

Abstract
p21-activated kinase 1 (PAK1) has attracted much attention as a potential therapeutic target due to its central role in many oncogenic signaling pathways, its frequent dysregulation in cancers and neurological disorders, and its tractability as a target for small-molecule inhibition. To date several PAK1-targeting compounds have been developed as preclinical agents, including one that has been evaluated in a clinical trial. A series of ATP-competitive inhibitors, allosteric inhibitors, and peptide inhibitors with distinct biochemical and pharmacokinetic properties represent useful laboratory tools for studies on the role of PAK1 in biology and in disease contexts, and could lead to promising therapeutic agents. Given the central role of PAK1 in vital signaling pathways, future clinical development of PAK1 inhibitors will require careful investigation of their safety and efficacy.
Introduction
PAK1 is a founding member of the Pak (p21-activated kinases) Ser/Thr protein kinase family. Initially identified as an interactor of the Rho GTPases RAC1 and CDC42 [1], PAK1 was later shown to play diverse role in cell signaling by means of its catalytic and scaffolding activities [2]. Signal transduction cascades modulated by PAK1 include proliferation and survival pathways such as MAPK, AKT, Wnt1/β-catenin, ERα, BAD and NF-κB [2]. PAK1 is also critically involved in regulation of cell motility, transmitting variety of signals controlling cytoskeleton dynamics, cell shape and adhesion [2–4].
While PAK1 shares functions with other family members, in particular PAK2 and PAK3 (which are, with PAK1, together referred to as group I Paks) much more is known of the function of PAK1 in terms of human biology and disease than any other isoform. PAK1 expression is dysregulated in several nervous system disorders, including Alzheimer disease and Fragile X syndrome [5], indicating a role in cognition. Gain-of-function alterations of PAK1 have been observed in a wide range of human malignancies, suggesting that this kinase plays a substantial role in tumor development and progression [2, 6]. Amplification of the PAK1 gene at 11q13, as well as elevated PAK1 protein levels, are often associated with aggressive tumor phenotypes, chemotherapy resistance, and poor outcome [2, 7–9]. Apart from gene amplification and protein overexpression, PAK1 can be hyperactivated by mutations in upstream regulators such as RAC1 [10], RAS [11] and Merlin [12], linking oncogenic signaling to cancer cell phenotypic changes.
For these reasons, targeting PAK1 may represent a promising therapeutic approach in certain disease contexts, and multiple efforts in identification of potent and selective PAK1 inhibitors have been made in the past decade [2, 13]. Here we discuss the suitability of PAK1 as a drug target and recent advances in the development of PAK1 inhibitors.
PAK1 structure and regulation
PAK1 is a 545 amino acid multidomain protein that contains an N-terminal regulatory region and a C-terminal kinase (catalytic) domain (Figure 1) [14, 15]. The PAK1 catalytic domain has the characteristic two-lobe kinase structure with a single phosphorylation site (Thr423) within the activation loop. The amino terminal end of PAK1 harbors several sequence motifs responsible for interacting with partner proteins. Residues 75–90 correspond to the CDC42/RAC1 interactive-binding (CRIB) domain, which partially overlaps the auto-inhibitory domain (AID, aa 83-149). Three Pro-rich N-terminal motifs interact with SH3-domain containing adaptor proteins, including GRB2 (aa 12–18), NCK (aa 40–45), and the exchange factor PIX (aa 186–203) [15]. A positively charged basic region adjacent to CRIB domain is critical for PAK1 binding to cell membrane phosphoinositides [16]. Several phosphorylation sites located in the regulatory region play role in enabling and stabilizing the active conformation of PAK1 (Figure 1A) [17–19].
Figure 1. PAK1 structure.
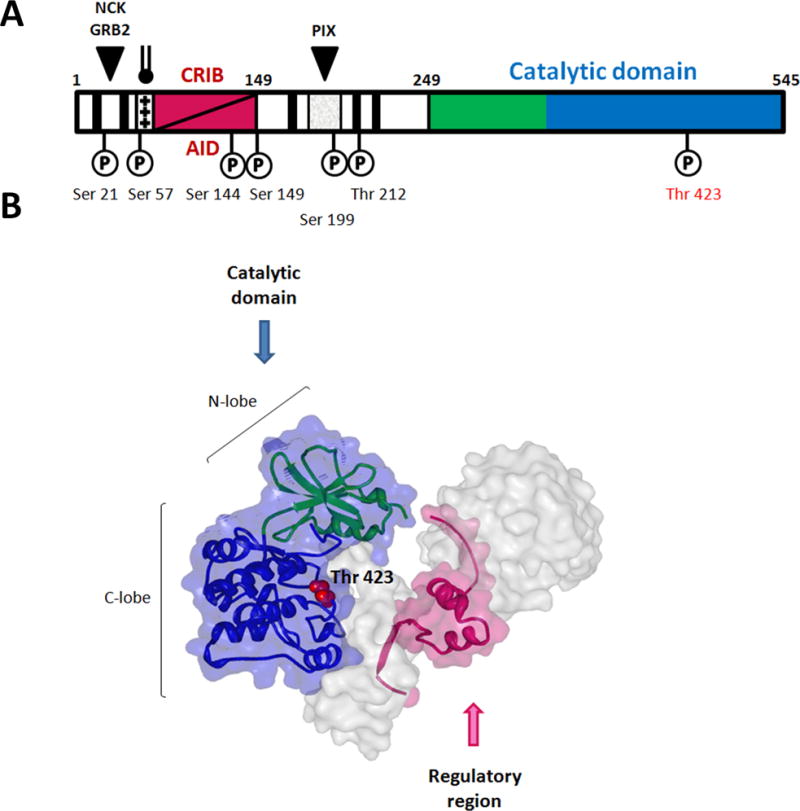
Organization of the PAK1 polypeptide chain highlighting sites of kinase phosphorylation. Numerals indicate residue numbers. PAK1 auto-regulatory region is in magenta, N-lobe of the catalytic domain is in green, and C-lobe is in blue. Proline-rich SH3-binding sites are shown as black bars. Phosphoinositide binding region enriched with basic residues is shown as srossed bar.
Diagram of dimeric PAK1 (PDB ID: 1F3M). One PAK1 complex is colored as in (A), Thr 423 is labeled. The other one is presented as surface diagram. Residues 1–77 and 148–248 are omitted.
PAK1 activity is regulated by a trans-autoinhibitory mechanism [20]. Inactive PAK1 folds into an asymmetric homodimer (Figure 1B), in which the AID of each molecule binds to the kinase domain of its counterpart. Interacting with the large lobe of kinase domain AID positions its C-terminal extension (aa 136–149) into catalytic cleft of the kinase domain (Figure 1B). This interaction prevents dimer deconstruction, opening of the catalytic site, and autophosphorylation that are essential for full PAK1 catalytic activity [17, 19]. Binding of GTP-loaded CDC42 and RAC1 to the CRIB motif causes an AID conformational change resulting in the dis-inhibition of the catalytic domain, dissociation of the dimer and phosphorylation of the regulatory region and the activation loop. Maximally active monomeric PAK1 can phosphorylate a variety of substrate proteins. PAK1 activity can also be regulated in a GTPase-independent manner. Such mechanisms of PAK1 activation include binding to phospholipids and SH3-containing proteins, as well as cross-phosphorylation by other kinases [21–25].
A detailed understanding of PAK1 regulation and catalysis has suggested two distinct strategies for pharmaceutical inhibition of PAK1: direct disabling of phosphotransfer in the active site (ATP-competitive inhibitors) and exploitation of unique PAK1 regulatory mechanisms (allosteric inhibitors).
ATP-competitive PAK1 inhibitors
ATP-competitive inhibitors are typically designed to occupy the ribose-binding pocket of the kinase active site, precluding ATP access [26]. Developing such ligands for PAK1 has proven challenging, as the PAK1 kinase domain is highly plastic and the ATP binding cleft is particularly open and flexible (Figure 2) [14, 27]. These challenges have not, however, prevented several serious attempts to create potent and specific inhibitors, and efforts to identify ATP-competitive PAK1 inhibitors with high affinity and kinase selectivity are ongoing. As the PAK1 catalytic domain shares strong sequence similarity with those of other Pak family members (~ 95% with PAK2 and PAK3, ~ 54% with PAK4,5,6), it is not surprising that the majority of described PAK1 targeting small molecules either inhibit all Paks (“pan-Pak”) or are group specific (e.g., PAK1/2/3 inhibitors) [2, 13] (Figure 3).
Figure 2. Co-crystal structures of ATP-competitive Pak inhibitors in complex with PAK1.
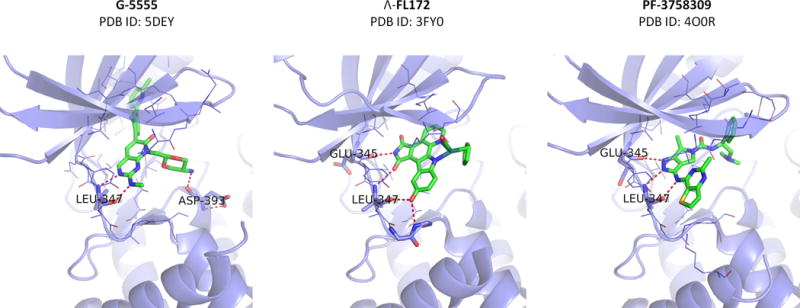
Hydrogen-bonding interactions are shown as dotted red lines.
Figure 3. Pak inhibitors.
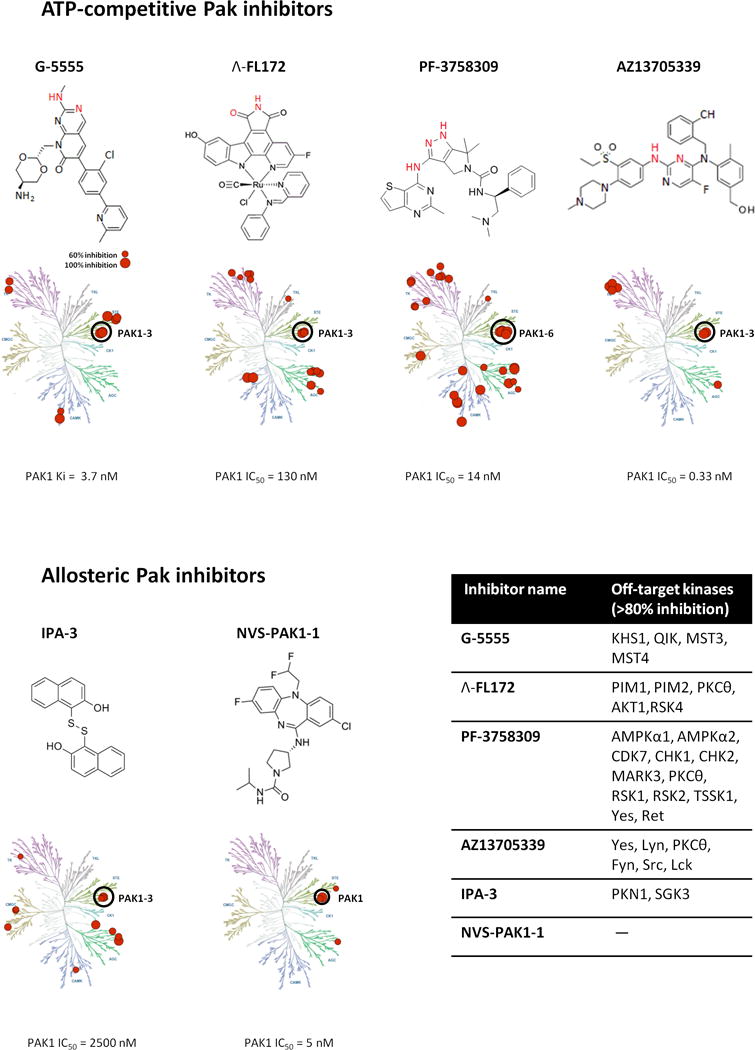
Biochemical structures and selectivity profiles (inhibition ≥ 50% shown) of select Pak inhibitors. The molecule portions of ATP-competitive inhibitors rendered in red indicate the atoms participating in kinase hinge hydrogen bonding contacts.
G-5555 (0.1 uM) screened aginst 235 kinases (Invitrogen) [49]
FL172 (3 uM) screened against 246 kinases (Millipore) [31]
PF-3758309 (1 uM) screened against 146 kinases at Pfizer, Invitrogen, and the University of Dundee.[32]
AZ13705339 (0.1 uM) screened against 125 kinases (Invitrogen), PAK3 activity was not tested in this assay.[51]
IPA-3 (10uM) screened against 214 kinases (Invitrogen) [53]
NVS-PAK1-1 (10 uM) tested against 442 kinases (DiscoverX).[57]
Illustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com)
Indolocarbazoles
Alkaloid staurosporine (and its analog K252) is a broad spectrum ATP-competitive kinase inhibitor with high affinity for STE20 family kinases, including PAK1 [28, 29]. The staurosporine structure mimics ATP and interacts with conservative catalytic domain residues, efficiently inhibiting >70% of human kinases [28–30]. Despite its high potency, the utility of staurosporine has been limited by its poor target selectivity, thus further derivatization of the indolocarbazole scaffold was needed to improve selectivity for PAK1. Attempting to exploit the capacious ATP binding pocket of PAK1, Meggers and colleagues designed a metal-containing scaffold with Pak inhibitory activities [31]. A bulky, octahedral ruthenium complex inhibitor (Λ-FL172) (Figure 2, 3) efficiently filled the large catalytic pocket (Figure 2), gaining high PAK1 inhibitory efficacy as well as reasonably high selectivity over related protein kinases (Figure 3). Among 264 kinases tested, only 15 kinases (5.7% of total) showed an inhibition of > 80%. However, no information on cell activity and pharmacokinetics has been reported for this organometal conjugate compound, therefore it is unclear whether further pursuit of this strategy will yield clinically useful inhibitors.
Pyrrolopyrazoles (Pfizer)
The Pfizer group has described and evaluated a series of pyrrolopyrazole-based Pak inhibitors [13], including compound PF-3758309, the only Pak inhibitor to date to proceed to human trials [32]. Though originally designed as a PAK4 inhibitor, PF-3758309 efficiently inactivates all Pak family kinases, as well as a number of off-targets (Figure 2, 3). This orally available compound with potent cellular activity (1.3–3.9 nM) was probed in a number of preclinical cancer models and exhibited marked antitumor efficacy [32–37]. PF-3758309 inhibited cellular proliferation in a panel of tumor cell lines [32–36], as well as tumor growth in xenograft models of melanoma (M24met, SK-MEL23, 537MEL) [10, 32], colorectal carcinoma (Colo-205, Collo-201, HCT116, RKO, PDT xenografts)[32, 36, 37], breast (MDA-MB231, BT-474)[32, 35], prostate (PC3)[34], lung cancer (A549, H358-S, H1299)[32, 33] and a transgenic Kras-driven squamous cell carcinoma model [38]. PF-3758309 was also suggested for the treatment of pigmentation disorders [39] and osteoclast-related disorders[40].
Given the relatively poor target selectivity of PF-3758309 it remains difficult to ascribe these desirable biological effects to Pak inhibition alone, since off-target inhibition most certainly contributes to the cellular activity of the compound. In any case, PF-3758309 was withdrawn from clinical investigation due to unexpectedly low oral bioavailability in humans, insignificant tumor responses, and adverse effects.
Aminopyrimidine-based series (Afraxis/Genentech)
Afraxis, Inc. developed a series of Pak-inhibiting compounds based on a pyrido[2,3-d]pyrimidine-7-one core for the treatment of neurological disorders. Subsequently FRAX compounds were utilized in various cancer preclinical studies. One such compound, FRAX597 (PDB ID: 4EQC), potently inhibits PAK1 (IC50 = 7.7 nM), while showing moderate selectivity against other kinases, particularly receptor tyrosine kinases [41]. This compound is orally available and was successfully used in a neurofibromatosis type 2 (NF2) orthotopic schwannoma model [41] and a meningioma model [42], as well as a transgenic KrasG12D squamous cell carcinoma mouse model [38].
Another compound of this chemical series, FRAX486 has been studied as a possible treatment of fragile X syndrome (FXS), a genetic disorder caused by inactivation of the fragile X mental retardation 1 (Fmr1) gene [43]. Fmr1 knockout (KO) mice recapitulate human FXS symptoms, including hyperactivity, repetitive behaviors, and seizures, as well as morphological synaptic abnormalities [43, 44]. FRAX486 has excellent PAK1 potency (IC50 = 8.25 nM) and pharmacokinetic properties upon subcutaneous injection, including effective blood–brain barrier penetration, allowed its exploitation in an Fmr1 KO model. Strikingly, single administration of FRAX486 was sufficient to ameliorate the FXS phenotype at both cellular and behavioral levels, in line with previous studies on genetic inactivation of Pak in this Fmr1 KO mouse model [45].
An advanced member of this series, FRAX1036 (PDB ID:5DFP), exhibits high PAK1 potency (PAK1 Ki = 23 nM), refined kinome selectivity [42, 46, 47], and represents a useful tool compound for single and combinatorial experimental therapeutics [42, 46–48]. However, all of these early FRAX compounds were found to have strong adverse inhibition of hERG potassium channels. Also, the compound permeability was far from ideal [49]. Addressing these concerns, Genentech designed a further compound based on FRAX1036, termed G-5555 (PDB ID: 5DEY, Figure 2) [49], with favorable cellular activity and permeability, as well as low hERG channel activity. To our knowledge, G-5555 is the most selective ATP-competitive inhibitor of PAK1 reported to date (Figure 3). Only 8 out of 235 kinases tested (other than PAK1) showed an inhibition of >70%. Additionally G-5555 showed desirable in vivo pharmacokinetic properties, suggesting a potential for good human exposure.
G-5555 was tested on a panel of 23 breast cancer cell lines and has shown greater growth inhibition in Pak-amplified cell lines [50]. In a non-small lung cancer xenograft model and in a PAK1-amplified breast cancer xenograft model tumor growth was significantly impaired when exposed to 25 mg/kg BID G-5555, however greater doses were not tolerated. Mouse tolerability studies revealed acute cardiovascular toxicity of aminopyrimidine-based inhibitors, including G-5555, which makes it unsuitable for clinical development, and also raised questions about the feasibility of targeting group I Paks in the clinical setting [50].
Bis-anilino pyrimidines (AstraZeneca)
AstraZeneca has recently disclosed ATP-competitive kinase inhibitors with exceptional potency against PAK1 [51]. Overlay of two chemotypes (bis-anilino pyrimidine and previously described 7-azaindole Pak inhibitor [52]) bound to PAK1 allowed the visualization and design small molecules with improved binding mode. One of the resulting in vitro probe compounds AZ13705339 inhibits PAK1 at IC50 <1 nM with only 8 off-target kinases giving >80% inhibition, mainly Src family kinases (Figure 3). Another compound of the series AZ13711265 displayed better pharmacokinetic properties and was proposed for in vivo studies.
Non-ATP-competitive PAK1 inhibitors
An alternative to ATP-competitive kinase inhibitors are allosteric inhibitors that bind outside of the ATP-binding cleft. PAK1 allosteric inhibitors achieve greater selectivity across the kinome relative to ATP-competitive inhibitors as they target less conserved regions. However such inhibitors have reduced potency compared to ATP-competitive inhibitors since protein pockets they target are not as deep and rich in inhibitor binding residues.
Naphthtols
IPA-3 (inhibitor p21-activated kinase-3), a sulfhydryl-containing compound that targets the N-terminal regulatory domain of PAK1, was discovered in a deliberate attempt to find non-competitive PAK1 inhibitors [53]. Reversible covalent binding of IPA-3 to the PAK1 regulatory domain prevents GTPase docking and the subsequent switch to a catalytically active state [54]. This unique mechanism of action accounts for the exceptional target specificity of IPA-3 (Figure 3), making it a useful tool compound for in vitro research. Unfortunately, cellular redox effects caused by the reduction of IPA-3 sulfhydryl moiety greatly limits its usefulness in cellular and animal models. This can be partially managed using structural inactive isomer of IPA-3 termed PIR-3.5 (PAK1 inhibitor-related 3.5) - a negative-control compound with similar redox effects [53]. Despite these limitations IPA-3 is widely used in cell culture and several attempts to evaluate IPA-3 potential in vivo have been made. In one such study encapsulation in sterically stabilized liposomes (SSL) has been used to improve IPA-3 metabolic stability and efficacy in inhibiting prostate cancer growth in xenograft model [55].
Overall IPA-3 represents a distinctive proof-of-concept compound with unique PAK1 binding mode and selectivity, and discovery of next generation IPA-3-like molecules with more suitable drug properties is desirable. In this regard, it is interesting to note that a second series of compounds have recently been identified that allosterically inhibit PAK1 by interacting with its regulatory domain [56]. Unlike IPA-3, these 1,4-naphthohydroquinone (1,4-NHQ)-based compounds do not appear to form covalent adducts with PAK1, and also display selectivity for PAK1 and PAK3 over PAK2 [56]. These features may be useful in limiting potential toxicities, as discussed in more detail below. As with IPA- 3, IC50 values for the 1,4-NHQ inhibitors are in the low micromolar range.
Dibenzodiazepines (Novartis)
Allosteric dibenzodiazepine PAK1 inhibitors emerged as hits in fragment-based screening performed by Novartis. These compounds bind to a novel allosteric site adjacent to ATP-binding pocket and interfere with ATP binding [57]. Optimized compound NVS-PAK1-1 (a.k.a. compound 3 [57]) showed exceptional selectivity across the kinome, including Pak intragroup isoform selectivity (Figure 3). Remarkably NVS-PAK1-1 demonstrates higher inhibitory activity on PAK1 than PAK2 as measured by in vitro kinase assays, Kd measurement, and immunoblot estimation of intracellular phospho-PAK1/2 levels. Thus NVS-PAK1-1 represents the first example of “PAK1 only” small molecule inhibitor. Despite high biochemical activity, NVS-PAK1-1 was not sufficiently potent to inhibit phosphorylation of the downstream substrate MEK1 Ser289 or cell proliferation. This phenomenon might hypothetically be explained by Pak isoforms functional redundancy [2]. In addition inhibition of PAK2 and/or PAK3 might be required for the functional activity in cells expressing these Pak isoforms. PAK1 amplified tumors that are highly dependent on PAK1 signaling might be chosen for further NVS-PAK1-1 validation studies.
Peptide inhibitors
Peptide inhibitors of PAK1 have been widely used as laboratory tools. For example, the PAK1 autoinhibitory domain (AID, aa 83–149) (Figure 1) effectively inhibits PAK1 activity. Interestingly, the induction of cell cycle arrest by the PAK1 AID can occur independent of inhibiting PAK1 kinase activity [58], most likely due to AID binding to the fragile-X proteins FMR1 and FRX1, which modulate the stability of the cyclin-dependent kinase inhibitor 1 p21waf1 [59, 60]. Contrariwise, the AID derived from PAK2 lacks FMR1/FXR1 binding and presumably exerts its biological effects mostly through Pak inhibition. Two other peptide inhibitors, comprising the cell permeant TAT peptide fused to the PIX-interacting motif (TAT-PAK18) or the NCK binding motif of PAK1, have also been described. These peptides are thought to prevent proper cellular localization (and activation) of PAK1 through disruption of PAK1-NCK or PAK1-PIX interactions. The Pak-mediated growth suppression effect of TAT-PAK18 has been shown on PAK1-dependent ovarian cancer cell lines [61], while a Pak-NCK inhibitory peptide affects endothelial cell migration and contractility[62, 63]. Issues regarding the delivery of peptide-based inhibitors into cells, however, present a challenge in terms of therapeutic use.
Conclusions and prospects
PAK1 represents a promising druggable therapeutic target for conditions with elevated PAK1 levels or deregulated Pak upstream/downstream signaling [2, 5, 6]. Such “signalopathies” include several tumorigenic disorders, primarily cancers associated with PAK1 amplification [8, 47]. Positioned downstream of difficult-to-target oncogenes such as RAS, PAK1 might open a new window for disruption of RAS-driven tumorigenesis [11, 38, 64]. PAK1 inhibition could also be beneficial for tumor growth targeting in neurofibromatoses type 1 (NF1) and type 2 (NF2) patients [41, 42, 64], as well as for management of NF1-associated social learning deficit [65]. Likewise, cognitive dysfunction in the context of FXS might be rescued upon PAK1 inactivation [43, 45].
Most described PAK1 inhibitors also bind related group I Pak family members PAK2 and PAK3. In our view, the major impediment to clinical development of Pak inhibitors relates to balancing efficacy and toxicity. To address this question, we need to better understand the functions of the three group I Paks in adult tissues. Genetic models have already established an essential role for PAK2 in cardiovascular function on adult mice, though the molecular underpinnings for this role are unclear [66]. In contrast, total PAK1 loss is well-tolerated in mice, with few obvious phenotypic consequences, though αMHC-Cre;Pak1f/f mice, which delete PAK1 in cardiac tissues, are prone to heart failure when subjected to pressure overload [67]. Whether this phenotype represents a developmental defect or a functional defect has not been established. PAK3 loss-of-function mutations in man are associated with mental retardation, but, as with PAK1, whether this represents a developmental or functional defect is not known. These genetic studies, coupled with the observed acute toxicities associated with several distinct classes of Pak inhibitors, necessarily introduce a note of caution into Pak inhibitor development.
Assuming it is safe to target PAK1, one path forward might be to develop isoform-specific inhibitors. In that regard, it is encouraging that the Novartis compound NVS-PAK1-1 displayed reasonable selectivity for PAK1 over PAK2. However, given that this compound had little effect on the growth of a panel of cancer cells, it is possible that blockade of PAK1 alone will be insufficient in many instances. To address this issue, better selection of tumor types may be required. Recent work from our group and others [46, 47, 68] suggests that using PAK1 gene amplification as a biomarker might select for tumor cells that are sensitive to PAK1 inhibition, and it will be instructive to test NVS-PAK1-1 and similar isoform-selective Pak inhibitors in this setting. Similarly, tumors bearing activating mutations in the small GTPase RAC1 (e.g., ~5% of melanoma) [69, 70], which encodes a direct activator of group I Paks, might be good candidates for consideration for future anti-Pak agents.
Acknowledgments
We thank Peter J. Huwe for the help Figure 2.
Abbreviations
- 1,4-NHQ
1,4-naphthohydroquinone
- AID
autoinhibitory domain
- CRIB
CDC42/RAC1 interactive-binding
- FMR1
fragile X mental retardation 1
- FXS
Fragile X syndrome
- IPA-3
inhibitor p21-activated kinase-3
- KO
knockout
- NF1
neurofibromatosis type 1
- NF2
neurofibromatosis type 2
- PAK1
p21-activated kinase 1
- MAPK
mitogen-activated protein kinase
- ERα
estrogen receptor alpha
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- BAD
Bcl-2-associated death promoter
- GRB2
growth factor receptor-bound protein 2
- hERG
human ether-à-go-go-related gene
Footnotes
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- 2.Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer. 2014;14:13–25. doi: 10.1038/nrc3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- 4.Delorme-Walker VD, Peterson JR, Chernoff J, Waterman CM, Danuser G, Dermardirossian C, Bokoch GM. Pak1 regulates focal adhesion strength, myosin IIA distribution, and actin dynamics to optimize cell migration. J Cell Biol. 2011;193:1289–1303. doi: 10.1083/jcb.201010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma QL, Yang F, Frautschy SA, Cole GM. PAK in Alzheimer disease, Huntington disease and X-linked mental retardation. Cell Logist. 2012;2:117–125. doi: 10.4161/cl.21602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ye DZ, Field J. PAK signaling in cancer. Cell Logist. 2012;2:105–116. doi: 10.4161/cl.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holm C, Rayala S, Jirstrom K, Stal O, Kumar R, Landberg G. Association between Pak1 expression and subcellular localization and tamoxifen resistance in breast cancer patients. J Natl Cancer Inst. 2006;98:671–680. doi: 10.1093/jnci/djj185. [DOI] [PubMed] [Google Scholar]
- 8.Ong CC, Jubb AM, Haverty PM, Zhou W, Tran V, Truong T, Turley H, O’brien T, Vucic D, Harris AL, Belvin M, Friedman LS, Blackwood EM, Koeppen H, Hoeflich KP. Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proc Natl Acad Sci U S A. 2011;108:7177–7182. doi: 10.1073/pnas.1103350108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamai T, Shirataki H, Nakanishi K, Furuya N, Kambara T, Abe H, Oyama T, Yoshida K. Increased Rac1 activity and Pak1 overexpression are associated with lymphovascular invasion and lymph node metastasis of upper urinary tract cancer. BMC Cancer. 2010;10:164. doi: 10.1186/1471-2407-10-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ong CC, Jubb AM, Jakubiak D, Zhou W, Rudolph J, Haverty PM, Kowanetz M, Yan Y, Tremayne J, Lisle R, Harris AL, Friedman LS, Belvin M, Middleton MR, Blackwood EM, Koeppen H, Hoeflich KP. P21-activated kinase 1 (PAK1) as a therapeutic target in BRAF wild-type melanoma. J Natl Cancer Inst. 2013;105:606–607. doi: 10.1093/jnci/djt054. [DOI] [PubMed] [Google Scholar]
- 11.Balbin OA, Prensner JR, Sahu A, Yocum A, Shankar S, Malik R, Fermin D, Dhanasekaran SM, Chandler B, Thomas D, Beer DG, Cao X, Nesvizhskii AI, Chinnaiyan AM. Reconstructing targetable pathways in lung cancer by integrating diverse omics data. Nat Commun. 2013;4:2617. doi: 10.1038/ncomms3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell. 2003;12:841–849. doi: 10.1016/s1097-2765(03)00382-4. [DOI] [PubMed] [Google Scholar]
- 13.Rudolph J, Crawford JJ, Hoeflich KP, Wang W. Inhibitors of p21-activated kinases (PAKs) J Med Chem. 2015;58:111–129. doi: 10.1021/jm501613q. [DOI] [PubMed] [Google Scholar]
- 14.Lei M, Lu W, Meng W, Parrini MC, Eck MJ, Mayer BJ, Harrison SC. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell. 2000;102:387–397. doi: 10.1016/s0092-8674(00)00043-x. [DOI] [PubMed] [Google Scholar]
- 15.Zhao ZS, Manser E. PAK family kinases: Physiological roles and regulation. Cell Logist. 2012;2:59–68. doi: 10.4161/cl.21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strochlic TI, Viaud J, Rennefahrt UE, Anastassiadis T, Peterson JR. Phosphoinositides are essential coactivators for p21-activated kinase 1. Mol Cell. 2010;40:493–500. doi: 10.1016/j.molcel.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pirruccello M, Sondermann H, Pelton JG, Pellicena P, Hoelz A, Chernoff J, Wemmer DE, Kuriyan J. A dimeric kinase assembly underlying autophosphorylation in the p21 activated kinases. J Mol Biol. 2006;361:312–326. doi: 10.1016/j.jmb.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 18.Chong C, Tan L, Lim L, Manser E. The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J Biol Chem. 2001;276:17347–17353. doi: 10.1074/jbc.M009316200. [DOI] [PubMed] [Google Scholar]
- 19.Buchwald G, Hostinova E, Rudolph MG, Kraemer A, Sickmann A, Meyer HE, Scheffzek K, Wittinghofer A. Conformational switch and role of phosphorylation in PAK activation. Mol Cell Biol. 2001;21:5179–5189. doi: 10.1128/MCB.21.15.5179-5189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parrini MC, Lei M, Harrison SC, Mayer BJ. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol Cell. 2002;9:73–83. doi: 10.1016/s1097-2765(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 21.Bokoch GM, Reilly AM, Daniels RH, King CC, Olivera A, Spiegel S, Knaus UG. A GTPase-independent mechanism of p21-activated kinase activation. Regulation by sphingosine and other biologically active lipids. J Biol Chem. 1998;273:8137–8144. doi: 10.1074/jbc.273.14.8137. [DOI] [PubMed] [Google Scholar]
- 22.King CC, Gardiner EM, Zenke FT, Bohl BP, Newton AC, Hemmings BA, Bokoch GM. p21-activated kinase (PAK1) is phosphorylated and activated by 3-phosphoinositide-dependent kinase-1 (PDK1) J Biol Chem. 2000;275:41201–41209. doi: 10.1074/jbc.M006553200. [DOI] [PubMed] [Google Scholar]
- 23.Shin YJ, Kim YB, Kim JH. Protein kinase CK2 phosphorylates and activates p21-activated kinase 1. Mol Biol Cell. 2013;24:2990–2999. doi: 10.1091/mbc.E13-04-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou GL, Zhuo Y, King CC, Fryer BH, Bokoch GM, Field J. Akt phosphorylation of serine 21 on Pak1 modulates Nck binding and cell migration. Mol Cell Biol. 2003;23:8058–8069. doi: 10.1128/MCB.23.22.8058-8069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fryer BH, Wang C, Vedantam S, Zhou GL, Jin S, Fletcher L, Simon MC, Field J. cGMP-dependent protein kinase phosphorylates p21-activated kinase (Pak) 1, inhibiting Pak/Nck binding and stimulating Pak/vasodilator-stimulated phosphoprotein association. J Biol Chem. 2006;281:11487–11495. doi: 10.1074/jbc.M600279200. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 27.Jha RK, Strauss CE. 3D structure analysis of PAKs: A clue to the rational design for affinity reagents and blockers. Cell Logist. 2012;2:69–77. doi: 10.4161/cl.21883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci. 1989;10:218–220. doi: 10.1016/0165-6147(89)90263-0. [DOI] [PubMed] [Google Scholar]
- 29.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 30.Tanramluk D, Schreyer A, Pitt WR, Blundell TL. On the origins of enzyme inhibitor selectivity and promiscuity: a case study of protein kinase binding to staurosporine. Chem Biol Drug Des. 2009;74:16–24. doi: 10.1111/j.1747-0285.2009.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maksimoska J, Feng L, Harms K, Yi C, Kissil J, Marmorstein R, Meggers E. Targeting large kinase active site with rigid, bulky octahedral ruthenium complexes. J Am Chem Soc. 2008;130:15764–15765. doi: 10.1021/ja805555a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray BW, Guo C, Piraino J, Westwick JK, Zhang C, Lamerdin J, Dagostino E, Knighton D, Loi CM, Zager M, Kraynov E, Popoff I, Christensen JG, Martinez R, Kephart SE, Marakovits J, Karlicek S, Bergqvist S, Smeal T. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc Natl Acad Sci U S A. 2010;107:9446–9451. doi: 10.1073/pnas.0911863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raja R, Sahasrabuddhe NA, Radhakrishnan A, Syed N, Solanki HS, Puttamallesh VN, Balaji SA, Nanjappa V, Datta KK, Babu N, Renuse S, Patil AH, Izumchenko E, Prasad TS, Chang X, Rangarajan A, Sidransky D, Pandey A, Gowda H, Chatterjee A. Chronic exposure to cigarette smoke leads to activation of p21 (RAC1)-activated kinase 6 (PAK6) in non-small cell lung cancer cells. Oncotarget. 2016 doi: 10.18632/oncotarget.11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang N, Hjorth-Jensen K, Hekmat O, Iglesias-Gato D, Kruse T, Wang C, Wei W, Ke B, Yan B, Niu Y, Olsen JV, Flores-Morales A. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene. 2015;34:2764–2776. doi: 10.1038/onc.2014.206. [DOI] [PubMed] [Google Scholar]
- 35.Arias-Romero LE, Villamar-Cruz O, Huang M, Hoeflich KP, Chernoff J. Pak1 kinase links ErbB2 to beta-catenin in transformation of breast epithelial cells. Cancer Res. 2013;73:3671–3682. doi: 10.1158/0008-5472.CAN-12-4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pitts TM, Kulikowski GN, Tan AC, Murray BW, Arcaroli JJ, Tentler JJ, Spreafico A, Selby HM, Kachaeva MI, Mcphillips KL, Britt BC, Bradshaw-Pierce EL, Messersmith WA, Varella-Garcia M, Eckhardt SG. Association of the epithelial-to-mesenchymal transition phenotype with responsiveness to the p21-activated kinase inhibitor, PF-3758309, in colon cancer models. Front Pharmacol. 2013;4:35. doi: 10.3389/fphar.2013.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bradshaw-Pierce EL, Pitts TM, Tan AC, Mcphillips K, West M, Gustafson DL, Halsey C, Nguyen L, Lee NV, Kan JL, Murray BW, Eckhardt SG. Tumor P-Glycoprotein Correlates with Efficacy of PF-3758309 in in vitro and in vivo Models of Colorectal Cancer. Front Pharmacol. 2013;4:22. doi: 10.3389/fphar.2013.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chow HY, Jubb AM, Koch JN, Jaffer ZM, Stepanova D, Campbell DA, Duron SG, O’farrell M, Cai KQ, Klein-Szanto AJ, Gutkind JS, Hoeflich KP, Chernoff J. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012;72:5966–5975. doi: 10.1158/0008-5472.CAN-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yun CY, You ST, Kim JH, Chung JH, Han SB, Shin EY, Kim EG. p21-activated kinase 4 critically regulates melanogenesis via activation of the CREB/MITF and beta-catenin/MITF pathways. J Invest Dermatol. 2015;135:1385–1394. doi: 10.1038/jid.2014.548. [DOI] [PubMed] [Google Scholar]
- 40.Choi SW, Yeon JT, Ryu BJ, Kim KJ, Moon SH, Lee H, Lee MS, Lee SY, Heo JC, Park SJ, Kim SH. Repositioning Potential of PAK4 to Osteoclastic Bone Resorption. J Bone Miner Res. 2015;30:1494–1507. doi: 10.1002/jbmr.2468. [DOI] [PubMed] [Google Scholar]
- 41.Licciulli S, Maksimoska J, Zhou C, Troutman S, Kota S, Liu Q, Duron S, Campbell D, Chernoff J, Field J, Marmorstein R, Kissil JL. FRAX597, a small molecule inhibitor of the p21-activated kinases, inhibits tumorigenesis of NF2-associated schwannomas. J Biol Chem. 2013 doi: 10.1074/jbc.M113.510933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chow HY, Dong B, Duron SG, Campbell DA, Ong CC, Hoeflich KP, Chang LS, Welling DB, Yang ZJ, Chernoff J. Group I Paks as therapeutic targets in NF2-deficient meningioma. Oncotarget. 2015;6:1981–1994. doi: 10.18632/oncotarget.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dolan BM, Duron SG, Campbell DA, Vollrath B, Shankaranarayana Rao BS, Ko HY, Lin GG, Govindarajan A, Choi SY, Tonegawa S. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc Natl Acad Sci U S A. 2013;110:5671–5676. doi: 10.1073/pnas.1219383110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bakker CE, Verheij C, van der Helm R, Oerlemans F, Vermey M, Bygrave A, et al. Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- 45.Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci U S A. 2007;104:11489–11494. doi: 10.1073/pnas.0705003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ong CC, Gierke S, Pitt C, Sagolla M, Cheng CK, Zhou W, Jubb AM, Strickland L, Schmidt M, Duron SG, Campbell DA, Zheng W, Dehdashti S, Shen M, Yang N, Behnke ML, Huang W, Mckew JC, Chernoff J, Forrest WF, Haverty PM, Chin SF, Rakha EA, Green AR, Ellis IO, Caldas C, O’brien T, Friedman LS, Koeppen H, Rudolph J, Hoeflich KP. Small molecule inhibition of group I p21-activated kinases in breast cancer induces apoptosis and potentiates the activity of microtubule stabilizing agents. Breast Cancer Res. 2015;17:59. doi: 10.1186/s13058-015-0564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prudnikova TY, Villamar-Cruz O, Rawat SJ, Cai KQ, Chernoff J. Effects of p21-activated kinase 1 inhibition on 11q13-amplified ovarian cancer cells. Oncogene. 2015 doi: 10.1038/onc.2015.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prudnikova TY, Chernoff J. The Group I Pak inhibitor Frax-1036 sensitizes 11q13-amplified ovarian cancer cells to the cytotoxic effects of Rottlerin. Small GTPases. 2016:1–6. doi: 10.1080/21541248.2016.1213089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ndubaku CO, Crawford JJ, Drobnick J, Aliagas I, Campbell D, Dong P, Dornan LM, Duron S, Epler J, Gazzard L, Heise CE, Hoeflich KP, Jakubiak D, La H, Lee W, Lin B, Lyssikatos JP, Maksimoska J, Marmorstein R, Murray LJ, O’brien T, Oh A, Ramaswamy S, Wang W, Zhao X, Zhong Y, Blackwood E, Rudolph J. Design of Selective PAK1 Inhibitor G-5555: Improving Properties by Employing an Unorthodox Low-pK a Polar Moiety. ACS Med Chem Lett. 2015;6:1241–1246. doi: 10.1021/acsmedchemlett.5b00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rudolph J, Murray LJ, Ndubaku CO, O’brien T, Blackwood E, Wang W, Aliagas I, Gazzard L, Crawford JJ, Drobnick J, Lee W, Zhao X, Hoeflich KP, Favor DA, Dong P, Zhang H, Heise CE, Oh A, Ong CC, La H, Chakravarty P, Chan C, Jakubiak D, Epler J, Ramaswamy S, Vega R, Cain G, Diaz D, Zhong Y. Chemically Diverse Group I p21-Activated Kinase (PAK) Inhibitors Impart Acute Cardiovascular Toxicity with a Narrow Therapeutic Window. J Med Chem. 2016;59:5520–5541. doi: 10.1021/acs.jmedchem.6b00638. [DOI] [PubMed] [Google Scholar]
- 51.Mccoull W, H EJ, Blades K, Chuaqui C, Dowling JE, Ferguson AD, Goldberg FW, Howe N, Jones CR, Kemmitt PD, Lamont G, Varnes JG, Ward RA, Yang B. Optimization of Highly Kinase Selective Bis-anilino Pyrimidine PAK1 Inhibitors. ACS Med Chem Lett. 2016 doi: 10.1021/acsmedchemlett.6b00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mccoull W, Hennessy EJ, Blades K, Box MR, Chuaqui C, Dowling JE, Davies CD, Ferguson AD, Goldberg FW, Howe NJ, Kemmitt PD, Lamont GM, Madden K, Mcwhirter C, Varnes JG, Ward RA, Williams JD, Yang B. Identification and optimisation of 7-azaindole PAK1 inhibitors with improved potency and kinase selectivity. Medchemcomm. 2014;5:1533–1539. doi: 10.1039/c4md00280f. [DOI] [Google Scholar]
- 53.Deacon SW, Beeser A, Fukui JA, Rennefahrt UE, Myers C, Chernoff J, Peterson JR. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem Biol. 2008;15:322–331. doi: 10.1016/j.chembiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viaud J, Peterson JR. An allosteric kinase inhibitor binds the p21-activated kinase autoregulatory domain covalently. Mol Cancer Ther. 2009;8:2559–2565. doi: 10.1158/1535-7163.MCT-09-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Al-Azayzih A, Missaoui WN, Cummings BS, Somanath PR. Liposome-mediated delivery of the p21activated kinase-1 (PAK-1) inhibitor IPA-3 limits prostate tumor growth in vivo. Nanomedicine. 2016;12:1231–1239. doi: 10.1016/j.nano.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim DJ, Choi CK, Lee CS, Park MH, Tian X, Kim ND, Lee KI, Choi JK, Ahn JH, Shin EY, Shin I, Kim EG. Small molecules that allosterically inhibit p21-activated kinase activity by binding to the regulatory p21-binding domain. Exp Mol Med. 2016;48:e229. doi: 10.1038/emm.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karpov AS, Amiri P, Bellamacina C, Bellance MH, Breitenstein W, Daniel D, Denay R, Fabbro D, Fernandez C, Galuba I, Guerro-Lagasse S, Gutmann S, Hinh L, Jahnke W, Klopp J, Lai A, Lindvall MK, Ma S, Mobitz H, Pecchi S, Rummel G, Shoemaker K, Trappe J, Voliva C, Cowan-Jacob SW, Marzinzik AL. Optimization of a Dibenzodiazepine Hit to a Potent and Selective Allosteric PAK1 Inhibitor. ACS Med Chem Lett. 2015;6:776–781. doi: 10.1021/acsmedchemlett.5b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thullberg M, Gad A, Beeser A, Chernoff J, Stromblad S. The kinase-inhibitory domain of p21-activated kinase 1 (PAK1) inhibits cell cycle progression independent of AK1 kinase activity. Oncogene. 2007;26:1820–1828. doi: 10.1038/sj.onc.1209983. [DOI] [PubMed] [Google Scholar]
- 59.Say E, Tay HG, Zhao ZS, Baskaran Y, Li R, Lim L, Manser E. A functional requirement for AK1 binding to the KH(2) domain of the fragile X protein-related FXR1. Mol Cell. 2010;38:236–249. doi: 10.1016/j.molcel.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 60.Davidovic L, Durand N, Khalfallah O, Tabet R, Barbry P, Mari B, Sacconi S, Moine H, Bardoni B. A novel role for the RNA-binding protein FXR1P in myoblasts cell-cycle progression by modulating p21/Cdkn1a/Cip1/Waf1 mRNA stability. PLoS Genet. 2013;9:e1003367. doi: 10.1371/journal.pgen.1003367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hashimoto H, Sudo T, Maruta H, Nishimura R. The direct AK1 inhibitor, TAT- AK18 blocks preferentially the growth of human ovarian cancer cell lines in which AK1 is abnormally activated by autophosphorylation at Thr 423. Drug Discov Ther. 2010;4:1–4. [PubMed] [Google Scholar]
- 62.Kiosses WB, Hood J, Yang S, Gerritsen ME, Cheresh DA, Alderson N, Schwartz MA. A dominant-negative p65 PAK peptide inhibits angiogenesis. Circ Res. 2002;90:697–702. doi: 10.1161/01.res.0000014227.76102.5d. [DOI] [PubMed] [Google Scholar]
- 63.Orr AW, Stockton R, Simmers MB, Sanders JM, Sarembock IJ, Blackman BR, Schwartz MA. Matrix-specific p21-activated kinase activation regulates vascular permeability in atherogenesis. J Cell Biol. 2007;176:719–727. doi: 10.1083/jcb.200609008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang Y, Marwaha S, Rutkowski JL, Tennekoon GI, Phillips PC, Field J. A role for Pak protein kinases in Schwann cell transformation. Proc Natl Acad Sci U S A. 1998;95:5139–5144. doi: 10.1073/pnas.95.9.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Molosh AI, Johnson PL, Spence JP, Arendt D, Federici LM, Bernabe C, Janasik SP, Segu ZM, Khanna R, Goswami C, Zhu W, Park SJ, Li L, Mechref YS, Clapp DW, Shekhar A. Social learning and amygdala disruptions in Nf1 mice are rescued by blocking p21-activated kinase. Nat Neurosci. 2014;17:1583–1590. doi: 10.1038/nn.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Radu M, Lyle K, Hoeflich KP, Villamar-Cruz O, Koeppen H, Chernoff J. p21 -Activated Kinase 2 Regulates Endothelial Development and Function through the Bmk1/Erk5 Pathway. Mol Cell Biol. 2015;35:3990–4005. doi: 10.1128/MCB.00630-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu W, Zi M, Naumann R, Ulm S, Jin J, Taglieri DM, Prehar S, Gui J, Tsui H, Xiao RP, Neyses L, Solaro RJ, Ke Y, Cartwright EJ, Lei M, Wang X. Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation. 2011;124:2702–2715. doi: 10.1161/CIRCULATIONAHA.111.048785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shrestha Y, Schafer EJ, Boehm JS, Thomas SR, He F, Du J, Wang S, Barretina J, Weir BA, Zhao JJ, Polyak K, Golub TR, Beroukhim R, Hahn WC. PAK1 is a breast cancer oncogene that coordinately activates MAPK and MET signaling. Oncogene. 2012;31:3397–3408. doi: 10.1038/onc.2011.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, Mccusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, Sznol M, Kluger HM, Brash DE, Stern DF, Materin MA, Lo RS, Mane S, Ma S, Kidd KK, Hayward NK, Lifton RP, Schlessinger J, Boggon TJ, Halaban R. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nature genetics. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]