

Abstract
Protein S (PS), a γ-carboxyglutamate-containing serum protein, was unexpectedly discovered in 1977. Soon after its discovery, PS gained the attention of researchers because of its physiological importance, acting as a multifunctional protein at the intersection of blood coagulation, inflammation, and other cellular processes. Protein S functions as an anticoagulant by directly inhibiting procoagulants, such as Factor Xa (FXa), FVa, and FIXa, while also serving as a cofactor for anticoagulants such as Activated Protein C and Tissue Factor Pathway Inhibitor. By associating with C4b binding protein (C4BP), PS has also been shown to minimize the effect of inflammation. Finally, PS promotes efferocytosis through TAM family protein kinase receptors. Mutations in the PS gene cause pathological conditions such as deep vein thrombosis and hereditary ischemia. In this review, we summarize studies regarding the multiple functions of PS.
Keywords: Protein S, Anticoagulant, Inflammation, Signaling, Apoptosis, Factor IXa
INTRODUCTION
In 1977, while purifying Protein C from bovine plasma investigators discovered a new γ-carboxyglutamate (Gla)- containing protein which they named Protein S (PS) after Seattle the city of its discovery [1]. In the ensuing years, PS has been established as a protein with a wide array of functions.
PS is secreted primarily by endothelial cells, megakaryocytes, hepatocytes, and Leydig cells [2]. These cells initially synthesize PS as a 676 amino acid precursor protein [3], which, upon cleavage of the signal and pro peptides, matures to a 635 amino acid protein composed of an N-terminal Gla domain, a thrombinsensitive region (TSR), four Epidermal Growth Factor (EGF)-like domains, and a sex hormone binding globule (SHBG) [3]. Protein S circulates in the blood at a concentration of 350 nM, 60% of which is bound to complement component 4 binding protein (C4BP), with the other 40% denoted as ‘free’ [4]. Both forms of Protein S can act as anticoagulants, and their deficiency is associated with deep vein thrombosis and ischemic stroke [8, 9]; furthermore, murine PS knockouts have been shown to result in embryonic lethality [5–7].
DISCUSSION AND CONCLUSION
Anticoagulant function of Protein S
Protein S is an anticoagulant that prevents clot formation, while aiding in clot localization [10]. The anticoagulant activity of PS is exerted, either, by its direct interaction with procoagulants and their active forms, or by its co-enzymatic function with other anticoagulants (Figure 1).
Figure 1.
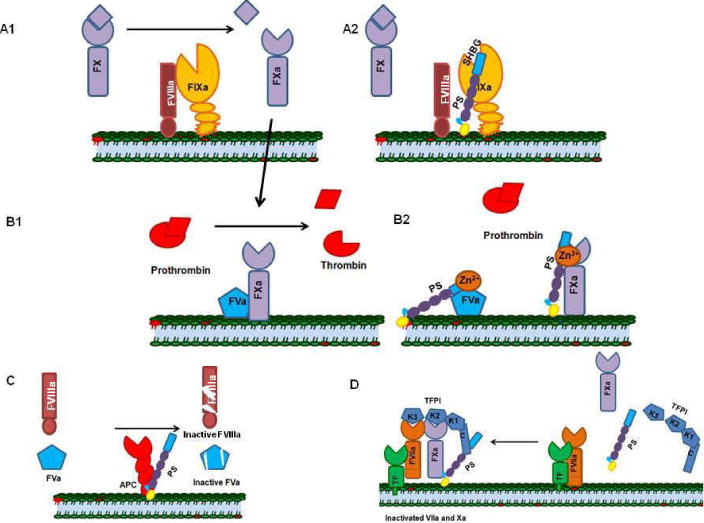
Mechanisms of PS-mediated anticoagulation. A1) Factor IXa, in the presence of its co-factor FVIIIa, activates FX to FXa. A2) Protein S directly binds to activated FIXa and inhibits the activation of FX. B1) FXa and FVa form the prothrombinase complex that converts prothrombin to thrombin. B2) Protein S in the presence of zinc directly binds FVa and FXa and inhibits their combined activity. C) Protein S serves as a cofactor for Activated Protein C and enhances the cleavage of FVIIIa and FVa. D) Protein S enhances TFPI-FX complex formation by serving as a cofactor for TFPI; the TFPI-FX complex inhibits TF- FVIIa.
Co-enzymatic function of Protein S
Protein S is reported to be a cofactor for activated protein C (APC) and for Tissue Factor Pathway Inhibitor (TFPI) [10]. Protein S enhances the function of APC by a factor of 3–10, and PS stimulates the activity of TFPI by a factor of 3–5 [11–13].
Cofactor for Activated Protein C
Protein C is a zymogen, which is activated on the surface of endothelial cell membranes by cleavage by thrombin in the presence of thrombomodulin [14]. Activation of Protein C results in a negative feedback mechanism that regulates the coagulation cascade, because Activated Protein C (APC) inhibits Factor Va (FVa) and Factor Villa (FVIIIa) by cleavage [15–17]. APC cleaves the FVa heavy chain at Arg506, producing a partially inactive intermediate; further cleavage of FVa at Arg306 results in complete inactivation [16, 17].
When not bound to PS, the proteolytic activity of free APC is weaker [18]. Studies of the interaction between PS and APC have revealed that the PS TSR, EGF1, and EGF2 domains are essential for its APC cofactor function; PS lacking its LG domains still functions as an APC cofactor in vitro but not in plasma [20]. Furthermore, during coagulation, the APC cofactor activity of PS is down- regulated by FXa-mediated cleavage of PS within the TSR [19]. Ultimately the significance of the cofactor function of PS in regulating APC activity is undermined by the fact that APC can function without PS.
The 40% of free plasma PS was considered to be the only PS form that could act as a cofactor for APC, but later studies showed that PS bound to C4BP could also function in this manner [21]. The PS-C4BP complex is a weak cofactor for APC in vitro, whereas the same complex acts more effectively in plasma. By demonstrating that PS-Heerlen mutant (Ser460Pro) disrupts a consensus glycosylation site of PS, thereby destabilizing its free form, it was shown that the PS-Heerlen mutant bound to C4BP is stable and continues to retain the anticoagulant function of PS. Further work also challenged the APC cofactor function of PS in baboon models, wherein antibody-mediated blockage of APC did not inhibit the antithrombotic function of PS [22]; this suggested that PS functions in ways other than as a cofactor of APC.
PS as a cofactor for Tissue Factor Pathway Inhibitor (TFPI)
Tissue Factor Pathway Inhibitor is a single-chain glycoprotein with Kunitz- type proteinase inhibitor domains; TFPI circulates at trace amounts in plasma (2.5 nM) [23]. Tissue Factor Pathway Inhibitor employs a two-step mechanism to modulate the tissue factor-mediated coagulation cascade [24]. The Kunitz-2 domain of TFPI binds directly to the active site of FXa, and this complex forms a quaternary complex with a TF-FVIIa complex [24]. PS interaction with the Kunitz- 3 domain and/or the C-terminal tail of TFPI enhances the TFPI and FXa interaction in the initiation phase of coagulation. However, the PS-TFPI complex inhibits only the FXa that escapes the VIIa-TF-Xa-TFPI complex, [25, 26].
Moreover, cofactor activity of PS in regulating TFPI could not be verified in vivo since mouse plasma lacks TFPI.
Direct inhibition of Protein S with procoagulants and their active forms
Plasma contains a number of procoagulants and their cofactors, which are vital for proper clot formation [27, 28]. FIX, FVII and FX are the major procoagulant zymogens, whereas FVa and FVIIIa act as cofactors for FXa and FIXa, respectively [29–33]. Recent evidence shows that PS can interact directly with some procoagulants and their cofactors, thereby inhibiting thrombin generation and clot formation [34–38].
PS interaction with FV, FVa and FXa
Factor V is a 330-kDa single-chain polypeptide secreted by hepatocytes, circulating in plasma as a procoagulant at a concentration of 20 nM [32, 33].
Thrombin activates FV to FVa by cleaving at FV Arg709, Arg1018, and Arg1545 [32, 39–41]. Factor Va binds with high affinity to FXa and forms the prothrombinase complex [41]. Interestingly, the zymogen form of FV can act as an anticoagulant by forming a complex with Protein S to enhance the PS-APC-mediated proteolytic inactivation of FVa [38]. In addition to working cooperatively with FV, Heeb et al., demonstrated that FVa directly binds in a calcium-dependent manner to immobilized zinc-containing PS through the heavy chain of FVa, with a Kd of 33 nM [36].
Factor X is a 59-kDa glycoprotein composed of a Gla domain, two EGF- like domains, an activation peptide, and a catalytic domain [42, 43]. Factor X circulates in the plasma at a concentration ~130 nM [42, 43] and upon clotting, FIXa-FVIIIa or FVII-TF complexes activate FX [43–45]. Upon activation, Factor Xa binds to FVa and this ‘prothrombinase’ complex converts prothrombin to thrombin [46, 47]. Heeb et al., demonstrated that FX can directly interact with immobilized, zinc-containing PS; furthermore, competitive binding studies demonstrated that zinc-containing PS inhibits FX with a Ki of ~25 nM [37].
However, physiologically, zinc is not associated with plasma PS, and a novel isolation procedure was used to prepare the zinc-containing Protein S, suggesting that the physiological importance of zinc-containing PS requires further evaluation [37, 48].
PS directly interacts with FIXa and inhibits its activity
Factor IX is a 55-kDa single-chain polypeptide zymogen that circulates in the plasma at a concentration of 70–90 nM and is activated by FXIa [49, 50]. In the presence of FVIIIa, FIXa activates FX to FXa, and this activation step plays a key role in the intrinsic pathway of blood coagulation [28].
Chattopadhyay et al., demonstrated that FIXa in the absence or presence of FVIIIa interacts with PS with a Kd of 40 nM, and this interaction inhibits the activation of FX. Low levels (0.4 pM) of TF directs thrombin generation through the intrinsic pathway, and thrombin generation is inhibited (~92%) by the addition of PS to the PS-deficient plasma, revealing the physiological significance of PS- mediated FIX inhibition [51]. Further, Chattopadhyay et al., showed that PS inhibits the intrinsic Xase complex (FIXa-FVIIIa complex) by inhibiting FIXa in the presence or absence of FVIIIa [51]. This work explained the anticoagulant mechanism of PS in inhibiting one of the most important complexes in blood coagulation, the intrinsic Xase.
PS-C4BP COMPLEX
Complement component 4 binding protein is a 570-kDa glycoprotein that circulates in the plasma with a concentration of ~350 nM [4, 52]. C4BP exists in several isoforms that differ in their combination of alpha and β chains; ~80% of circulating C4BP consists of seven identical alpha chains (70 kDa) and one β chain (45 kDa) [52]; C4BP forms a high affinity complex (Kd ~ 100 pM) with PS in 1:1 stoichiometric ratio by means of an interaction mediated by its β-chain [4, 53, 54].
The PS-C4BP complex links the coagulation cascade and the inflammation pathway by interfering with the anticoagulant function of PS by modulating its APC cofactor affinity [4, 53, 54]. PS enhances C4BP binding to the phosphatidylserine moieties of apoptotic cell membranes, by means of its Gla domain; this results in the inhibition of inflammation in the vicinity of dying cells (Figure 2) [4, 53, 54].
Figure 2.
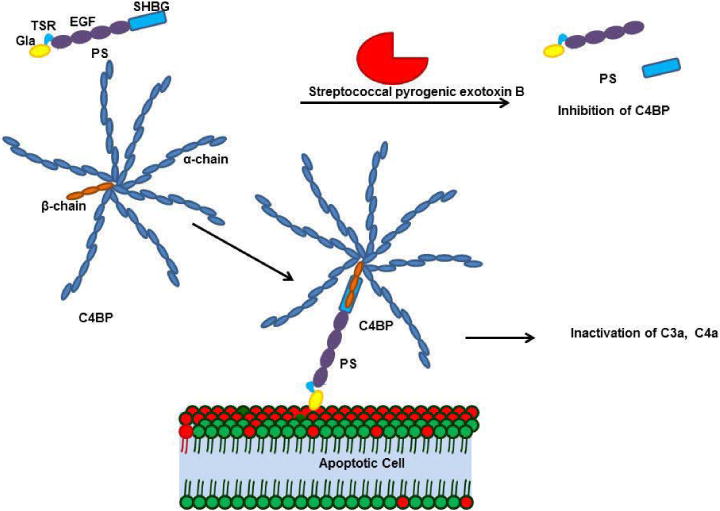
The PS-C4BP complex is essential for inhibiting inflammation. C4BP is a multimer formed by seven alpha chains and one beta chain crosslinked by cysteine disulfide bonds; the C4BP beta chain binds to the sex hormone binding globulin domain (SHBG) of PS. PS-C4BP further binds to the phosphatidylserine in the apoptotic cell membrane and inactivates complements such as C3a and C4a. Streptococcal pyrogenic exotoxin B (SPEB) inactivates the PS-C4BP complex during streptococcal infection. SPEB cleaves PS within the SHBG domain, preventing the PS-C4BP complex from binding the membrane.
PROTEIN S IN THE TYRO-3, AXL, MER (TAM) FAMILY RECEPTOR SIGNALING CASCADE
Tyro-3, Axl, and Mer receptors are tyrosine kinases that are grouped in a family based on their sequence similarities [61]. These receptors share a common architecture having N-terminal immunoglobulin-like (IG) domains followed by fibronectinlike domains and a single-pass alpha helical domain [61]. The receptors also contain an extracellular domain that binds ligands such as PS and the growth arrest-specific 6 (GAS-6) protein [62]. TAM family receptors are expressed in a wide variety of cells, including cells of the immune, nervous, reproductive, and vascular systems [2]. These receptors transduce a wide range of signals, e.g., signals for cell survival, metastasis, apoptosis, and differentiation. PS-TAM receptor signaling is involved in processes such as regulation of inflammation, vasculogenesis, angiogenesis, and metastasis (Figure 4) [63–67].
Figure 4.
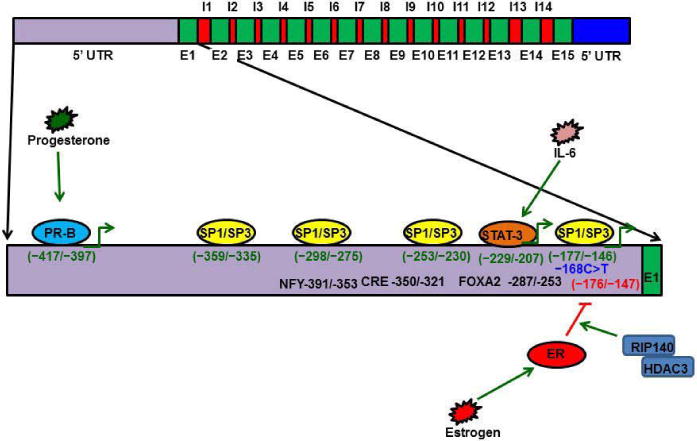
Protein S gene regulation. The PS gene is composed of 15 exons and 14 introns. The PS promoter has four Sp1 and Sp3 transcription factor binding sites (−359/−335; −298/−275; −253/−230; −177/−146). A C>T mutation at −168 has been identified in a PS-deficient individual, and later studies have reported that this −168 substitution disrupts Sp1 binding and reduces PS expression. During inflammation, IL-6 promotes PS expression through the STAT-3 transcription factor. STAT3 binds the PS promoter at a consensus sequence located at −229/−207. Progesterone promotes PS expression through Progesterone isoform B (PR-B). PR-B binds the PS promoter at −417/−397. Estrogen activates Estrogen receptor (ER) and the activated ER binds to the PS promoter at −176/−146 to recruit RIP-140 and HDAC3, which, in turn, deacetylate histones and inactivate PS gene expression.
The Gla domain of PS binds to the phosphatidylserine present in the membranes of apoptotic cells and, simultaneously, the PS SHBG domain forms a bridge with a TAM receptor on macrophages [68, 69]. This bridge enhances phagocytosis of the apoptotic bodies, thereby preventing exposure of antigens from the apoptotic cells [67–69]. PS-TAM signaling inhibits inflammation by inhibiting p38 mitogen activated protein kinase (MAPK), extracellular signal regulated kinase-1 (ERK1)/ERK2, nuclear factor-kB (NFkB), tumor necrosis factor (TNF)-receptor-associated factor 3 (TRAF3) and TRAF6, while also activating protein-1 interferon regulatory factors such as TNFa and IL6 [70]. Toll- like receptor signaling is blocked by inhibition of the activities of all these kinases and factors [2, 71]. TAM signaling-mediated inhibition of inflammation further enhances expression of suppressors of cytokine signaling (SOCSs), and SOCSs play a key role in regulating inflammation [2, 71]. PS-TAM signaling also protects the integrity of the blood-brain barrier (BBB) during ischemic/hypoxic stroke [72]. By activating the PI3/AKT pathway, PS-Tyro-3 signaling protects neurons from excitotoxic injury, and this signaling enhances phosphorylation of antiapoptotic proteins such as Bcl-2 and Bcl-XL [73]. Further, the association of PS deficiency with ischemic stroke indicates the importance of PS in protecting the BBB and neurons [9].
Protein S knockout mice die due to defective extension and remodeling of the primary vascular network, which suggests that PS plays a vital role in vasculogenesis [5]. Similarly, PS-Axl expression in vascular smooth muscle cells (VSMCs) is essential for VEGF-α induced vascular permeability and angiogenesis [74, 75]. In contrast to the role of PS in VSMCs, PS-Mer signaling antagonizes VEGF-α signaling on endothelial cells and further inhibits angiogenesis [76].
The role for PS in tumor metastasis is controversial [67]. PS deficiency caused by an A > G nucleotide substitution at position 1907 is associated with colorectal cancer, whereas PS and TAM receptor expression are unregulated in a variety of cancers [67, 77, 78]. A role for PS over expression in cancer cells is yet to be determined [67].
GENE REGULATION AND PS DEFICIENCY
The PS gene is located on Chromosome 3 at 3q11.2, and the gene spans 101 kb of genomic DNA [79, 80]. The PS gene is composed of 15 exons and 14 introns [80, 81]. The 5′ promoter region of the PS gene contains binding sites for transcription factors such as Sp1, estrogen receptor (ER), progesterone isoform B (PR-B), FOXA2, CRE/ATF, NFY, STAT3, and C/EBPb (Figure 3) [67, 82–86].
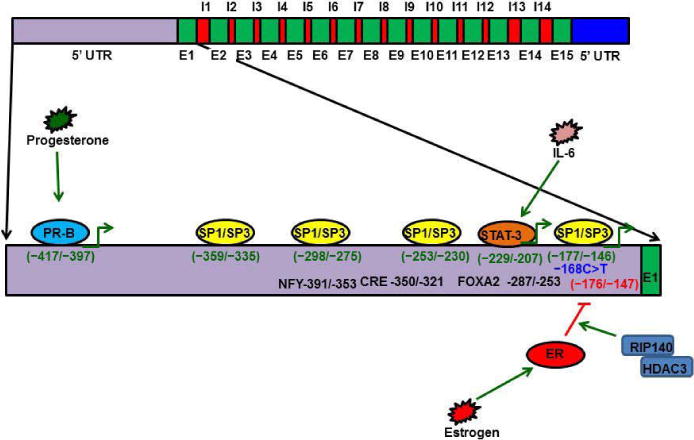
Figure 3.
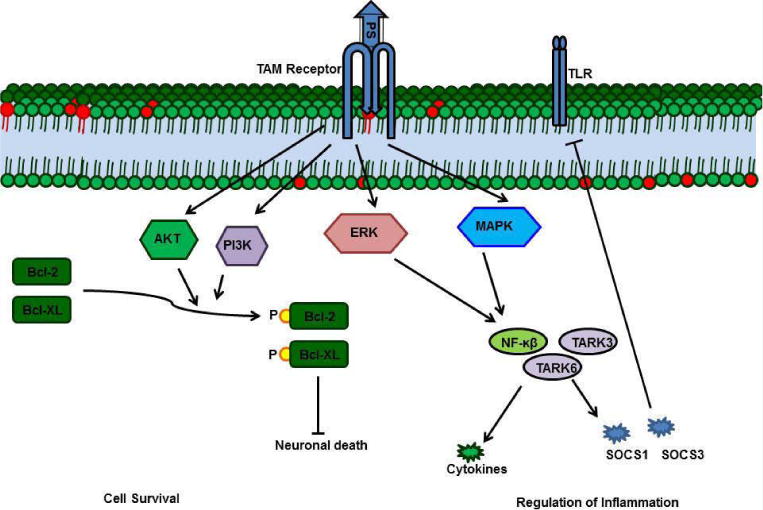
Functions of PS-TAM receptor signaling. A) The Gla domain of PS interacts with phosphatidylserine on the apoptotic cell membrane, and the SHBG domain of PS forms a crosslink with a TAM receptor on a macrophage. This crosslink promotes phagocytosis of the apoptotic cell.
PS-TAM signaling activates the MERK/ERK pathway, which, in turn, activates NFkb, TARK6, and TARK3 to promote expression of cytokines and suppressors of cytokine signaling (SOCSs). Enhanced SOCSs expression blocks Toll-like receptors. In neurons, PS-TAM signaling activates the PI3 and Akt pathways, which, in turn, phosphorylate Bcl2 and Bcl-XL; phosphorylated Bcl2 and Bcl-XL promote cell survival.
PS-Axl enhances VEGFla signaling and enhances angiogenesis in vascular smooth muscle cells, whereas PS-MER antagonizes VEGF signaling in endothelial cells.
Transcription factor Sp1 has four binding sites within 370 bp proximal to the PS promoter [83]. Sp3 also binds to the Sp1 DNA consensus sequence of the PS gene promoter [83]. Thus, it has been hypothesized that dysregulation of Sp1 in cancer cells causes PS over expression [83, 86, 87].
PS is also regulated by hormones such as progesterone and estrogen [87]. Progesterone promotes PS expression by 25% [67, 84], while Estrogen down-regulates PS expression by promoting ER binding to promoter-distal GC-rich motifs of the PS gene [85]. ER binding to the GC rich motif recruits the RIP140 and HDA3 complexes that deacetylate histones; in turn, expression of PS ceases, and this repression is responsible for the deep vein thrombosis (DVT) that occurs in pregnant women and women using estrogen-containing contraceptives [67, 85]. PR-B promotes PS expression by binding directly to the PS promoter at a consensus sequence between bp −397 and −417 [84].
Interleukins play major roles in regulating inflammation. Interleukin 6 (IL6) stimulates transcription factor STAT3; STAT3 binds to the PS promoter at −220 bp and promotes PS expression, which plays a vital role in regulating inflammation [67, 88]. PS expression and function are altered by mutations in the PS gene [79].
To date, more than 200 mutations in PS have been mapped; these mutations have been found to cause three classes of PS deficiency [89, 90]. In type I deficiency, the total amount of serum PS is decreased, in type II deficiency, the total PS amount is unchanged, but the APC cofactor function of PS is altered, and, in type III deficiency, the total PS amount is unaltered, but both free PS and its cofactor activity are decreased [79].
Several mutations in the PS gene, which are responsible for PS deficiency and altered PS function, are shown in Figure (5). A C >T mutation at −168 in the promoter region alters Sp1 transcription factor binding, which is responsible for a 38% reduction in free PS and a 68% reduction in total PS [91]. Mutations pPro15His and pVal18Glu in the signal peptide prevent PS secretion and lead to degradation of PS in the cytosol [92, 93]. Mutations in the propeptide (Arg40Leu and Arg40Leu) cause type II PS deficiency because the mutations prevent maturation of the protein [94]. Mutations in the Gla domain alter PS function and cause misfolding of the protein, thereby destabilizing PS [79]. An example of a Gla domain mutation is Phe72Cys that creates a destabilization cavity in the Gla domain and promotes PS degradation [95–97]. The Glu67Ala mutation prevents carboxylation of Glu and causes type I PS deficiency [96]. The pGly52Asp mutation causes a 5-fold loss of affinity for phospholipids and a similar reduction in PS anticoagulant activity [98]. Similarly, the pAla68Asp mutation causes loss of C2+-induced phospholipid interaction [96]. Mutations Arg111Ser, Arg101Cys, p.Arg90His, and Arg90Cys prevent thrombin-mediated cleavage of the PS TSR, and these mutations impair the APC cofactor activity of PS [19, 95, 99, 100].
Mutations
Cys121Tyr, Cys154Phe, Cys175Phe, Cys186Tyr, Cys228Ser, Cys241Ser, Cys265Trp, and Cys267Ser cause destabilization of PS by impairing disulfide bond formation in the EGF domains [79]. The Asn258Ser mutation impairs Ca2+ binding to the EGF1 domain, causing misfolding of the protein such that PS secretion is reduced by 70%. Similarly, the APC cofactor function of PS is reduced by 50% in Glu204Gly, Asp245Gly, and Thr144Asn mutants [101].
Mutations in the LG domains are also common; for example, mutations in Cys288–Cys568 of the LG domains destabilize PS and lead to impaired secretion of PS [79].
CONCLUSION
We began this review by noting that the "S" in Protein S stands for the city in which the protein was discovered. We conclude this review suggesting that "S" also stands for survival because knockout of the PS gene is embryonic lethal, and mutations in the PS gene cause pathological conditions such as DVT and hereditary ischemic stroke. Protein S is involved in many cellular activities, including inhibition of the blood coagulation cascade, inhibition of inflammation, clearance of apoptotic bodies, angiogenesis and cell survival. Protein S functions as an anticoagulant by inhibiting FIXa and by serving as a cofactor for APC and TFPI. Although the minimal domain(s) of PS required for its anticoagulant function is/are not known, identification of such a minimal domain will enable pursuit of therapy for thrombophilic patients. PS-Axl enhances VEGF signaling in VSMCs, whereas PS-Mer antagonizes the VEGF signaling in endothelial cells; these opposite effects highlight the existence of functional diversity in PS-TAM receptor signaling. This diversity is waiting to be probed in greater detail.
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute, National Institutes of Health grant, 5R01HL118557-02 (R.M).
ABBREVIATIONS
- SHBG
Sex Hormone Binding Globule
- TFPI
Tissue Factor Pathway Inhibitor
- APC
Activated Protein C PS- Protein S
- TAM
Tyro-3-Axl, Mer
- VSMCs
Vascular Smooth Muscle Cells C4BP-C4b Binding Protein
References
- 1.Di Scipio RG, Hermodson MA, Yates SG, Davie EW. A comparison of human prothrombin, factor IX (Christmas factor), factor X (Stuart factor), and protein S. Biochemistry. 1977;16:698–706. doi: 10.1021/bi00623a022. [DOI] [PubMed] [Google Scholar]
- 2.Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–336. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stenflo J. Contributions of Gla and EGF-like domains to the function of vitamin K-dependent coagulation factors. Crit Rev Eukaryot Gene Expr. 1999;9:59–88. [PubMed] [Google Scholar]
- 4.Dahlbäck B. The tale of protein S and C4b-binding protein, a story of affection. Thromb Haemost. 2007;98:90–96. [PubMed] [Google Scholar]
- 5.Burstyn-Cohen T, Heeb MJ, Lemke G. Lack of protein S in mice causes embryonic lethal coagulopathy and vascular dysgenesis. J Clin Invest. 2009;119:2942–2953. doi: 10.1172/JCI39325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comp PC, Esmon CT. Recurrent venous thromboembolism in patients with a partial deficiency of protein S. N Engl J Med. 1984;311:1525–1528. doi: 10.1056/NEJM198412133112401. [DOI] [PubMed] [Google Scholar]
- 7.Comp PC, Nixon RR, Cooper MR, Esmon CT. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest. 1984;74:2082–2088. doi: 10.1172/JCI111632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayakawa T, Morimoto A, Nozaki Y, Kashii Y, Aihara T, Maeda K, et al. Mesenteric venous thrombosis in a child with type 2 protein S deficiency. J Pediatr Hematol Oncol. 2011;33:141–143. doi: 10.1097/MPH.0b013e3181fce4d4. [DOI] [PubMed] [Google Scholar]
- 9.Wang ZH, Zhao ZJ, Xu K, Sun GB, Song L, Yin HX, et al. Hereditary protein S deficiency leads to ischemic stroke. Mol Med Rep. 2015;12:3279–3284. doi: 10.3892/mmr.2015.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castoldi E, Hackeng TM. Regulation of coagulation by protein S. Curr Opin Hematol. 2008;15:529–536. doi: 10.1097/MOH.0b013e328309ec97. [DOI] [PubMed] [Google Scholar]
- 11.Hackeng TM, Seré KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci USA. 2006;103:3106–3113. doi: 10.1073/pnas.0504240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosing J, Hoekema L, Nicolaes GA, Thomassen MC, Hemker HC, Varadi K, et al. Effects of protein S and factor Xa on peptide bond cleavages during inactivation of factor Va and factor VaR506Q by activated protein C. J Biol Chem. 1995;270:27852–27858. doi: 10.1074/jbc.270.46.27852. [DOI] [PubMed] [Google Scholar]
- 13.Bakker HM, Tans G, Janssen-Claessen T, Thomassen MC, Hemker HC, Griffin JH, et al. The effect of phospholipids, calcium ions and protein S on rate constants of human factor Va inactivation by activated human protein C. Eur J Biochem. 1992;208:171–178. doi: 10.1111/j.1432-1033.1992.tb17171.x. [DOI] [PubMed] [Google Scholar]
- 14.Esmon CT. The protein C pathway. Chest. 2003;124:26–32. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 15.Fay PJ, Smudzin TM, Walker FJ. Activated protein C-catalyzed inactivation of human factor VIII and factor VIIIa. Identification of cleavage sites and correlation of proteolysis with cofactor activity. J Biol Chem. 1991;266:20139–20145. [PubMed] [Google Scholar]
- 16.Kalafatis M, Rand MD, Mann KG. The mechanism of inactivation of human factor V and human factor Va by activated protein C. J Biol Chem. 1994;269:31869–31880. [PubMed] [Google Scholar]
- 17.Nicolaes GA, Tans G, Thomassen MC, Hemker HC, Pabinger I, Varadi K, et al. Peptide bond cleavages and loss of functional activity during inactivation of factor Va and factor VaR506Q by activated protein C. J Biol Chem. 1995;270:21158–21166. doi: 10.1074/jbc.270.36.21158. [DOI] [PubMed] [Google Scholar]
- 18.Walker FJ. Regulation of activated protein C by a new protein. A possible function for bovine protein S. J Biol Chem. 1980;255:5521–5524. [PubMed] [Google Scholar]
- 19.Villoutreix BO, Teleman O, Dahlbäck B. A theoretical model for the Gla-TSR-EGF-1 region of the anticoagulant cofactor protein S: from biostructural pathology to species-specific cofactor activity. J Comput Aided Mol Des. 1997;11:293–304. doi: 10.1023/a:1007912929828. [DOI] [PubMed] [Google Scholar]
- 20.Van Wijnen M, Stam JG, Chang GT, Meijers JC, Reitsma PH, Bertina RM, et al. Characterization of mini-protein S, a recombinant variant of protein S that lacks the sex hormone binding globulin-like domain. Biochem J. 1998;330:389–396. doi: 10.1042/bj3300389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maurissen LF, Thomassen MC, Nicolaes GA, Dahlbäck B, Tans G, Rosing J, et al. Re-evaluation of the role of the protein S-C4b binding protein complex in activated protein C-catalyzed factor Va-inactivation. Blood. 2008;111:3034–3041. doi: 10.1182/blood-2007-06-089987. [DOI] [PubMed] [Google Scholar]
- 22.Heeb MJ, Marzec U, Gruber A, Hanson SR. Antithrombotic activity of protein S infused without activated protein C in a baboon thrombosis model. Thromb Haemost. 2012;107:690–698. doi: 10.1160/TH11-10-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broze GJ. Tissue factor pathway inhibitor. Thromb Haemost. 1995;74:90–93. [PubMed] [Google Scholar]
- 24.Girard TJ, Warren LA, Novotny WF, Likert KM, Brown SG, Miletich JP, et al. Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature. 1989;338:518–520. doi: 10.1038/338518a0. [DOI] [PubMed] [Google Scholar]
- 25.Hackeng TM, Maurissen LF, Castoldi E, Rosing J. Regulation of TFPI function by protein S. J Thromb Haemost. 2009;7:165–168. doi: 10.1111/j.1538-7836.2009.03363.x. [DOI] [PubMed] [Google Scholar]
- 26.Hackeng TM, Rosing J. Protein S as cofactor for TFPI. Arterioscler Thromb Vasc Biol. 2009;29:2015–2020. doi: 10.1161/ATVBAHA.108.177436. [DOI] [PubMed] [Google Scholar]
- 27.Green D. Coagulation cascade. Hemodial Int. 2006;10:2–4. doi: 10.1111/j.1542-4758.2006.00119.x. [DOI] [PubMed] [Google Scholar]
- 28.Palta S, Saroa R, Palta A. Overview of the coagulation system. Indian J Anaesth. 2014;58:515–523. doi: 10.4103/0019-5049.144643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bardelle C, Furie B, Furie BC, Gilbert GE. Membrane binding kinetics of factor VIII indicate a complex binding process. J Biol Chem. 1993;268:8815–8824. [PubMed] [Google Scholar]
- 30.Giles AR, Mann KG, Nesheim ME. A combination of factor Xa and phosphatidylcholine-phosphatidylserine vesicles bypasses factor VIII in vivo. Br J Haematol. 1988;69:491–497. doi: 10.1111/j.1365-2141.1988.tb02405.x. [DOI] [PubMed] [Google Scholar]
- 31.Dahlbäck B. Blood coagulation. Lancet. 2000;355:1627–1632. doi: 10.1016/S0140-6736(00)02225-X. [DOI] [PubMed] [Google Scholar]
- 32.Dahlbäck B. Pro- and anticoagulant properties of factor V in pathogenesis of thrombosis and bleeding disorders. Int J Lab Hematol. 2016;38:4–11. doi: 10.1111/ijlh.12508. [DOI] [PubMed] [Google Scholar]
- 33.Segers K, Dahlbäck B, Nicolaes GA. Coagulation factor V and thrombophilia: background and mechanisms. Thromb Haemost. 2007;98:530–542. [PubMed] [Google Scholar]
- 34.Chang GT, Aaldering L, Hackeng TM, Reitsma PH, Bertina RM, Bouma BN. Construction and characterization of thrombin-resistant variants of recombinant human protein S. Thromb Haemost. 1994;72:693–697. [PubMed] [Google Scholar]
- 35.Hackeng TM, van't Veer C, Meijers JC, Bouma BN. Human protein S inhibits prothrombinase complex activity on endothelial cells and platelets via direct interactions with factors Va and Xa. J Biol Chem. 1994;269:21051–21058. [PubMed] [Google Scholar]
- 36.Heeb MJ, Mesters RM, Tans G, Rosing J, Griffin JH. Binding of protein S to factor Va associated with inhibition of prothrombinase that is independent of activated protein C. J Biol Chem. 1993;268:2872–2877. [PubMed] [Google Scholar]
- 37.Heeb MJ, Rosing J, Bakker HM, Fernandez JA, Tans G, Griffin JH. Protein S binds to and inhibits factor Xa. Proc Natl Acad Sci USA. 1994;91:2728–2732. doi: 10.1073/pnas.91.7.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nyberg P, Dahlbäck B, García de Frutos P. The SHBG-like region of protein S is crucial for factor V-dependent APC-cofactor function. FEBS Lett. 1998;433:28–32. doi: 10.1016/s0014-5793(98)00877-1. [DOI] [PubMed] [Google Scholar]
- 39.Huang JN, Koerper MA. Factor V deficiency: a concise review. Haemophilia. 2008;14:1164–1169. doi: 10.1111/j.1365-2516.2008.01785.x. [DOI] [PubMed] [Google Scholar]
- 40.Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 41.Steen M, Dahlbäck B. Thrombin-mediated proteolysis of factor V resulting in gradual B-domain release and exposure of the factor Xa-binding site. J Biol Chem. 2002;277:38424–38430. doi: 10.1074/jbc.M204972200. [DOI] [PubMed] [Google Scholar]
- 42.Greipp PR, Kyle RA, Bowie EJ. Factor-X deficiency in amyloidosis: a critical review. Am J Hematol. 1981;11:443–450. doi: 10.1002/ajh.2830110414. [DOI] [PubMed] [Google Scholar]
- 43.Hertzberg M. Biochemistry of factor X. Blood Rev. 1994;8:56–62. doi: 10.1016/0268-960x(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 44.Fujikawa K, Legaz ME, Davie EW. Bovine factors X 1 and X 2 (Stuart factor). Isolation and characterization. Biochemistry. 1972;11:4882–4891. doi: 10.1021/bi00776a002. [DOI] [PubMed] [Google Scholar]
- 45.Fujikawa K, Titani K, Davie EW. Activation of bovine factor X (Stuart factor): conversion of factor Xaalpha to factor Xabeta. Proc Natl Acad Sci USA. 1975;72:3359–3363. doi: 10.1073/pnas.72.9.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lechtenberg BC, Murray-RustTA, Johnson DJ, Adams TE, Krishnaswamy S, Camire RM, et al. Crystal structure of the prothrombinase complex from the venom of Pseudonaja textilis. Blood. 2013;122:2777–2783. doi: 10.1182/blood-2013-06-511733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mann KG, Nesheim ME, Tracy PB, Hibbard LS, Bloom JW. Assembly of the prothrombinase complex. Biophys J. 1982;37:106–107. doi: 10.1016/S0006-3495(82)84624-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pedersen AH, Lund-Hansen T, Komiyama Y, Petersen LC, Oestergård PB, Kisiel W. Inhibition of recombinant human blood coagulation factor VIIa amidolytic and proteolytic activity by zinc ions. Thromb Haemost. 1991;65:528–534. [PubMed] [Google Scholar]
- 49.Di Scipio RG, Kurachi K, Davie EW. Activation of human factor IX (Christmas factor) J Clin Invest. 1978;61:1528–1538. doi: 10.1172/JCI109073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taran LD. Factor IX of the blood coagulation system: a review. Biochemistry (Mosc) 1997;62:685–693. [PubMed] [Google Scholar]
- 51.Chattopadhyay R, Sengupta T, Majumder R. Inhibition of intrinsic Xase by protein S: a novel regulatory role of protein S independent of activated protein C. Arterioscler Thromb Vasc Biol. 2012;32:2387–2393. doi: 10.1161/ATVBAHA.112.250928. [DOI] [PubMed] [Google Scholar]
- 52.Blom AM, Villoutreix BO, Dahlbäck B. Complement inhibitor C4b-binding protein-friend or foe in the innate immune system? Mol Immunol. 2004;40:1333–1346. doi: 10.1016/j.molimm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 53.Dahlbäck B, Stenflo J. High molecular weight complex in human plasma between vitamin K-dependent protein S and complement component C4b-binding protein. Proc Natl Acad Sci USA. 1981;78:2512–2516. doi: 10.1073/pnas.78.4.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hillarp A, Hessing M, Dahlbäck B. Protein S binding in relation to the subunit composition of human C4b-binding protein. FEBS Lett. 1989;259:53–56. doi: 10.1016/0014-5793(89)81492-9. [DOI] [PubMed] [Google Scholar]
- 55.Ermert D, Blom AM. C4b-binding protein: The good, the bad and the deadly. Novel functions of an old friend. Immunol Lett. 2016;169:82–92. doi: 10.1016/j.imlet.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 56.Kask L, Trouw LA, Dahlbäck B, Blom AM. The C4b-binding protein-protein S complex inhibits the phagocytosis of apoptotic cells. J Biol Chem. 2004;279:23869–23873. doi: 10.1074/jbc.C400159200. [DOI] [PubMed] [Google Scholar]
- 57.Webb JH, Villoutreix BO, Dahlback B, Blom AM. Localization of a hydrophobic binding site for anticoagulant protein S on the beta -chain of complement regulator C4b-binding protein. J Biol Chem. 2001;276:4330–4337. doi: 10.1074/jbc.M006541200. [DOI] [PubMed] [Google Scholar]
- 58.Braunschweig A, Józsi M. Human pentraxin 3 binds to the complement regulator c4b-binding protein. PLoS One. 2011;6:23991. doi: 10.1371/journal.pone.0023991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mihlan M, Blom AM, Kupreishvili K, Lauer N, Stelzner K, Bergström F, et al. Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J. 2011;25:4198–4210. doi: 10.1096/fj.11-186460. [DOI] [PubMed] [Google Scholar]
- 60.Chen CL, Wu YY, Lin CF, Kuo CF, Han CL, Wang S. Streptococcal pyrogenic exotoxin B inhibits apoptotic cell clearance by macrophages through protein S cleavage. Sci Rep. 2016;6:26026. doi: 10.1038/srep26026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 62.Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80:661–670. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 63.Angelillo-Scherrer A, Burnier L, Flores N, Savi P, DeMol M, Schaeffer P, et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J Clin Invest. 2005;115:237–246. doi: 10.1172/JCI22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5:009076. doi: 10.1101/cshperspect.a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prasad D, Rothlin CV, Burrola P, Burstyn-Cohen T, Lu Q, Garcia de Frutos P, et al. TAM receptor function in the retinal pigment epithelium. Mol Cell Neurosci. 2006;33:96–108. doi: 10.1016/j.mcn.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 67.Suleiman L, Négrier C, Boukerche H. Protein S: A multifunctional anticoagulant vitamin K-dependent protein at the crossroads of coagulation, inflammation, angiogenesis, and cancer. Crit Rev Oncol Hematol. 2013;88:637–654. doi: 10.1016/j.critrevonc.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 68.Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, Shacter E. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol. 2003;4:87–91. doi: 10.1038/ni871. [DOI] [PubMed] [Google Scholar]
- 69.Uehara H, Shacter E. Auto-oxidation and oligomerization of protein S on the apoptotic cell surface is required for Mer tyrosine kinasemediated phagocytosis of apoptotic cells. J Immunol. 2008;180:2522–2530. doi: 10.4049/jimmunol.180.4.2522. [DOI] [PubMed] [Google Scholar]
- 70.Deng T, Zhang Y, Chen Q, Yan K, Han D. Toll-like receptor-mediated inhibition of Gas6 and ProS expression facilitates inflammatory cytokine production in mouse macrophages. Immunology. 2012;135:40–50. doi: 10.1111/j.1365-2567.2011.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 72.Zhu D, Wang Y, Singh I, Bell RD, Deane R, Zhong Z, et al. Protein S controls hypoxic/ischemic blood-brain barrier disruption through the TAM receptor Tyro3 and sphingosine 1-phosphate receptor. Blood. 2010;115:4963–4972. doi: 10.1182/blood-2010-01-262386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhong Z, Wang Y, Guo H, Sagare A, Fernández JA, Bell RD, et al. Protein S protects neurons from excitotoxic injury by activating the TAM receptor Tyro3-phosphatidylinositol 3-kinase-Akt pathway through its sex hormone-binding globulin-like region. J Neurosci. 2010;30:15521–15534. doi: 10.1523/JNEUROSCI.4437-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benzakour O, Formstone C, Rahman S, Kanthou C, Dennehy U, Scully MF, et al. Evidence for a protein S receptor(s) on human vascular smooth muscle cells. Analysis of the binding characteristics and mitogenic properties of protein S on human vascular smooth muscle cells. Biochem J. 1995;308:481–485. doi: 10.1042/bj3080481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gasic GP, Arenas CP, Gasic TB, Gasic GJ. Coagulation factors X, Xa, and protein S as potent mitogens of cultured aortic smooth muscle cells. Proc Natl Acad Sci USA. 1992;89:2317–2320. doi: 10.1073/pnas.89.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fraineau S, Monvoisin A, Clarhaut J, Talbot J, Simonneau C, Kanthou C, et al. The vitamin K-dependent anticoagulant factor, protein S, inhibits multiple VEGF-A-induced angiogenesis events in a Mer- and SHP2-dependent manner. Blood. 2012;120:5073–5083. doi: 10.1182/blood-2012-05-429183. [DOI] [PubMed] [Google Scholar]
- 77.Ji M, Yoon SN, Lee W, Jang S, Park SH, Kim DY, et al. Protein S deficiency with a PROS1 gene mutation in a patient presenting with mesenteric venous thrombosis following total colectomy. Blood Coagul Fibrinolysis. 2011;22:619–621. doi: 10.1097/MBC.0b013e32834a0421. [DOI] [PubMed] [Google Scholar]
- 78.Saraon P, Musrap N, Cretu D, Karagiannis GS, Batruch I, Smith C, et al. Proteomic profiling of androgen-independent prostate cancer cell lines reveals a role for protein S during the development of high grade and castration-resistant prostate cancer. J Biol Chem. 2012;287:34019–34031. doi: 10.1074/jbc.M112.384438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.García de Frutos P, Fuentes-Prior P, Hurtado B, Sala N. Molecular basis of protein S deficiency. Thromb Haemost. 2007;98:543–556. [PubMed] [Google Scholar]
- 80.Ploos van Amstel JK, van der Zanden AL, Bakker E, Reitsma PH, Bertina RM. Two genes homologous with human protein S cDNA are located on chromosome 3. Thromb Haemost. 1987;58:982–987. [PubMed] [Google Scholar]
- 81.Ploos van Amstel HK, Reitsma PH, Bertina RM. The human protein S locus: identification of the PS alpha gene as a site of liver protein S messenger RNA synthesis. Biochem Biophys Res Commun. 1988;157:1033–1038. doi: 10.1016/s0006-291x(88)80978-1. [DOI] [PubMed] [Google Scholar]
- 82.Blanco-Molina A, Monreal M. Venous thromboembolism in women taking hormonal contraceptives. Expert Rev Cardiovasc Ther. 2010;8:211–215. doi: 10.1586/erc.09.175. [DOI] [PubMed] [Google Scholar]
- 83.de Wolf CJ, Cupers RM, Bertina RM, Vos HL. The constitutive expression of anticoagulant protein S is regulated through multiple binding sites for Sp1 and Sp3 transcription factors in the protein S gene promoter. J Biol Chem. 2006;281:17635–17643. doi: 10.1074/jbc.M603094200. [DOI] [PubMed] [Google Scholar]
- 84.Hughes Q, Watson M, Cole V, Sayer M, Baker R, Staton J. Upregulation of protein S by progestins. J Thromb Haemost. 2007;5:2243–2249. doi: 10.1111/j.1538-7836.2007.02730.x. [DOI] [PubMed] [Google Scholar]
- 85.Suzuki A, Sanda N, Miyawaki Y, Fujimori Y, Yamada T, Takagi A, et al. Down-regulation of PROS1 gene expression by 17beta-estradiol via estrogen receptor alpha (ERalpha)-Sp1 interaction recruiting receptor-interacting protein 140 and the corepressor-HDAC3 complex. J Biol Chem. 2010;285:13444–13453. doi: 10.1074/jbc.M109.062430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tatewaki H, Tsuda H, Kanaji T, Yokoyama K, Hamasaki N. Characterization of the human protein S gene promoter: a possible role of transcription factors Sp1 and HNF3 in liver. Thromb Haemost. 2003;90:1029–1039. doi: 10.1160/TH03-07-0443. [DOI] [PubMed] [Google Scholar]
- 87.Sankpal UT, Goodison S, Abdelrahim M, Basha R. Targeting Sp1 transcription factors in prostate cancer therapy. Med Chem. 2011;7:518–525. doi: 10.2174/157340611796799203. [DOI] [PubMed] [Google Scholar]
- 88.Sriuranpong V, Park JI, Amornphimoltham P, Patel V, Nelkin BD, Gutkind JS. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003;63:2948–2956. [PubMed] [Google Scholar]
- 89.Gandrille S, Borgel D, Sala N, Espinosa-Parrilla Y, Simmonds R, Rezende S, et al. Protein S deficiency: a database of mutations–summary of the first update. Thromb Haemost. 2000;84:918. [PubMed] [Google Scholar]
- 90.Jang MA, Kim SH, Kim DK, Kim HJ. A novel nonsense mutation Tyr301* of PROS1 causing protein S deficiency. Blood Coagul Fibrinolysis. 2015;26:223–224. doi: 10.1097/MBC.0000000000000217. [DOI] [PubMed] [Google Scholar]
- 91.Sanda N, Fujimori Y, Kashiwagi T, Takagi A, Murate T, Mizutani E, et al. An Sp1 binding site mutation of the PROS1 promoter in a patient with protein S deficiency. Br J Haematol. 2007;138:663–665. doi: 10.1111/j.1365-2141.2007.06668.x. [DOI] [PubMed] [Google Scholar]
- 92.Espinosa-Parrilla Y, Morell M, Borrell M, Souto JC, Fontcuberta J, Estivill X, et al. Optimization of a simple and rapid single-strand conformation analysis for detection of mutations in the PROS1 gene: identification of seven novel mutations and three novel, apparently neutral, variants. Hum Mutat. 2000;15:463–473. doi: 10.1002/(SICI)1098-1004(200005)15:5<463::AID-HUMU8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 93.Rezende SM, Lane DA, Zoller B, Mille-Baker B, Laffan M, Dalhback B, et al. Genetic and phenotypic variability between families with hereditary protein S deficiency. Thromb Haemost. 2002;87:258–265. doi: 10.1055/s-0037-1612982. [DOI] [PubMed] [Google Scholar]
- 94.Gandrille S, Aiach M. Identification of mutations in 90 of 121 consecutive symptomatic French patients with a type I protein C deficiency. The French INSERM Network on Molecular Abnormalities Responsible for Protein C and Protein S deficiencies. Blood. 1995;86:2598–2605. [PubMed] [Google Scholar]
- 95.Biguzzi E, Razzari C, Lane DA, Castaman G, Cappellari A, Bucciarelli P, et al. Molecular diversity and thrombotic risk in protein S deficiency: the PROSIT study. Hum Mutat. 2005;25:259–269. doi: 10.1002/humu.20136. [DOI] [PubMed] [Google Scholar]
- 96.Razzari C, Franchi F, Biguzzi E. PROSIT. Protein S binding to phospholipids: evaluation of eight variants of recombinant human protein S from the PROSIT study. J Thromb Haemost. 2006;4:273–274. doi: 10.1111/j.1538-7836.2005.01739.x. [DOI] [PubMed] [Google Scholar]
- 97.Simmonds RE, Ireland H, Kunz G, Lane DA. Identification of 19 protein S gene mutations in patients with phenotypic protein S deficiency and thrombosis. Protein S Study Group. Blood. 1996;88:4195–4204. [PubMed] [Google Scholar]
- 98.Rezende SM, Lane DA, Mille-Baker B, Samama MM, Conard J, Simmonds RE. Protein S Gla-domain mutations causing impaired Ca(2+)-induced phospholipid binding and severe functional protein S deficiency. Blood. 2002;100:2812–2819. doi: 10.1182/blood-2002-03-0909. [DOI] [PubMed] [Google Scholar]
- 99.Boinot C, Borgel D, Kitzis A, Guicheteau M, Aiach M, Alhenc-Gelas M. Familial thrombophilia is an oligogenetic disease: involvement of the prothrombin G20210A, PROC and PROS gene mutations. Blood Coagul Fibrinolysis. 2003;14:191–196. doi: 10.1097/01.mbc.0000046180.72384.39. [DOI] [PubMed] [Google Scholar]
- 100.Franchi F, Viscardi Y, Razzari C, Faioni EM, Bonara P, Biguzzi E, et al. c.301C > T (p.Arg101Cys): a novel mutation in the thrombin-sensitive region of protein S associated with a dysfunctional protein. Thromb Haemost. 2006;96:381–383. doi: 10.1160/TH06-01-0053. [DOI] [PubMed] [Google Scholar]
- 101.Persson KE, Stenflo J, Linse S, Stenberg Y, Preston RJ, Lane DA, et al. Binding of calcium to anticoagulant protein S: role of the fourth EGF module. Biochemistry. 2006;45:10682–10689. doi: 10.1021/bi0601151. [DOI] [PMC free article] [PubMed] [Google Scholar]