

Abstract
Glycan permethylation was introduced as a tool to facilitate the study of glycans in 1903. Since that time, permethylation procedures have been continually modified to improve permethylation efficiency and qualitative applicability. Typically, however, either laborious preparation steps or cumbersome and uneconomical spin columns have been needed to obtain decent permethylation yields on small glycan samples. Here we describe a spin column-free (SCF) glycan permethylation procedure that is applicable to both O- and N-linked glycans and can be employed upstream to intact glycan analysis by MALDI-MS, ESI-MS, or glycan linkage analysis by GC-MS. The SCF procedure involves neutralization of NaOH beads by acidified phosphate buffer, which eliminates the risk of glycan oxidative degradation and avoids the use of spin columns. Optimization of the new permethylation procedure provided high permethylation efficiency for both hexose (>98%) and HexNAc (>99%) residues—yields which were comparable to (or better than) those of some widely-used spin column-based procedures. A light vs. heavy labelling approach was employed to compare intact glycan yields from a popular spin-column based approach to the SCF approach. Recovery of intact N-glycans was significantly better with the SCF procedure (p < 0.05), but overall yield of O-glycans was similar or slightly diminished (p < 0.05 for tetrasaccharides or smaller). When the SCF procedure was employed upstream to hydrolysis, reduction and acetylation for glycan linkage analysis of pooled glycans from unfractionated blood plasma, analytical reproducibility was on par with that from previous spin column-based “glycan node” analysis results. When applied to blood plasma samples from stage III–IV breast cancer patients (n = 20) and age-matched controls (n = 20), the SCF procedure facilitated identification of three glycan nodes with significantly different distributions between the cases and controls (ROC c-statistics > 0.75; p < 0.01). In summary, the SCF permethylation procedure expedites and economizes both intact glycan analysis and linkage analysis of glycans from whole biospecimens.
Introduction
Glycans are complex carbohydrates that play important roles in numerous biological processes, such as intercellular trafficking, signal transduction, cell adhesion, endocytosis, immunosurveillance, and immune response initiation.1–3 Recently, much attention has been focused on the structural analysis and quantification of glycans in order to elucidate their biological roles in chronic human diseases, most notably cancer.4–6
Permethylation is a useful derivatization method that confers several advantages with regard to the analysis of glycans—especially when combined with mass spectrometry (MS). It helps facilitate the determination of sequence and composition of monosaccharides in glycans, branching position and interglycosidic linkage information, and the presence of configurational and conformational isomers. Additionally, permethylation stabilizes sialic acid residues in glycan structures, improves measurement sensitivity, enhances separation of glycans by reversed-phase HPLC, and generates more predictable spectral patterns when subjected to tandem mass spectral (MS) analysis.7,8 Moreover, permethylation helps facilitate quantitative reproducibility when glycans are analyzed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS.9
The first carbohydrate permethylation methodology was reported by Purdie and Irvine in 1903.10 Since that time, several improved permethylation procedures have been developed. In 1964, Hakomori introduced a procedure with enhanced permethylation yield compared to previously described procedures by utilizing sodium methylsulfinyl carbanion (Na dimsyl) and iodomethane.11 However, even with some modifications,12–14 the Hakomori procedure was still laborious and hazardous because the dimsyl reagent is extremely sensitive to moisture, air and carbon dioxide. In 1984, to circumvent these problems with the dimsyl reagent, Ciucanu and Kerek developed a procedure based on the reaction of glycans with iodomethane in dimethyl sulfoxide (DMSO) containing powdered sodium hydroxide (NaOH).15 This procedure has been successfully applied in various glycan structural studies and adopted or adapted by numerous laboratories.16 The per-methylation yield of this procedure for glycans containing N-acetylhexosamine (HexNAc) units, however, was not examined—perhaps because, as we have found, the yield of HexNAc residues is rather low with this procedure (unpublished data). More recently, a solid-phase spin column-based permethylation procedure was pioneered by Mechref, Novotny and coworkers7,17 for the analysis of small quantities of glycans, which could not be satisfactorily achieved by the Ciucanu procedure. In this procedure, the reaction of glycans with iodomethane is proposed to take place on the surface of NaOH beads that are packed in microspin columns. Since the solution-phase permethylated glycans are separated from the NaOH beads by centrifugation following the reaction, the issues associated with peeling reactions and oxidative degradation of glycans due to the presence of NaOH during the subsequent liquid/liquid extraction step are avoided. With its high derivatization efficiency and good analytical reproducibility, this procedure has demonstrated its utility for model glycoproteins and glycans derived from biomedical samples—even with low picomole to femtomole sample quantities. N-Glycans, however, still need to be pre-isolated from glycoproteins and complex biological samples prior to permethylation with the spin column procedure. In 2013, we adapted this permethylation method and incorporated trifluoroacetic acid (TFA) hydrolysis, reduction, and acetylation steps, to perform glycan linkage analysis directly from whole biofluids (such as serum and plasma) and homogenized tissue samples (Fig. 1).18,19 With the additional steps, O-linked glycans, N-linked glycans, and glycolipids are all released and ultimately converted into partially methylated alditol acetates (PMAAs, Fig. 2) which are highly amenable to analysis by gas chromatography-mass spectrometry (GC-MS). Relative quantification of the resulting PMAAs (as surrogates for monosaccharide-and-linkage specific “glycan nodes”) facilitates detection of changes in unique glycan features due to disease pathobiology.18 Based on the unique branch points and linkages that they represent, some glycan nodes serve as 1 : 1 molecular surrogates of corresponding glycosyltransferases—the enzymes that facilitate construction of glycan polymers. Moreover, by analytically pooling together each chemically distinct glycan node from across all intact glycans in a complex sample, the approach condenses unique glycan structural features such as “core fucosylation”, “α2–6 sialylation”, “bisecting GlcNAc”, and “β1–6 branching” into single analytical signals (Fig. 1).
Fig. 1.

Conceptual overview of glycan linkage analysis from whole biospecimens. Intact normal and abnormal glycans including O-glycans, N-glycans and glycolipids, are processed and transformed into partially methylated alditol acetates (PMAAs, Fig. 2), each of which corresponds to a particular monosaccharide-and-linkage-specific glycan “node” in the original polymer. As illustrated, analytically pooling together the glycan nodes from amongst all the aberrant intact glycan structures provides a more direct surrogate measurement of abnormal glycosyltransferase activity than any individual intact glycan, while simultaneously converting unique glycan features such as “core fucosylation”, “α2-6 sialylation”, “bisecting GlcNAc”, and “β1–6 branching” into single analytical signals. Actual extracted ion chromatograms from 9 μL blood plasma samples are shown. Numbers adjacent to monosaccharide residues in glycan structures indicate the position at which the higher residue is linked to the lower residue. Figure adapted with permission from C. R. Borges, et al., Anal. Chem., 2013, 85(5), 2927–2936. Copyright 2013 American Chemical Society.
Fig. 2.
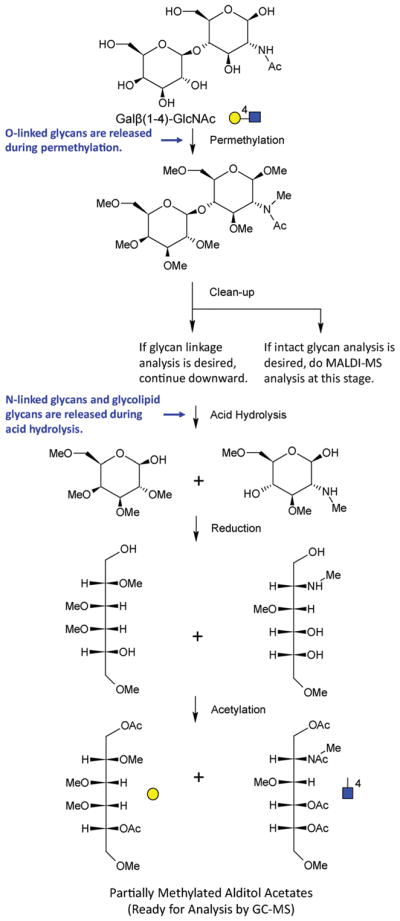
Molecular overview of the glycan linkage analysis procedure. The Gal-4-GlcNAc disaccharide is illustrated. For glycans from blood plasma and other biofluids, O-linked glycans are released during permethylation, while N-linked glycans and glycolipids are released during acid hydrolysis. The unique stereochemistry and pattern of methylation and acetylation in the final partially methylated alditol acetates (PMAAs) corresponds to the unique “glycan node” in the original intact polymer and provides the molecular basis for separation and quantification by GC-MS.
Herein we describe and document the performance characteristics of a new procedure that eliminates the need for a spin column when permethylating glycans from complex biological samples. Glycans containing HexNAc units and small glycan-containing samples from whole biofluids are readily permethylated by the new procedure. Permethylation efficiency is compared to standard spin column-based approaches based on both PMAA yield as well as intact permethylated O- and N-glycan yields. Finally, we evaluated the reproducibility of the spin column-free procedure toward relative quantification of glycan nodes in blood plasma and assessed its clinical performance with regard to its ability to generate glycan nodes that distinguish breast cancer patient plasma from that of healthy, aged-matched controls.
Experimental
Materials and samples
Materials
Galactopyranosyl-β-1,4-N-acetyl-D-glucosamine and 3-O-(2-acetamido-2-deoxy-β-D-galactopyranosyl)-D-galacto-pyranose were obtained from Carbosynth (Compton, Berkshire, UK). Peptide-N-glycosidase F (500 000 units per mL) was acquired from New England Biolabs (Ipswich, MA). Fetuin from fetal bovine serum and Bovine Ribonuclease B were purchased from Sigma-Aldrich (St Louis, MO). Acetone was purchased from Avantor Performance Materials (Center Valley, PA). Methanol was obtained from Honeywell Burdick & Jackson (Muskegon, Ml). Acetonitrile and methylene chloride were acquired from Fisher Scientific (Fair Lawn, NJ). DMSO, iodomethane (99%, Cat. No. I8507), iodomethane-d3 (99%, Cat. No. 176036), chloroform, trifluoroacetic acid (TFA), ammonium hydroxide, sodium borohydride, acetic anhydride, 2,5-dihydroxybenzoic acid (DHB), sodium acetate and sodium hydroxide beads (20–40 mesh, Cat. No. 367176) were purchased from Sigma-Aldrich. Pierce spin columns (900 μL volume) used in the 10 min permethylation procedure were obtained from ThermoFisher Scientific (Waltham, MA, Cat. No. 69705). Micro-spin columns (500 μL volume) for extended spin column permethylation procedure were purchased from Harvard Apparatus (Holliston, MA, Cat. No. 74-4420). GC-MS autosampler vials and Teflon-lined pierceable caps were obtained from ThermoFisher Scientific. GC consumables were acquired from Agilent (Santa Clara, CA); MS consumables were obtained from Waters (Milford, MA).
Samples
A set of 40 blood plasma samples, half from stage III–IV breast cancer patients and half from age-matched healthy women, were purchased from Bioreclamation IVT (Hicksville, NY). A 300-mL plasma sample from an individual donor was also obtained from Bioreclamation, which served as a quality control sample to ensure batch-to-batch quantitative reproducibility.
Permethylation procedures
Spin column-free (SCF) permethylation procedure
Approximately 625 mg of sodium hydroxide (NaOH) beads were collected in a silanized 13 × 100 mm glass test tube, and then conditioned by one rinse with 350 μL of acetonitrile (ACN) followed by two rinses with 350 μL of DMSO. Thirty microliters (30 μL) of aqueous sample solution and 270 μL of DMSO were added to the preconditioned NaOH beads, followed by the addition of 105 μL iodomethane. The solution was then mixed thoroughly on a shaker at 650 rpm for 11 min. Next, to dissolve the NaOH beads and create a neutral to slightly acidic solution, 3.5 mL of acidified phosphate buffer was slowly added to each sample with continual mixing by manual swirling. To make the acidified buffer, concentrated hydrochloric acid (HCl) was mixed with 0.2 M phosphate buffer (composed of half 0.2 M Na2HPO4 and half 0.2 M NaH2PO4, pH 7) at a 1.3 : 2.2 volume ratio. Then 1.2 mL of chloroform was added and mixed thoroughly with the aqueous layer. After brief centrifugation (30 s at 3000g), the top layer (aqueous layer) of each sample was discarded, and replaced by a fresh 3.5 mL aliquot of 0.5 M NaCl solution in a 0.2 M sodium phosphate buffer (pH 7). Two additional liquid/liquid extraction cycles with 0.2 M sodium phosphate buffer (pH 7) were then performed. At last, the chloroform layer of each sample was collected and then dried under vacuum (Speed Vac) with no heat applied (for further intact glycan analysis) or under a gentle stream of nitrogen (if the sample was to undergo glycan linkage analysis).
10 min spin column (SC 10 min) permethylation procedure
The following procedure was employed by Borges et al. in 201318 and 2016.19 Sodium hydroxide beads were collected in a Pierce spin column (900 μL volume) up to 5 mm below the rim, and washed once with 350 μL of acetonitrile (ACN) followed by two rinses with 350 μL of DMSO. Nine microliters (9 μL) of aqueous glycan standard solution or a whole biofluid sample (e.g., blood plasma, serum, homogenized tissue, etc.) were mixed with 270 μL of DMSO in a 1.5 mL Eppendorf tube. The solution was added to the pre-conditioned NaOH beads in the plugged microfuge spin column, followed by the addition of 105 μL of iodomethane and immediate stirring. The sample mixture was then allowed to stand for 11 min with occasional gentle stirring. After reaction, the microfuge spin column was unplugged and spun for 30 s at 5000 rpm (1000g in a fixed-angle rotor) to collect the sample solution, which was quickly transferred into 3.5 mL of 0.5 M NaCl solution in 0.2 M sodium phosphate buffer (pH 7) within a silanized 13 × 100 glass test tube. NaOH beads were then washed twice by 300 μL of ACN, and the spin-throughs were all immediately transferred into the same silanized glass test tube. To each sample, 1.2 mL of chloroform was added to perform liquid/ liquid extraction. With brief centrifugation (as above), the top layer (aqueous layer) was discarded and then replaced by a fresh aliquot of 3.5 mL of 0.5 M NaCl solution in 0.2 M sodium phosphate buffer (pH 7). In total, three L/L extractions were performed for each sample, and finally the chloroform layer was extracted and then dried under vacuum (Speed Vac) with no heat applied (for further intact glycan analysis) or under a gentle stream of nitrogen (if the sample was to undergo glycan linkage analysis).
Extended spin column (SC 50 min) permethylation procedure
The following procedure was adapted from the protocol of Mechref et al.20 Sodium hydroxide beads were added to a Harvard Apparatus micro-spin column (500 μL volume) up to 3 cm in height and washed twice with 50 μL of DMSO. In a 0.5 mL Eppendorf tube, 1.2 μL of aqueous glycan standard solution was mixed with 30 μL of DMSO. After mixing with 20 μL of iodomethane, the sample mixture was applied to the sodium hydroxide beads packed in a spin column. The sample mixture was allowed to stand for 30 min, followed by the addition of another 20 μL of iodomethane and 20 min incubation. The sample was collected by centrifugation at 200g for 1 min after reaction, and the NaOH beads were washed twice by 50 μL of DMSO. The sample and two eluted volumes of DMSO were collected in a silanized 13 × 100 mm glass test tube. To perform L/L extractions, 3.5 mL of 0.5 M NaCl solution in 0.2 M sodium phosphate buffer (pH 7) and 1.2 mL chloroform were added to each sample. The chloroform layer was washed 3 times with the aqueous solution, then extracted and dried under vacuum (Speed Vac) with no heat applied (for further intact glycan analysis) or under a gentle stream of nitrogen (if the sample was to undergo glycan linkage analysis).
As carried out here, the final L/L extraction steps were necessary in order to facilitate subsequent analysis by GC-MS or MALDI-MS. As recently described elsewhere,21,22 however, the effluent from the extended spin column procedure can simply be dried and reconstituted in LC-MS mobile phase provided that online desalting is carried out on the LC prior to elution of the permethylated glycans into the mass spectrometer. LC-MS was not employed in the studies described here, but it is unlikely that the product of the SCF procedure described above would be amenable to this L/L extraction-free, dry, dilute and shoot approach due to the high concentration of salts that are present following dissolution of the NaOH beads with acidified phosphate buffer.
Glycan linkage analysis
Permethylation
Permethylation was carried out following one of the procedures listed above.
Hydrolysis, reduction, and acetylation
Hydrolysis, reduction and acetylation were carried out as we have previously described.18,19 Two molar TFA was prepared and added (325 μL) to each test tube and allowed to react for 2 h at 120 °C. Samples were dried under a gentle stream of nitrogen at 73 °C. Next, 10 mg mL−1 sodium borohydride in 1 M ammonium hydroxide was freshly prepared and added (475 μL) to each sample, which was allowed to stand for 1 h in room temperature. To remove excess borate, 100 μL of methanol (MeOH) was added and dried under nitrogen, followed by 125 μL of 9 : 1 (v/v) MeOH : acetic acid and drying under nitrogen. To achieve efficient acetylation, 18 μL of deionized water was added to each test tube, thoroughly mixed and sonicated for 2 min to dissolve any precipitates. Then each sample was mixed with 250 μL acetic anhydride and heated at 50 °C for 10 min, followed by adding 230 μL of concentrated TFA and heating again at 50 °C for 10 min. To clean up the sample mixture, 1.8 mL of dichloromethane and 2 mL of deionized water were added to perform liquid/liquid extraction twice, with the aqueous layer discarded for each cycle. The organic layer of each sample was transferred to a silanized autosampler vial, and then dried under a gentle stream of nitrogen at 40 °C.
Gas chromatography-mass spectrometry
An Agilent Model A7890 gas chromatograph (equipped with a CTC PAL auto-sampler) coupled to a Waters GCT (time-of-flight) mass spectrometer was employed for analysis of PMAAs by GC-MS. To each sample vial, 64 μL of acetone was added to reconstitute the PMAAs. One microliter (1 μL) was then injected onto a hot (280 °C), silanized glass liner containing a plug of silanized glass wool (Agilent Cat. No. 5183-4647) at a split ratio of 20. Samples were separated via chromatography over a 30 m DB-5 ms GC column, facilitated by helium as carrier gas with a 0.8 mL min−1 constant flow rate. The GC oven was held at an initial temperature of 165 °C for 0.5 min, ramped to 265 °C at a rate of 10 °C min−1, followed by immediately ramping to 325 °C at a rate of 30 °C min−1 and holding at 325 °C for 3 min. Sample components eluted from GC column into the TOF mass spectrometer equipped with an electron ionization source (70 eV, 250 °C). MS spectra were collected in positive-ion mode. Spectra from individual TOF pulses over a m/z range of 40–800 were summed every 0.1 s. The mass spectrometer was tuned and calibrated daily using perfluorotributylamine to ensure reproducible relative abundances of EI ions and mass accuracy within 10 ppm.
Intact glycan analysis
Enzymatic release and reduction of N-glycans
The following procedure, including the amount of glycoprotein employed, was adapted from Hu et al. for release and reduction of N-glycans.20 A 10 mg mL−1 stock solution of N-glycoprotein (Bovine Ribonuclease B) was prepared. A 1 μL aliquot of the stock solution was mixed with 9 μL GlycoBuffer 2 (10 ×, 50 mM sodium phosphate buffer, pH 7.5). Next, the mixture was heated in boiling water for 10 min, and then chilled on ice. Peptide-N-glycosidase F (PNGase F) stock solution (500 000 units per mL) was diluted ten times with GlycoBuffer 2, and added (1 μL) to each sample. The glycoprotein samples were then incubated at 37 °C for 18 h, and dried under vacuum with assisted heat at approximately 55 °C.
A 10 mg mL−1 solution of sodium borohydride in 1 M ammonium hydroxide solution was freshly prepared and added (10 μL) to each sample, which was then incubated in a 60 °C water bath for 1 h. Samples were mixed with 10 μL of 5% acetic acid and dried under vacuum with assisted heat. Next, HPLC-grade methanol was added (100 μL) to each sample and removed by drying under vacuum. To eliminate residual borate in samples, the last two steps were repeated five times.
Permethylation
Permethylation was carried out following one of the procedures listed above.
MALDI-TOF mass spectrometry
The dried permethylated samples were reconstituted in 6 μL of ethanol. A 10 mg mL−1 2,5-dihydroxybenzoic acid (DHB) matrix solution was made in 1 mM sodium acetate solution. To directly compare samples derivatized with the SCF vs. SC 50 min procedures, 1 μL of sample permethylated by the SC 50 min procedure with iodo-methane and 1 μL of sample permethylated by the SCF procedure with iodomethane-d3 were mixed into 2 μL of DHB matrix solution, making a solution with a total volume of 4 μL. Next, 2 μL of the mixed solution was transferred onto a MALDI target spot, and then dried under vacuum for 10 min
A Bruker Daltonics, Autoflex III L200 MALDI-TOF mass spectrometer was utilized for intact glycan analysis. The instrument was used in linear, positive ion, delayed-extraction mode, with ‘ionsource 1’ at 20 kV, ‘ionsource 2’ at 18.45 kV, lens at 7.7 kV, 50 ns delayed extraction, deflection signal suppression up to m/z 500, and 1 GS per s sample rate. To ensure good ion counting statistics, at least 2500 laser-shots were signal averaged for each mass spectrum. The MALDI-TOF mass spectrometer was externally calibrated with peptide calibration standard II, a mixture of 9 peptides supplied by Bruker Daltonics (Cat. No. NC9349683).
Data analysis
GC-MS data
Masslynx 4.1 program and Quanlynx 4.1 software were utilized to process all GC-MS data for glycan linkage analysis. For routine relative quantification of each glycan “node” from glycan standards or blood plasma, the 2–4 most abundant and/or diagnostic fragment ions18,19 were summed using a 0.15 Da extracted ion chromatogram mass window for quantification. The summed extracted ion chromatogram (XIC) peak areas were automatically integrated and quantified, then exported to a spreadsheet for further calculation. All integration events were evaluated manually to ensure accuracy.
Permethylation efficiency was determined by the fraction of ion signal derived from PMAAs representing undermethylated form(s) of a specific glycan node divided by this same value plus the ion signal derived from the PMAA representing the fully permethylated form of that particular glycan node. (For example, for analysis of the Gal-4-GlcNAc standard, ion signal from the PMAA representing terminal galactose (t-Gal) represented the fully permethylated hexose while all other galactose-related ion signals such as those from PMAAs representing 2-Gal, 3-Gal, 4-Gal, etc. represented undermethylated forms of the t-Gal residue.) Because the total ion current chromatogram (TIC) contained a few chromatographic signals from sample preparation-related non-glycan contaminants that, in some cases, co-eluted with PMAAs of interest, extracted ion chromatograms had to be employed for this analysis. As such, we ensured that ions extracted for analysis of glycan undermethylation (see ESI Table 1†) covered greater than 90% of the ion current derived from the corresponding glycan node (ESI Fig. S1†).
MALDI-TOF MS data
MALDI MS data for intact glycan analysis were processed by Bruker FlexAnalysis 3.0 software. Mass spectra were baseline subtracted using the TopHat algorithm, and MS peaks were integrated baseline-to-baseline. Integration events were inspected manually and, if necessary, adjusted to baseline as appropriate. Peak intensities were exported to a spreadsheet for further calculation.
To directly compare the permethylation efficiency of the spin column-free procedure (SCF) and extended spin column procedure (SC 50 min), the intensity ratio of mass spectral peaks from the two procedures (SCF/SC 50 min) was calculated for each intact glycan. Every experiment was also run with the labeling reagents switched (i.e., iodomethane used for the SCF procedure and iodomethane-d3 used for the SC 50 min procedure), then all results were pooled together to ensure a lack of labeling bias.
Statistical analysis software
Software employed for statistical analysis included Microsoft Excel for the two-tailed Student’s t-test to compare the experimentally determined mean SCF/SC ratio for each glycoform with the theoretical value of 1. Excel was also used for the Benjamini–Hochberg false discovery rate adjustment of p-values for the Wilcoxon rank sum test. GraphPad Prism 5 was utilized for Kruskal–Wallis followed by Dunn’s post hoc tests. And SAS 9.4 was employed for the Wilcoxon rank sum test.
Results and discussion
Optimization of permethylation reagent volumes and order of addition for the spin column-free (SCF) permethylation procedure
To maximize permethylation efficiency of the SCF procedure, several parameters were assessed, including total volume of water in the original sample and order of reagent addition. Holding the volume of DMSO and iodomethane constant, the effect of sample water volume on permethylation yield was investigated at total water volumes of 10 μL, 20 μL, 30 μL, 40 μL and 80 μL. These volumes are given assuming that the original sample is aqueous in nature and constitutes a fraction of the stated volumes. N-Acetylactosamine (LacNAc), composed of terminal galactose (t-Gal) linked to the 4-position of N-acetylglucosamine (4-GlcNAc), was chosen as the glycan standard for the assessment of permethylation efficiency, since the permethylation yields of both hexose and N-acethylhexosamine (HexNAc) monomer units can be evaluated from it simultaneously. For each sample, 5 μg of LacNAc was dissolved into the volumes of water listed above, and then processed by spin column-free permethylation followed by hydrolysis, reduction and acetylation steps.
As shown in Fig. 3, the highest permethylation efficiencies for both the hexose and HexNAc units, were found in samples with total original-sample water volumes of more than 30 μL. For samples with 30 μL total volume, approximately 2% of hexose and 1% of HexNAc residues were undermethylated. But for samples with total original-sample water volumes under 30 μL, the permethylation efficiencies were not as good, evidenced by higher mean values and significantly higher undermethylation fractions (especially for 10 μL) based on Kruskal–Wallis followed by Dunn’s post hoc tests. Based on previous work published by Ciucanu and coworkers,23 a small amount of water added to the permethylation system improves the solubility of glycans in organic solvents, such as DMSO, which is critical for the permethylation reaction. However, too much water added to the sample can lead to undesired effects on the permethylation efficiency, due to the oxidative degradation of glycans in basic solution. In accordance with the previous work by Ciucanu et al.,23 a paste of sodium hydroxide was found on the walls of the test tubes immediately following the 11-minute incubation with NaOH beads for samples with a total volume above 40 μL, which could potentially result in inconsistent permethylation yields. Therefore, 30 μL was selected as the optimal original-sample water volume and was used in subsequent experiments.
Fig. 3.

Effect of original-aqueous-sample volume on permethylation efficiency: (a) univariate distribution of the undermethylated fraction of t-Gal, (b) univariate distribution of the undermethylated fraction of 4-GlcNAc. N = 6 to 15 for each volume. Undermethylated fractions were determined as described in the Methods section. Kruskal–Wallis followed by Dunn’s post hoc tests demonstrated significant differences in the degree of under-methylation at the tested volumes of water in the original samples. Error bars indicate mean ± standard error of the mean (SEM). Single asterisk (*) indicates significant differences between groups with p ≤ 0.05. Three asterisks (***) indicate significant differences between groups with p ≤ 0.001.
The order in which water/DMSO-solubilized glycan samples were exposed to either iodomethane or the NaOH beads was also evaluated with regard to its impact on permethylated glycan yield. Generally, iodomethane was added to samples before the NaOH beads, even though sometimes a color change appeared suggesting oxidative degradation of glycans might be taking place.23 To evaluate the possibility of completely avoiding oxidative degradation, samples were added to NaOH beads first, followed by addition of iodo-methane. Five micrograms (5 μg) of LacNAc were permethylated by the SCF permethylation procedure, followed by hydrolysis, reduction and acetylation (n = 6 each). To compare the degrees of oxidative degradation for the two orders of reagent addition, the total ion signals (raw summed peak areas of XICs representing >90% of the total ion PMAA ion signal) of t-Gal and 4-GlcNAc, as well as the ratios of 4-GlcNAc to t-Gal were tested by Wilcoxon rank sum test. No statistically significant differences were found for the degree of oxidative degradation of hexose and HexNAc units between the two orders of reagent addition (p = 0.7922 for t-Gal, and p = 0.6623 for 4-GlcNAc); neither was the HexNAc/hexose ratio significantly different. Even so, to minimize the risk of oxidative degradation it is preferable to add water/DMSO-solubilized samples to NaOH beads first followed by iodomethane.
Permethylation efficiency of the spin column-free procedure relative to spin column-based approaches
To validate the permethylation efficiency of our spin column-free procedure, we compared it with two other published and prevalent spin column-based permethylation procedures. One spin column permethylation procedure with 10 min reaction time (SC 10 min) was developed by Kang et al.,7 Goetz et al.24 and further modified by Borges et al.,19 the details of which can be found in their recently published work. A second spin column permethylation procedure with 50 min reaction time (SC 50 min) was summarized in recent work of Hu et al.20 2-Acetamido-2-deoxy-D-lactose (Gal-4GlcNc, a.k.a. LacNAc) and 3-O-(2-acetamido-2-deoxy-β-D-galactopyranosyl)-D-galactopyranose (GalNAc-3Gal) were chosen as the model glycan standards for the investigation, both of which are disaccharides composed of hexose and HexNAc residues but with switched positions (reducing or non-reducing end) of the two classes of monosaccharides. We processed 5 μg samples of glycan standard with the three different permethylation procedures in parallel, then performed the hydrolysis, reduction and acetylation procedures together (i.e., with all samples in the same batch) to reduce variability in steps other than permethylation.
The undermethylated fraction of each monosaccharide was calculated (refer back to Method section) and results were evaluated by Kruskal–Wallis followed by Dunn’s post hoc tests (Fig. 4). The data revealed that the spin column-free procedure has comparable or slightly better permethylation efficiency for hexoses and HexNAcs in comparison with the two spin column procedures (Fig. 4). According to our previous work,19 HexNAc residues are more susceptible to pH change in the aqueous solution before liquid/liquid extraction and thus have a higher risk of undergoing oxidative degradation during permethylation than hexoses. But as shown here (Fig. 4), the undermethylation of HexNAc residues by the spin column-free procedure can reach as low as 1.4% (for 4-GlcNAc) and 0.4% (for t-GalNAc), demonstrating optimal control of permethylation efficiency by the spin column-free procedure. The peak areas under the summed extracted ion chromatograms (XICs) of hexose and HexNAc residues (refer to Method section and ESI Table 1†) were compared by evaluating their ratios, as shown in Fig. 4e and f. Even with apparently lower relative yields of HexNAc residues for SCF procedure compared to the other two spin column procedures, the HexNAc/hexose ratios for the SCF procedure were sufficiently consistent and reproducible, which is critical during the routine analysis of biological samples.
Fig. 4.

Evidence that the new spin column-free procedure has comparable permethylation efficiency for terminal/non-terminal hexose and terminal/non-terminal HexNAc relative to other prevalently used spin column procedures.18–20 Undermethylated fractions were calculated and shown for (a) t-Gal from Galactopyranosyl-β-1,4-N-acetyl-D-glucosamine (Gal-4-GlcNAc), (b) 4-GlcNAc from Gal-4-GlcNAc, (c) 3-Gal from 3-O-(2-acet-amido-2-deoxy-b-D-galactopyranosyl)-D-galactopyranose (GalNAc-3Gal), and (d) t-GalNAc from GalNAc-3Gal. Peak areas under summed extracted ion chromatograms (XICs) of the HexNAc species were compared with that of hexose species, for both glycan standards, and the ratios were shown in (e) 4-GlcNAc/t-Gal, and (f) t-GalNAc/3-Gal. N = 12 for each permethylation procedure. Kruskal–Wallis followed by Dunn’s post hoc tests demonstrated significant differences in the permethylation procedures. The SCF procedure produces a slightly higher undermethylated fraction than the extended SC procedure (SC 50 min) for t-Gal (a), but similar or lower undermethylated fractions than the two SC procedures for 4-GlcNAc (b), 3-Gal (c) and t-GalNAc (d). Error bars indicate mean ± standard error of the mean (SEM). Single asterisk (*) indicates significant differences between groups with p ≤ 0.05. Two asterisks (**) indicate significant differences between groups with p ≤ 0.01. Three asterisks (***) indicate significant differences between groups with p ≤ 0.001.
Analysis of intact glycans following SCF permethylation
Generally, permethylation is utilized by most researchers to facilitate analysis of intact glycans. The spin column-free permethylation procedure can also be applied to intact O-glycan analysis, as well as intact N-glycan analysis if coupled with the release of glycans from N-glycoproteins. Therefore, we characterized the efficiency of SCF vs. spin column permethylation procedures for both intact O- and N-linked glycans.
Fetuins are heavily glycosylated proteins that carry both O- and N-linked glycans. Within the permethylation step, O-glycans are cleaved from the protein by β-elimination, while N-glycans remain attached.24 Three samples of 145 μg of Fetuin were processed by the extended spin column permethylation procedure (SC 50 min), a commonly used spin column-based procedure.20,25–29 In parallel, 3 samples were processed with the SCF procedure but were permethylated with iodomethane-d3. Samples treated by the two procedures were reconstituted in 6 μL ethanol, mixed at a 1 : 1 ratio in a DHB matrix solution, and 2 μL was spotted onto a MALDI target. All three samples derivatized by the SCF procedure with iodomethane-d3 were mixed in all possible combinations with the three samples processed by the SC 50 min procedure that employed unlabeled iodomethane, leading to a total of 9 combinations (3 × 3). To eliminate the possibility of labeling reagent bias, the two procedures were carried out again on the same samples but with the labeling reagents switched.
The SCF procedure effectively permethylated all three major types of O-glycans from Fetuin-A (Fig. 5; overlaid with a background spectrum in ESI Fig. S2†), with O-linked tetrasaccharides (NeuAc2-Hex-HexNAc) and hexasaccharides (NeuAc2-Hex2-HexNAc2) permethylated at nearly the same efficiency as the more commonly employed spin column-based procedure.20 Highly averaged MALDI-MS peak intensities of identical glycoforms processed with the two procedures were compared directly and are shown as the SCF/SC ratio in Fig. 6. An SCF/SC ratio of 1 would indicate identical permethylation efficiency and yield for the two procedures. As such, we used a two-tailed Student’s t-test to compare the experimentally determined mean SCF/SC ratio for each glycoform with the theoretical ratio of 1 (which would indicate an equal yield from the two procedures), using eqn (1).30
Fig. 5.

Permethylation of intact glycans by the spin column-free procedure (SCF) and extended spin column procedure (SC 50 min).20 (a) O-glycans of fetuin from fetal bovine serum; (b) N-glycans of bovine Ribonuclease B. Glycan structure symbols indicate the most likely (isomerically ambiguous) corresponding glycan permethylated by the SC 50 min procedure with unlabelled iodomethane, or by the SCF procedure with iodomethane-d3 (as indicated by “-dn” labels where the subscripts represent the number of hydrogen atoms replaced by deuterium atoms in each glycan structure). Red lines under the peaks representing permethylated glycans indicate integration baselines. Calculated masses of the permethylated glycans depicted are provided in ESI Table 2.†
Fig. 6.

Ratio of intact, permethylated glycan yields produced by the SCF procedure relative to a commonly employed SC procedure (SC 50 min):20 (a) O-glycans from Fetuin and (b) N-glycans from bovine ribonuclease B. For each glycoform, the highly-averaged intensities of the two MALDI-MS peaks corresponding to the two permethylation procedures conducted with light and heavy labeling reagents were compared and presented here as the SCF/SC ratio. Two-tailed Student’s t-test comparing the SCF/SC ratios to the theoretical value of 1 demonstrated significant differences between the two procedures as indicated (p < 0.05). Error bars indicate mean ± standard error of the mean (SEM). † indicates significant differences between a group mean and the theoretical value of 1.
| (1) |
where t is the t-statistic, x̄ is the sample mean, μ is the theoretical population mean of 1, s is the sample standard deviation, and N is the number of observations. For the case of tetra- and hexasaccharides, the SCF/SC ratios were nearly 1, despite the fact that the t-statistic for the tetrasaccharide was slightly higher than the critical value of t (tcrit) at the 99% confidence level (CL). Relative to the SC procedure, the SCF procedure was found to result in significantly lower yields of O-linked tri-saccharides (NeuAc-Hex-HexNAc), mean SCF/SC ratio = 0.50 (Fig. 6). The mechanism underlying this phenomenon is not known, but it is not likely caused by less efficient permethylation in the SCF procedure (cf. Fig. 4). It could, however, be related to relatively higher solubility of the permethylated tri-saccharide in the aqueous layers of SCF samples, which have a lower pH during extraction—potentially resulting in decreased extraction efficiency. For larger O-linked glycans (tetra- and hexasaccharides), the comparison demonstrated that the SCF procedure produces nearly the same permethylated glycan yield as the widely employed spin column procedure (SC 50 min).20
To investigate the effectiveness of the SCF procedure on permethylation of N-glycans in comparison with the spin column procedure, 10 μg of bovine ribonuclease B (RNase B), a well-known N-glycoprotein with only one N-glycosylation site, which bears one of four major high-mannose N-glycans was employed31 (Fig. 5; overlaid with a background spectrum in ESI Fig. S2†). Since only O-glycans are released from proteins by β-elimination during the permethylation step,24 traditional enzymatic cleavage steps were adopted to release N-glycans from the glycoprotein (see Methods section). Using the comparison strategy described above for O-glycans, the SCF/SC ratios for the four major RNase B N-glycans were compared to the theoretical value of 1 by a two-tailed Student’s t-test. Calculated t-statistics were larger than tcrit at the 99% CL, indicating that the SCF procedure produces a higher yield of per-methylated high-mannose N-glycans compared to a widely used-spin column procedure (SC 50 min) (Fig. 6). Notably, while low-microgram quantities of RNase B were required to facilitate consistently high signal-to-noise (S/N) levels for this quantitative comparison, the SCF-based analysis was also run with 100 ng of RNAse B to demonstrate the sensitivity of the new approach. The S/N ratio for all four glycans was greater than 5 (ESI Fig. S3†).
Application of the SCF procedure to human blood plasma
As shown by Borges et al. in 2013,18 spin-column based permethylation of whole biofluids and homogenized tissues followed by hydrolysis, reduction and acetylation can be used to generate composite glycan linkage analysis data from all major classes of glycans—including not only O-linked glycans, but N-linked and lipid-linked glycans as well—without the need for glycan-releasing enzymes. Relative quantification of each hexose is carried out by dividing its summed XIC peak area by the sum of all hexose XIC peak areas; likewise, relative quantification of each HexNAc is carried out by dividing its summed XIC peak area by the sum of all HexNAc XIC peak areas. These normalized abundances (NAs) are then used for further evaluation of biological differences between samples from donors with and without disease.19 To determine the reproducibility of the SCF permethylation procedure as applied to blood plasma, 9-μL aliquots of an EDTA blood plasma sample from an individual donor were processed by SCF permethylation followed by hydrolysis, reduction, acetylation and analysis by GC-MS as we have previously described.18 Six 9-μL aliquots were processed and analyzed three different times (i.e., 18 samples total in three separate batches) to evaluate intra- and interassay reproducibility. Shown in Table 1 are the intra- and inter-assay reproducibility of all glycan nodes for which the mean normalized abundances contribute more than 1% of the total hexose or HexNAc signal. Average intra-assay reproducibility was 9.7% and inter-assay reproducibility was 17.7%. Of these 16 routinely detected glycan nodes in plasma samples, 15 had intra- and inter-assay reproducibility of less than 20% for SCF procedure. These metrics of analytical precision are comparable to those of the 10 min spin column permethylation procedure, as reported by Borges et al. in 2016.19 The quantitatively inconsistent glycan node, 4-Glc, typically constitutes about 5% of the total hexose signal, but is a node that is primarily derived from glycolipids—which may behave differently than N- and O-linked glycans under the sample processing conditions applied.
Table 1.
Intra- and interassay reproducibility of permethylation procedures as applied to glycan linkage analysis of blood plasma
| Glycan nodes | Average intra-assay CV%a | Inter-assay CV%a | ||
|---|---|---|---|---|
|
|
|
|||
| SCFb | SC 10 minc | SCFb | SC 10 minc | |
| t-Fuc | 5.3 | 6.6 | 17.8 | 12.3 |
| t-Gal | 3.6 | 7.8 | 10.3 | 7.7 |
| 2-Man | 3.6 | 2.8 | 7.6 | 5.3 |
| 4-Glc | 45.2 | 13.5 | 54.7 | 15.6 |
| 3-Gal | 13.4 | 20.2 | 18.9 | 26.8 |
| 6-Gal | 3.8 | 2.7 | 10.8 | 9.8 |
| 2,4-Man | 7.6 | 6.4 | 13.2 | 11.2 |
| 2,6-Man | 5.2 | 5.8 | 8.9 | 6.5 |
| 3,6-Man | 8.9 | 13.2 | 16.5 | 15.7 |
| 3,4,6-Man | 4.6 | 7.4 | 18.7 | 19.0 |
| t-GlcNAc | 3.2 | 5.0 | 6.6 | 4.9 |
| 4-GlcNAc | 2.0 | 2.1 | 6.5 | 2.9 |
| 3-GalNAc | 10.1 | 7.0 | 16.5 | 8.7 |
| 3,4-GlcNAc | 5.7 | 7.3 | 14.8 | 11.5 |
| 4,6-GlcNAc | 6.1 | 3.3 | 15.8 | 6.1 |
| 3,6-GalNAc | 9.9 | 12.2 | 18.2 | 15.4 |
Values represent %CV of total hexose or total HexNAc-normalized individual glycan nodes. All glycan nodes contributing at least 1% of the total hexose or HexNAc signal are listed.
Data were acquired by one analyst on 3 separate days by the new spin column-free procedure.
Application of the SCF permethylation procedure to glycan node analysis of blood plasma from breast cancer patients
The new SCF permethylation procedure was coupled to hydrolysis, reduction and acetylation and applied to a cohort of 40 blood plasma samples from breast cancer patients (stages III–IV, n = 20) and age-matched female controls (n = 20). Samples were randomized and analyzed blindly in seven batches as we have previously described.19 Three glycan nodes with between-batch reproducibility of 16% CV or better (Table 1) were found to be significantly different in the late stage-breast cancer samples compared to controls, according to a Wilcoxon rank sum test and Benjamini–Hochberg false discovery rate adjustment procedure with p-values less than 0.05 (shown in Fig. 7). The receiver operating characteristic (ROC) c-statistics (AUCs) of the three glycan nodes were in the range of 0.75–0.85 (Fig. 7), demonstrating that 2,4-linked mannose (2,4-Man), 2,6-linked mannose (2,6-Man) and 4,6-linked GlcNAc (4,6-GlcNAc) may potentially be useful as markers for stage III–IV breast cancer. Interestingly, these three glycan nodes represent the sum total of β1-4 branching, β1-6 branching, and core fucosylation, respectively—as pooled together from all intact glycans within each sample.
Fig. 7.

Application of the SCF procedure to glycan node analysis18,19 of clinical samples. Data shown were from blood plasma acquired from 20 stage III–IV breast cancer patients and 20 age-matched healthy women. Receiver operating characteristic (ROC) analysis of 20 glycan nodes indicated that three glycan nodes have the potential to distinguish breast cancer patients from controls: (a) univariate distribution of 2,4-Man, (b) ROC curve for 2,4-Man, (c) univariate distribution of 2,6-Man, (d) ROC curve for 2,6-Man, (e) univariate distribution of 4,6-GlcNAc, and (f) ROC curve for 4,6-GlcNAc. Data are based on normalized abundance (NA), which is defined as the summed XIC peak area of each glycan node divided by the sum of all other hexose (or HexNAc) summed XIC areas. For all three glycan nodes, p < 0.01 and AUC (area under curve) >0.75 for ROC curves. Error bars indicate mean ± standard error of the mean (SEM).
Conclusions
A new spin column-free (SCF) permethylation procedure that reduces consumable costs and the number of experimental steps required for glycan permethylation was developed and applied to mass spectrometry-based intact-glycan analysis and linkage-analysis of glycans from whole biospecimens. The permethylation efficiency of the new SCF procedure averaged about 98% for hexose residues and 99% for HexNAc residues, which are values comparable to or slightly higher than those of some widely-used spin column-based procedures. With regard to intact glycan analysis, the SCF procedure provided almost identical yields for permethylated O-glycans composed of more than three monosaccharides, and higher yields for the permethylated N-glycans examined. The SCF permethylation approach, combined with hydrolysis, reduction and acetylation facilitated glycan linkage analysis of pooled glycans from unfractionated blood plasma with good quantitative reproducibility. Furthermore, glycan linkage analysis with the SCF procedure identified specific, previously unreported glycan “nodes” representative of β1–4 branching, β1–6 branching, and core fucosylation as potential markers of stage III–IV breast cancer.
Supplementary Material
Acknowledgments
Research reported in this publication was supported in part by the National Cancer Institute of the National Institutes of Health under Award Number R33CA191110. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/ c7an00396j
References
- 1.Ohtsubo K, Marth JD. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 2.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2. Cold Spring Harbor Laboratory Press; Cold SpringHarbor (NY): 2009. [PubMed] [Google Scholar]
- 3.Varki A. Glycobiology. 2017;27:3–49. doi: 10.1093/glycob/cww086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varki A, Kannagi R, Toole B. In: Essentials of Glycobiology. 2. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. ch. 44. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2009. [PubMed] [Google Scholar]
- 5.Dewald JH, Colomb F, Bobowski-Gerard M, Groux-Degroote S, Delannoy P. Cells. 2016;5(4):43. doi: 10.3390/cells5040043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adua E, Russell A, Roberts P, Wang Y, Song M, Wang W. OMICS: J Integr Biol. 2017;21:183–196. doi: 10.1089/omi.2017.0035. [DOI] [PubMed] [Google Scholar]
- 7.Kang P, Mechref Y, Novotny MV. Rapid Commun Mass Spectrom. 2008;22:721–734. doi: 10.1002/rcm.3395. [DOI] [PubMed] [Google Scholar]
- 8.Banazadeh A, Veillon L, Wooding KM, Zabet-Moghaddam M, Mechref Y. Electrophoresis. 2017;38:162–189. doi: 10.1002/elps.201600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wada Y, Azadi P, Costello CE, Dell A, Dwek RA, Geyer H, Geyer R, Kakehi K, Karlsson NG, Kato K. Glycobiology. 2007;17:411–422. doi: 10.1093/glycob/cwl086. [DOI] [PubMed] [Google Scholar]
- 10.Purdie T, Irvine JC. J Chem Soc, Trans. 1903;83:1021–1037. [Google Scholar]
- 11.Hakomori SI. J Biochem. 1964;55:205–208. [PubMed] [Google Scholar]
- 12.Sandford PA, Conrad H. Biochemistry. 1966;5:1508–1517. doi: 10.1021/bi00869a009. [DOI] [PubMed] [Google Scholar]
- 13.Phillips LR, Fraser BA. Carbohydr Res. 1981;90:149–152. [Google Scholar]
- 14.Blakeney AB, Stone BA. Carbohydr Res. 1985;140:319–324. [Google Scholar]
- 15.Ciucanu I, Kerek F. Carbohydr Res. 1984;131:209–217. [Google Scholar]
- 16.Ciucanu I. Anal Chim Acta. 2006;576:147–155. doi: 10.1016/j.aca.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun Mass Spectrom. 2005;19:3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borges CR, Rehder DS, Boffetta P. Anal Chem. 2013;85:2927–2936. doi: 10.1021/ac3035579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaare S, Aguilar JS, Hu Y, Ferdosi S, Borges CR. J Visualized Exp. 2016;111:e53961. doi: 10.3791/53961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y, Shihab T, Zhou S, Wooding K, Mechref Y. Electrophoresis. 2016;37:1498–1505. doi: 10.1002/elps.201500560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou S, Dong X, Veillon L, Huang Y, Mechref Y. Anal Bioanal Chem. 2017;409:453–466. doi: 10.1007/s00216-016-9996-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou S, Wooding KM, Mechref Y. Methods Mol Biol. 2017;1503:83–96. doi: 10.1007/978-1-4939-6493-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciucanu I, Costello CE. J Am Chem Soc. 2003;125:16213–16219. doi: 10.1021/ja035660t. [DOI] [PubMed] [Google Scholar]
- 24.Goetz JA, Novotny MV, Mechref Y. Anal Chem. 2009;81:9546–9552. doi: 10.1021/ac901363h. [DOI] [PubMed] [Google Scholar]
- 25.Mechref Y, Kang P, Novotny MV. In: Glycomics: Methods and Protocols. Nicolle PH, Niclas KG, editors. ch. 4. Humana Press; New York City, NY: 2009. pp. 53–64. [Google Scholar]
- 26.Harvey DJ. J Chromatogr B: Biomed Appl. 2011;879:1196–1225. doi: 10.1016/j.jchromb.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 27.Leymarie N, Zaia J. Anal Chem. 2012;84:3040–3048. doi: 10.1021/ac3000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Powers TW, Jones EE, Betesh LR, Romano PR, Gao P, Copland JA, Mehta AS, Drake RR. Anal Chem. 2013;85:9799–9806. doi: 10.1021/ac402108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai TH, Wang M, Di Poto C, Hu Y, Zhou S, Zhao Y, Varghese RS, Luo Y, Tadesse MG, Ziada DH. J Proteome Res. 2014;13:4859–4868. doi: 10.1021/pr500460k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.West WD, Skoog DA, Holler FJ, Crouch SR. Fundamentals of Analytical Chemistry. 9. ch. 7. Cengage - Brooks/ Cole; Belmont, CA: 2014. pp. 123–152. [Google Scholar]
- 31.An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Anal Chem. 2003;75:5628–5637. doi: 10.1021/ac034414x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.