

Abstract
The human preterm brain is particularly susceptible to cerebral white matter injury (WMI) that disrupts the normal progression of developmental myelination. Advances in the care of preterm infants have resulted in a sustained reduction in the severity of WMI that has shifted from more severe focal necrotic lesions to milder diffuse WMI. Nevertheless, WMI remains a global health problem and the most common cause of chronic neurological morbidity from cerebral palsy and diverse neurobehavioral disabilities. Diffuse WMI involves maturation dependent vulnerability of the oligodendrocyte (OL) lineage with selective degeneration of late oligodendrocyte progenitors (preOLs) triggered by oxidative stress and other insults. The magnitude and distribution of diffuse WMI is related to both the timing of appearance and regional distribution of susceptible preOLs. Diffuse WMI disrupts the normal progression of OL lineage maturation and myelination through aberrant mechanisms of regeneration and repair. PreOL degeneration is accompanied by early robust proliferation of OL progenitors that regenerates and augments the preOL pool available to generate myelinating OLs. However, newly generated preOLs fail to differentiate and initiate myelination along their normal developmental trajectory despite the presence of numerous intact-appearing axons. Disrupted preOL maturation is accompanied by diffuse gliosis and disturbances in the composition of the extracellular matrix and is mediated in part by inhibitory factors derived from reactive astrocytes. Signaling pathways implicated in disrupted myelination include those mediated by Notch, WNT-beta catenin and hyaluronan. Hence, there exists a potentially broad but still poorly defined developmental window for interventions to promote white matter repair and myelination and potentially reverse the widespread distrubances in cerebral gray matter growth that accompany WMI.
Significance of preterm WMI as a global health problem
Preterm birth continues to be a major global health problem associated with considerable mortality and morbidity. The world health organization estimates that about 15 million preterm infants (1 in 10 live births) are born less than 37 weeks post-conceptional age world wide each year. Preterm birth is the leading cause of death of babies less than five years old [114]. Rates of prematurity vary considerably even among countries with considerable resources and range from 5-18%. The rate of prematurity is markedly higher in the United States, for example, compared to many countries in Europe and reached a rate of 9.6% in 2015 [36]. In the U.S. alone each year, very low birth weight (VLBW) infants comprise about 1.5% of the 4 million live births. Hence, the world wide social and economic burden is considerable and relates in large part to the chronic sequelae associated with WMI. The average lifetime costs for each person with cerebral palsy (CP) is about 1 million dollars in the United States [1].
Although ongoing advances in the care of preterm infants have resulted in significant improvements in survival of very low birth weight (VLBW) infants (< 1.5 kg), improved survival has been accompanied by a persistent increase in pre-term survivors with chronic neurological disabilities [183]. Permanent motor impairment (i.e., CP) is reported in ~10% of preterm survivors and ranges from mild to profound spastic motor deficits [28, 80, 113, 128, 131, 179] Neurodevelopment is further comprised in 25-50% of preterm survivors by a broad spectrum of cognitive, visual, social-behavioral, attention and learning disabilities [7, 72, 95, 112, 166].
Cerebral white matter injury (WMI) is the major form of brain injury in survivors of premature birth [180]. The period of highest risk for WMI is ~23-32 weeks post-conceptional age. In the first months of life, MRI-defined WMI in preterm survivors manifests as abnormal movements that are predictive of CP [42, 169, 170]. The impact of WMI can be appreciated from one large population based study of children with CP. Perinatal WMI, including the necrotic lesions of periventricular leukomalacia (PVL), was the most common finding, seen in almost half (42.5%) of affected children [27]. Moreover, premature birth alone is associated with a greater risk for reduction in both cerebral white and gray matter volumes, which are associated with adverse cognitive development [2, 8, 48, 104, 117, 141, 156, 167].
Although largely associated with premature birth, WMI may also commonly occur some groups of full term infants where in utero insults appear to coincide with a susceptible period in white matter development prior to the onset of cerebral myelination [105, 107, 122, 140]. Such insults appear to occur, for example, in association with chronic placental insufficiency [119, 149, 191] or cyanotic congenital heart disease (CHD) [171] where a prolonged period of in utero ischemia or reduced cerebral oxygenation may contribute to antenatal WMI. Although the mechanism of WMI is unknown, WMI can precede surgical repair of heart lesions [126], which suggests that CHD itself may be a risk factor for WMI. This review focuses on the pathogenesis of preterm human WMI and will address its relevance to develop new strategies to define the cellular and molecular pathogenesis of acute and chronic WMI.
Spectrum of focal and diffuse human white matter injury
During human cerebral development, acquired WMI is a common lesion that can vary considerably in severity. Three major forms of pathology comprise the spectrum of human WMI: focal cystic necrosis, focal microscopic necrosis and diffuse non-necrotic lesions. The relative representation of these three types of pathology in a given patient population is related to a wide variety of clinical factors that affect the antenatal or postnatal hemodynamic stability and inflammatory status of the preterm fetus or infant. These include postnatal infections, hypoxia-ischemia, hypoxemia, hypocarbia, metabolic acidosis and hypoglycemia [19]. During intensive care, preterm infants are also often exposed routinely to multiple painful and stressful procedures that are associated with disturbances in postnatal growth [178] and brain maturation that involve both white and gray matter structures [32, 165].
Necrotic WMI evolves to macroscopic or microscopic focal lesions
The most severe form of WMI involves large regions of cystic necrosis that typically range from about 1 to 6 mm in diameter. These large foci of necrosis are the hallmark of PVL, which most commonly localizes deep in the white matter adjacent to the ventricular wall [142]. However, in its most severe forms, PVL can extend into the centrum semiovale and even the subcortical white matter (Fig. 1A, B). PVL has a particular predilection for the white matter anterior to the frontal horn, the external angles of the lateral ventricles at the level of the foramen of Monro and the lateral aspects of the trigone and occipital horns adjacent to the optic radiations. Severe white matter necrosis has decreased markedly in recent years. In several series, focal cystic lesions were detected by MRI in less than 5% of cases (Fig. 1C, D) [43, 78, 83, 92, 118, 130]. Both macroscopic and microscopic forms of white matter necrosis were found to decrease by ~10-fold in contemporary cohorts relative to retrospective cases from earlier decades [33].
Figure 1. Severe WMI results in focal macroscopic or microscopic necrosis.

(A) Gross pathology demonstrates the typical features of severe cystic necrotic WMI in an infant who died of complications of prematurity. Note the large foci of severe cystic necrosis (arrowheads) in frontal (upper specimen) and parietal (lower specimens) periventricular white matter. (B) Histological analysis of the frontal lesion (stained with hematoxylin and eosin) shows a large focus of necrosis (arrowheads) adjacent to the external angle of lateral ventricle. The inset shows a high power detail of the edge of the lesion (arrows) where marked rarefaction of the tissue can be appreciated adjacent to a region of gliosis at lower left. (C) Appearance of cystic necrotic WMI on MRI. Images from a preterm infant born at 33 weeks gestational age and scanned at 5 weeks of age (38 weeks adjusted gestational age). Small areas of cavitation (arrowheads) are appreciated as hypointensity on the sagittal T1 weighted image, and as hyperintensity on the axial T2 weighted image, (D, E) Low power image of the typical sparse and focal distribution of microscopic necrosis in the periventricular white matter from a human autopsy brain at 32 weeks post conceptional age. The microcyst was visualized with β-amyloid precursor protein, a marker of degenerating axons. (F, G) Panels F and G provide higher power detail of the degenerating axons in the microcyst seen in the box in E and demonstrate that axonal degeneration was present both within the core and periphery of the microcyst. Scale bars: E, 500 μm; F, 100 μm; G, 25 μm. (Images in A and B, Courtesy of Dr. Marjorie Grafe, Oregon Health & Science University. C, D, Courtesy of Dr. Ken Poskitt, Children’s and Women’s Hospital, University of British Columbia; E-F adapted from Back and Miller, 2014; [19]).
Cystic necrosis is initiated as coagulation necrosis that rapidly within hours leads to death of all cellular elements (glia, axons, blood vessels and neural progenitors), because of severe energy failure. Major features include nuclear pyknosis, cytotoxic edema with tissue vacuolation and focal axon swellings (axonal spheroids) that disrupt axonal trafficking. The latter can be appreciated with staining for neurofilament protein [18], beta-amyolid precursor protein [33] and the fractin antibody [33, 86]. Advanced axonal necrosis may be associated with mineralization of dystrophic axons that stain for calcium or iron. Necrosis progresses to the focal infiltration and reaction of macrophages and microglia, including lipid-laden macrophages visualized with markers that include CD45, CD68, Iba1 and the ricin and tomato lectins [16, 18, 30, 33]. The process of microglial reaction coincides with an early and progressive astrocyte reaction that delineates the borders of focal necrosis. These astrocytes can be visualized with GFAP, vimentin and the hyaluronan receptor CD44 [18, 33]. When severe necrosis progresses to tissue liquefaction and cavitation, large and sometimes bizarre-appearing astroglial somata and processes form a pronounced glial scar that is enriched in extracellular matrix molecules, notably hyaluronan, and stains palely for myelin markers, such as myelin basic protein (MBP) [17, 33]. When the cavitation is large and diffuse, it may be associated with a pronounced reduction in white matter volume, thinning of the corpus callosum and ventriculomegaly.
Although large necrotic lesions have decreased markedly, discrete small foci of microscopic necrosis (microcysts) less than 1 mm in diameter continue to occur commonly [142]. Similar to focal cystic necrosis, microcysts involve tissue destruction and are enriched in cellular debris, degenerating axons and phagocytic macrophages (Fig. 1D-F). Due to their smaller diameter, microcysts are not routinely detected on clinical MRI scans at field strengths of 3 Tesla, but can be detected experimentally at higher magnetic field strengths of 12 Tesla [150]. A study of human archival and contemporary autopsy cases found that microcysts occurred in at least 30% of cases, but comprised only ~1-5% of the total burden of WMI [33]. Hence, microscopic necrosis occurs with high incidence, but the burden is typically low. The clinical importance of these small necrotic lesions remains an inaccessible question, since microcysts are not readily visualized with clinical MRI. Hence, microcysts may be clinically silent lesions or a significant contributor to motor or cognitive disabilities, depending upon the number and distribution within functionally significant regions of white matter. The distribution of microcysts is similar to that of cystic necrosis, which suggests a similar vascular ischemic origin.
The basis for the preferential localization of necrotic WMI to deep cerebral white matter remains speculative. There are apparent vascular end and border zones in the preterm periventricular white matter that may predispose to more severe ischemia [91, 134, 137, 173]. Multiple studies in human [6, 75, 143, 145–147] and preterm fetal sheep [73, 151, 172] found that cerebral white matter has an intrinsically lower basal cerebral blood flow compared to other gray matter regions, which suggested that the susceptibility of the preterm infant to WMI is related to a heightened propensity for ischemia. Preterm infants studied by near-infrared spectroscopy displayed disturbances in cerebral cortical flow consistent with disturbances in cerebral auto-regulation and a heightened risk for ischemia-induced WMI [168, 175]. Measurements of global cortical or white matter flow in human lack, however, the regional sensitivity to quantify flow in discrete regions of deep cerebral white matter where necrosis preferentially localizes. Studies in preterm fetal sheep achieved spatially resolved quantitative measurements of fetal CBF in utero that were co-registered with histologically defined analyses of WMI. These studies found that there were no pathologically significant gradients of flow between fetal cerebral cortex and deep cerebral white matter during either ischemia or reperfusion [125, 151]. Even during moderately severe global ischemia, some regions of white matter were relatively spared, whereas other neighboring regions sustained significantly greater cell death. Moreover, histologically confirmed WMI did not localize to regions susceptible to greater ischemia. Nor did less vulnerable regions of white matter have greater blood flow during ischemia. Ischemia thus appeared to be necessary but not sufficient to generate necrotic or diffuse WMI. In preterm fetal sheep, the severity of WMI was significantly associated with the magnitude of hypotension, hypoxemia and hypoglycemia [152]. Collectively, these findings suggest that the propensity for more severe WMI is related to a combination of vascular anatomic immaturity, disturbances in cerebrovascular autoregulation and a variety of metabolic factors that may predispose to more severe energy failure in regions of focal white matter necrosis.
Diffuse WMI is the predominant lesion in most preterm survivors
Diffuse WMI is the most common form of injury in contemporary cohorts of preterm newborns [33]. It may exist in isolation or be associated with foci of necrosis [142]. Because of its diffuse nature and the lack of discrete borders for many large regions of the centrum semiovale, it has been difficult to estimate the boundaries of diffuse lesions. We provided an estimate by adapting stereological principles to quantify the area fraction of GFAP-labeled astrocytes and Iba1-labeled microglia within putative lesions defined independently by histopathological review of hematoxylin and eosin (H & E) stained sections [33]. Unexpectedly, when the burden of astrogliosis and microgliosis was compared within putative lesions and the peri-lesion surround, the burden of reactive glia was found to be significantly elevated within the peri-lesion areas that were predicted to be normal-appearing on the basis of review of H & E staining.
Given that quantitative morphometric approaches are required to estimate the magnitude and distribution of diffuse reactive gliosis, it is not surprising that clinical imaging modalities appear to underestimate these lesions. Cranial ultrasound is the preferred bed-side imaging technique for diagnosing necrotic WMI, but it is less reliable for detection of diffuse WMI [92, 118, 130]. MRI is the preferred method to visualize diffuse WMI. On diagnostic MRI scans, WMI is visualized as either discrete focal or more diffuse areas of MR signal abnormalities [39]. The spectrum of non-cystic WMI- or “punctate” lesions, has recently been described as linear or cluster lesions with different evolutions over time [103]. There is, however, unexplained variability in the nature of lesions detected at different centers, which may reflect differences in management, clinical acuity or modes of detection. In particular, MRI appears to underestimate early diffuse lesions. Disparities between clinical presentation and the distribution of lesions on MRI has been widely noted. Experimental studies that co-registered MRI and histopathology data demonstrated that early WMI is particularly well visualized at high magnetic field strength (12 T) [150], which suggests that currently employed clinical MRI field strengths of 1.5-3T may be a limiting factor to detect both diffuse WMI, as well as microcysts.
It should further be emphasized that preterm survivors frequently display diffuse abnormalities in gray and white matter maturation related to preterm birth that may be more common than focal or diffuse WMI [39] but are not well-defined pathologically. These diffuse abnormalities are apparent on MRI as enlarged subarachnoid spaces, a reduction in white matter, ventriculomegaly and impaired gyral development [59, 163]. However, many premature newborns do not have these dramatic abnormalities, and up to 20% with adverse outcomes do not have significant qualitative abnormalities on MRI [31, 131].
Diffuse WMI involves selective vulnerability of late oligodendrocyte (OL) progenitors
In contrast to necrotic injury, diffuse WMI is defined by selective degeneration of pre-oligodendrocytes (preOLs), whereas axons are mostly spared except in necrotic foci [86, 153]. Human preOLs are the predominant population of pre-myelinating OL progenitors in preterm human cerebral white matter [16]. They are particularly susceptible to significant oxidative damage of a magnitude comparable to cerebral cortical hypoxia-ischemic lesions in term infants [18]. The susceptibility to preterm WMI peaks at ~23-32 weeks post-conceptional age. During this protracted window in human pre-myelinating white matter development, human OL lineage cells develop according to a well-established sequence of maturational events, defined by stage-specific antibodies specific for sequentially expressed OL cell-surface and myelin-specific epitopes [16, 17, 96, 97].
Human preOLs are pre-myelinating late OL progenitors that are non-migratory but mitotically active (Fig. 2A-C). They label with the O4 but not the O1 antibody. They derive from a population of pre-myelinating, migratory and mitotically active early OL progenitors that label for the NG2 proteoglycan and the platelet-derived growth factor-alpha receptor (PDGFRα). Although both of these OL progenitor populations are typically subsumed under the designation “OL progenitors cells” or OPCs when studied in vitro, these two successive OL stages can be differentiated, for example, by the lack of O4 labeling of the PDGFRα-labeled OL progenitors. Moreover, the OL progenitors are highly resistant to hypoxia-ischemia, whereas preOLs are very susceptible to degeneration [14]. Premyelinating immature OLs (O4+O1+) (Fig. 2D) and mature myelinating OLs (Fig. 2E, F) also display greater resistance to cell death from hypoxia ischemia or kainate toxicity in vitro [14, 155].
Figure 2. The differentiation of human oligodendrocyte (OL) progenitors and myelination progresses according to a well-defined lineage defined by specific markers.

(A) Diagram depicting the maturation of the OL lineage. The four principal stages of the OL lineage are depicted together with their corresponding morphological features and potential for proliferation, migration or myelination. Each stage is uniquely defined by a combination of marker genes or antibodies. A2B5, O4, O1 refer to mouse monoclonal antibodies. Olig 2 and Sox10 are genes that are highly enriched in premyelinating OL progenitors. Olig 2 is also expressed at later stages in the OL lineage. Abbreviations: CNP (CNPase), 2′:3′-cyclic nucleotide-3′-phosphodiesterase; GalC, galactocerebroside; MAG, myelin associated glycoprotein; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; NG2, chondroitin sulfate proteoglycan 4; PDGFRα, platelet-derived growth factor-alpha; PLP, proteolipid protein. (B, C) Human preOLs (green) visualized with the O4 antibody. In C, note the GFAP-labeled astrocyte (red) with distinctly different morphology from the preOLs (green). (D) Human immature OLs visualized with the O4 antibody (arrowheads), which appear much more ramified compared to adjacent preOLs (arrows). (E) An early myelinating human immature OL (green; O1 antibody-labeled) extends several fine processes that make apparent contacts with individual axons visualized with the pan-axonal neurofilament protein marker, SMI312. (F) A 3-dimensional reconstruction of an axon in the early stages of myelination that was visualized in the optic radiation from a case at 30-weeks post-conceptional age. An early myelinating OL, visualized with the O4 antibody, generates early loose axonal contacts (yellow) and myelin sheath wrappings (green) that spiral around an axon visualized with SMI312 (red). Two views of the axon are shown. The portion of the axon visualized at lower right demonstrates that the myelin sheath appears to be laid down in a segmental fashion (arrowheads). A portion of the O4-labeled OL soma is seen at lower left (s). The images in E and F were adapted from Back et. al., 2002 [17].
An extensive human and experimental literature supports that preOLs are particularly susceptible to cell death, whereas other OL lineage stages are markedly more resistant [14, 18, 158]. Immature neurons in the preterm human cerebral cortex (Fig. 3A) and subcortical gray matter are also very resistant to oxidative stress as defined by cell death markers and quantitative measurements of F4-neuroprostanes, a sensitive and stable marker of oxidative damage to neuronal membranes [18]. By contrast, the preterm cerebral white matter sustains widespread preOL death under conditions that do not trigger significant degeneration of cortical neurons (Fig. 3B,C). In response to moderately severe global cerebral ischemia, immature neurons in the cerebral cortex and caudate nucleus of preterm fetal sheep were similarly found to be strikingly resistant to cell death in contrast to preOLs [45, 124].
Figure 3. Acute human diffuse WMI is accompanied by selective preOL death.
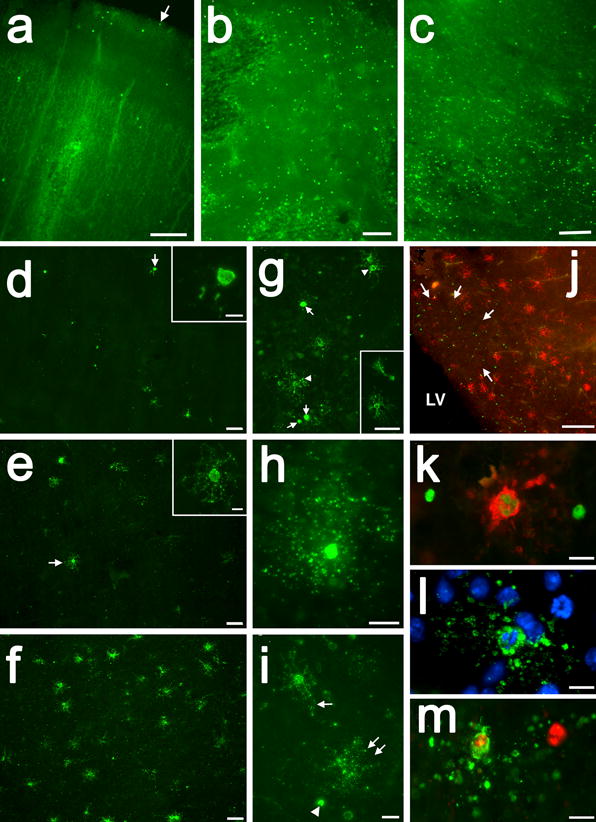
(A-C) Cell death is visualized in A-C by TUNEL staining, which detects DNA fragmentation of apoptotic and necrotic cells. A, Rare cell death was seen in the overlying cerebral cortex, shown here from analysis of a preterm infant with diffuse WMI at autopsy. The arrow marks the pial surface. (B, C) The periventricular white matter from the same case showed extensive cell death that was confirmed below to be due to preOL degeneration. The individual degenerating cells appear as bright green. (D-F) Typical example of a gradient of early WMI that preferentially localized to deeper areas of cerebral white matter in D and E with more normal appearing white matter in F. Note the marked reduction in staining for preOLs in D and E compared to F. Also note the severely damaged cell remnants in D (arrow; detail in inset) and the paucity of normal appearing preOLs in E (arrow; detail in inset). (G) Typical appearance of shrunken degenerating preOLs (arrows) scattered among more normal-appearing cells (arrowheads), also shown in the inset. (H, I) Typical morphology of a degenerating preOLs with a halo of fragmented degenerating processes. (J) Numeous preOLs (red) also seen adjacent to but not within a focal lesion (arrows) in the subventricular zone deep to the periventricular white matter. Note the many degenerating TUNEL-labeled cells (green). (K-M) Examples of apoptotic preOLs with a typical fragmented condensed nucleus. Adapted from reference: [18] Scale bars: A, 200 μm; B, 100 μm; C, 200 μm; D-F, 50 μm; Inset in D and E, 10 μm; G and inset in G, 50 μm; H, 10μm; I, 20 μm; J, 200 μm; K-M, 10μm.
When preOLs undergo apparent necrosis (Fig. 3D-J), they display a progressive series of morphological changes that include focal swelling of processes (Fig. 3E), followed by fragmentation, which appears as a halo around the shrunken cell body (Fig. 3G-I). With the collapse of membrane integrity, membrane associated markers display intracellular localization (Fig. 3 D H), as can be observed with staining with the O4 antibody [18]. A striking example of terminal preOL degeneration is also shown, for example, from a PVL case in figure 8A, as reported by Billiards et. al., 2008 [29].
Caspase-mediated mechanisms of apoptosis (Fig. 3K-M) also may contribute to acute preOL death from oxidative stress in human WMI [18] and hypoxia-ischemia in experimental models [14, 35, 38, 138]. The magnitude of caspase-activation appears to be related to the severity of the hypoxic-ischemic insult. In response to moderate perinatal hypoxia-ischemia, most preOLs in rodents degenerated acutely by apparent necrosis that did not involve caspase-3 activation [14, 158]. By contrast, in the preterm sheep fetus, preOL degeneration more commonly involved activation of caspase-3, consistent with a milder insult [151]. Similar observations were made in late-gestation sheep [35, 38]. Hence, a less severe insult may result in a combination of apoptosis and necrosis, whereas more severe insults mostly triggers necrosis [40, 84]. The relative extent of apoptotic versus necrotic preOL degeneration is typically difficult to establish due to the lack of histological markers of necrosis. Ultrastructural studies from neonatal rodents in vitro and in vivo demonstrated that oxidative stress or moderate hypoxia-ischemia caused a form of oligodendroglial degeneration that displayed mixed features of necrosis and apoptosis [12, 13].
Maturation-dependent vulnerability of the human OL lineage to oxidative stress an hypoxia-ischemia
The major period of vulnerability for human WMI coincides with a developmental window when the white matter is at increased risk for oxidative stress from hypoxia-ischemia or maternal-fetal infection [68, 81]. The high risk period for WMI thus precedes the onset of myelination [16, 148]. As noted above, the high risk period occurs when the white matter is populated primarily by preOLs that are the principal target for cell death in white matter lesions. The decline in risk for WMI coincides with the onset of a wave of differentiation of preOLs to immature OLs that initiate myelination [17]. PreOLs are particularly susceptible to maturation-dependent mechanisms of free radical-mediated injury, whereas earlier and later OL lineage stages are much more resistant to intrinsic and extrinsic sources of oxidative stress both in vitro and in vivo [13, 14, 25, 67, 70, 111].
A role for oxidative stress in preOL degeneration is supported by in vitro, experimental and human neuropathology studies. During fetal brain development, the cerebral white matter is already enriched in membrane lipids that are readily oxidized [85, 98, 133]. The preterm white matter may be more vulnerable to oxidative damage than at term, because of a delay in expression of the antioxidant enzymes, superoxide dismutases-1 and -2, catalase, and glutathione peroxidase [25, 26, 69]. However, expression data in rat and human have not been confirmed with enzyme activity data. A fetal rabbit model of reperfusion-reoxygenation demonstrated increased superoxide generation with immediate ischemia-reperfusion that was associated with a doubling in the rate of hypertonia observed in neonatal pups delivered at term. Notably, there was a signifcant reduction in hypertonia in response to early delivery of a superoxide dismutase mimic or anti-oxidants [55].
Although controversy exists regarding the role of reactive nitrogen species in the pathogenesis of early WMI, human preterm white matter is especially prone to lipid peroxidation-mediated injury [18, 87]. Both the biochemical and immunohistochemical detection of protein, lipid and DNA adducts generated by reaction oxygen and nitrogen species (e.g., protein carbonyls, 4-hydroxynonenal and 8-oxo-deoxyguanosine) is unreliable in human preterm WMI [18]. Sensitive detection of the aldehydes formed from lipid hydroperoxides in glia is specifically achieved by quantification of F2-isoprostanes, which are stable adducts in postmortem human tissue [135]. F2-isoprostanes were significantly elevated in histopathologically confirmed cases of early WMI [18] and were similar to the levels detected in the cerebral cortex where hypoxic ischemic encephalopathy was diagnosed. This oxidative damage was accompanied by a pronounced increase in degenerating preOLs in the periventricular white matter. F4-neuroprostanes, a specific marker of oxidative injury to neuronal elements, were not detected in white matter or cerebral cortex, consistent with the notion that neurons and axons were spared. Hence, oxidative damage selectively triggers cell death in preterm human white matter. Moreover, during the early phases of WMI, the magnitude of oxidative damage is comparable to that sustained in the cerebral cortex after severe perinatal asphyxia.
As noted above, it was shown experimentally that ischemia is necessary but not sufficient to cause WMI. Even under conditions of moderately severe ischemia, some regions of white matter may be relatively spared. Consistent with observations that preOLs are selectively vulnerable to oxidative stress, the timing of appearance [34] and spatial distribution of susceptible preOLs [151] has provided an explanation for the magnitude and distribution of acute hypoxic-ischemic WMI in several experimental models of WMI. The white matter of two day old preterm-equivalent rat pups, for example, displays a much greater susceptibility to hypoxia-ischemia than in 7 day-old near-term equivalent animals [14]. A major developmental difference at these two ages is the extent of differentiation of the OL lineage. At postnatal day two, the rat white matter contains predominantly preOLs and, thus, resembles human cerebral white matter during the high-risk period for WMI [44]. By day 5, in three different strains of rat, the white matter is, in fact, populated mainly by a more differentiated population of immature OLs, and thus resembles near-term human [46]. PreOLs are very susceptible to hypoxia-ischemia, whereas earlier and later OL lineage stages are much more resistant [14, 151, 158]. The enhanced susceptibility of preOLs in vivo is, thus, a stage-specific property that is independent of the developmental age of the animal or the location of these cells in cerebral white matter.
As the white matter matures, it displays greater developmental resistance to hypoxia-ischemia that is related to the onset of preOL differentiation. As noted above, premyelinating immature OLs and mature myelinating OLs display reduced susceptibility to oxidative stress and hypoxia-ischemia. Similar to reactive astrocytes, immature OLs in the white matter often display a robust reactive response to cerebral injury, which is common after hypoxia-ischemia at postnatal day 7 in the rat [14]. This response coincides with gray matter injury and secondary injury to axons in the adjacent callosal white matter. Cerebral hypoxic-ischemic injury in rodents thus differs markedly from human due to substantial necrotic neuro-axonal injury. Gray matter necrosis uncommonly occurs in association with human WMI unless the latter injury also involves necrosis [22, 47, 88, 120, 142]. In preterm fetal sheep, more selective WMI is generated by global ischemia and severe necrotic WMI is typically observed under conditions of prolonged global cerebral ischemia [151]. When selective WMI is generated, the regional distribution of WMI is closely associated with the distribution of vulnerable preOLs and other neural cell types (astrocytes, microglia and axons) are markedly more resistant to injury.
The notion that preOLs define the susceptibility to WMI is further supported by studies that asked whether WMI would occur in fetal animals that lacked preOLs in cerebral white matter tracts. In a fetal rabbit model of placental insufficiency, reduced susceptibility of the white matter to hypoxia-ischemia at embyronic day 22 (E22) was accounted for by a paucity of preOLs. During fetal rabbit white matter development, a coordinate increase in the generation of preOLs occurs in most white matter tracts around E25. By E25, a pronounced increase in preOLs coincided with a marked increase in WMI and significant degeneration of susceptible preOLs. Taken together, these findings support that perturbations in cerebral blood flow are necessary but not sufficient for WMI. The developmental predilection for WMI is related to both the timing of appearance and regional distribution of susceptible preOLs. These findings predict that some near-term infants with delayed OL differentiation and myelination may also be more susceptible to WMI. As noted above, near term and term infants with congenital heart disease are at high risk for WMI [126]. Several clinical and MRI studies support the notion that this susceptibility relates to a delay in brain maturation [41, 51, 109, 110, 132]. Hence, the targeted death of preOLs from hypoxia-ischemia or inflammatory mediators may contribute to the pathogenesis of acute human WMI across a broad range of gestational ages and regions of white matter.
Mechanisms of myelination disturbances in chronic focal or diffuse WMI Myelin loss in chronic focal WMI involves axon degeneration
Disturbances in OL lineage maturation and myelination are central features that distinguish chronic WMI in human preterm survivors from other forms of neonatal cerebral injury. Myelination disturbances arise from two related mechanisms (Fig. 4A, upper pathway). The first involves axonal degeneration in focal WMI. Early histopathological descriptions of myelination disturbances included many cases where focal cystic necrotic WMI (PVL) with axonal degeneration was associated with diffuse white matter astrogliosis [29, 90].
Figure 4. Chronic WMI displays myelination disturbances related to arrested preOL maturation.

(A) Distinctly different pathogenetic mechanisms mediate abnormal myelination in focal necrotic lesions (periventricular leukomalacia (PVL); upper pathway) versus lesions with diffuse WMI (lower pathway). When more severe, hypoxia–ischemia triggers white matter necrosis (upper pathway) with pancellular degeneration that depletes the white matter of glia and axons. Severe necrosis results in cystic PVL, whereas milder necrosis results in microcysts. Milder hypoxia–ischemia (lower pathway) selectively triggers early preOL death, but preOLs are rapidly regenerated in chronic lesions enriched in reactive astrocytes that contribute to a block in preOL differentiation to myelinating OLs. Myelination failure in diffuse WMI thus results from preOL arrest rather than axonal degeneration, as occurs with white matter necrosis. Note that the lower pathway is the dominant one for many contemporary preterm survivors, whereas the minor upper pathway reflects the declining burden of white matter necrosis. (B) Typical appearance of normal early myelination in neonatal rodents. Axons are visualized in red and early myelination of axons is in green. (C) Arrested maturation of preOLs in a chronic white matter lesion arising from hypoxia-ischemia (HI) where numerous preOLs (green; arrowheads) are seen, but the axons (red; arrows) are diffusely unmyelinated. (D) Normal early myelination (O1-antibody; green) in control subcortical white matter (corpus callosum/external capsule) at postnatal day10 (1 week after HI). There are low levels of GFAP-labeled astrocytes (red) in the white matter, adjacent cortex (CTX) and caudate putamen (CPu). (E) Absence of myelin (green) in the contralateral post-ischemic lesion coincided with a diffuse glial scar that stained diffusely for GFAP-labeled astrocytes (red). (F) Early myelination in control white matter at P10 with sheaths (yellow) double-labeled for O4 and O1 antibodies. (G) Absence of myelin in the contralateral lesion coincided with clusters of preOLs (O4+O1-) in maturation arrest (red; arrowheads). Such dense clusters of preOLs are not normally seen in control white matter and are consistent with the pronounced proliferative state that is triggered in response to injury. Apparent oligodendrocytes (yellow; arrows; O4+O1+) are rarely seen in the lesions. Panels A-C adapted from Back and Miller, 2014 [19]. Panels D-G adapted from Back and Rosenberg, 2014 [20]. Scale bars: B, C 100μm.
Focal necrotic lesions often contain dystrophic axons and axonal spheroids, which degenerate during the early phase of coagulative necrosis [22, 47, 88, 120]. Small premyelinated axons are particularly susceptible to glutamate-mediated excitotoxicity at sites of contact with OL processes and involve both N-methyl-D-aspartic acid (NMDA) and non-NMDA glutamate receptors [4, 66]. Axons also display maturation-dependent vulnerability to oxidative stress and hypoxia-ischemia [5]. Larger caliber axons, which are preparing to myelinate, are particularly susceptible to injury via axolemma-associated voltage-gated calcium channels, in contrast to smaller caliber unmyelinated axons, which are more resistant. More diffuse axonal dysfunction (e.g. trafficking disturbances) that does not involve overt degeneration remains speculative and difficult to study due to lack of useful histopathological markers.
The second mechanism of myelination disturbances (Fig. 4A, lower pathway) involves preOL death in diffuse WMI, which disrupts OL lineage maturation and myelination (see below). Primary axonal injury does not appear to be a major feature of diffuse human WMI during the pre-myelinating phase of white matter development. Rather, diffuse preOL death predominates but may be accompanied by axonal degeneration in sporadic focal necrotic lesions. Focal axonal loss was found to be a minor component of WMI in both sheep and human studies, where they comprised about 5% of the total burden of WMI [33, 150]. During the chronic phase of diffuse WMI in preterm fetal sheep, no significant axonal degeneration, axonal loss or shift in the distribution of axon calibers was observed by light [150] or quantitative electron microscopy [153] studies.
In contemporary cohorts of patients, MRI more commonly defines focal or diffuse signal abnormalities that do not involve large cystic lesions. Although these diffuse MRI abnormalities are often attributed to abnormal “myelination,” these studies lack histopathological correlation and the cellular basis for the white matter signal abnormalities is unclear. For example, in subjects with these milder forms of diffuse WMI, fractional anisotropy (FA) did not increase in central white matter tracts relative to normal infants [89, 127, 129], and this was attributed to abnormal myelination. However, findings from a preterm fetal rabbit model of global cerebral ischemia [49, 53, 54] identified that the most rapid rise in FA normally occurs prior to myelination at a time when pre-myelinating immature OLs increase in number in central white matter tracts and axons show maturational changes [56]. These experimental findings suggest that MRI-defined WMI in contemporary cohorts of patients may be related to disturbances in OL lineage maturation rather than axonal degeneration, but histopathological confirmation in human is needed.
Myelination disturbances in chronic diffuse WMI are related to aberrant preOL maturation
As discussed above, focal necrotic WMI results in myelination failure, which is likely to be permanent, because it typically involves necrotic degeneration of all cell types in the lesion. The pathogenesis of diffuse chronic WMI is distinct from the process that leads to focal necrosis and may be potentially reversible. Diffuse WMI involves two major overlapping events that disrupt the normal progression of preOL differentiation and myelination in developing white matter. The initial response to injury involves an early proliferative response to WMI characterized by a significant increase in OL progenitors in subacute lesions. This plasticity response results in expansion of the pool of OL progenitors that are available to generate new preOLs to replace those that degenerate during acute WMI. The second overlapping event occurs over days to weeks after WMI and involves a disruption in the normal differentiation of newly generated preOLs to myelinating OLs. Despite the pronounced selective degeneration of preOLs in early diffuse WMI (see above), disturbances in myelination in chronic lesions do not appear to arise from a loss of myelinating OLs. Rather newly generated OL progenitors fail to differentiate beyond the preOL stage to mature myelinating OLs. These two key events in disrupted white matter regeneration and repair are discussed in the following sections.
OL progenitor proliferative response
In several experimental studies, the total pool of preOLs in neonatal rat white matter lesions increased several fold in subacute lesions [158, 184, 190]. This robust response is initiated rapidly. Within 24 hours after hypoxia-ischemia in neonatal rats, the total preOL pool was already increased 2-3 fold and continued to increased further for several days [158]. Although preOLs are a mitotically active population of cells in vitro, this robust expansion of the preOL pool appears to be mostly driven by a large reservoir of OL progenitors that proliferate locally in white matter lesions. A potential additional site for OL progenitor proliferation is the neural stem cell rich subventricular zone (SVZ). However, consistent with prior studies [65, 187, 189], we found that local proliferation of OL progenitors in the SVZ was less robust than within the lesions [158].
Human studies similarly found that the total pool of OL lineage cells was not depleted in chronic lesions [29, 33]. Although a pronounced loss of preOLs occurs during the acute phase of WMI [18], a significant diffuse increase in preOLs was found in chronic lesions from subjects that survived for at least 2-3 weeks after birth [33]. Interestingly, in age-related cognitive decline, human vascular WMI also displays a significant increase in total OL lineage cells [15] that is similar to that reported in human preterm WMI [33]. Hence, both in the developing and aging white matter, injury may trigger an initial robust OL progenitor proliferative response. Also common to both forms of WMI is a diffuse activation of microglia and astrocytes [33, 142, 177]. This diffuse inflammatory response thus accompanies the robust proliferative response of early OL progenitors and points to a generalized activation of multiple glial cell populations in response to WMI.
PreOL dysmaturation in chronic WMI
A major consequence of the chronic glial inflammatory response in chronic WMI is a disruption of the normal progression of preOL maturation and myelination. Although a large pool of preOLs are regenerated and survive in chronic lesions, they do not follow the normal trajectory for differentiation and myelination. This disruption in myelination occurs even though experimentally generated lesions contain numerous intact axons [153, 158]. In response to focal cerebral hypoxia-ischemia in the preterm-equivalent neonatal rat, for example, the control hemisphere displays normal myelination (Fig. 4B), whereas preOLs in white matter lesions fail to differentiate or myelinate despite the presence of numerous intact-appearing axons (Fig. 4C). Notably, in diffuse WMI in preterm fetal sheep, no significant axonal degeneration, axonal loss or shift in the distribution of axon sizes was identified by light or quantitative electron microscopy studies [150, 151, 153]. Morphometric and ultrastructural studies provide no information about the long-term functional integrity of axons, however, which may be disrupted due to lack of trophic support from oligodendrocytes [106].
In preterm equivalent rodents and preterm fetal sheep, chronic hypoxic-ischemic WMI was accompanied by disrupted maturation of preOLs to myelinating OLs. In diffuse WMI in preterm sheep, a near doubling in preOL density coincided with about a fifty percent decrease in OLs [150]. Similarly, in human diffuse WMI, a significant increase in preOLs coincided with a reduction in the total percentage of OLs [33]. Moreover, in both human and preterm fetal sheep, the accumulation of preOLs in chronic lesions was significantly associated with the magnitude of astrogliosis [33, 150]. The striking lack of myelination in gliotic lesions can also be appreciated in the neonatal rat where myelinating white matter (O4+O1+) is associated with modest levels of GFAP-labeled astrocytes (Fig. 4D) compared to injured white matter (Fig. 4E), which displays profound reductions in myelination. This lack of myelination is related to the pronounced accumulation of preOLs (O4+O1−) that fail to differentiate to myelinating OLs (Fig. 4F, G). As discussed below, a mechanistic role for several astrocyte-derived factors has been linked to preOL dysmaturation and myelination failure.
Since preOLs are highly susceptible to oxidative stress, an important unresolved question is whether the accumulation of preOLs in chronic WMI renders these lesions persistently more susceptible to recurrent injury from hypoxia-ischemia. To determine whether preOL dysmaturation extended the developmental window for susceptibility to WMI, we analyzed the severity of the WMI in response to recurrent hypoxia-ischemia. In response to recurrent hypoxia-ischemia, preterm-equivalent rodents displayed a pronounced increase in apoptotic preOL death that may have been related to loss of trophic support from diffuse neuro-axonal degeneration [158]. However, recurrent hypoxia-ischemia in preterm fetal sheep did not trigger pronounced preOL death or neuroaxonal degeneration [82]. This suggests that when only dysmature preOLs populate a chronic lesion, they may confer protection against recurrent WMI. An important future direction would be to determine if preOL dysmaturation confers upon these cells a greater resistance to oxidative stress via an ischemic tolerance-like mechanism.
Chronic WMI: What is the potential for regeneration and repair?
The persistence of a substantial pool of dysmature preOLs in WMI, suggests the potential to promote repair of chronic lesions through strategies that promote preOL differentiation. Disrupted maturation of the OL progenitor pool in chronic adult WMI has been studied in experimental models of myelination failure relevant to neonatal WMI, multiple sclerosis and vascular dementias. These studies have resulted in the identification of intrinsic, extrinsic and epigenetic factors that regulate the OL progenitor cell cycle, preOL differentiation and myelination [23, 58, 62, 161, 162]. Oxidative stress activates multiple genes, which regulate OL maturation, and oxidative stress promotes global histone acetylation, which can block OL differentiation [21, 50, 71, 108, 159]. Post-transcriptional control by microRNAs regulates OL differentiation and OL progenitors that lack mature microRNAs display arrested maturation [57, 160].
Disruption of the extracellular matrix is a prominent feature of chronic WMI in both the developing and aging white matter and is linked to diffuse reactive astrogliosis [15, 33, 37]. In diffuse preterm human WMI, the extracellular matrix is a particularly rich source of hyaluronic acid and one of its receptors, CD44 [33]. CD44-positive reactive astrocytes are significantly associated with the magnitude of WMI. Reactive astrocytes synthesize megadalton forms of hyaluronic acid that are associated with lesions [9]. In preterm fetal sheep hyaluronic acid was robustly elevated in the first week after acute WMI from hypoxia-ischemia and was still modestly elevated in chronic WMI at two weeks [82]. A block in preOL maturation is stimulated both in vitro and in vivo by hyaluronic acid and is related to hyaluronic acid digestion products generated by CNS enriched hyaluronidases that orient to the extracellular matrix [11, 82, 144, 164]. Pharmacological inhibition of hyaluronidases promotes preOL maturation in vitro and myelination in vivo, which is accompanied by enhanced nerve conduction [144]. Reactive astrocytes also display enhanced expression of bone morphogenetic proteins that inhibit OL progenitor differentiation and promote astrocyte differentiation [182]. Reactive astrocytes display enhance expression of the Notch ligand Jagged1 which is elevated in demyelinating lesions and activates Notch signaling in OL progenitors to prevent their maturation [100].
Dysregulation of WNT-beta catenin signaling also disrupts OL progenitor maturation, delays normal myelination and disrupts remyelination [60, 61, 64, 123, 174, 188]. Constitutive expression of the epidermal growth factor receptor (EGFR) in neonatal white matter promotes proliferation of OL progenitors [93], and enhanced EGFR signaling stimulates adult CNS myelination and remyelination [3]. EGFR activation via an intranasally administered form of EGF reduced OL death from chronic neonatal hypoxia, promoted OL maturation and myelination and led to functional improvement [157].
Since multiple signaling pathways are implicated in the pathogenesis of myelination failure, one therapeutic strategy has targeted multiple signaling pathways via the pleiotropic growth factor, erythropoietin (EPO), which promote angiogenesis, neurogenesis and gliogenesis during normal brain maturation [74, 94, 99, 101, 102, 115, 186]. A randomized trial of high dose recombinant EPO to preterm infants in the first two day of life was safe and associated with enhanced MRI-defined white matter maturation [63, 139], but two year neurodevelopmental outcomes were not improved [136]. A recent double-blinded, placebo-controlled trial of EPO administered with therapeutic hypothermia showed potential benefit for neonatal encephalopathy in term infants. MRI-defined brain injury in the perinatal period was significantly reduced and improved motor function was observed at 1 year of age [185]. Other agents that may provide at least partial protection against preterm WMI include melatonin [76], thyroxine [181] and magnesium sulfate [52]. Another potentially promising approach to block multiple cell death pathways is to combine cerebral hypothermia with an agent that protects against preOL death. In preclinical studies, cerebral hypothermia rendered greater global protection against WMI and preOL degeneration [24]. However there is insufficient contemporary clinical data regarding the safety of hypothermia in the human preterm neonate [79]. Moreover, although a variety of agents have shown promising preclinical neuro-protection for cerebral injury in full term neonates, many of these agents have not been extensively evaluated for preterm WMI [154].
Taken together, these studies suggest multiple potential strategies to stimulate regeneration and repair of WMI to circumvent disrupted myelination in neonates. An important future direction will be to define the evolution of cerebral WMI over months to years to identify the relative contributions of disrupted myelination and axonal dysfunction to functional disabilities in preterm survivors. What is the period over which the glial scar remodels and preOL arrest persists? Such information is of critical importance to define the window of opportunity for interventions to promote repair of WMI. Definition of this therapeutic window may be achieved by more sensitive neuroimaging that reliably discriminates disrupted myelination from other pathological processes such as reactive gliosis or disruption of the extraceullar matrix that progresses in tandem in the chronic injury environment.
As recently reviewed [20], it is evident from studies of human WMI that preterm infants sustain a unique pattern of injury responses that differ substantially from that seen in rodent models. Some rodent models of hypoxia-ischemia, hyperoxia or chronic severe hypoxemia generate markedly different patterns of WMI from human. In some instances, these models do not trigger preOL degeneration or reactive gliosis. Diffuse human WMI is also not associated with significant injury to cortical and subcortical gray matter structures, in contrast to some hypoxia-ischemia models in neonatal rodents [116, 121, 176] or fetal rabbits [34] where widespread necrotic and apoptotic neuronal degeneration occurs.
To better define the translational relevance of mechanisms to promote regeneration and repair of WMI, more widespread application of large pre-clinical models is needed. Despite the greater expense and challenges of nonhuman primate or preterm fetal sheep models, they provide access to forms of white matter pathology that more closely approximate that seen in human preterm survivors [10, 77]. Large pre-clinical animal models also provide access to study gyrencephalic white matter, which more closely approximates the human connectome. Recent studies from preterm fetal sheep, for example, support that WMI is accompanied by widespread disturbances in gray matter growth and maturation that resemble those defined in human by volumetric MRI studies [45, 124]. Such findings support that neonatal WMI occurs within the broader scope of global disturbances in cerebral gray matter maturation. It is presently unclear if disturbances in gray matter growth are related to WMI or occur due to independent mechanisms of neuronal dysmaturation. It is also unclear what the relative contributions of WMI and gray matter dysmaturation are to the complex neurobehavioral disabilities sustained by many preterm survivors. It nevertheless seems likely that disturbances in early myelination will have wide-ranging deleterious consequences for the establishment of neuronal connectivity during a critical window in brain development. Hence, developing optimal therapeutic strategies to lessen the impact of WMI may have significant impact on both preterm white and gray matter functional development.
Acknowledgments
Dr. Back is supported by the NIH (National Institutes of Neurological Disorders and Stroke: 1RO1NS054044, R37NS045737; National Institute on Aging: 1R01AG031892-01) and the American Heart Association (17GRNT33370058).
References
- 1.(CDC) CfDCaP. Economic costs associated with mental retardation, cerebral palsy, hearing loss and vision impairment–United States, 2003. MMWR Morb Mortal Wkly Rep. 2004;53:57–59. [PubMed] [Google Scholar]
- 2.Aarnoudse-Moens CS, Weisglas-Kuperus N, van Goudoever JB, Oosterlaan J. Meta-analysis of neurobehavioral outcomes in very preterm and/or very low birth weight children. Pediatrics. 2009;124:717–728. doi: 10.1542/peds.2008-2816. [DOI] [PubMed] [Google Scholar]
- 3.Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- 4.Alix JJ, Fern R. Glutamate receptor-mediated ischemic injury of premyelinated central axons. Ann Neurol. 2009;66:682–693. doi: 10.1002/ana.21767. [DOI] [PubMed] [Google Scholar]
- 5.Alix JJ, Zammit C, Riddle A, Meshul CK, Back SA, Valentino M, Fern R. Central axons preparing to myelinate are highly sensitivity to ischemic injury. Ann Neurol. 2012;72:936–951. doi: 10.1002/ana.23690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altman DI, Powers WJ, Perlman JM, Herscovitch P, Volpe SL, Volpe JJ. Cerebral blood flow requirement for brain viability in newborn infants is lower than in adults. Ann Neurol. 1988;24:218–226. doi: 10.1002/ana.410240208. [DOI] [PubMed] [Google Scholar]
- 7.Anderson PJ, De Luca CR, Hutchinson E, Spencer-Smith MM, Roberts G, Doyle LW. Attention problems in a representative sample of extremely preterm/extremely low birth weight children. Dev Neuropsychol. 2011;36:57–73. doi: 10.1080/87565641.2011.540538. [DOI] [PubMed] [Google Scholar]
- 8.Anderson PJ, Doyle LW. Cognitive and educational deficits in children born extremely preterm. Semin Perinatol. 2008;32:51–58. doi: 10.1053/j.semperi.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Asher R, Perides G, Vanderhaeghen J, Bignami A. Extracellular matrix of central nervous system white matter: demonstration of an hyaluronan-protein complex. J Neurosci Res. 1991;28:410–421. doi: 10.1002/jnr.490280314. [DOI] [PubMed] [Google Scholar]
- 10.Back S, Riddle A, Hohimer A. The instrumented fetal sheep as a model of cerebral white matter injury in the preterm infant. Neurotherapeutics. 2012;9:359–370. doi: 10.1007/s13311-012-0108-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Back S, Tuohy T, Chen H, Wallingford N, Craig A, Struve J, Luo N, Banine F, Liu Y, Chang A, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nature Medicine. 2005;9:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- 12.Back SA, Craig A, Kayton R, Luo NL, Meshul C, Allcock N, Fern R. Hypoxia-Ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. J Cereb Blood Flow Metab. 2007;27:334–347. doi: 10.1038/sj.jcbfm.9600344. [DOI] [PubMed] [Google Scholar]
- 13.Back SA, Gan X-D, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Back SA, Han BH, Luo NL, Chrichton CA, Tam J, Xanthoudakis S, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Back SA, Kroenke CD, Sherman LS, Lawrence G, Gong X, Taber EN, Sonnen JA, Larson EB, Montine TJ. White matter lesions defined by diffusion tensor imaging in older adults. Ann Neurol. 2011;70:465–476. doi: 10.1002/ana.22484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. [Google Scholar]
- 17.Back SA, Luo NL, Borenstein NS, Volpe JJ, Kinney HC. Arrested oligodendrocyte lineage progression during human cerebral white matter development: dissociation between the timing of progenitor differentiation and myelinogenesis. J Neuropathol Exp Neurol. 2002;61:197–211. doi: 10.1093/jnen/61.2.197. [DOI] [PubMed] [Google Scholar]
- 18.Back SA, Luo NL, Mallinson RA, O’Malley JP, Wallen LD, Frei B, Morrow JD, Petito CK, Roberts JCT, Murdoch GH, et al. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann Neurol. 2005;58:108–120. doi: 10.1002/ana.20530. [DOI] [PubMed] [Google Scholar]
- 19.Back SA, Miller SP. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann Neurol. 2014;75:469–486. doi: 10.1002/ana.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Back SA, Rosenberg PA. Pathophysiology of glia in perinatal white matter injury. Glia. 2014;62:1790–1815. doi: 10.1002/glia.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baltan S, Murphy SP, Danilov CA, Bachleda A, Morrison RS. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci. 2011;31:3990–3999. doi: 10.1523/JNEUROSCI.5379-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banker B, Larroche J. Periventricular leukomalacia of infancy. A form of neonatal anoxic encephalopathy. Arch Neurol. 1962;7:386–410. doi: 10.1001/archneur.1962.04210050022004. [DOI] [PubMed] [Google Scholar]
- 23.Barres B, Schmid R, Sendnte M, Raff M. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118:283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- 24.Barrett RD, Bennet L, Naylor A, George SA, Dean JM, Gunn AJ. Effect of cerebral hypothermia and asphyxia on the subventricular zone and white matter tracts in preterm fetal sheep. Brain Res. 2012;1469:35–42. doi: 10.1016/j.brainres.2012.06.018. [DOI] [PubMed] [Google Scholar]
- 25.Baud O, Greene A, Li J, Wang H, Volpe JJ, Rosenberg PA. Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J Neurosci. 2004;24:1531–1540. doi: 10.1523/JNEUROSCI.3989-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baud O, Haynes R, Wang H, Folkerth RD, Li J, Volpe J, Rosenberg PA. Developmental up-regulation of MnSOD in rat oligodendrocytes confers protection against oxidative injury. Eur J Neurosci. 2004;19:2669–2681. doi: 10.1111/j.0953-816X.2004.03451.x. [DOI] [PubMed] [Google Scholar]
- 27.Bax M, Tydeman C, Flodmark O. Clinical and MRI correlates of cerebral palsy: the European Cerebral Palsy Study. JAMA. 2006;296:1602–1608. doi: 10.1001/jama.296.13.1602. [DOI] [PubMed] [Google Scholar]
- 28.Beaino G, Khoshnood B, Kaminski M, Pierrat V, Marret S, Matis J, Ledesert B, Thiriez G, Fresson J, Roze JC, et al. Predictors of cerebral palsy in very preterm infants: the EPIPAGE prospective population-based cohort study. Dev Med Child Neurol. 2010;52:e119–125. doi: 10.1111/j.1469-8749.2010.03612.x. [DOI] [PubMed] [Google Scholar]
- 29.Billiards S, Haynes R, Folkerth R, NS B, Trachtenberg F, Rowitch D, Ligon K, Volpe J, Kinney H. Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathology. 2008;18:153–163. doi: 10.1111/j.1750-3639.2007.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Billiards SS, Haynes RL, Folkerth RD, Trachtenberg FL, Liu LG, Volpe JJ, Kinney HC. Development of microglia in the cerebral white matter of the human fetus and infant. J Comp Neurol. 2006;497:199–208. doi: 10.1002/cne.20991. [DOI] [PubMed] [Google Scholar]
- 31.Boardman JP, Counsell SJ, Rueckert D, Hajnal JV, Bhatia KK, Srinivasan L, Kapellou O, Aljabar P, Dyet LE, Rutherford MA, et al. Early growth in brain volume is preserved in the majority of preterm infants. Ann Neurol. 2007;62:185–192. doi: 10.1002/ana.21171. [DOI] [PubMed] [Google Scholar]
- 32.Brummelte S, Grunau RE, Chau V, Poskitt KJ, Brant R, Vinall J, Gover A, Synnes AR, Miller SP. Procedural pain and brain development in premature newborns. Ann Neurol. 2012;71:385–396. doi: 10.1002/ana.22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buser J, Maire J, Riddle A, Gong X, Nguyen T, Nelson K, Luo N, Ren J, Struve J, Sherman L, et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol. 2012;71:93–109. doi: 10.1002/ana.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buser J, Segovia K, Dean J, Nelson K, Beardsley D, Gong X, Luo N, Ren J, Wan Y, Riddle A, et al. Timing of appearance of late oligodendrocyte progenitors coincides with enhanced susceptibility of preterm rabbit cerebral white matter to hypoxia-ischemia. J Cereb Blood Flow Metab. 2010;30:1053–1065. doi: 10.1038/jcbfm.2009.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao Y, Gunn A, Bennet L, Wu D, George S, Gluckman PD, Shao X-M, Guan J. Insulin-like growth factor (IGF)-1 suppresses oligodendrocyte caspase-3 activation and increases glial proliferation after ischemia in near-term fetal sheep. J Cereb Blood Flow Metab. 2003;23:739–747. doi: 10.1097/01.WCB.0000067720.12805.6F. [DOI] [PubMed] [Google Scholar]
- 36.March of Dimes Premature Birth Report Card. 2016 http://www.multivu.com/players/English/7945951-march-of-dimes-premature-birth-report/docs/premature-report-149208847.pdf. (2/07/17; date of search)
- 37.Cargill R, Kohama SG, Struve J, Su W, Banine F, Witkowski E, Back SA, Sherman LS. Astrocytes in aged nonhuman primate brain gray matter synthesize excess hyaluronan. Neurobiology of aging. 2012;33:830 e813–824. doi: 10.1016/j.neurobiolaging.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castillo-Melendez M, Chow J, Walker D. Lipid peroxidation, caspase-3 immunoreactivity, and pyknosis in late-gestation fetal sheep brain after umbilical cord occlusion. Pediatr Res. 2004;55:864–871. doi: 10.1203/01.PDR.0000115679.86566.C4. [DOI] [PubMed] [Google Scholar]
- 39.Chau V, Synnes A, Grunau R, Poskitt K, Brant R, Miller S. Abnormal brain maturation in preterm neonates associated with adverse developmental outcomes. Neurology. 2013;81:2082–20889. doi: 10.1212/01.wnl.0000437298.43688.b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng Y, Deshmukh M, D’Costa A, Demaro J, Gidday J, Shah A, Sun Y, Jacquin M, Johnson E, Jr, Holtzman D. Caspase inhibitor affords neuroprotection with delayed adminstration in a rat model of neonatal hypoxic-ischemic brain injury. The Journal of clinical investigation. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clouchoux C, du Plessis AJ, Bouyssi-Kobar M, Tworetzky W, McElhinney DB, Brown DW, Gholipour A, Kudelski D, Warfield SK, McCarter RJ, et al. Delayed cortical development in fetuses with complex congenital heart disease. Cereb Cortex. 2013;23:2932–2943. doi: 10.1093/cercor/bhs281. [DOI] [PubMed] [Google Scholar]
- 42.Constantinou JC, Adamson-Macedo EN, Mirmiran M, Fleisher BE. Movement, imaging and neurobehavioral assessment as predictors of cerebral palsy in preterm infants. J Perinatol. 2007;27:225–229. doi: 10.1038/sj.jp.7211664. [DOI] [PubMed] [Google Scholar]
- 43.Counsell S, Allsop J, Harrison M, Larkman D, Kennea N, Kapellou O, Cowan F, Hajnal J, Edwards A, Rutherford M. Diffusion-weighted imaging of the brain in preterm infants with focal and diffuse white matter abnormality. Pediatrics. 2003;112:176–180. doi: 10.1542/peds.112.1.1. [DOI] [PubMed] [Google Scholar]
- 44.Craig A, Luo NL, Beardsley DJ, Wingate-Pearse N, Walker DW, Hohimer AR, Back SA. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp Neurol. 2003;181:231–240. doi: 10.1016/s0014-4886(03)00032-3. [DOI] [PubMed] [Google Scholar]
- 45.Dean J, McClendon E, Hansen K, Azimi-Zonooz A, Chen K, Riddle A, Gong X, Sharifnia E, Hagen M, Ahmad T, et al. Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci Transl Med. 2013;5:101–111. doi: 10.1126/scitranslmed.3004669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dean J, Moravec M, Grafe M, Abend N, Ren J, Gong X, Volpe J, Jensen F, Hohimer A, Back S. Strain-specific differences in perinatal rodent oligodendrocyte lineage progression and its correlation with human. Dev Neurosci. 2011;33:251–260. doi: 10.1159/000327242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deguchi K, Oguchi K, Takashima S. Characteristic neuropathology of leukomalacia in extremely low birth weight infants. Pediatr Neurol. 1997;16:296–300. doi: 10.1016/s0887-8994(97)00041-6. [DOI] [PubMed] [Google Scholar]
- 48.Delobel-Ayoub M, Arnaud C, White-Koning M, Casper C, Pierrat V, Garel M, Burguet A, Roze JC, Matis J, Picaud JC, et al. Behavioral problems and cognitive performance at 5 years of age after very preterm birth: the EPIPAGE Study. Pediatrics. 2009;123:1485–1492. doi: 10.1542/peds.2008-1216. [DOI] [PubMed] [Google Scholar]
- 49.Derrick M, L NL, Bregman JC, Jilling T, Ji X, Fisher K, Gladson CL, Beardsley DJ, Murdoch GA, Back SA, et al. Preterm fetal hypoxia causes hypertonia and motor deficits in the neonatal rabbit: a model for human cerebral palsy? J Neurosci. 2004;24:24–34. doi: 10.1523/JNEUROSCI.2816-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dewald LE, Rodriguez JP, Levine JM. The RE1 binding protein REST regulates oligodendrocyte differentiation. J Neurosci. 2011;31:3470–3483. doi: 10.1523/JNEUROSCI.2768-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dimitropoulos A, McQuillen PS, Sethi V, Moosa A, Chau V, Xu D, Brant R, Azakie A, Campbell A, Barkovich AJ, et al. Brain injury and development in newborns with critical congenital heart disease. Neurology. 2013;81:241–248. doi: 10.1212/WNL.0b013e31829bfdcf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doyle L. Antenatal magnesium sulfate and neuroprotection. Curr Opin Pediatr. 2012;24:154–159. doi: 10.1097/MOP.0b013e3283504da1. [DOI] [PubMed] [Google Scholar]
- 53.Drobyshevsky A, Derrick M, Prasad P, Ji X, Englof I, Tan S. Fetal brain magnetic resonance imaging response acutely to hypoxia-ischemia predicts postnatal outcome. Ann Neurol. 2007;61:307–314. doi: 10.1002/ana.21095. [DOI] [PubMed] [Google Scholar]
- 54.Drobyshevsky A, Derrick M, Wyrwicz A, Ji X, Englof I, Ullman L, Zelaya M, Northington F, Tan S. White matter injury correlates with hypertonia in an animal model of cerebral palsy. J Cereb Blood Flow Metab. 2007;27:270–281. doi: 10.1038/sj.jcbfm.9600333. [DOI] [PubMed] [Google Scholar]
- 55.Drobyshevsky A, Luo K, Derrick M, Yu L, Du H, Prasad PV, Vasquez-Vivar J, Batinic-Haberle I, Tan S. Motor deficits are triggered by reperfusion-reoxygenation injury as diagnosed by MRI and by a mechanism involving oxidants. J Neurosci. 2012;32:5500–5509. doi: 10.1523/jneurosci.5986-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Drobyshevsky A, Song S-K, Gamkrelidze G, Wyrwicz A, Derrick M, Meng F, Li L, Ji X, Trommer D, Beardsley D, et al. Developmental changes in diffusion anisotropy coincide with immature oligodendrocyte progression and maturation of compound action potential. J Neurosci. 2005;25:5988–5997. doi: 10.1523/JNEUROSCI.4983-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dugas JC, Cuellar TL, Scholze A, Ason B, Ibrahim A, Emery B, Zamanian JL, Foo LC, McManus MT, Barres BA. Dicer1 and miR-219 Are required for normal oligodendrocyte differentiation and myelination. Neuron. 2010;65:597–611. doi: 10.1016/j.neuron.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, Barres BA. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009;138:172–185. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Engelhardt E, Inder TE, Alexopoulos D, Dierker DL, Hill J, Van Essen D, Neil JJ. Regional impairments of cortical folding in premature infants. Ann Neurol. 2015;77:154–162. doi: 10.1002/ana.24313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fancy SP, Kotter MR, Harrington EP, Huang JK, Zhao C, Rowitch DH, Franklin RJ. Overcoming remyelination failure in multiple sclerosis and other myelin disorders. Exp Neurol. 2010;225:18–23. doi: 10.1016/j.expneurol.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 63.Fauchere JC, Koller BM, Tschopp A, Dame C, Ruegger C, Bucher HU. Safety of Early High-Dose Recombinant Erythropoietin for Neuroprotection in Very Preterm Infants. J Pediatr. 2015;167:52–57.e51. 53. doi: 10.1016/j.jpeds.2015.02.052. [DOI] [PubMed] [Google Scholar]
- 64.Feigenson K, Reid M, See J, Crenshaw EB, 3rd, Grinspan JB. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol Cell Neurosci. 2009;42:255–265. doi: 10.1016/j.mcn.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 65.Felling RJ, Snyder MJ, Romanko MJ, Rothstein RP, Ziegler AN, Yang Z, Givogri MI, Bongarzone ER, Levison SW. Neural stem/progenitor cells participate in the regenerative response to perinatal hypoxia/ischemia. J Neurosci. 2006;26:4359–4369. doi: 10.1523/JNEUROSCI.1898-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fern R, Davis P, Waxman S, Ransom B. Axon conduction and survival in CNS white matter during energy deprivation: a developmental study. J Neurophysiol. 1998;79:95–105. doi: 10.1152/jn.1998.79.1.95. [DOI] [PubMed] [Google Scholar]
- 67.Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferriero DM, Miller SP. Imaging selective vulnerability in the developing nervous system. J Anat. 2010;217:429–435. doi: 10.1111/j.1469-7580.2010.01226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Folkerth R, Haynes R, Borenstein NS, Volpe JJ, Kinney HC. Developmental lag in superoxide dismutases relative to other antioxidant enzymes in premyelinated human telencephalic white matter. J Neuropathol Exp Neurol. 2004;63:990–999. doi: 10.1093/jnen/63.9.990. [DOI] [PubMed] [Google Scholar]
- 70.Fragoso G, Martinez-Bermudez A, Lui H-N, Khorchid A, Chemtob S, Mushynski W, Almazan G. Developmental differences in H2O2-induced oligodendrocyte cell death: role of glutathione, mitogen-activated protein kinases and caspase 3. J Neurochem. 2004;90:392–404. doi: 10.1111/j.1471-4159.2004.02488.x. [DOI] [PubMed] [Google Scholar]
- 71.French HM, Reid M, Mamontov P, Simmons RA, Grinspan JB. Oxidative stress disrupts oligodendrocyte maturation. Journal of neuroscience research. 2009;87:3076–3087. doi: 10.1002/jnr.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Glass HC, Fujimoto S, Ceppi-Cozzio C, Bartha AI, Vigneron DB, Barkovich AJ, Glidden DV, Ferriero DM, Miller SP. White-matter injury is associated with impaired gaze in premature infants. Pediatr Neurol. 2008;38:10–15. doi: 10.1016/j.pediatrneurol.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gleason CA, Hamm C, Jones MD., Jr Cerebral blood flow, oxygenation, and carbohydrate metabolism in immature fetal sheep in utero. Am J Physiol. 1989;256:R1264–1268. doi: 10.1152/ajpregu.1989.256.6.R1264. [DOI] [PubMed] [Google Scholar]
- 74.Gonzalez FF, Larpthaveesarp A, McQuillen P, Derugin N, Wendland M, Spadafora R, Ferriero DM. Erythropoietin increases neurogenesis and oligodendrogliosis of subventricular zone precursor cells after neonatal stroke. Stroke. 2013;44:753–758. doi: 10.1161/STROKEAHA.111.000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Greisen G. Cerebral blood flow in preterm infants during the first week of life. Acta Paediatr Scand. 1986;75:43–51. doi: 10.1111/j.1651-2227.1986.tb10155.x. [DOI] [PubMed] [Google Scholar]
- 76.Gressens P, Schwendimann L, Husson I, Sarkozy G, Mocaer E, Vamecq J, Spedding M. Agomelatine, a melatonin receptor agonist with 5-HT(2C) receptor antagonist properties, protects the developing murine white matter against excitotoxicity. Eur J Pharmacol. 2008;588:58–63. doi: 10.1016/j.ejphar.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 77.Griffith JL, Shimony JS, Cousins SA, Rees SE, McCurnin DC, Inder TE, Neil JJ. MR imaging correlates of white-matter pathology in a preterm baboon model. Pediatr Res. 2012;71:185–191. doi: 10.1038/pr.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Groenendaal F, Termote JU, van der Heide-Jalving M, van Haastert IC, de Vries LS. Complications affecting preterm neonates from 1991 to 2006: what have we gained? Acta Paediatr. 2010;99:354–358. doi: 10.1111/j.1651-2227.2009.01648.x. [DOI] [PubMed] [Google Scholar]
- 79.Gunn A, Bennet L. Brain cooling for preterm infants. Clin Perinatal. 2008;35:735–748. doi: 10.1016/j.clp.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 80.Hack M, Taylor H, Drotar D, Schluchter M, Cartar L, Andreias L, Wilson-Costello D, Klein N. Chronic conditions, functional limitations, and special health care needs of school-aged children born with extremely low-birth-weight in the 1990’s. JAMA. 2005;294:318–325. doi: 10.1001/jama.294.3.318. [DOI] [PubMed] [Google Scholar]
- 81.Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. MRDD Res Rev. 2002;8:30–38. doi: 10.1002/mrdd.10007. [DOI] [PubMed] [Google Scholar]
- 82.Hagen MW, Riddle A, McClendon E, Gong X, Shaver D, Srivastava T, Dean JM, Bai JZ, Fowke TM, Gunn AJ, et al. Role of recurrent hypoxia-ischemia in preterm white matter injury severity. PloS One. 2014;9:e112800. doi: 10.1371/journal.pone.0112800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hamrick S, Miller SP, Leonard C, Glidden D, Goldstein R, Ramaswamy V, Piecuchi R, Ferriero DM. Trends in severe brain injury and neurodevelopmental outcome in premature newborn infants: the role of cystic periventricular leukomalacia. J Pediatr. 2004;145:593–599. doi: 10.1016/j.jpeds.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 84.Han B, D’Costa A, Back SA, Parsadian M, Patel S, Shah A, Gidday J, Srinvasan A, Deshmukh M, Holtzman D. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis. 2000;7:38–53. doi: 10.1006/nbdi.1999.0275. [DOI] [PubMed] [Google Scholar]
- 85.Hasegawa K, Yoshioka H, Sawada T, Nishikawa H. Lipid peroxidation in neonatal mouse brain subjected to two different types of hypoxia. Brain Dev. 1991;13:101–103. doi: 10.1016/s0387-7604(12)80115-x. [DOI] [PubMed] [Google Scholar]
- 86.Haynes RL, Billiards SS, Borenstein NS, Volpe JJ, Kinney HC. Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res. 2008;63:656–661. doi: 10.1203/PDR.0b013e31816c825c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, Volpe JJ, Kinney HC. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- 88.Hirayama A, Okoshi Y, Hachiya Y, Ozawa Y, Ito M, Kida Y, Imai Y, Kohsaka S, Takashima S. Early immunohistochemical detection of axonal damage and glial activation in extremely immature brains with periventricular leukomalacia. Clin Neuropathol. 2001;20:87–91. [PubMed] [Google Scholar]
- 89.Huppi PS, Murphy B, Maier SE, Zientara GP, Inder TE, Barnes PD, Kikinis R, Jolesz FA, Volpe JJ. Microstructural brain development after perinatal cerebral white matter injury assessed by diffusion tensor magnetic resonance imaging. Pediatrics. 2001;107:455–460. doi: 10.1542/peds.107.3.455. [DOI] [PubMed] [Google Scholar]
- 90.Iida K, Takashima S, Ueda K. Immunohistochemical study of myelination and oligodendrocyte in infants with periventricular leukomalacia. Pediatr Neurol. 1995;13:296–304. doi: 10.1016/0887-8994(95)00192-1. [DOI] [PubMed] [Google Scholar]
- 91.Inage YW, Itoh M, Takashima S. Correlation between cerebrovascular maturity and periventricular leukomalacia. Pediatr Neurol. 2000;22:204–208. doi: 10.1016/s0887-8994(99)00153-8. [DOI] [PubMed] [Google Scholar]
- 92.Inder TE, Anderson NJ, Spencer C, Wells S, Volpe JJ. White matter injury in the premature infant: a comparison between serial cranial sonographic and MR findings at term. AJNR AmJNeuroradiol. 2003;24:805–809. [PMC free article] [PubMed] [Google Scholar]
- 93.Ivkovic S, Canoll P, Goldman JE. Constitutive EGFR signaling in oligodendrocyte progenitors leads to diffuse hyperplasia in postnatal white matter. J Neurosci. 2008;28:914–922. doi: 10.1523/JNEUROSCI.4327-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iwai M, Stetler RA, Xing J, Hu X, Gao Y, Zhang W, Chen J, Cao G. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke. 2010;41:1032–1037. doi: 10.1161/STROKEAHA.109.570325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jacobson LK, Dutton GN. Periventricular leukomalacia: an important cause of visual and ocular motility dysfunction in children. Surv Ophthalmol. 2000;45:1–13. doi: 10.1016/s0039-6257(00)00134-x. [DOI] [PubMed] [Google Scholar]
- 96.Jakovcevski I, Filipovic R, Mo Z, Rakic S, Zecevic N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front Neuroanat. 2009;3:5. doi: 10.3389/neuro.05.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jakovcevski I, Zecevic N. Sequence of oligodendrocyte development in the human fetal telencephalon. Glia. 2005;49:480–491. doi: 10.1002/glia.20134. [DOI] [PubMed] [Google Scholar]
- 98.Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med. 1990;9:515–540. doi: 10.1016/0891-5849(90)90131-2. [DOI] [PubMed] [Google Scholar]
- 99.Jantzie LL, Miller RH, Robinson S. Erythropoietin signaling promotes oligodendrocyte development following prenatal systemic hypoxic-ischemic brain injury. Pediatr Res. 2013;74:658–667. doi: 10.1038/pr.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.John G, Shankar S, Shafit-Zagardo B, Massimi A, Lee S, Raine C, Brosnan C. Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nature Medicine. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- 101.Juul SE, Mayock DE, Comstock BA, Heagerty PJ. Neuroprotective potential of erythropoietin in neonates; design of a randomized trial. Maternal health, neonatology and perinatology. 2015;1:27. doi: 10.1186/s40748-015-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kako E, Kaneko N, Aoyama M, Hida H, Takebayashi H, Ikenaka K, Asai K, Togari H, Sobue K, Sawamoto K. Subventricular zone-derived oligodendrogenesis in injured neonatal white matter in mice enhanced by a nonerythropoietic erythropoietin derivative. Stem cells (Dayton, Ohio) 2012;30:2234–2247. doi: 10.1002/stem.1202. [DOI] [PubMed] [Google Scholar]
- 103.Kersbergen KJ, Benders MJ, Groenendaal F, Koopman-Esseboom C, Nievelstein RA, van Haastert IC, de Vries LS. Different patterns of punctate white matter lesions in serially scanned preterm infants. PloS one. 2014;9:e108904. doi: 10.1371/journal.pone.0108904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kesler SR, Reiss AL, Vohr B, Watson C, Schneider KC, Katz KH, Maller-Kesselman J, Silbereis J, Constable RT, Makuch RW, Ment LR. Brain volume reductions within multiple cognitive systems in male preterm children at age twelve. J Pediatr. 2008;152:513–520. 520 e511. doi: 10.1016/j.jpeds.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lasry O, Shevell MI, Dagenais L. Cross-sectional comparison of periventricular leukomalacia in preterm and term children. Neurology. 2010;74:1386–1391. doi: 10.1212/WNL.0b013e3181dad62d. [DOI] [PubMed] [Google Scholar]
- 106.Lee Y, Morrison B, Li Y, Lengacher S, Farah M, Hoffman P, Liu Y, Tsinalia A, Jin L, Zhang P-W, et al. Oligodendrolia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li AM, Chau V, Poskitt KJ, Sargent MA, Lupton BA, Hill A, Roland E, Miller SP. White matter injury in term newborns with neonatal encephalopathy. Pediatr Res. 2009;65:85–89. doi: 10.1203/PDR.0b013e31818912d2. [DOI] [PubMed] [Google Scholar]
- 108.Li W, Zhang B, Tang J, Cao Q, Wu Y, Wu C, Guo J, Ling EA, Liang F. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J Neurosci. 2007;27:2606–2616. doi: 10.1523/JNEUROSCI.4181-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Licht DJ, Shera DM, Clancy RR, Wernovsky G, Montenegro LM, Nicolson SC, Zimmerman RA, Spray TL, Gaynor JW, Vossough A. Brain maturation is delayed in infants with complex congenital heart defects. J Thorac Cardiovasc Surg. 2009;137:529–536. doi: 10.1016/j.jtcvs.2008.10.025. discussion 536-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Limperopoulos C, Tworetzky W, McElhinney DB, Newburger JW, Brown DW, Robertson RL, Jr, Guizard N, McGrath E, Geva J, Annese D, et al. Brain volume and metabolism in fetuses with congenital heart disease: evaluation with quantitative magnetic resonance imaging and spectroscopy. Circulation. 2010;121:26–33. doi: 10.1161/CIRCULATIONAHA.109.865568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin S, Rhodes P, Lei M, Zhang F, Cai Z. a-Phenyl-n-tert-butyl-nitrone attenuates hypoxic-ischemic white matter injury in the neonatal rat brain. Brain Res. 2004;1007:132–141. doi: 10.1016/j.brainres.2004.01.074. [DOI] [PubMed] [Google Scholar]
- 112.Litt J, Taylor H, Klein N, Hack M. Learning disabilities in children with very low birthweight:prevalence, neuropsychological correlates and educational interventions. J Learn Disabil. 2005;8:130–141. doi: 10.1177/00222194050380020301. [DOI] [PubMed] [Google Scholar]
- 113.Liu J, Li J, Qin GL, Chen YH, Wang Q. Periventricular leukomalacia in premature infants in mainland China. Am J Perinatol. 2008;25:535–540. doi: 10.1055/s-0028-1083841. [DOI] [PubMed] [Google Scholar]
- 114.Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, Lawn JE, Cousens S, Mathers C, Black RE. Global, regional, and national causes of under-5 mortality in 2000-15: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet. 2017;388:3027–3035. doi: 10.1016/S0140-6736(16)31593-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu W, Shen Y, Plane JM, Pleasure DE, Deng W. Neuroprotective potential of erythropoietin and its derivative carbamylated erythropoietin in periventricular leukomalacia. Exp Neurol. 2011;230:227–239. doi: 10.1016/j.expneurol.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lodygensky GA, West T, Moravec MD, Back SA, Dikranian K, Holtzman DM, Neil JJ. Diffusion characteristics associated with neuronal injury and glial activation following hypoxia-ischemia in the immature brain. Magn Reson Med. 2011;66:839–845. doi: 10.1002/mrm.22869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Loeliger M, Inder T, Cain S, Ramesh RC, Camm E, Thomson MA, Coalson J, Rees SM. Cerebral outcomes in a preterm baboon model of early versus delayed nasal continuous positive airway pressure. Pediatrics. 2006;118:1640–1653. doi: 10.1542/peds.2006-0653. [DOI] [PubMed] [Google Scholar]
- 118.Maalouf E, Duggan P, Counsell SJ, Rutherford MA, Cowan FM, Azzopardi D, Edwards A. Comparison of findings on cranial ultrasound and magnetic resonance imaging in preterm infants. Pediatrics. 2001;107:719–727. doi: 10.1542/peds.107.4.719. [DOI] [PubMed] [Google Scholar]
- 119.Mallard E, Rees S, Stringer M, Cock M, Harding R. Effects of chronic placental insufficiency on brain development in fetal sheep. Pediatr Res. 1998;43:262–270. doi: 10.1203/00006450-199802000-00018. [DOI] [PubMed] [Google Scholar]
- 120.Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. II: white matter lesions of the neocortex. J Neuropathol Exp Neurol. 1997;56:219–235. doi: 10.1097/00005072-199703000-00001. [DOI] [PubMed] [Google Scholar]
- 121.Marret S, Mukendi R, Gadisseux J-F, Gressens P, Evrard P. Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic-like lesions. J Neuropathol Exp Neurol. 1995;54:358–370. doi: 10.1097/00005072-199505000-00009. [DOI] [PubMed] [Google Scholar]
- 122.Martinez-Biarge M, Madero R, Gonzalez A, Quero J, Garcia-Alix A. Perinatal morbidity and risk of hypoxic-ischemic encephalopathy associated with intrapartum sentinel events. Am J Obstet Gynecol. 2012;206:148.e141–147. doi: 10.1016/j.ajog.2011.09.031. [DOI] [PubMed] [Google Scholar]
- 123.McClain CR, Sim FJ, Goldman SA. Pleiotrophin suppression of receptor protein tyrosine phosphatase-beta/zeta maintains the self-renewal competence of fetal human oligodendrocyte progenitor cells. J Neurosci. 2012;32:15066–15075. doi: 10.1523/JNEUROSCI.1320-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McClendon E, Chen K, Gong X, Sharifnia E, Hagen M, Cai V, Shaver DC, Riddle A, Dean JM, Gunn AJ, et al. Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann Neurol. 2014;75:508–524. doi: 10.1002/ana.24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McClure M, Riddle A, Manese M, Luo N, Rorvik D, Kelly K, Barlow C, Kelly JJEJ, Bernard SL, Glenny RW, Barlow CH, Vinecore K, Roberts C, et al. Cerebral blood flow heterogeneity in preterm sheep: lack of physiological support for vascular boundary zones in fetal cerebral white matter. J Cereb Blood Flow Metab. 2008;28:995–1008. doi: 10.1038/sj.jcbfm.9600597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McQuillen P, Miller S. Congenital heart disease and brain development. Ann N Y Acad Sci. 2010;1184:68–86. doi: 10.1111/j.1749-6632.2009.05116.x. [DOI] [PubMed] [Google Scholar]
- 127.Ment LR, Hirtz D, Huppi PS. Imaging biomarkers of outcome in the developing preterm brain. Lancet Neurol. 2009;8:1042–1055. doi: 10.1016/S1474-4422(09)70257-1. [DOI] [PubMed] [Google Scholar]
- 128.Mercier CE, Dunn MS, Ferrelli KR, Howard DB, Soll RF. Neurodevelopmental outcome of extremely low birth weight infants from the Vermont Oxford network: 1998-2003. Neonatology. 2010;97:329–338. doi: 10.1159/000260136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Miller S, Vigneron D, Henry R, Bohland M, Ceppi-Cozzio C, Hoffman C, Newton N, Partridge JC, Ferriero DM, Barkovich AJ. Serial quantitative diffusion tensor MRI of the premature brain: development in newborns with and without injury. J Magn Reson Imaging. 2002;16:621–632. doi: 10.1002/jmri.10205. [DOI] [PubMed] [Google Scholar]
- 130.Miller SP, Cozzio CC, Goldstein RB, Ferriero DM, Partridge JC, Vigneron DB, Barkovich AJ. Comparing the diagnosis of white matter injury in premature newborns with serial MR imaging and transfontanel ultrasonagraphy findings. AJNR Am J Neuroradiol. 2003;24:1661–1669. [PMC free article] [PubMed] [Google Scholar]
- 131.Miller SP, Ferriero DM, Leonard C, Piecuch R, Glidden D, Partridge JC, Perez M, Mukherjee P, Vigneron D, Barkovich AJ. Early brain injury in premature newborns detected with magnetic resonance imaging is associated with adverse neurodevelopmental outcome. J Pediatr. 2005;147:609–616. doi: 10.1016/j.jpeds.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 132.Miller SP, McQuillen PS, Hamrick S, Xu D, Glidden DV, Charlton N, Karl T, Azakie A, Ferriero DM, Barkovich AJ, et al. Abnormal brain development in newborns with congenital heart disease. N Engl J Med. 2007;357:1928–1938. doi: 10.1056/NEJMoa067393. [DOI] [PubMed] [Google Scholar]
- 133.Mishra OP, Delivoria-Papadopoulos M. Lipid peroxidation in developing fetal guinea pig brain during normoxia and hypoxia. Brain Res Developmental Brain Research. 1989;45:129–135. doi: 10.1016/0165-3806(89)90014-x. [DOI] [PubMed] [Google Scholar]
- 134.Miyawaki T, Matsui K, Takashima S. Developmental characteristics of vessel density in the human fetal and infant brains. Early Hum Dev. 1998;53:65–72. doi: 10.1016/s0378-3782(98)00043-7. [DOI] [PubMed] [Google Scholar]
- 135.Montine K, Quinn J, Zhang J, Fessel J, Roberts LJ, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation. Chem Phy Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 136.Natalucci G, Latal B, Koller B, Ruegger C, Sick B, Held L, Bucher HU, Fauchere JC. Effect of Early Prophylactic High-Dose Recombinant Human Erythropoietin in Very Preterm Infants on Neurodevelopmental Outcome at 2 Years: A Randomized Clinical Trial. Jama. 2016;315:2079–2085. doi: 10.1001/jama.2016.5504. [DOI] [PubMed] [Google Scholar]
- 137.Nelson MD, Jr, Gonzalez-Gomez I, Gilles FH. Dyke Award. The search for human telencephalic ventriculofugal arteries. AJNR Am J Neuroradiol. 1991;12:215–222. [PMC free article] [PubMed] [Google Scholar]
- 138.Ness JK, Romanko MJ, Rothstein RP, Wood TL, Levison SW. Perinatal hypoxia-ischemia induces apoptotic and excitotoxic death of periventricular white matter oligodendrocyte progenitors. Dev Neurosci. 2001;23:203–208. doi: 10.1159/000046144. [DOI] [PubMed] [Google Scholar]
- 139.O’Gorman RL, Bucher HU, Held U, Koller BM, Huppi PS, Hagmann CF. Tract-based spatial statistics to assess the neuroprotective effect of early erythropoietin on white matter development in preterm infants. Brain. 2015;138:388–397. doi: 10.1093/brain/awu363. [DOI] [PubMed] [Google Scholar]
- 140.Pagliano E, Fedrizzi E, Erbetta A, Bulgheroni S, Solari A, Bono R, Fazzi E, Andreucci E, Riva D. Cognitive profiles and visuoperceptual abilities in preterm and term spastic diplegic children with periventricular leukomalacia. J Child Neurol. 2007;22:282–288. doi: 10.1177/0883073807300529. [DOI] [PubMed] [Google Scholar]
- 141.Peterson BS, Vohr B, Staib LH, Cannistraci CJ, Dolberg A, Schneider KC, Katz KH, Westerveld M, Sparrow S, Anderson AW, et al. Regional brain volume abnormalities and long-term cognitive outcome in preterm infants. JAMA. 2000;284:1939–1947. doi: 10.1001/jama.284.15.1939. [DOI] [PubMed] [Google Scholar]
- 142.Pierson CR, Folkerth RD, Billiards SS, Trachtenberg FL, Drinkwater ME, Volpe JJ, Kinney HC. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. 2007;114:619–631. doi: 10.1007/s00401-007-0295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Powers WJ, Grubb RL, Darriet D, Raichle ME. Cerebral blood flow and cerebral metabolic rate of oxygen requirements for cerebral function and viability in humans. J Cereb Blood Flow Metab. 1985;5:600–608. doi: 10.1038/jcbfm.1985.89. [DOI] [PubMed] [Google Scholar]
- 144.Preston M, Gong X, Su W, Matsumoto SG, Banine F, Winkler C, Foster S, Xing R, Struve J, Dean J, et al. Digestion products of the PH20 hyaluronidase inhibit remyelination. Ann Neurol. 2013;73:266–280. doi: 10.1002/ana.23788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pryds O, Andersen GE, Friis-Hansen B. Cerebral blood flow reactivity in spontaneously breathing, preterm infants shortly after birth. Acta Paediatr Scand. 1990;79:391–396. doi: 10.1111/j.1651-2227.1990.tb11482.x. [DOI] [PubMed] [Google Scholar]
- 146.Pryds O, Greisen G, Friis-Hansen B. Compensatory increase of CBF in preterm infants during hypoglycaemia. Acta Paediatr Scand. 1988;77:632–637. doi: 10.1111/j.1651-2227.1988.tb10721.x. [DOI] [PubMed] [Google Scholar]
- 147.Pryds O, Greisen G, Friis-Hansen B. Heterogeneity of cerebral vasoreactivity in preterm infants supported by machanical ventilation. J Pediatr. 1989;115:638–645. doi: 10.1016/s0022-3476(89)80301-4. [DOI] [PubMed] [Google Scholar]
- 148.Rakic S, Zecevic N. Early oligodendrocyte precursor cells in the human fetal telencephalon. Glia. 2003;41:117–127. doi: 10.1002/glia.10140. [DOI] [PubMed] [Google Scholar]
- 149.Rees S, Mallard C, Breen S, Stringer M, Cock M, Harding R. Fetal brain injury following prolonged hypoxemia and placental insufficiency: a review. Comp Biochem Physiol A Mol Integr Physiol. 1998;119:653–660. doi: 10.1016/s1095-6433(98)01001-0. [DOI] [PubMed] [Google Scholar]
- 150.Riddle A, Dean J, Buser J, Gong X, Maire J, Chen K, Ahmad T, Chen V, Nguyen T, Kroenke C, et al. Histopathological correlates of MRI-defined chronic perinatal white matter injury. Ann Neurol. 2011;70:493–507. doi: 10.1002/ana.22501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Riddle A, Luo N, Manese M, Beardsley D, Green L, Rorvik D, Kelly K, Barlow C, Kelly J, Hohimer A, et al. Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci. 2006;26:3045–3055. doi: 10.1523/JNEUROSCI.5200-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Riddle A, Maire J, Cai V, Nguyen T, Gong X, Hansen K, Grafe MR, Hohimer AR, Back SA. Hemodynamic and metabolic correlates of perinatal white matter injury severity. PloS one. 2013;8:e82940. doi: 10.1371/journal.pone.0082940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Riddle A, Maire J, Gong X, Chen KX, Kroenke CD, Hohimer AR, Back SA. Differential susceptibility to axonopathy in necrotic and non-necrotic perinatal white matter injury. Stroke. 2012;43:178–184. doi: 10.1161/STROKEAHA.111.632265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Robertson NJ, Tan S, Groenendaal F, van Bel F, Juul SE, Bennet L, Derrick M, Back SA, Valdez RC, Northington F, et al. Which neuroprotective agents are ready for bench to bedside translation in the newborn infant? J Pediatr. 2012;160:544–552 e544. doi: 10.1016/j.jpeds.2011.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rosenberg PA, Weimin D, Gan XD, Ali S, Back SA, Sanchez RM, Segal MM, Follet PE, Jensen FE, Volpe JJ. Mature myelin basic protein expressing oligodendrocytes are insensitive to kainate toxicity. J Neurosci Res. 2003;71:237–245. doi: 10.1002/jnr.10472. [DOI] [PubMed] [Google Scholar]
- 156.Scafidi J, Fagel DM, Ment LR, Vaccarino FM. Modeling premature brain injury and recovery. Int J Dev Neurosci. 2009;27:863–871. doi: 10.1016/j.ijdevneu.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Scafidi J, Hammond TR, Scafidi S, Ritter J, Jablonska B, Roncal M, Szigeti-Buck K, Coman D, Huang Y, McCarter RJ, Jr, et al. Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature. 2014;506:230–234. doi: 10.1038/nature12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Segovia K, McClure M, Moravec M, Luo N, Wang Y, Gong X, Riddle A, Craig A, Struve J, Sherman L, et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol. 2008;63:517–526. doi: 10.1002/ana.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J Cell Biol. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shin D, Shin JY, McManus MT, Ptacek LJ, Fu YH. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann Neurol. 2009;66:843–857. doi: 10.1002/ana.21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Silbereis J, Huang E, Back S, Rowitch D. Toward improved animal models of neonatal white matter injury associated with cerebral palsy. Dis Model Mech. 2010;3:678–688. doi: 10.1242/dmm.002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sizonenko SV, Bednarek N, Gressens P. Growth factors and plasticity. Semin Fetal Neonatal Med. 2007;12:241–249. doi: 10.1016/j.siny.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 163.Skranes J, Lohaugen GC, Martinussen M, Haberg A, Brubakk AM, Dale AM. Cortical surface area and IQ in very-low-birth-weight (VLBW) young adults. Cortex. 2013;49:2264–2271. doi: 10.1016/j.cortex.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 164.Sloane J, Batt C, Ma Y, Harris Z, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci USA. 2010;107:11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Smith GC, Gutovich J, Smyser C, Pineda R, Newnham C, Tjoeng TH, Vavasseur C, Wallendorf M, Neil J, Inder T. Neonatal intensive care unit stress is associated with brain development in preterm infants. Ann Neurol. 2011;70:541–549. doi: 10.1002/ana.22545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Soria-Pastor S, Gimenez M, Narberhaus A, Falcon C, Botet F, Bargallo N, Mercader JM, Junque C. Patterns of cerebral white matter damage and cognitive impairment in adolescents born very preterm. Int J Dev Neurosci. 2008;26:647–654. doi: 10.1016/j.ijdevneu.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 167.Soria-Pastor S, Padilla N, Zubiaurre-Elorza L, Ibarretxe-Bilbao N, Botet F, Costas-Moragas C, Falcon C, Bargallo N, Mercader JM, Junque C. Decreased regional brain volume and cognitive impairment in preterm children at low risk. Pediatrics. 2009;124:e1161–1170. doi: 10.1542/peds.2009-0244. [DOI] [PubMed] [Google Scholar]
- 168.Soul J, Hammer P, Tsuji M, Saul J, Bassan H, Limperopoulous C, Disalvo D, Moore M, Akins P, Ringer S, et al. Fluctuating pressure-passivity is common in the cerebral circulation of sick premature infants. Pediatr Res. 2007;61:467–473. doi: 10.1203/pdr.0b013e31803237f6. [DOI] [PubMed] [Google Scholar]
- 169.Spittle AJ, Boyd RN, Inder TE, Doyle LW. Predicting motor development in very preterm infants at 12 months’ corrected age: the role of qualitative magnetic resonance imaging and general movements assessments. Pediatrics. 2009;123:512–517. doi: 10.1542/peds.2008-0590. [DOI] [PubMed] [Google Scholar]
- 170.Spittle AJ, Brown NC, Doyle LW, Boyd RN, Hunt RW, Bear M, Inder TE. Quality of general movements is related to white matter pathology in very preterm infants. Pediatrics. 2008;121:e1184–1189. doi: 10.1542/peds.2007-1924. [DOI] [PubMed] [Google Scholar]
- 171.Sun L, Macgowan CK, Sled JG, Yoo SJ, Manlhiot C, Porayette P, Grosse-Wortmann L, Jaeggi E, McCrindle BW, Kingdom J, et al. Reduced fetal cerebral oxygen consumption is associated with smaller brain size in fetuses with congenital heart disease. Circulation. 2015;131:1313–1323. doi: 10.1161/CIRCULATIONAHA.114.013051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Szymonowicz W, Walker AM, Cussen L, Cannata J, Yu VY. Developmental changes in regional cerebral blood flow in fetal and newborn lambs. Am J Physiol. 1988;254:H52–H58. doi: 10.1152/ajpheart.1988.254.1.H52. [DOI] [PubMed] [Google Scholar]
- 173.Takashima S, Tanaka K. Development of cerebrovascular architecture and its relationship to periventricular leukomalacia. Arch Neurol. 1978;35:11–16. doi: 10.1001/archneur.1978.00500250015003. [DOI] [PubMed] [Google Scholar]
- 174.Tawk M, Makoukji J, Belle M, Fonte C, Trousson A, Hawkins T, Li H, Ghandour S, Schumacher M, Massaad C. Wnt/beta-catenin signaling is an essential and direct driver of myelin gene expression and myelinogenesis. J Neurosci. 2011;31:3729–3742. doi: 10.1523/JNEUROSCI.4270-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tsuji M, Saul J, du Plessis A, Eichenwald E, Sobh J, Crocker R, Volpe J. Cerebral intravascular oxygenation correlates with mean arterial pressure in critically ill premature infants. Pediatrics. 2000;106:625–632. doi: 10.1542/peds.106.4.625. [DOI] [PubMed] [Google Scholar]
- 176.Vannucci RC, Vannucci SJ. Perinatal hypoxic-ischemic brain damage: evolution of an animal model. Dev Neurosci. 2005;27:81–86. doi: 10.1159/000085978. [DOI] [PubMed] [Google Scholar]
- 177.Verney C, Pogledic I, Biran V, Adle-Biassette H, Fallet-Bianco C, Gressens P. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J Neuropathol Exp Neurol. 2012;71:251–264. doi: 10.1097/NEN.0b013e3182496429. [DOI] [PubMed] [Google Scholar]
- 178.Vinall J, Miller SP, Chau V, Brummelte S, Synnes AR, Grunau RE. Neonatal pain in relation to postnatal growth in infants born very preterm. Pain. 2012;153:1374–1381. doi: 10.1016/j.pain.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 179.Vohr BR. Neurodevelopmental outcomes of extremely preterm infants. Clin Perinatol. 2014;41:241–255. doi: 10.1016/j.clp.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 180.Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110–124. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Vose LR, Vinukonda G, Jo S, Miry O, Diamond D, Korumilli R, Arshad A, Zia MT, Hu F, Kayton RJ, et al. Treatment with thyroxine restores myelination and clinical recovery after intraventricular hemorrhage. J Neurosci. 2013;33:17232–17246. doi: 10.1523/JNEUROSCI.2713-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wang Y, Cheng X, He Q, Zheng Y, Kim D, Whittemore S, Cao Q. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J Neurosci. 2011;31:6053–6058. doi: 10.1523/JNEUROSCI.5524-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wilson-Costello D, Fridedman H, Minich N, Fanaroff A, Hack M. Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics. 2005;115:997–1003. doi: 10.1542/peds.2004-0221. [DOI] [PubMed] [Google Scholar]
- 184.Wright J, Zhang G, Yu T-S, Kernie S. Age-related changes in the oligodendrocyte progenitor pool influence brain remodeling after injury. Dev Neurosci. 2010;32:499–509. doi: 10.1159/000322081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wu YW, Mathur AM, Chang T, McKinstry RC, Mulkey SB, Mayock DE, Van Meurs KP, Rogers EE, Gonzalez FF, Comstock BA, et al. High-Dose Erythropoietin and Hypothermia for Hypoxic-Ischemic Encephalopathy: A Phase II Trial. Pediatrics. 2016;137 doi: 10.1542/peds.2016-0191. [DOI] [PubMed] [Google Scholar]
- 186.Yamada M, Burke C, Colditz P, Johnson DW, Gobe GC. Erythropoietin protects against apoptosis and increases expression of non-neuronal cell markers in the hypoxia-injured developing brain. The Journal of pathology. 2011;224:101–109. doi: 10.1002/path.2862. [DOI] [PubMed] [Google Scholar]
- 187.Yang Z, Levison S. Hypoxia/ischemia expands the regenerative capacity of progenitors in the perinatal subventricular zone. Neuroscience. 2006;139:555–564. doi: 10.1016/j.neuroscience.2005.12.059. [DOI] [PubMed] [Google Scholar]
- 188.Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zaidi A, Bessert D, Ong J, Xu H, Barks J, Silverstein F. New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia. 2004;46:380–390. doi: 10.1002/glia.20013. [DOI] [PubMed] [Google Scholar]
- 190.Zhiheng H, Liu J, Cheung P-Y, Chen C. Long-term cognitive impairment and myelination deficiency in a rat model of perinatal hypoxic-ischemia brain injury. Brain Res. 2009;1301:100–109. doi: 10.1016/j.brainres.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 191.Zhu MY, Milligan N, Keating S, Windrim R, Keunen J, Thakur V, Ohman A, Portnoy S, Sled JG, Kelly E, et al. The hemodynamics of late-onset intrauterine growth restriction by MRI. Am J Obstet Gynecol. 2016;214:367.e361–367.e317. doi: 10.1016/j.ajog.2015.10.004. [DOI] [PubMed] [Google Scholar]