

Abstract
Williams syndrome (WS) is a rare neurodevelopmental disorder with a well-described, known genetic etiology. In contrast to Autism Spectrum Disorders (ASD), WS has a unique phenotype characterized by global reductions in IQ and visuospatial ability, with relatively preserved language function, enhanced reactivity to social stimuli and music, and an unusual eagerness to interact socially with strangers. A duplication of the deleted region in WS has been implicated in a subset of ASD cases, defining a spectrum of genetic and behavioral variation at this locus defined by these opposite extremes in social behavior. The hypersociability characteristic of WS may be linked to abnormalities of frontostriatal circuitry that manifest as deficits in inhibitory control of behavior. Here, we examined the density of neurons and glia in associative and limbic territories of the striatum including the caudate, putamen, and nucleus accumbens regions in Nissl stained sections in five pairs of age, sex, and hemisphere-matched WS and typically-developing control (TD) subjects. In contrast to what is reported in ASD, no significant increase in overall neuron density was observed in this study. However, we found a significant increase in the density of glia in the dorsal caudate nucleus, and in the ratio of glia to neurons in the dorsal and medial caudate nucleus in WS, accompanied by a significant increase in density of oligodendrocytes in the medial caudate nucleus. These cellular abnormalities may underlie reduced frontostriatal activity observed in WS, with implications for understanding altered connectivity and function in ASD.
Introduction
Autism Spectrum Disorders (ASD) are characterized by an extremely complex and heterogenous genetic (Sebat et al., 2007; Geschwind, 2011; Miles, 2011; Persico and Napolioni, 2013; Jeste and Geschwind, 2014) and phenotypic (Tager-Flusberg and Joseph, 2003; Ring et al., 2008; Lombroso et al., 2009) profile. By contrast, Williams syndrome (WS) is known to be caused by a hemizygous microdeletion of 25-28 genes on chromosome band 7q11.23. It is a rare disorder (<1 in 7500, Strømme et al., 2002) characterized by multiple somatic effects, including distinctive facial morphology, slowed growth, and cardiac abnormalities (Williams et al., 1961; Beuren et al., 1962). Individuals with WS additionally display a relatively predictable and consistent cognitive and behavioral phenotype, including global reductions in total IQ, particularly with respect to visuospatial abilities (Meyer-Lindenberg et al., 2006). Curiously, individuals with WS have been shown to demonstrate linguistic capacity characterized by relatively broad vocabulary and unusually expressive use of phrases (Karmiloff-Smith et al., 1997; Reilly et al., 2004; Udwin and Yule, 2005), but may struggle with temporal, spatial, and relational elements (Thomas et al., 2001; Phillips et al., 2004; Brock, 2007; Mervis and Becerra, 2007) of language use.
In stark contrast to ASD, perhaps the best-described feature of individuals with WS includes what has been described as hypersociability, and the intense drive to engage in social interactions with strangers (Jones et al., 2000; Doyle et al., 2004; Järvinen-Pasley et al., 2008, 2010; Järvinen et al., 2013). This unique trait may be best viewed as a deficit in inhibitory control of socially-directed behavior (Little et al., 2013). Evidence from functional imaging studies has suggested that frontostriatal circuitry may be differentially affected by developmental abnormalities characteristic of the disorder (Mobbs et al., 2007). Hypersociality and increased affiliative drive in Williams syndrome patients may represent selected deficits in the ability to suppress behavioral responses (Frigerio et al., 2006), and attention deficit hyperactivity disorder is a frequent comorbid diagnosis in these individuals (Carrasco et al., 2005). Mobbs and colleagues (2007) showed slower reaction times among WS patients in a go/No-go task as compared with typically-developing control participants, and substantially reduced blood-oxygen level dependent contrast (BOLD) signal suggesting reduced activity in the striatum was observed. Given the role of frontostriatal circuits in controlling behavioral responses, microstructural abnormalities in the striatum (Fan et al., 2017), and particularly the caudate (Reiss et al., 2004) may underlie behavioral deficits in response inhibition and the control of behavioral responses.
Brains of individuals with WS tend to be smaller overall than in typically-developing individuals (Jernigan and Bellugi, 1990), including reductions in cortical and subcortical grey matter. Reductions in grey matter volume have been noted in the caudate in individuals with Williams syndrome (Reiss et al., 2004) and recent structural imaging studies have found the greatest volumetric reductions occur in the orbitofrontal cortex and striatum in WS (Fan et al., 2017). Microstructural analyses of Brodmann areas 10 and 11 of the orbitofrontal cortex have demonstrated a reduction in neuronal density in these regions (Lew et al., 2016), further supporting the assertion that neural systems implicated in emotional processes are affected in WS at the cellular level. No histological studies have yet aimed to identify the microstructural changes underlying volume differences in the striatum in WS.
Here we examined the caudate, putamen, and nucleus accumbens regions corresponding to the associative and limbic territories within the striatum, a key part of the reward-processing system in the primate striatum associated with learning, flexibility, and behavioral control (Fudge and Haber, 2002; Haber et al., 2006) to examine microstructural features of the striatum that may underlie cognitive and behavioral features of the disorder. In these regions in ASD, the caudate has been shown to involve structural increases in size (Langen et al., 2007, 2012) and reductions of total neuronal density (Wegiel et al., 2014). We utilized postmortem tissue from ten adult individuals including a unique sample of five WS and five age-, sex-, and hemisphere-matched typically-developing (TD) control subjects, to measure the density of neurons and glia in the rostral striatum.
Methods
Materials
Subjects were adults, ranging in age from 18 to 45 years at time of death and included two males and two females with genetically confirmed diagnoses of WS, and one male (WS1) with a behaviorally confirmed diagnosis of WS with significant cardiac history consistent with the genetic disorder’s biomedical profile, for which no genetic information was available (Table 1). These five specimens in the Ursula Bellugi Williams Syndrome Brain Collection met the criteria for inclusion based on completeness of the targeted ROIs. Another set of five age, sex, and right hemisphere-matched specimens from five adult TD cases were provided by University of Maryland Brain and Tissue Bank (UMBTB), a repository of the NIH NeuroBioBank.
Table 1.
Background information for matched pairs of subjects with Williams syndrome (WS) and typically developing (TD) individuals. PMI = postmortem interval. Asterisk indicates specimen for whom no genetic information was available.
| Subject | Age at Death | Sex | Hemisphere | PMI (hours) | Cause of death |
|---|---|---|---|---|---|
| WS 10 | 18y | M | R | 24 | WS/Cardiac complications |
| TD 4916 | 19y | M | R | 5 | Drowning |
|
| |||||
| WS 1* | 31y | M | R | 26 | WS/Cardiac complications |
| TD 5539 | 31y | M | R | 24 | Acute Drug Intoxication |
|
| |||||
| WS 14 | 42y | F | R | 18 | WS/Cardiac complications |
| TD 5445 | 42y | F | R | 10 | Pulmonary Thromboembolism |
|
| |||||
| WS 9 | 43y | F | R | 12 | WS/Cardiac complications |
| TD 4636 | 43y | F | R | 19 | Pulmonary Thromboembolism |
|
| |||||
| WS 12 | 45y | M | R | 24 | WS/Cardiac complications |
| TD 5875 | 45y | M | R | 19 | Aortic Dissection |
Tissue Processing
Blocks including the rostral portion of the striatum were used in our analyses owed to the close association of these territories with functional connectivity with regions of the prefrontal cortex (Fudge and Haber, 2002; Haber, 2003; Haber et al., 2006; Delmonte et al., 2013; Averbeck et al., 2014; Jarbo and Verstynen, 2015). For each WS brain, 3-4 cm thick blocks of tissue from the right hemisphere were removed and cryoprotected using successive concentrations of 10, 20, and 30% buffered sucrose solutions, and sectioned at 40μm on a Leica SM2010 freezing microtome. TD subjects were dissected following standard procedures at the UMBTB, which include blocking of hemispheres into 1 cm thick slabs. These blocks were also cryoprotected using sucrose solutions and sectioned at 40μm. Following cryosectioning procedures, a 1-in-10 series of tissue sections was stored for 48 hours in a neutral phosphate buffer solution to rehydrate the tissue. Sections were then mounted on gelatin-coated slides, dried at room temperature for 48 hours, and stained using a 0.25% thionine stain for Nissl substance to visualize cytoarchitecture and quantify neuron and glia densities in WS and TD subjects. Remaining sections were stored in cryoprotectant at −20°C for use in later immunohistochemical and other analyses.
Anatomical Regions of Interest (ROIs)
The striatum is organized topographically by the overlapping projections it receives from cortical territories, which have been categorized on the basis of functionally significant loops (Holt et al., 1997; Nakano et al., 2000; Fudge and Haber, 2002; Haber, 2003; Choi et al., 2012). The regions of interest targeted were in the associative and limbic territories in the dorsal (dC) and medial caudate nucleus (mC), associative putamen (aP), and nucleus accumbens (NA) regions (Figure 1), comprised of areas corresponding to cognitive and limbic loops (Holt et al., 1997; Nakano et al., 2000; Haber, 2003) and reward systems (Balleine et al., 2007; Delgado, 2007; Tanaka et al., 2015). Stereological quantification took place in a consistent manner between sections and cases, within these nuclei as described below.
Figure 1.
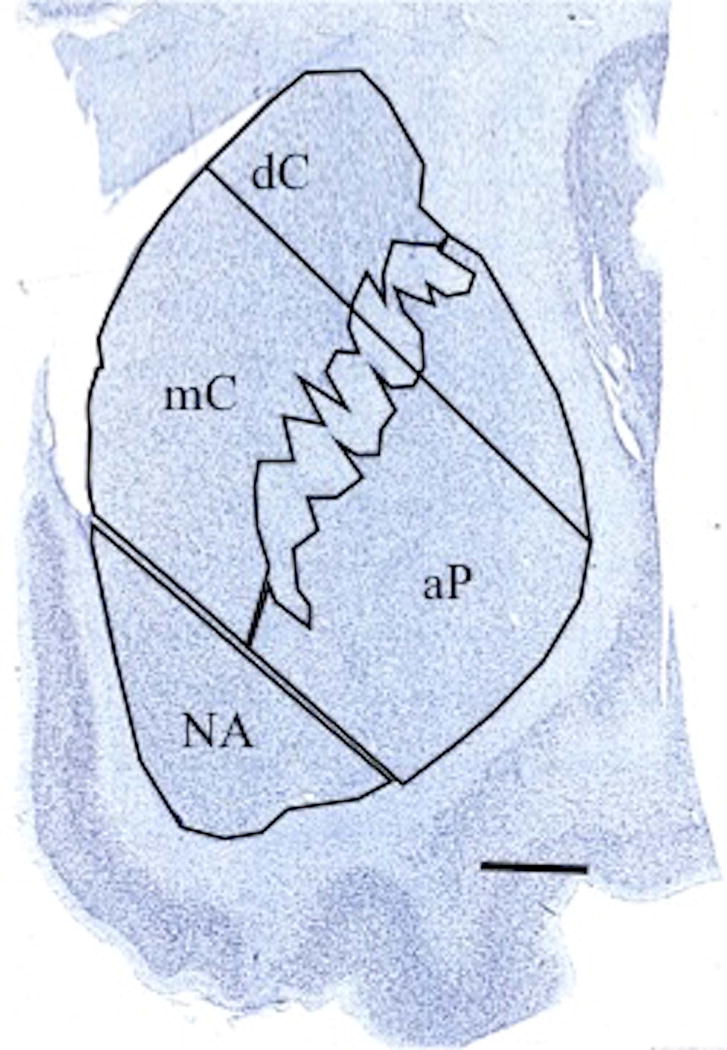
Anatomical regions of interest in the associative territories of the dorsal (dC) and medial caudate nucleus (mC), associative putamen (aP), and nucleus accumbens (NA) regions. The dorsal portion of the putamen, which receives primarily sensorimotor projections, was not measured. Scale bar = 5mm.
Dorsal caudate nucleus (dC)
The caudate head emerges anterior to the appearance of the lateral ventricle and caudal to the genu of the corpus callosum in coronal sections. It is bounded laterally by the emerging fibers of the internal capsule, which separates it from the putamen. Medially, the caudate is bounded by the lateral ventricle, and functional territories receiving projections primarily from prefrontal cortical regions were targeted rostral to the anterior commissure (Haber et al., 2006; Choi et al., 2012). The dorsal territories of the caudate nucleus share overlapping projections from premotor areas and the dorsal prefrontal cortex (Brodmann area 9/46) forming part of a frontal executive loop (Haber, 2003).
Medial caudate nucleus (mC)
To target morphology associated with areas connecting to orbitofrontal cortices, we specifically targeted the medial regions of the caudate nucleus (Haber, 2003; Stephenson et al., 2017) which shares connectivity with areas 11, 12, 13, 14, 24b, and 25 of the orbitofrontal and anterior cingulate cortex (Haber, 2003; Haber et al., 2006). These regions form an important site of convergence for inputs related to cognitive processes including decision-making, reward processing, and cognitive control (Averbeck et al., 2014).
Associative putamen (aP)
The rostral associative territory of the putamen is bounded laterally by the external capsule, which separates it from the claustrum, and medially by the fibers of the internal capsule in coronal sections anterior to the emergence of the globus pallidus. Posterior boundaries of the rostral putamen were delimitated by the emergence of the anterior commissure and rostral to the external medullary lamina of the globus pallidus (Choi et al., 2012). We targeted the medioventral region of the rostral putamen that receives projections from the dorsal prefrontal (PFC) and anterior cingulate cortices (Haber et al., 2006) as part of a reward network defined by its connectivity with the PFC. In each coronal section we sampled within the associative putamen found in the lower two thirds of the nucleus of the putamen, to the exclusion of its dorsal territory (Haber, 2003; Haber et al., 2006), which receives primarily sensorimotor projections.
Nucleus accumbens (NA)
Previous studies have noted that core and shell regions of the human NA show a difference in the distribution of calbindin D28k (Meredith et al., 1996); however, consistent with our own observations in our sample, these differences in staining represent a gradient of CB distribution that do not allow for absolute delineations of boundaries between these regions (Selden et al., 1994, Parent et al., 1996). Since there is cytoarchitectonic continuity of the dorsal striatum and NA region, we have defined our region of interest consistent with previous investigations in TD and ASD subjects (Wegiel et al., 2014), superiorly bounded by the inferior limit of the internal capsule, by drawing a line perpendicular to its orientation. The most rostral limit of the NA is its emergence inferior to the internal capsule. Posteriorly, the region is bounded by the termination of the anterior commissure. The NA is bounded by white matter along the inferior extent of the NA, separating it from cortical regions and the olfactory tubercle.
Stereological Analyses
Unbiased stereological methods were utilized to quantify the density of neurons and glia and neuronal soma area in each ROI, consistently defined by cytoarchitectonic and anatomical criteria across specimens as outlined above, using StereoInvestigator software (v.10, MBF Bioscience, Williston, VT). The rater (KLH) was blinded to diagnosis and subject ID at the time of data collection and no apparent qualitative pathological changes were noted between subject groups that distinguished WS from TD subjects. Intrarater reliability was insured through repeated measures of neuron and glia counting probes in 3 out of the 10 subjects in each ROI with less than 2% difference in estimated counts.
In each region of interest, 8-10 sections were selected in a 1-in-20 series (800 μm apart) spanning its rostral to caudal extent anteriorly from the emergence of the lateral medullary lamina of the globus pallidus in the coronal plane, with care taken to sample consistently across subjects from the region of interest. Stereological analyses were performed on a Dell workstation receiving live video feed from an Optronics MicroFire color video camera (East Muskogee, OK) attached to a Nikon Eclipse 80i microscope equipped with a Ludl MAC5000 stage (Hawthorn, NY) and a Heidenhain z-axis encoder (Plymouth, MN). Regions of interest were hand-traced at 2x magnification, and a square grid was superimposed over the tracing of the region of interest. Grid size was adjusted to optimize counting based on the size of the regions of interest, and measured 2000μm 2 for the nucleus accumbens and dorsal caudate regions, and 3000μm2 for the caudate and putamen regions. The height of the disector measured 9μm, with a 1μm top guard zone. For the quantification of neurons and glia, cells were counted by placing a software marker on each neuron visible in the given counting frame. As white matter inclusions are common within the striatum, care was taken to avoid sampling within white matter by drawing boundaries of the region of interest well outside the boundaries of the internal capsule. Additionally, no markers for glia were counted where neurons were not observed within the field of view, though not necessarily in the counting frame itself, to ensure that counts were not performed within white matter inclusions in striatal territories.
Neurons and glia were counted at 100x magnification using an oil lens (NA 1.4) using the Optical Fractionator probe in StereoInvestigator. Neurons and glial cells were counted using different software markers within the same frame, and distinguished on the basis of their morphology (Figure 2). Briefly, neurons are characterized by their large size, distinct nucleus, and the presence of a darkly-stained nucleolus (Barger et al., 2012; Wegiel et al., 2014). Soma area was also measured in one out of every three neurons counted using the Nucleator probe in StereoInvestigator, using a 4-point array.
Figure 2.
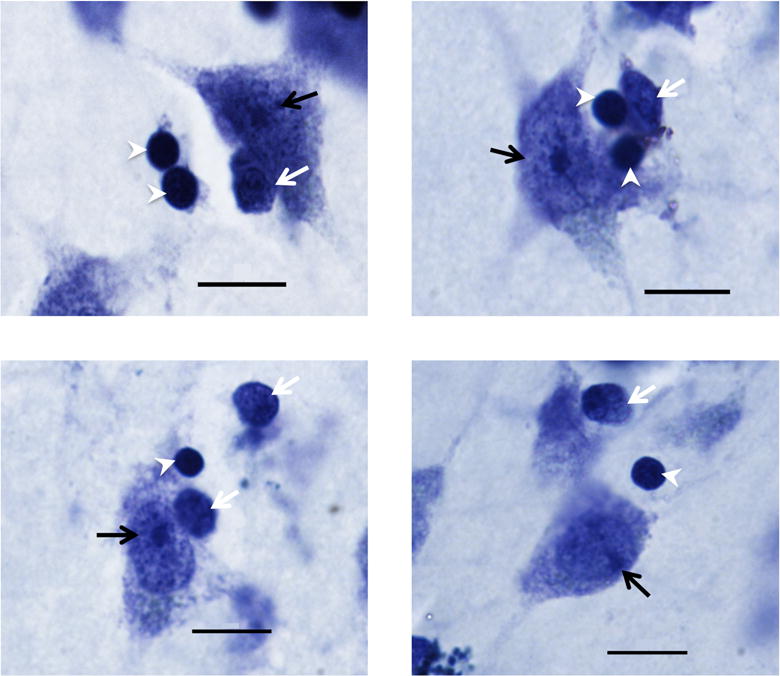
Photomicrographs of Nissl stained cells in the dorsal caudate (top left), medial caudate (top right), putamen (bottom left), and nucleus accumbens (bottom right) regions. Neurons (black arrow) were distinguished from oligodendrocytes (white arrowheads) and other glia (white arrows) by their large size and distinctively stained nucleolus. Image taken at 100x (1.4 NA). Scale bar = 10 μm.
Glia were distinguished from neurons and non-cellular artifacts by the presence of darkly stained cytoplasm, smaller size, and the lack of a distinctive nucleolus (Sherwood et al., 2006; Kreczmanski et al., 2007). Following initial glia counts, we also chose to focus specifically on oligodendrocytes due to their relative abundance within the caudate nucleus, and their distinctive morphology. Oligodendrocytes were identified using criteria described in the literature; as smaller and rounder than astrocytes and microglia, and more darkly-stained, with a more compact nucleus and less granular-appearing cytoplasm, and lacking a distinctive nucleolus in contrast to immature neurons (Baumann and Pham-Dinh, 2001; Hamidi et al., 2004; Pelvig et al., 2008; Karlsen and Pakkenberg, 2011; Morgan et al., 2014) (Figure 2). Given the heterogeneity in morphotypes of activated microglia (Morgan et al., 2014) and the difficulty in distinguishing these from astrocytes in Nissl-stained tissue, only oligodendrocytes were quantified. Additional immunostaining will be implemented in future series to quantify these elements independently. Endothelial cells were sparsely present within the tissue, but were not quantified.
Neuron and glia density for the regions measured was estimated by dividing the number of cells by the projected volume of the sampled region in each ROI using planimetric sampling in StereoInvestigator. A target of 100-200 cells of each type was counted per region of interest with a coefficient of error (Gundersen m=1) less than 0.1.
Statistical Analysis
Independent 2-tailed t-tests (P< 0.05) with Welch’s correction were calculated using GraphPad Prism software to compare means for neuron density, glia density, glia-to-neuron ratio, and soma volume. Grubb’s Outlier test was applied to all data to determine if any single value was a statistically significant outlier, but no outliers were detected and no data points were removed.
Results
Results for all regions of interest are summarized in Table 2 and Figure 3. Table 3 and Figure 4 summarize results for oligodendrocytes in the mC and dC only. A significantly higher density of glia was found in the dC (Figure 3B), as well as an increase in the ratio of glia to neurons (Figure 3C). In the mC, an increased ratio of glia to neurons (Figure 3C), and a significant increase in oligodendrocyte density (Figure 4) was found. Overall, neuron density did not vary significantly between WS and TD for any regions sampled. Total glia density was elevated significantly in the dC, and mean density of glia was also higher in the mC and aP, but these values did not reach statistical significance.
Table 2.
Summary of results, statistical analyses, and percent change for average neuron and glia density, glia to neuron ratio, and neuronal soma area in the dorsal (dC) and medial (mC) caudate nucleus, associative putamen (aP), and nucleus accumbens (NA) regions of the striatum in typically developing (TD) individuals and individuals with Williams syndrome (WS).
| dC | mC | aP | NA | ||
|---|---|---|---|---|---|
| Average neuron density | WS | 19,496±1,471 | 22,935±843 | 26,629±2610 | 27,736±2632 |
| TD | 21,061±776 | 24,931±1,210 | 26,509±1,129 | 28,734±3,455 | |
| Average percent change | −6.51% | −6.85% | +2.37% | +6.32% | |
| t-statistics | t=0.9408 df=6.065 |
t=1.354 df=7.144 |
t=0.422 df=5.447 |
t=0.2299 df=7.473 |
|
| P-value | 0.382 | 0.217 | 0.967 | 0.824 | |
| Average glia density | WS | 58,304±3,595 | 65,990±4,266 | 73,106±8,075 | 60,597±4,502 |
| TD | 47,156±2,482 | 55,046±4,213 | 59,100±4,754 | 58,750±7,743 | |
| Average percent change | +25.42% | +22.63% | +28.92% | +12.24% | |
| t-statistics |
t=2.552 df=7.108 |
t=1.826 df=7.999 |
t=1.495 df=6.476 |
t=0.2062 df=6.427 |
|
| P-value | 0.038* | 0.105 | 0.182 | 0.843 | |
| Glia per neuron | WS | 2.92±0.21 | 2.81±0.01 | 2.70±0.19 | 2.23±0.18 |
| TD | 2.24±0.10 | 2.18±0.08 | 2.22±0.12 | 2.03±0.04 | |
| Average percent change | +31.97% | +29.23% | +23.61% | +9.67% | |
| t-statistics |
t=2.947 df=5.647 |
t=4.286 df=6.896 |
t=2.108 df=6.782 |
t=1.094 df=4.443 |
|
| P-value | 0.028* | 0.004** | 0.074 | 0.330 | |
| Average soma area (μm2) | WS | 128±8 | 121±12 | 101±11 | 112±11 |
| TD | 154±4 | 158±8 | 132±11 | 143±12 | |
| Average percent change | −16.62% | −21.76% | −23.27% | −20.14% | |
| t-statistics |
t=2.964 df=5.755 |
t=2.603 df=7.275 |
t=2.029 df=7.952 |
t=1.952 df=7.919 |
|
| P-value | 0.026* | 0.034* | 0.077 | 0.087 | |
Figure 3.
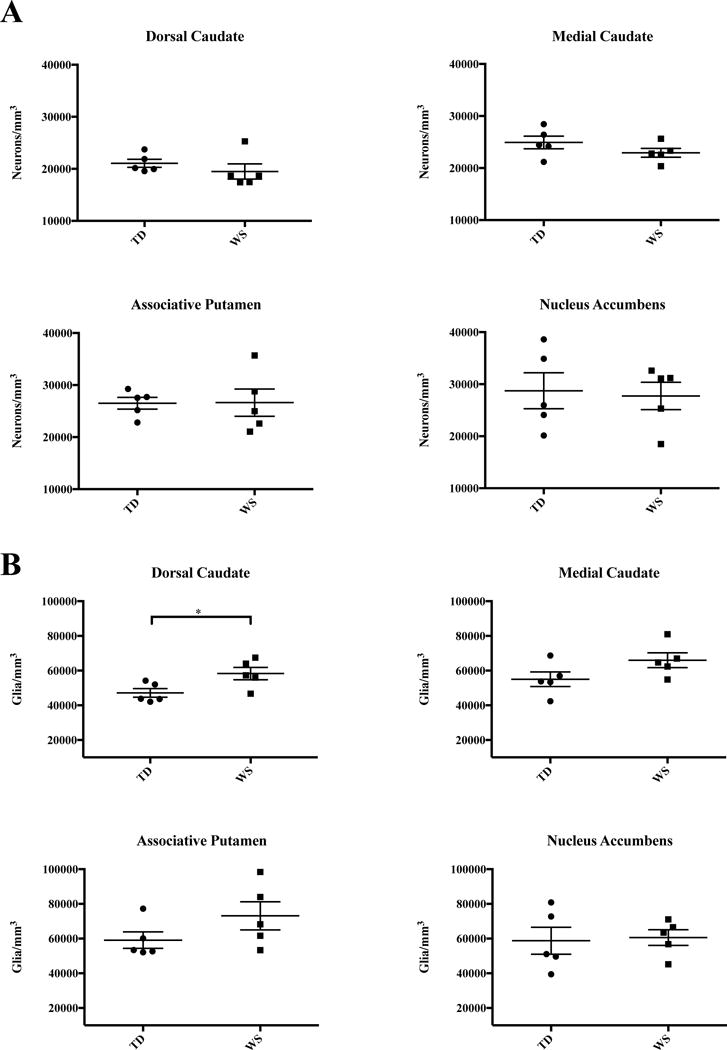
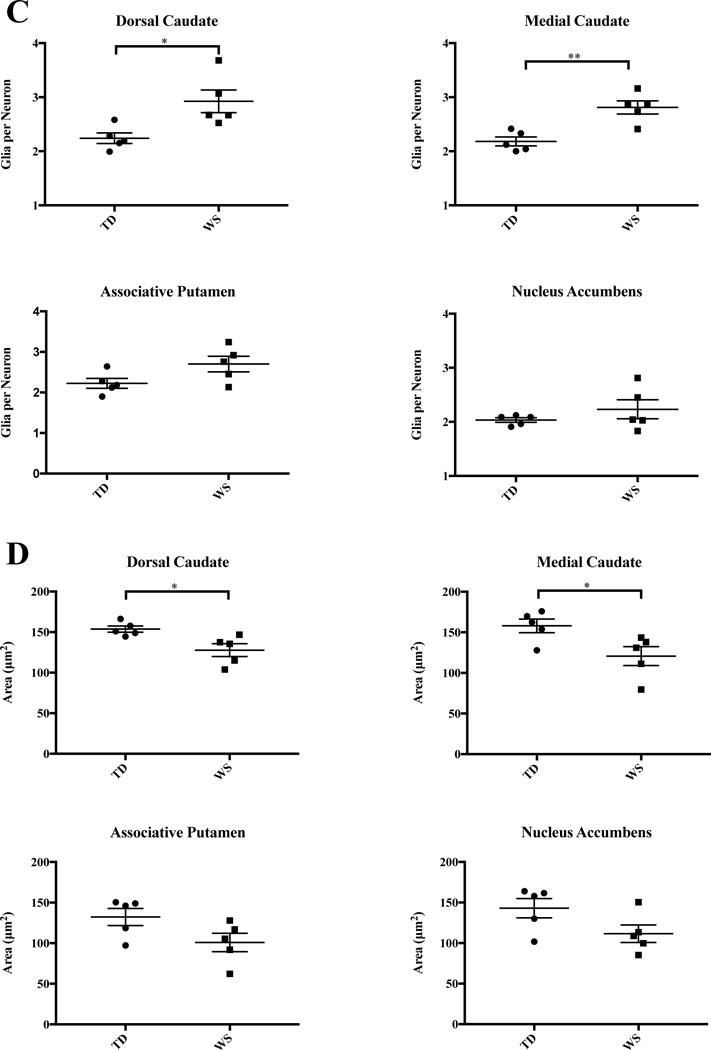
Results of stereological analyses in the four ROIs in WS and TD A. Neuron density. B. Density of glia. C. Glia-to-neuron ratio. D. Average neuronal soma area.
Table 3.
Summary of results and statistical analyses for density of oligodendrocytes and percent change in the medial (mC) and dorsal (dC) regions of the caudate nucleus in typically developing (TD) individuals and individuals with Williams syndrome (WS).
| dC | mC | ||
|---|---|---|---|
| Oligodendrocyte density | WS | 33,810±3,742 | 40,605±3,161 |
| TD | 25,281±1,343 | 27,375±2,566 | |
| t-statistics |
t=2.145 df=5.014 |
t=3.249 df=7.675 |
|
| P-value | 0.085 | 0.012* | |
| Average percent increase | +35.63% | +51.35% | |
| Oligodendrocytes: percent of total glia | WS | 57.41% | 62.20% |
| TD | 53.77% | 50.55% | |
Figure 4.
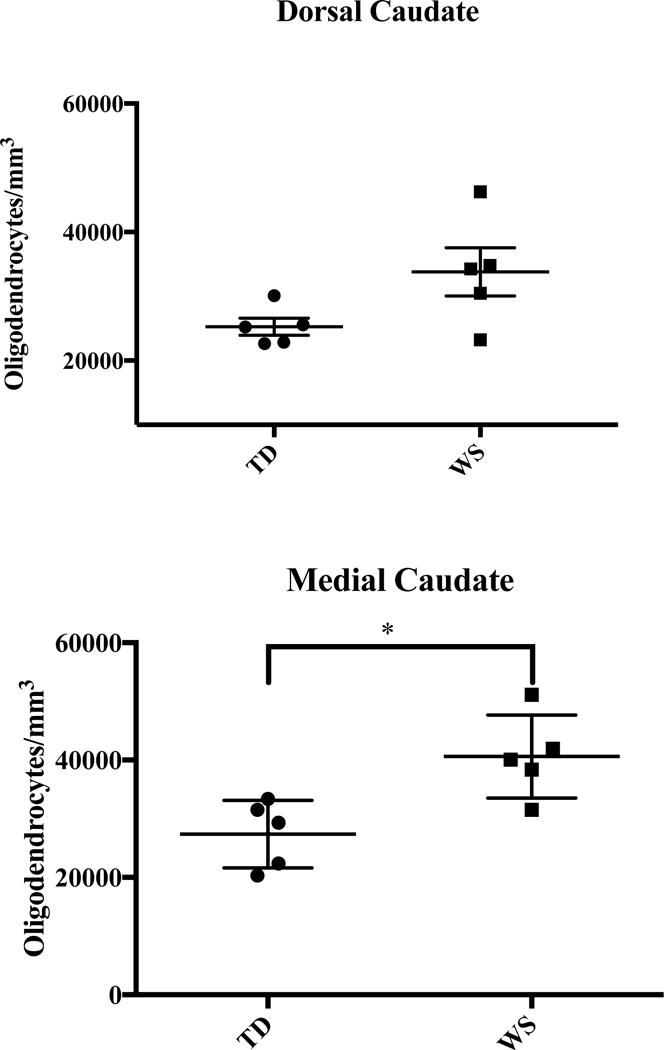
Density of oligodendrocytes in the dorsal and medial caudate nucleus.
Dorsal Caudate Nucleus
A significant increase in the total density of glia was found in the dC (58,304±3,595 WS vs. 47,156±2,482 glia/mm3; P=0.038). The ratio of glia to neurons was also significantly elevated (2.92 WS vs. 2.24 TD glia/neuron, P=0.028). Oligodendrocyte density was higher on average in WS (33,810±3,742 cells/mm3, Figure 4) than in TD subjects (25,281±1,343 cells/mm3), though these results did not reach statistical significance (P=0.085). Though no significant difference in neuron density was found (19,496±1,471 WS vs. 21,061±776 cells/mm3 TD), mean neuronal soma area was smaller in WS (128±8 μm2) than in TD subjects (154±4 μm2, P=0.026).
Medial Caudate Nucleus
Average density of glia was slightly elevated in the WS as compared to TD subjects (65,990±4,266 vs. 55,046±4,213 glia/mm3) though this did not reach significance (P=0.105). However, the ratio of glia to neurons in the medial caudate was significantly elevated in WS as compared to TD subjects (2.81 vs. 2.18 glia/neuron, P=0.004). Additionally, a significant increase of oligodendrocyte density was found in the caudate in WS subjects (40,605±3,161 cells/mm3) as compared to TD controls (27,375±2,566 cells/mm3; P=0.012). No significant difference in overall neuron density was observed between TD and WS subjects in the mC (22,935±843 WS vs. 24,931±1,210 TD neurons/mm3), though average soma area was significantly smaller in WS (121±12 μm2) as compared to TD (158±8 μm2, P=0.034).
Associative Putamen
Glia density was higher on average in WS as compared to TD cases (73,106±8,075 vs. 59,100±4,754glia/mm3), but this did not reach statistical significance (P=0.182). The ratio of glia to neurons in the associative putamen was slightly elevated in WS as compared to TD subjects (2.70 vs. 2.22 glia per neuron), though this also did not reach statistical significance (P=0.074). Mean soma area was also slightly decreased in the associative putamen, but did not reach statistical significance (P=0.077).
Nucleus Accumbens
No significant differences were observed in the nucleus accumbens region for neuron density (27,736 WS vs. 28,734 TD, P=0.824), glia density (60,597 WS vs. 58,750 TD, P=0.843), or glia-to-neuron ratio (2.23 WS vs. 2.03 TD, P=0.330) between WS and TD subjects. Average soma area was modestly smaller in WS (112±11 μm2) than in TD (143±12 μm2) subjects, but this did not reach significance (P=0.087).
Discussion
Summary
Mean glia density was significantly elevated in the dC, and increased glia density was observed across all ROIs of the caudate and putamen, but not in the NA. The glia-to-neuron ratio was significantly elevated in subjects with WS in the medial caudate (P=0.004), and although average glia-to-neuron ratio was higher in the associative putamen, this did not reach significance (P=0.068). (Figure 3C). Significant differences in the density of neurons in territories of the striatum were not observed between WS and TD subjects. Average values of glia density were greater in WS as compared to TD controls in the associative territories of the caudate and putamen but not in the limbic nucleus accumbens region. Average neuronal soma area was consistently smaller across all regions of interest in WS (Figure 3D).
Specific cognitive and neurological impairments in Williams Syndrome have been noted in individuals diagnosed with the disorder. Brains of individuals with Williams Syndrome tend to be smaller overall than in typically-developing individuals (Jernigan and Bellugi, 1990), with notable size reductions found in the parietal lobule (Eckert et al., 2005), occipital grey matter (Reiss et al., 2000), intraparietal sulcus and orbitofrontal cortex (Meyer-Lindenberg et al., 2005; Fan et al., 2017). Functional abnormalities have been demonstrated with respect to primary auditory (Levitin et al., 2003) and visual perception (Galaburda et al., 2002; Atkinson et al., 2007). Additionally, deficits in object-focused and spatial cognition and memory have implicated the dorsal visual stream (Atkinson et al., 1997, 2003) and the hippocampal formation (Meyer-Lindenberg et al., 2005).
Previous histological studies in Williams Syndrome have targeted cellular density and soma size in cortical areas (Galaburda and Bellugi, 2000), with a specific focus on sensory territories. Primary visual cortex in a postmortem sample of Williams Syndrome patients has shown abnormalities in cell size and packing density (Galaburda et al., 2002). Additionally, primary auditory cortex shows no overall significant difference in cell density compared to controls, but enlarged neuronal size in layer IV suggests that there may be differences in connectivity with limbic regions that reflect differences in emotional reactivity to sound (Holinger et al., 2005), particularly music. Lew and colleagues (2016) found a decrease in neuron density specific to BAs 10 and 11 of the prefrontal cortex, specifically in the infragranular layers, that was not found in somatosensory (BA 3), motor (BA 4), or visual cortex (BA 18), further implicating dysfunction of subcortical connectivity.
Analyzing specific neural networks that are affected in WS serves the dual purpose of identifying structures and systems that contribute to selected behavioral and cognitive deficits in WS, and informing the spectrum of neuroanatomical variation that could be compromised in various other neurodevelopmental disorders, including ASD. Given that a portion of the deleted region at chromosome band 7q11.23 is frequently seen as a duplication in the closely-related Dup7 syndrome, which presents universally with an ASD-like social and cognitive phenotype (Berg et al., 2007; Sanders et al., 2012) examining the neuroanatomical phenotype in WS offers the unique opportunity to contrast phenotypic features underlying this unique disorder as a counterpoint to pathological processes in ASD.
Comparisons of the Striatum in TD, WS and ASD
Our results for neuron density in TD subjects fell within expected ranges based on previously reported data (Kreczmanski et al., 2007; Khundakar et al., 2011; Wegiel et al., 2014). Average glia density in TD controls measured was somewhat higher than previously reported for the entire caudate nucleus (Khundakar et al., 2011), but within the range of variation defined by previous studies (Karlsen and Pakkenberg, 2011). This likely results from the specificity of our region of interest as a high-integration area, or perhaps due to the average age of our subjects being somewhat higher than in the sample reported by Khundakar (2011), as older age may be a factor in increased glia densities (Pelvig et al., 2008).
Significant volumetric (Langen et al., 2009, Langen et al., 2014) and microstructural (Wegiel et al., 2014) differences have been noted in the striatum in ASD. Specifically, reductions in total neuronal density were observed in the caudate and nucleus accumbens in ASD as compared to TD controls, with ASD subjects displaying overall increases in the volume of the caudate (Wegiel et al., 2014). In contrast, we found no significant differences in the density of neurons in four striatal regions of interest in WS. We found an increase in the density of glia in the dorsal caudate nucleus, in addition to increased glia-to-neuron ratio in the medial caudate nucleus, which our results suggest is driven by a significant overall increase in oligodendrocytes. Similar quantitative data for the striatum in ASD are not published, but glial pathologies have been noted in several cortical areas (Edmonson et al., 2014) and in the amygdala (Morgan et al., 2014). Specifically, the abundance of microglia, and particularly activated microglia, in the prefrontal cortex in ASD (Morgan et al., 2010) suggests that neuroinflammation may account in part for dysfunction in the PFC. The methods used in the present study do not allow for these comparisons to be made in WS, but future research will utilize similar techniques to examine additional populations of glia, including microglia, in our sample.
Here, we targeted oligodendrocytes due to their consistent morphology and abundant appearance in all cases, finding an increase in their density in WS. The mechanism for the observed increase in oligodendrocytes in the gray matter of the caudate nucleus is not known, but one can offer some speculation for this important difference. It is possible that an excess of oligodendrocytes is being produced, either early in development, or by oligodendrocyte progenitor cells (OPCs), which continue to differentiate and produce additional oligodendrocytes throughout adulthood. Successive waves of migrating oligodendrocyte precursor cells replace embryonic-derived and postnatally generated lineages of oligodendrocytes, competing for space and replacing older lineages (Kessaris et al., 2006). Failure of newly derived oligodendrocytes to eliminate older lineages through apoptosis may result in their excess in the adult brain. Another possible mechanism may include abnormalities in migration. OPCs migrate along vasculature, guided by endothelial cells (Tsai et al., 2016). Thus, disruptions in vasculature and associated endothelial cells may lead to poor migration. Animal models of the deletion of GTF2i and GTF2iRD1, two genes that are part of the hemizygous gene deletion observed in WS, have shown abnormalities in cerebral angiogenesis, as well as disrupted development of endothelial cells (Enkhmandakh et al., 2009), attributed to the resulting downregulation of endothelial growth factor receptor-2 gene. These disruptions in vasculature and endothelial cell development likely have important consequences for the fate of migrating oligodendrocytes. In particular, oligodendrocytes may not effectively migrate into white matter, leading to their excessive numbers in grey matter. This likely has important implications for understanding hypoactivity in frontostriatal circuitry that underlies deficits in behavioral control seen in WS. Specifically, the functional role of oligodendrocytes in myelination may point to mechanisms underlying dysfunction of frontostriatal connectivity related to hypomyelination and associated white matter abnormalities.
Significant reductions in white matter volume that outpace deficits in gray matter are reported in structural imaging studies of WS (Faria et al., 2012). Studies of white matter architecture using diffusion tensor imaging (DTI) have demonstrated aberrations in fiber tract directionality and coherence (Marenco et al., 2007). Increased fractional anisotropy (FA) was also observed in the superior longitudinal fasciculus in WS, which may reflect deficits in myelination (Alba-Ferrara and de Erausquin, 2013). The role of oligodendrocytes in the complex relationship between myelination in gray and white matter, and white matter architecture in WS, bears further study. Future research will utilize advanced techniques to examine fiber densities and myelination in frontal and striatal regions of interest.
Developmental considerations
A major limitation in postmortem studies of many neurodevelopmental disorders includes lack of a sufficient developmental sample. This is particularly true in WS, given the rarity of the disorder. Though there is no comprehensive data available examining average lifespan in individuals with WS, death in early middle age is not uncommon due to cardiac complications of the disorder, and may represent a peak in mortality. Accordingly, three of our subjects (WS9, WS 12, and WS 14) are very close in age, and similar patterns can be observed in these individuals across our measures for glia. Figure 5 summarizes glia and oligodendrocyte densities comparing across matched pairs. Though no clear pattern emerges for total glia density (Figure 5A/B) or oligodendrocyte density in the mC (Figure 5D), oligodendrocyte density increases slightly with age in the dC (Figure 5C) in WS (P=0.125) but not in TD (P=0.713) subjects when older subjects (age 42-45) are compared to younger individuals (ages 18-31). Additional data from a larger sample may help to clarify this pattern.
Figure 5.
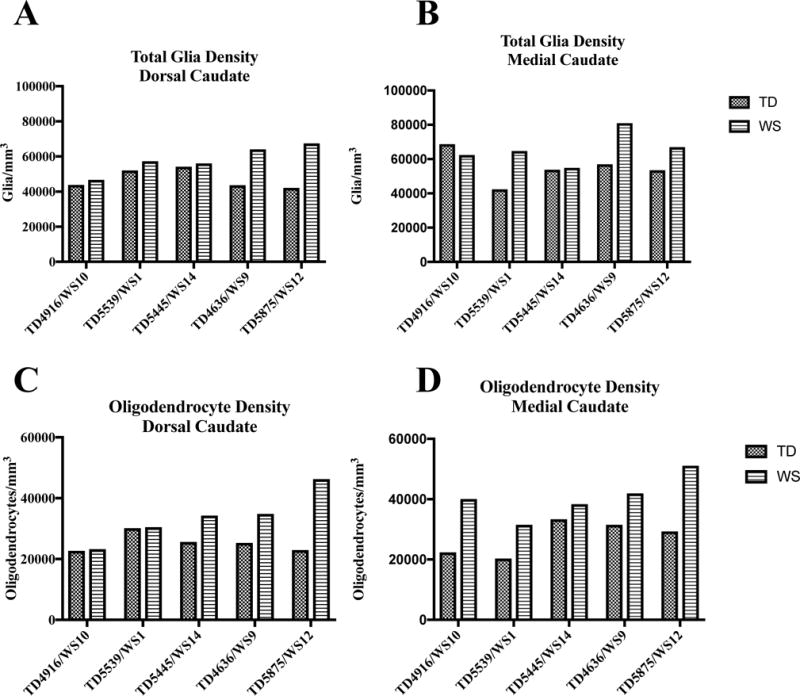
Individual values for density of total glia (A/B) and oligodendrocytes (C/D) in the dorsal and medial caudate nucleus in matched pairs.
Frontostriatal Dysfunction in ASD and WS
Targeting psychiatric endophenotypes common to both disorders may help to elucidate patters of striatal neuroanatomy, particularly with reference to territories that share extensive connectivity with the PFC, in a way that contributes to deficits of executive function. This may be particularly true with respect to features of behavioral control that are impaired in both ASD (Christ et al., 2007; Kana et al., 2007; Langen et al., 2012) and WS (Carrasco et al., 2005; Mobbs et al., 2007) as well.
Previous research has shown anatomical abnormalities of the PFC in both ASD and WS. Significant overgrowth in the PFC in early development is reflected in increased neuron number in dorsal and medial prefrontal cortical regions in ASD (Courchesne et al., 2011), which corresponds to narrower spacing of cortical minicolumns in the PFC of ASD (Buxhoeveden et al., 2006; Casanova et al., 2006). This may relate to local overconnectivity in the PFC to the detriment of long-range connectivity (Courchesne and Pierce, 2005) with subcortical structures. Conversely, reduced neuronal density in lower layers V/VI of BA 10 was observed in WS (Lew et al., 2016), which implicates a differential decrease in PFC neurons that share connectivity with subcortical structures. Additionally, layer III pyramidal neurons of BA 10 in WS may not display the pattern of longer and more branched basal dendrites as observed in TDs (Hrvoj-Mihic et al., 2017) relatively to primary processing areas, suggesting compromised circuitry in high-integration PFC areas.
Important connectivity between prefrontal cortical areas and territories of the striatum observed in primates (Haber, 2003; Haber et al., 2006; Averbeck et al., 2014) investigated here has been established in human subjects (Choi et al., 2012; Delmonte et al., 2013; Jarbo and Verstynen, 2015) suggesting that abnormalities of this network may account for downstream dysfunction of striatal territories. Alterations in striatal morphology and function have been noted as a characteristic of both disorders. Structural findings consistently report alterations of striatal morphology in ASD, including increased volume of the caudate (Hollander et al., 2005; Langen et al., 2007; 2009). Functionally, reduced frontostriatal activity in ASD subjects as compared to controls has been observed, particularly in response to social stimuli (DelMonte et al., 2012; Kohls et al., 2013). Deficits of behavioral control in response inhibition tasks have also been attributed to frontostriatal hypoactivity in WS generally (Mobbs et al., 2007) though specifically social stimuli were not utilized. Repetitive behaviors are also a common feature of ASD related to deficits in behavioral control thought to relate to striatal abnormalities (Hollander et al., 2005), though it is not a common behavioral feature associated with WS (Rodgers et al., 2012). These differences may be rooted in the different patterns of functional connectivity in frontostriatal territories in WS and ASD: whereas connectivity with associative regions of the caudate may be reflected in reduced activity in response inhibition tasks, connectivity between frontal regions and the putamen has shown significant impairment (Balsters et al., 2013) in ASD.
Overall, dysfunction in frontostriatal territories in WS contributes to deficits in the ability to inhibit the drive for social interaction (Mobbs et al., 2007) ultimately characterizing WS as a disorder of inhibitory behavioral control in distinctively social domains. Here, we reported abnormalities at the cellular level that may underlie frontostriatal dysfunction in the caudate nucleus that complements previous evidence for reduced neuron density in prefrontal cortical regions (Lew et al., 2016). It is likely that dysfunction in other systems in WS, including impaired frontolimbic connectivity between the prefrontal cortex and amygdala (Ng et al., 2016), is similarly reflected in pathologies within the amygdala in WS, which is an additional target of ongoing investigations given known aberrations in neuron numbers in ASD (Schumann et al., 2006) in discrete amygdala nuclei. Frontostriatal (Fan et al., 2016) and frontolimbic (Ng et al., 2016) systems contribute to distinctive functional deficits in WS in different patterns that affect the cognitive and emotional profiles of this unique disorder in characteristic ways providing an interesting contrast to patterns seen in ASD.
Acknowledgments
This research was supported by the National Institutes of Health P01 NICHD033113 and 5R03MH103697. We are grateful to the individuals with WS and their families, past and present, who have participated in research characterizing this disorder’s unique cognitive and social phenotype. In particular, we thank the families who have graciously donated WS human tissue, which was obtained under the Ursula Bellugi WS Brain Collection, curated by KS at UCSD. Human TD tissue was obtained from the NIH Neurobiobank at the University of Maryland, Baltimore, MD. We additionally thank Mary Ann Raghanti, Chelsea Brown, Deion Cuevas, Valerie Judd, Hailee Orfant, and Linnea Wilder for assistance with tissue processing and feedback in the development of this manuscript.
References
- Alba-Ferrara LM, de Erausquin GA. What does anisotropy measure? Insights from increased and decreased anisotropy in selective fiber tracts in schizophrenia. Front Integr Neurosci. 2013;7:9. doi: 10.3389/fnint.2013.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson J, Anker S, Braddick O, Nokes L, Mason A, Braddick F. Visual and visuospatial development in young children with Williams syndrome. Dev Med Child Neurol. 2007;43:330–337. doi: 10.1017/s0012162201000615. [DOI] [PubMed] [Google Scholar]
- Atkinson J, Braddick O, Anker S, Curran W, Andrew R, Wattam-Bell J, Braddick F. Neurobiological models of visuospatial cognition in children with Williams syndrome: measures of dorsal-stream and frontal function. Dev Neuropsychol. 2003;23:139–72. doi: 10.1080/87565641.2003.9651890. [DOI] [PubMed] [Google Scholar]
- Atkinson J, King J, Braddick OJ, Nokes L, Anker S, Braddick F. A specific deficit of dorsal stream function in Williams’ syndrome. Neuroreport. 1997;8:1919–1922. doi: 10.1097/00001756-199705260-00025. [DOI] [PubMed] [Google Scholar]
- Averbeck BB, Lehman J, Jacobson M, Haber SN. Estimates of Projection Overlap and Zones of Convergence within Frontal-Striatal Circuits. J Neurosci. 2014;34:9497–9505. doi: 10.1523/JNEUROSCI.5806-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine BW, Delgado MR, Hikosaka O. The Role of the Dorsal Striatum in Reward and Decision-Making. J Neurosci. 2007;27:8161–8165. doi: 10.1523/JNEUROSCI.1554-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger N, Stefanacci L, Schumann CM, Sherwood CC, Annese J, Allman JM, Buckwalter JA, Hof PR, Semendeferi K. Neuronal populations in the basolateral nuclei of the amygdala are differentially increased in humans compared with apes: a stereological study. J Comp Neurol. 2012;520:3035–54. doi: 10.1002/cne.23118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Berg JS, Brunetti-Pierri N, Peters SU, Kang S-HL, Fong C-T, Salamone J, Freedenberg D, Hannig VL, Prock LA, Miller DT, Raffalli P, Harris DJ, et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007;9:427–441. doi: 10.1097/gim.0b013e3180986192. [DOI] [PubMed] [Google Scholar]
- Beuren AJ, Apitz J, Harman D. Supravalvular Aortic Stenosis in Association with Mental Retardation and a Certain Facial Appearance. Circulation. 1962;26:1235–1240. doi: 10.1161/01.cir.26.6.1235. [DOI] [PubMed] [Google Scholar]
- Brock J. Language abilities in Williams syndrome: A critical review. Dev Psychopathol. 2007;19:97–127. doi: 10.1017/S095457940707006X. [DOI] [PubMed] [Google Scholar]
- Buxhoeveden DP, Semendeferi K, Buckwalter J, Schenker N, Switzer R, Courchesne E. Reduced minicolumns in the frontal cortex of patients with autism. Neuropathol Appl Neurobiol. 2006;32:483–91. doi: 10.1111/j.1365-2990.2006.00745.x. [DOI] [PubMed] [Google Scholar]
- Carrasco X, Castillo S, Aravena T, Rothhammer P, Aboitiz F. Williams syndrome: pediatric, neurologic, and cognitive development. Pediatr Neurol. 2005;32:166–72. doi: 10.1016/j.pediatrneurol.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Casanova MF, Kooten IAJ, van Switala AE, Engeland H, van Heinsen H, Steinbusch HWM, Hof PR, Trippe J, Stone J, Schmitz C. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006;112:287–303. doi: 10.1007/s00401-006-0085-5. [DOI] [PubMed] [Google Scholar]
- Choi EY, Yeo BTT, Buckner RL. The organization of the human striatum estimated by intrinsic functional connectivity. J Neurophysiol. 2012;108:2242–2263. doi: 10.1152/jn.00270.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ SE, Holt DD, White DA, Green L. Inhibitory control in children with autism spectrum disorder. J Autism Dev Disord. 2007;37:1155–1165. doi: 10.1007/s10803-006-0259-y. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, Barnes CC, Pierce K. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306:2001–10. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Pierce K. Why the frontal cortex in autism might be talking only to itself: Local over-connectivity but long-distance disconnection. Current Opinion in Neurobiology. 2005;15:225–230. doi: 10.1016/j.conb.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Delgado MR. Reward-related responses in the human striatum. Annals NY Acad Sci. 2007;1104:70–88. doi: 10.1196/annals.1390.002. [DOI] [PubMed] [Google Scholar]
- Delmonte S, Gallagher L, O’Hanlon E, McGrath J, Balsters JH. Functional and structural connectivity of frontostriatal circuitry in Autism Spectrum Disorder. Front Hum Neurosci. 2013;7:430. doi: 10.3389/fnhum.2013.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle TF, Bellugi U, Korenberg JR, Graham J. “Everybody in the world is my friend” hypersociability in young children with Williams syndrome. Am J Med Genet A. 2004;124A:263–273. doi: 10.1002/ajmg.a.20416. [DOI] [PubMed] [Google Scholar]
- Eckert MA, Hu D, Eliez S, Bellugi U, Galaburda A, Korenberg J, Mills D, Reiss AL. Evidence for superior parietal impairment in Williams syndrome. Neurology. 2005;64:152–3. doi: 10.1212/01.WNL.0000148598.63153.8A. [DOI] [PubMed] [Google Scholar]
- Enkhmandakh B, Makeyev AV, Erdenechimeg L, Ruddle FH, Chimge N-O, Tussie-Luna MI, Roy AL, Bayarsaihan D. Essential functions of the Williams-Beuren syndrome-associated TFII-I genes in embryonic development. Proc Natl Acad Sci U S A. 2009;106:181–186. doi: 10.1073/pnas.0811531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan CC, Brown TT, Bartsch H, Kuperman JM, Hagler DJ, Schork A, Searcy Y, Bellugi U, Halgren E, Dale AM. Williams syndrome-specific neuroanatomical profile and its associations with behavioral features. NeuroImage Clin. 2017;15:343–347. doi: 10.1016/j.nicl.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria AV, Landau B, O’Hearn KM, Li X, Jiang H, Oishi K, Zhang J, Mori S. Quantitative analysis of gray and white matter in Williams syndrome. Neuroreport. 2012;23:283–289. doi: 10.1097/WNR.0b013e3283505b62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigerio E, Burt DM, Gagliardi C, Cioffi G, Martelli S, Perrett DI, Borgatti R. Is everybody always my friend? Perception of approachability in Williams syndrome. Neuropsychologia. 2006;44:254–9. doi: 10.1016/j.neuropsychologia.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Fudge JL, Haber SN. Defining the Caudal Ventral Striatum in Primates: Cellular and Histochemical Features. J Neurosci. 2002;22:10078–10082. doi: 10.1523/JNEUROSCI.22-23-10078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaburda AM, Bellugi U. V. Multi-level analysis of cortical neuroanatomy in Williams syndrome. J Cogn Neurosci. 2000;12(Suppl 1):74–88. doi: 10.1162/089892900561995. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Holinger DP, Bellugi U, Sherman GF. Williams syndrome: neuronal size and neuronal-packing density in primary visual cortex. Arch Neurol. 2002;59:1461–1467. doi: 10.1001/archneur.59.9.1461. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Genetics of autism spectrum disorders. Trends in Cognitive Sciences. 2011;9:409–416. doi: 10.1016/j.tics.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN. The primate basal ganglia: parallel and integrative networks. J Chem Neuroanat. 2003;26:317–330. doi: 10.1016/j.jchemneu.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Haber SN, Kim K-S, Mailly P, Calzavara R. Reward-Related Cortical Inputs Define a Large Striatal Region in Primates That Interface with Associative Cortical Connections, Providing a Substrate for Incentive-Based Learning. J Neurosci. 2006;26:8368–8376. doi: 10.1523/JNEUROSCI.0271-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi M, Drevets WC, Price JL. Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol Psychiatry. 2004;55:563–569. doi: 10.1016/j.biopsych.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Holinger DP, Bellugi U, Mills DL, Korenberg JR, Reiss AL, Sherman GF, Galaburda AM. Relative sparing of primary auditory cortex in Williams Syndrome. Brain Res. 2005;1037:35–42. doi: 10.1016/j.brainres.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Holt DJ, Graybiel ANNM, Saper CB. Neurochemical Architecture of the Human Striatum. J Comp Neurol. 1997;384:1–25. doi: 10.1002/(sici)1096-9861(19970721)384:1<1::aid-cne1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Jarbo K, Verstynen TD. Converging structural and functional connectivity of orbitofrontal, dorsolateral prefrontal, and posterior parietal cortex in the human striatum. J Neurosci. 2015;35:3865–3878. doi: 10.1523/JNEUROSCI.2636-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järvinen-Pasley A, Adolphs R, Yam A, Hill KJ, Grichanik M, Reilly J, Mills D, Reiss AL, Korenberg JR, Bellugi U. Affiliative behavior in Williams syndrome: Social perception and real-life social behavior. Neuropsychologia. 2010;48:2110–2119. doi: 10.1016/j.neuropsychologia.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järvinen-Pasley A, Bellugi U, Reilly J, Mills DL, Galaburda A, Reiss AL, Korenberg JR. Defining the social phenotype in Williams syndrome: a model for linking gene, the brain, and behavior. Dev Psychopathol. 2008;20:1–35. doi: 10.1017/S0954579408000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järvinen A, Korenberg JR, Bellugi U. The social phenotype of Williams syndrome. Current Opinion in Neurobiology. 2013;23:414–422. doi: 10.1016/j.conb.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernigan TL, Bellugi U. Anomalous Brain Morphology on Magnetic Resonance Images in Williams Syndrome and Down Syndrome. Arch Neurol. 1990;47:529–533. doi: 10.1001/archneur.1990.00530050049011. [DOI] [PubMed] [Google Scholar]
- Jeste SS, Geschwind DH. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat Rev Neurol. 2014;10:74–81. doi: 10.1038/nrneurol.2013.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones W, Bellugi U, Lai Z, Chiles M, Reilly J, Lincoln A, Adolphs R. Hypersociability in Williams Syndrome. J Cogn Neurosci. 2000;12:30–46. doi: 10.1162/089892900561968. [DOI] [PubMed] [Google Scholar]
- Kana RK, Keller TA, Minshew NJ, Just MA. Inhibitory Control in High-Functioning Autism: Decreased Activation and Underconnectivity in Inhibition Networks. Biol Psychiatry. 2007;62:198–206. doi: 10.1016/j.biopsych.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsen AS, Pakkenberg B. Total numbers of neurons and glial cells in cortex and basal ganglia of aged brains with down syndrome-a stereological study. Cereb Cortex. 2011;21:2519–2524. doi: 10.1093/cercor/bhr033. [DOI] [PubMed] [Google Scholar]
- Karmiloff-Smith A, Grant J, Berthoud I, Davies M, Howlin P, Udwin O. Language and Williams Syndrome: How Intact Is “Intact”? Child Dev. 1997;68:246–262. [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khundakar A, Morris C, Oakley A, Thomas AJ. Morphometric analysis of neuronal and glial cell pathology in the caudate nucleus in late-life depression. Am J Geriatr Psychiatry. 2011;19:132–41. doi: 10.1097/JGP.0b013e3181df4642. [DOI] [PubMed] [Google Scholar]
- Kreczmanski P, Heinsen H, Mantua V, Woltersdorf F, Masson T, Ulfig N, Schmidt-Kastner R, Korr H, Steinbusch HWM, Hof PR, Schmitz C. Volume, neuron density and total neuron number in five subcortical regions in schizophrenia. Brain. 2007;130:678–92. doi: 10.1093/brain/awl386. [DOI] [PubMed] [Google Scholar]
- Langen M, Durston S, Staal WG, Palmen SJMC, van Engeland H. Caudate Nucleus Is Enlarged in High-Functioning Medication-Naive Subjects with Autism. Biol Psychiatry. 2007;62:262–266. doi: 10.1016/j.biopsych.2006.09.040. [DOI] [PubMed] [Google Scholar]
- Langen M, Leemans A, Johnston P, Ecker C, Daly E, Murphy CM, dell’Acqua F, Durston S, Murphy DGM. Fronto-striatal circuitry and inhibitory control in autism: Findings from diffusion tensor imaging tractography. Cortex. 2012;48:183–193. doi: 10.1016/j.cortex.2011.05.018. [DOI] [PubMed] [Google Scholar]
- Levitin DJ, Menon V, Schmitt JE, Eliez S, White CD, Glover GH, Kadis J, Korenberg JR, Bellugi U, Reiss AL. Neural Correlates of Auditory Perception in Williams Syndrome: An fMRI Study. Neuroimage. 2003;18:74–82. doi: 10.1006/nimg.2002.1297. [DOI] [PubMed] [Google Scholar]
- Lew CH, Brown C, Bellugi U, Semendeferi K. Neuron density is decreased in the prefrontal cortex in Williams syndrome. Autism Res. 2017;10:99–112. doi: 10.1002/aur.1677. [DOI] [PubMed] [Google Scholar]
- Little K, Riby DM, Janes E, Clark F, Fleck R, Rodgers J. Heterogeneity of social approach behaviour in Williams syndrome: The role of response inhibition. Res Dev Disabil. 2013;34:959–967. doi: 10.1016/j.ridd.2012.11.020. [DOI] [PubMed] [Google Scholar]
- Lombroso PJ, Ogren MP, Jones W, Klin A. Heterogeneity and Homogeneity Across the Autism Spectrum: The Role of Development. J Am Acad Child Adolesc Psychiatry. 2009;48:471–473. doi: 10.1097/CHI.0b013e31819f6c0d. [DOI] [PubMed] [Google Scholar]
- Marenco S, Siuta MA, Kippenhan JS, Grodofsky S, Chang W-L, Kohn P, Mervis CB, Morris CA, Weinberger DR, Meyer-Lindenberg A, Pierpaoli C, Berman KF. Genetic contributions to white matter architecture revealed by diffusion tensor imaging in Williams syndrome. Proc Natl Acad Sci. 2007;104:15117–15122. doi: 10.1073/pnas.0704311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith GE, Pattiselanno A, Groenewegen HJ, Haber SN. Shell and core in monkey and human nucleus accumbens identified with antibodies to calbindin-D28k. J Comp Neurol. 1996;365:628–639. doi: 10.1002/(SICI)1096-9861(19960219)365:4<628::AID-CNE9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Mervis CB, Becerra AM. Language and communicative development in Williams syndrome. Mental Retardation and Developmental Disabilities Research Reviews. 2007;13:3–15. doi: 10.1002/mrdd.20140. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Hariri AR, Munoz KE, Mervis CB, Mattay VS, Morris CA, Berman KF. Neural correlates of genetically abnormal social cognition in Williams syndrome. Nat Neurosci. 2005;8:991–3. doi: 10.1038/nn1494. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Mervis CB, Berman KF. Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat Rev Neurosci. 2006;7:380–93. doi: 10.1038/nrn1906. [DOI] [PubMed] [Google Scholar]
- Miles JH. Autism spectrum disorders—A genetics review. Genet Med. 2011;13:278–294. doi: 10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- Mobbs D, Eckert MA, Mills D, Korenberg J, Bellugi U, Galaburda AM, Reiss AL. Frontostriatal dysfunction during response inhibition in Williams syndrome. Biol Psychiatry. 2007;62:256–61. doi: 10.1016/j.biopsych.2006.05.041. [DOI] [PubMed] [Google Scholar]
- Morgan JT, Barger N, Amaral DG, Schumann CM. Stereological study of amygdala glial populations in adolescents and adults with autism spectrum disorder. PLoS One. 2014;9 doi: 10.1371/journal.pone.0110356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, Courchesne E, Everall IP. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry. 2010;68:368–376. doi: 10.1016/j.biopsych.2010.05.024. [DOI] [PubMed] [Google Scholar]
- Nakano K, Kayahara T, Tsutsumi T, Ushiro H. Neural circuits and functional organization of the striatum. J Neurol. 2000;247:V1–V15. doi: 10.1007/pl00007778. [DOI] [PubMed] [Google Scholar]
- Parent A, Fortin M, Côté PY, Cicchetti F. Calcium binding proteins in primate basal ganglia. Neuroscience Research. 1996;4:309–334. doi: 10.1016/0168-0102(96)01065-6. [DOI] [PubMed] [Google Scholar]
- Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiol Aging. 2008;29:1754–1762. doi: 10.1016/j.neurobiolaging.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Persico AM, Napolioni V. Autism genetics. Behavioural Brain Research. 2013;251:95–112. doi: 10.1016/j.bbr.2013.06.012. [DOI] [PubMed] [Google Scholar]
- Phillips CE, Jarrold C, Baddeley AD, Grant J, Karmiloff-Smith A. Comprehension of Spatial Language Terms in Williams Syndrome: Evidence for an Interaction Between Domains of Strength and Weakness. Cortex. 2004;40:85–101. doi: 10.1016/s0010-9452(08)70922-5. [DOI] [PubMed] [Google Scholar]
- Reilly J, Losh M, Bellugi U, Wulfeck B. “Frog, where are you?” Narratives in children with specific language impairment, early focal brain injury, and Williams syndrome. Brain Lang. 2004;88:229–47. doi: 10.1016/S0093-934X(03)00101-9. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Eliez S, Schmitt JE, Straus E, Lai Z, Jones W, Bellugi U. Neuroanatomy of Williams syndrome: A high-resolution MRI study. J Cogn Neurosci. 2000;12(Suppl 1):65–73. doi: 10.1162/089892900561986. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Eckert MA, Rose FE, Karchemskiy A, Kesler S, Chang M, Reynolds MF, Kwon H, Galaburda A. An experiment of nature: brain anatomy parallels cognition and behavior in Williams syndrome. J Neurosci. 2004;24:5009–15. doi: 10.1523/JNEUROSCI.5272-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring H, Woodbury-Smith M, Watson P, Wheelwright S, Baron-Cohen S. Clinical heterogeneity among people with high functioning autism spectrum conditions: evidence favouring a continuous severity gradient. Behav Brain Funct. 2008;4:11. doi: 10.1186/1744-9081-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–41. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–9. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood CC, Stimpson CD, Raghanti MA, Wildman DE, Uddin M, Grossman LI, Goodman M, Redmond JC, Bonar CJ, Erwin JM, Hof PR. Evolution of increased glia-neuron ratios in the human frontal cortex. Proc Natl Acad Sci U S A. 2006;103:13606–11. doi: 10.1073/pnas.0605843103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson AR, Edler MK, Erwin JM, Jacobs B, Hopkins WD, Hof PR, Sherwood CC, Raghanti MA. Cholinergic innervation of the basal ganglia in humans and other anthropoid primates. J Comp Neurol. 2017;525:319–332. doi: 10.1002/cne.24067. [DOI] [PubMed] [Google Scholar]
- Strømme P, Bjørnstad PG, Ramstad K. Prevalence Estimation of Williams Syndrome. J Child Neurol. 2002;17:269–271. doi: 10.1177/088307380201700406. [DOI] [PubMed] [Google Scholar]
- Tager-Flusberg H, Joseph RM. Identifying neurocognitive phenotypes in autism. Philos Trans R Soc B Biol Sci. 2003;358:303–314. doi: 10.1098/rstb.2002.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Pan X, Oguchi M, Taylor JE, Sakagami M. Dissociable functions of reward inference in the lateral prefrontal cortex and the striatum. Front Psychol. 2015;6 doi: 10.3389/fpsyg.2015.00995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MSC, Grant J, Barham Z, Gsödl M, Laing E, Lakusta L, Tyler LK, Grice S, Paterson S, Karmiloff-Smith A. Past tense formation in Williams syndrome. Lang Cogn Process. 2001;16:143–176. [Google Scholar]
- Tsai H-H, Niu J, Munji R, Davalos D, Chang J, Zhang H, Tien A-C, Kuo CJ, Chan JR, Daneman R, Fancy SPJ. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science (80-) 2016;351:379–384. doi: 10.1126/science.aad3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udwin O, Yule W. Expressive language of children with Williams syndrome. Am J Med Genet. 2005;37:108–114. doi: 10.1002/ajmg.1320370620. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Flory M, Kuchna I, Nowicki K, Ma SY, Imaki H, Wegiel J, Cohen IL, London E, Wisniewski T, Brown WT. Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol Commun. 2014;2:141. doi: 10.1186/s40478-014-0141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JCP, Barratt-Boyd BG, Lowe JB. Supravalvular Aortic Stenosis. Circulation. 1961;24:1311–1318. doi: 10.1161/01.cir.24.6.1311. [DOI] [PubMed] [Google Scholar]