blank NC, Zhang Y, Marto JA. Multiplierz: an extensible API based desktop environment for proteomics data analysis. BMC Bioinformatics. 2009;10:364. doi: 10.1186/1471-2105-10-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry. 2007;46:350–358. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol. 2011;18:699–710. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennel E, Waine E, Guan H, Schuler Y, Leenders W, Woolard J, Sugiono M, Gillatt D, Kleinerman E, Bates D, et al. The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumor growth in mice. Br J Cancer. 2008;98:1250–1257. doi: 10.1038/sj.bjc.6604309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards L. Targeted therapies: disappointing outcomes for anti-VEGF therapy. Nat Rev Clin Oncol. 2011;8:194. doi: 10.1038/nrclinonc.2011.28. [DOI] [PubMed] [Google Scholar]
- Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- Roth MB, Murphy C, Gall JG. A monoclonal antibody that recognizes a phosphorylated epitope stains lampbrush chromosome loops and small granules in the amphibian germinal vesicle. J Cell Biol. 1990;111:2217–2223. doi: 10.1083/jcb.111.6.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, Gray NK, Beckmann K, Caceres JF. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004;18:755–768. doi: 10.1101/gad.286404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Chen CA. The epidemiology of age-related macular degeneration. Int Ophthalmol Clin. 2004;44:17–39. doi: 10.1097/00004397-200404440-00004. [DOI] [PubMed] [Google Scholar]
- Shima C, Sakaguchi H, Gomi F, Kamei M, Ikuno Y, Oshima Y, Sawa M, Tsujikawa M, Kusaka S, Tano Y. Complications in patients after intravitreal injection of bevacizumab. Acta Ophthalmol. 2008;86:372–376. doi: 10.1111/j.1600-0420.2007.01067.x. [DOI] [PubMed] [Google Scholar]
- Siebel CW, Feng L, Guthrie C, Fu XD. Conservation in budding yeast of a kinase specific for SR splicing factors. Proc Natl Acad Sci USA. 1999;96:5440–5445. doi: 10.1073/pnas.96.10.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Siqueira RP, Barbosa Éde A, Polêto MD, Righetto GL, Seraphim TV, Salgado RL, Ferreira JG, Barros MV, de Oliveira LL, Laranjeira AB, et al. Potential antileukemia effect and structural analyses of SRPK inhibition by N-(2-(piperidin-1-yl)-5-(trifluoromethyl)phenyl)isonicotinamide (SRPIN340) PLoS One. 2015;10:e0134882. doi: 10.1371/journal.pone.0134882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Székelyhidi Z, Pató J, Wáczek F, Bánhegyi P, Hegymegi-Barakonyi B, Erös D, Mészáros G, Hollósy F, Hafenbradl D, Obert S, et al. Synthesis of selective SRPK-1 inhibitors: novel tricyclic quinoxaline derivatives. Bioorg Med Chem Lett. 2005;15:3241–3246. doi: 10.1016/j.bmcl.2005.04.064. [DOI] [PubMed] [Google Scholar]
- Ventrice P, Leporini C, Aloe JF, Greco E, Leuzzi G, Marrazzo G, Scorcia GB, Bruzzichesi D, Nicola V, Scorcia V. Anti-vascular endothelial growth factor drugs safety and efficacy in ophthalmic diseases. J Pharmacol Pharmacother. 2013;4:S38–S42. doi: 10.4103/0976-500X.120947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC, Fu XD. SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of premRNA splicing factors in mammalian cells. J Cell Biol. 1998;140:737–750. doi: 10.1083/jcb.140.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- Woolard J, Wang WY, Bevan HS, Qiu Y, Morbidelli L, Pritchard-Jones RO, Cui TG, Sugiono M, Waine E, Perrin R, et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64:7822–7835. doi: 10.1158/0008-5472.CAN-04-0934. [DOI] [PubMed] [Google Scholar]
- Zhang K, Zhang L, Weinreb RN. Ophthalmic drug discovery: novel targets and mechanisms for retinal diseases and glaucoma. Nat Rev Drug Discov. 2012;11:541–559. doi: 10.1038/nrd3745. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Marshall AG. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J Am Soc Mass Spectrom. 1998;9:225–233. doi: 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]
- Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009;23:482–495. doi: 10.1101/gad.1752109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma. 2013;122:191–207. doi: 10.1007/s00412-013-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZH, Qiu JS, Wen L, Zhou Y, Plocinik RM, Li H, Hu QD, Ghosh G, Adams JA, Rosenfeld MG, et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell. 2012;47:422–433. doi: 10.1016/j.molcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.