

Abstract
Galectin-3 is a biomarker of heart disease. However, it remains unknown whether increase in galectin-3 levels is dependent on aetiology or disease-associated conditions and whether diseased heart releases galectin-3 into the circulation. We explored these questions in mouse models of heart disease and in patients with cardiomyopathy. All mouse models (dilated cardiomyopathy, DCM; fibrotic cardiomyopathy, ischemia-reperfusion, I/R; treatment with β-adrenergic agonist isoproterenol) showed multi-fold increases in cardiac galectin-3 expression and preserved renal function. In mice with fibrotic cardiomyopathy, I/R or isoproterenol treatment, plasma galectin-3 levels and density of cardiac inflammatory cells were elevated. These models also exhibited parallel changes in cardiac and plasma galectin-3 levels and presence of trans-cardiac galectin-3 gradient, indicating cardiac release of galectin-3. DCM mice showed no change in circulating galectin-3 levels nor trans-cardiac galectin-3 gradient or myocardial inflammatory infiltration despite a 50-fold increase in cardiac galectin-3 content. In patients with hypertrophic cardiomyopathy or DCM, plasma galectin-3 increased only in those with renal dysfunction and a trans-cardiac galectin-3 gradient was not present. Collectively, this study documents the aetiology-dependency and diverse mechanisms of increment in circulating galectin-3 levels. Our findings highlight cardiac inflammation and enhanced β-adrenoceptor activation in mediating elevated galectin-3 levels via cardiac release in the mechanism.
Introduction
Galectin-3 (Gal-3) is a β-galactoside-binding lectin that binds to glycoproteins thereby regulating their activities1. Clinical studies have shown that high circulating Gal-3 levels are indicative of severity of heart diseases or associated with increased risk of major adverse cardiovascular events including heart failure (HF)2, arrhythmias3–5, arterial stiffening6, re-hospitalization post-HF discharge7, diastolic dysfunction8, severity of atrial fibrosis5,9 or mortality10,11. However, a lack of association of Gal-3 levels and disease severity has also been reported in some studies12–15.
The mechanism(s) responsible for increased blood levels of Gal-3 remains incompletely defined. While increased cardiac Gal-3 expression was observed in human cardiac biopsies5,9,16,17, cardiac release of Gal-3 is not evident in patients with atrial fibrillation or severe HF indicated by the absence of a trans-cardiac Gal-3 gradient18,19. A positive correlation between blood and myocardial levels of Gal-3 was reported in one study20, but not in another study16. Clinical studies have consistently reported a strong and negative correlation between circulating Gal-3 levels and estimated glomerular filtration rate (eGFR), indicating that renal dysfunction is a determinant of blood Gal-3 levels13,21–23. Indeed, Gal-3 levels are markedly elevated in patients with end-stage renal failure24.
There is an increasing appreciation of the presence of inflammation contributing to HF25. In patients with dilated (DCM) or inflammatory cardiomyopathy, cardiac expression of Gal-3 correlated with inflammatory cell density16. In this context, myocardial infarction (MI) is known to trigger intense regional and systemic inflammation26, and in patients with acute MI, blood levels of inflammatory biomarkers including Gal-3 are increased27. Another hallmark of heart disease is activation of the sympatho-β-adrenergic system28. So far, there has been no study that has addressed whether activation of β-adrenoceptors (β-AR) affects circulating Gal-3 levels.
We have investigated factors responsible for increased circulating Gal-3 in cardiomyopathy by addressing the following questions: First, whether changes in circulating Gal-3 levels is aetiology-dependent; Second, whether the inflammatory state of heart disease is associated with increased circulating Gal-3; Third, whether activation of β-ARs alters cardiac and circulating Gal-3 levels; Finally, whether there is Gal-3 release from the heart into the circulation. We studied mouse models of cardiomyopathies and mice with ischemia/reperfusion (I/R) or treatment with the β-AR agonist isoproterenol. Two cohorts of patients with hypertrophic cardiomyopathy (HCM) or DCM and ischemic cardiomyopathy (ICM) were also studied.
Results
Blood and cardiac levels of Gal-3 in mouse models of cardiomyopathy or ischemia/reperfusion
In Mst1-TG mice aged at 3 and 6 months, blood levels of Gal-3 were comparable to that of nTG littermates (Fig. 1A) despite a 50-fold increase in cardiac Gal-3 expression at mRNA and protein levels (P < 0.05 vs. nTG, Fig. 1A). The β2-TG and nTG mice were studied at 3, 6 and 9 months, respectively. Relative to age-matched nTG levels, plasma Gal-3 concentrations were age-dependently elevated in β2-TG mice (Fig. 1B). In β2-TG mice, cardiac Gal-3 expression was age-dependently increased by 5∼20-fold at mRNA and protein levels (Fig. 1B).
Figure 1.

Plasma and myocardial levels of galectin-3 in the mouse models of cardiomyopathy and ischemia-reperfusion (I/R). Changes in plasma concentrations of Gal-3 and cardiac expression of Gal-3 at mRNA and protein levels in Mst1-TG and non-TG (nTG) mice at 3 and 6 months of age (n = 6∼8/group, A) in β2-TG (n = 5∼9/group) and nTG mice (n = 4∼6/group) at 3, 6 and 9 months of age (B) and in mice subjected to 1-hour ischemia followed by reperfusion of 24 or 48 hours (n = 5∼7/group) or sham-operation as control (SH, n = 5, C). In hearts from mice subjected to I/R (1/48 h, n = 5) or SH (n = 4), the infarct zone (IZ) was separated from the non-infarct zone (NIZ) and the assays were performed separately. Mean ± SEM. *P < 0.05 vs. nTG or SH, and †P < 0.05 vs. 3-month-old β2-TG or NIZ.
For comparison with the cardiomyopathic models, I/R model was similarly studied. Mice with I/R (1/24 h or 1/48 h) showed a 2-fold increase in plasma Gal-3 levels (P < 0.05 vs. SH, Fig. 1C). In the ischemic myocardium (i.e. infarct zone, IZ), Gal-3 was upregulated by 7-fold at mRNA level and 30-fold at protein level (Fig. 1C). In the non-infarct zone (NIZ), Gal-3 increased by 5-fold at protein level whilst mRNA level were not significantly different versus SH group (Fig. 1C).
In all three models, circulating levels of renal biomarker cystatin-C were comparable between diseased and control animals (Fig. 2A–C).
Figure 2.

Plasma levels of the renal biomarker cyctatin-C in mouse models of heart disease. Mouse models consist of transgenic cardiomyopathy due to cardiac overexpression of Mst1 (A) or β2-AR (B) and mice subjected to ischemia/reperfusion (I/R, C) or treatment with isoproterenol (ISO, D). N = 5∼9 per group in all experiments.
Inflammatory parameters in mouse models of cardiomyopathy or ischemia/reperfusion
The degree of inflammation was estimated by determining the density of leukocytes (CD45+) and macrophages (CD68+) in the myocardium and also by gene expression of Gal-3, MMP-9 and TNF-α in isolated circulating leukocytes. Figure 3 show representative images of immunohistochemical staining of CD45+ and CD68+ cells all animal models studied.
Figure 3.

Representative histological images of CD45 and CD68 cells by immunohistochemistry of the left ventricles. Images were from of two transgenic mouse models of cardiomyopathy, mice with ischemia/reperfusion (I/R, 1/48 h) or treated with isoproterenol (ISO, 6 mg/kg for 48 hours). Arrows indicate positively stained cells. Bar = 50 μm.
In Mst1-TG hearts, densities of CD45+ and CD68+ cells were marginally increased versus nTG controls (Fig. 4A). In circulating leukocytes of Mst1-TG, Gal-3 gene expression was increased but expression of MMP-9 and TNF-α was unchanged relative to nTG values (Fig. 4A). β2-TG hearts exhibited a significant and age-dependent increase in the density of CD45+ and CD68+ cells versus nTG hearts (Fig. 4B). In circulating leukocytes, gene expression of Gal-3, MMP-9 and TNF-α was 40% lower in β2-TG than nTG mice (Fig. 4B). The I/R model was characterised by a robust increase in the densities of CD45+ and CD68+ cells within the ischemic region (P < 0.05 vs. SH, Fig. 4C). Related to SH group, there was a 2∼3-fold upregulation in the expression of Gal-3 (P < 0.05), MMP-9 (P = 0.2) and TNF-α (P < 0.05) in circulating leukocytes from I/R mice (Fig. 4C).
Figure 4.

Inflammatory parameters of the myocardium and circulating leukocytes in mouse models of cardiomyopathy and ischemia-reperfusion (I/R). Cardiac and systemic inflammation was assessed by immunohistochemistry-based quantification of leukocytes (CD45+) or macrophages (CD68+) in the LV myocardium and by gene expression of Gal-3, matrix metalloproteinase-9 (MMP-9) and tumour necrosis factor-α (TNF-α) in circulating leukocytes. Results were from (A) Mst1-TG and nTG mice (n = 6/group for immunohistochemistry and n = 9/group for gene expression, 3–6 months of age). Data were combined from mice aged 3 and 6 months of age; (B) β2-TG and nTG mice aged at 3, 6 and 9 months (n = 5∼8 per age group for immunohistochemistry and n = 6∼12/group for gene expression of circulating leukocytes from 6-month-old mice); and (C) mice subjected to I/R (1/48 h) or sham-operation (SH) (n = 7/group for immunohistochemistry and n = 5∼8/group for gene expression). Mean ± SEM. *P < 0.05 vs. nTG or SH, †P < 0.05 vs. 3-month-old β2-TG, #P < 0.05 vs. 6-month-old β2-TG.
Upregulation of circulating and cardiac Gal-3 by β-AR activation with isoproterenol
ISO treatment was used to simulate an enhanced sympatho-β-adrenergic activity in the setting of heart disease. In C57Bl/6 mice, treatment with ISO for 48 h increased plasma Gal-3 levels in a dose-dependent fashion (Fig. 5A) without change in cystatin-C levels (Fig. 2D). Cardiac expression of Gal-3 also dose-dependent increased at mRNA and protein levels (Fig. 5B). In the hearts of mice treated with ISO at 6 mg/kg/day for 48 h, densities of CD45+ and CD68+ cells were modestly but significantly increased relative to control (P < 0.05, Fig. 5C). Gene expression of Gal-3 and MMP-9 in circulating leukocytes was unaffected by ISO treatment whilst TNF-α was significantly reduced (Fig. 5D). Concomitant treatment with ISO (6 mg/kg/day) and either β1-AR antagonist atenolol (2 mg/kg/day) or β2-AR antagonist ICI-118551 (1 mg/kg/day) partially and significantly suppressed ISO-induced Gal-3 increase in the heart and plasma (P < 0.05 vs. 6 mg ISO, Fig. 5E).
Figure 5.

Effect of the β-adrenoceptor agonist isoproterenol (ISO) on circulating and cardiac levels of galectin-3 and inflammatory parameters. Plasma levels of Gal-3 (A) and cardiac expression of Gal-3 (B) at the mRNA and protein levels in C57Bl/6 mice treated with the β-agonist ISO for 48 h at 2, 6 or 30 mg/kg/day (via subcutaneous osmotic minipump) or control without treatment (CTL). (C) Density of leukocytes (CD45+) or macrophages (CD68+) quantified by immunohistochemistry in hearts of mice without and with treatment of ISO at 6 mg/kg/day for 48 h. (D) Expression of Gal-3, matrix metalloproteinase-9 (MMP-9) and tumour necrosis factor-α (TNF-α) genes in circulating leukocytes from mice without and with ISO treatment (6 mg/kg/day for 48 hours). (E) ISO (6 mg/kg/day) stimulated Gal-3 upregulation was partially inhibited by atenolol (AT, β1-antagonist, 2 mg/kg/day) or by ICI-118551 (ICI, β2-antagonist, 1 mg/kg/day), both P < 0.05 vs. ISO alone. (F) Effect of treatment with ISO (6 mg/kg/day for 48 hours) on circulating level of Gal-3 in Mst1-TG mice. *P < 0.05 vs. control (CTL) or untreated Mst1-TG, #P < 0.05 vs. 2 mg/kg/day ISO group, and †P < 0.05 vs. 6 mg/kg/day ISO group. Mean ± SEM. N = 5∼8/group in all experiments.
To test ISO treatment as stimulus is able to increase circulating Gal-3 levels in the Mst1-TG cardiomyopathy model, Mst1-TG mice were treated with ISO (6 mg/kg/day for 48 h). ISO treatment led to a significant increase in circulating Gal-3 levels over untreated Mst1-TG mice (Fig. 5F).
Analysis of circulating Gal-3 gradients in mice
Differences in Gal-3 levels of blood sampled from the RA, LV and IVC was determined in Mst1-TG mice and mice treated with ISO (6 mg/kg/day for 48 h) or subjected to I/R (1/48 h). Respective control mice were similarly studied. There was no Gal-3 gradient detected among samples collected from control groups, except for a significant RA-IVC gradient in nTG mice of Mst1-TG colony (Fig. 6A). In Mst1-TG mice, there was no significant Gal-3 gradient (P = NS by paired t-test, Fig. 6A). Mice with ISO treatment or with I/R showed higher Gal-3 levels compared to respective controls (P < 0.05 by two-way ANOVA, Fig. 6B,C), and Gal-3 concentration was higher in the RA than that of IVC or LV samples (P < 0.05 by paired t-test), implying Gal-3 release from the heart into the circulation. Gal-3 gradient was not observed in their respectively control groups (Fig. 6B,C).
Figure 6.
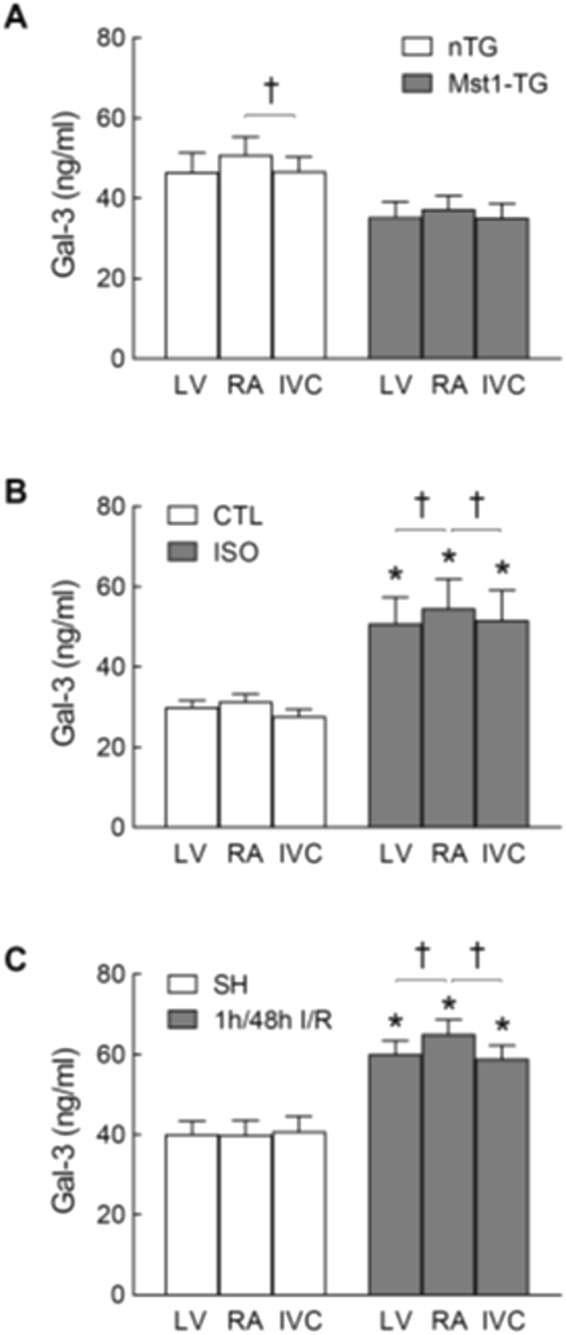
Galectin-3 gradients of plasma samples from three sites of the heart in different mouse models. In each mouse, blood was sampled from the right atrium (RA), left ventricle (LV) and inferior vena cava (IVC), respectively. Blood was collected from Mst1-TG and nTG mice (n = 9 and 6, respectively; A) C57Bl/6 mice treated with isoproterenol (ISO, 6 mg/kg/day for 48 h, n = 9) untreated control (CTL, n = 6, B) or subjected to ischemia/reperfusion (I/R, 1/48 h, n = 8) or sham-operation (SH, n = 5, C). Mean ± SEM. *P < 0.05 vs. CTL, SH or nTG, respectively, by two-way ANOVA. †P < 0.05 by paired t-test.
Blood levels of Gal-3 and trans-organ gradients in HF patients
Table 1 shows basic clinical data of cardiomyopathy patients and their respective control subjects. In HF patients due to DCM or ICM, eGFR was 51 ± 5 ml/min/m2, indicating impaired renal function. These patients were assessed and catheterised as a part of heart transplant evaluation and hence they all had severe HF (NYHA III∼IV) with poor LV ejection fraction (LVEF, Table 1). Compared to non-HF subjects, plasma Gal-3 concentrations were consistently higher in DCM/ICM patients with HF, irrespective of the site of blood sampling (P < 0.05 vs. non-HF, Fig. 7A). Trans-cardiac or trans-hepatic gradient of Gal-3 concentration was not observed. However, a significant trans-renal reduction in Gal-3 level was evident in both HF and control groups, indicating renal clearance of circulating Gal-3 (P < 0.05 by paired t-test, Fig. 7A).
Table 1.
Basic characteristics of patient cohorts and controls.
| HCM | Cohort | Control | DCM/ICM | Conhort | |
|---|---|---|---|---|---|
| Non-obstructive | Obstructive | DCM/ICM | Control | ||
| Number | 27 | 21 | 20 | 15 | 5 |
| Age (year) | 45 ± 14 | 53 ± 13 | 46 ± 13 | 56 ± 3 | 46 ± 10 |
| Male (%) | 74 | 81 | 70 | 75 | 80 |
| NYHF class | I∼II | I∼II | — | III∼IV | — |
| LVEF (%) | 67 ± 8* | 72 ± 5*† | 61 ± 6 | 25 ± 2 | — |
| β-blocker use (%) | 44 | 67 | — | 73 | — |
HCM: hypertrophic cardiomyopathy; DCM: dilated cardiomyopathy; ICM: ischemic cardiomyopathy. LVEF: left ventricular ejection fraction. *P < 0.05 vs. control, †P < 0.05 vs. non-obstrutive group.
Figure 7.

Plasma levels of galectin-3 in cardiomyopathy patients with and without heart failure. (A) Arterial and venous plasma levels of Gal-3 across the heart, kidney and liver from cardiomyopathy patients with HF (n = 15) and non-HF subjects (n = 5). Arterial blood was collected from the radial or brachial artery. Venous blood across the heart, kidney and liver was collected from the coronary sinus, renal vein and hepatic vein, respectively. Paired t-test was used to determine trans-organ effect of plasma Gal-3. Mean ± SD. *P < 0.05 vs. non-HF control. †P < 0.05 by paired-t-test. (B) Left ventricular mass index (LVMI), plasma level of Gal-3 and estimated glomerular filtration rate (eGFR) in healthy controls (n = 20) and patients with non-obstructive (n = 27) or obstructive (n = 21) hypertrophic cardiomyopathy (HCM). Mean ± SD. *P < 0.05 vs. control group and #P < 0.05 vs. non-obstructive HCM group.
Circulating Gal-3 levels in HCM patients
Basic clinical data of HCM patients (n = 48) and healthy controls (n = 20) were shown in Table 1. HCM patients had disease duration of 4.5 ± 5.7 years and 25% had a family history of HCM. HCM patients were divided into non-obstructive (n = 27) and obstructive (n = 21) groups. LV mass index (LVMI) was increased by 35% and 55%, respectively, relative to healthy controls (Fig. 7B). LVEF was typically higher in HCM patients compared with control subjects, particularly in obstructive HCM group (Table 1). Plasma levels of Gal-3 were unchanged in the non-obstructive HCM group. Patients with obstructive HCM had a 20% increase in Gal-3 level together with a significant reduction in eGFR (Fig. 7B).
Discussion
In this joint pre-clinical and clinical study, several major findings have been made regarding the factors responsible for increased circulating Gal-3 levels. First, elevated cardiac content of Gal-3 is a common phenomenon of heart disease whilst increase in circulating level of Gal-3 is inconsistent depending on the aetiology of cardiomyopathy. Second, cardiac and systemic inflammation, measured by inflammatory infiltration and upregulated Gal-3 expression in circulating leukocytes, was associated with elevated circulating Gal-3 levels. In these models, cardiac release of Gal-3 was indicated by parallel changes in cardiac and blood Gal-3 levels and by the presence of Gal-3 gradient. Third, activation of β-ARs leads to increased cardiac and circulating levels of Gal-3 in healthy or cardiomyopathy hearts. Finally, in obstructive HCM or DCM/ICM patients, the increased Gal-3 level was associated with reduction in eGFR, whilst the trans-cardiac Gal-3 gradient was no evidence in cardiomyopathy patients with severe HF.
Elevation of circulating Gal-3 levels in patients with heart disease has been well documented, but an increase in cardiac Gal-3 level, relative to appropriate controls, has been rarely studied, except one report showing a significant increase in cardiac Gal-3 expression in patients with hypertensive heart disease20. In contrast, increased cardiac expression of Gal-3 has been consistently observed by a number of animal studies29–34, and yet blood levels of Gal-3 were not determined in these animal studies. Here, we reported multi-fold increases in cardiac Gal-3 expression at mRNA and protein levels in all four experimental models, which permits the examination of parallel changes of cardiac and circulating levels of Gal-3 as well as cardiac release of Gal-3 (Fig. 8). In the models of I/R, β2-TG cardiomyopathy or ISO-treatment, increased plasma Gal-3 levels were in parallel to that of cardiac Gal-3 expression. Furthermore, treatment with selective β-antagonists simultaneously suppressed cardiac and circulating levels of Gal-3 in ISO-treated mice. In sharp contrast, the Mst1-TG mice showed no change in circulating levels of Gal-3 nor Gal-3 gradient, albeit cardiac content of Gal-3 is the highest among the models studied. The reason for this dissociation remains unclear but does indicate that changes in circulating Gal-3 levels are aetiology-dependent. To replicate clinical conditions of stress or acute decompensation of heart disease, in Mst1-TG mice without change in blood level of Gal-3, treatment with ISO significantly increased circulating Gal-3. This finding further highlights the significance of β-AR stimulation as a key factor mediating increment of circulating Gal-3.
Figure 8.

Determinants of elevated circulating levels of galectin-3 in cardiomyopathy and heart disease. Changes in circulating Gal-3 level is dependent on the aetiology of cardiomyopathy, inflammatory status and renal function. While cardiac release of Gal-3 is not observed in Mst1-TG DCM mice and HF patients (hDCM for human DCM, hHCM for human HCM), this mechanism is strongly indicated by findings from animal models of β2-TG cardiomyopathy, ischemia/reperfusion (I/R) or treatment with isoproterenol (ISO). Two factors closely associated with heart disease, i.e. cardiac and/or systemic inflammation and activation of the sympatho-β-adrenergic system, contribute cardiac Gal-3 release and increased blood levels of Gal-3. Red arrows indicate potential sources whilst blue arrow denotes renal clearance of circulating Gal-3 pool.
Absence of a trans-cardiac Gal-3 gradient was reported by two clinical studies in patients with atrial fibrillation18 or with hypertensive heart disease20. We observed in patients with advanced HF due to DCM/ICM or in Mst1-TG mice with DCM no sign of Gal-3 release from the heart. However, our findings from other animal models strongly indicate cardiac Gal-3 release as a mechanism for increased circulating Gal-3 levels. In mouse models of β2-TG cardiomyopathy, I/R and ISO treatment, there are parallel changes in cardiac and blood Gla-3 levels. Furthermore, mice with ISO treatment or I/R exhibited significantly higher circulating Gal-3 levels in the RA relative to LV or IVC sample, indicating cardiac release of Gal-3 into the circulation. Due to technical difficulty in sampling from the coronary sinus in the mouse, we elected to collect blood from the RA instead. It is likely that by this method, the trans-cardiac gradient of Gal-3 concentration is underestimated due to dilution of coronary sinus blood by systemic venous blood. The I/R and ISO models represent acute cardiac damage, inflammation and/or β-AR activation. Interestingly, ISO treatment to Mst1-TG cardiomyopathy mice increased circulating Gal-3. Thus it is worthwhile to test whether trans-cardiac Gal-3 gradient exists in patients in acute HF or acutely decompensated HF, acute MI or other inflammatory heart disease.
The significance of inflammation in heart disease and HF has been well appreciated by clinical findings of higher levels of inflammatory biomarkers25 or cardiac inflammatory cell infiltration in inflammatory cardiomyopathy, myocarditis, acute MI or I/R16,25,26,35–38. In the present study, we observed in the β2-AR TG model an age-dependent increase in cardiac density of CD45+ and CD68+ cells. Interestingly, Gal-3 levels in the LV and blood were also age-dependently increased in this model. In sharp contrast, inflammatory cell infiltration was only marginally increased in Mst1-TG hearts and yet cardiac Gal-3 level was extremely high. There are multiple cellular sources of the overall cardiac expression of Gal-3. In addition to macrophages commonly regarded as the major cell type expressing Gal-316,34,39, other cardiac cells such as cardiomyocytes and fibroblasts also express Gal-3 in diseased settings31,34,40. In both TG models, transgene expression is restricted to cardiomyocytes and hence the high cardiac expression of Gal-3 in both models might be largely attributable to cardiomyocytes. Thus, coexistence of elevated cardiac Gal-3 expression and inflammatory status is a prerequisite for an increase in circulating Gal-3 level.
We previously showed in mice and human patients that acute MI not only leads to cardiac but also systemic inflammation26,36. Following MI, circulating leukocytes are activated with enhanced expression of inflammatory molecules such as MMP-9 and interleukins26,36. We therefore speculated that another mechanism for increased circulating Gal-3 is systemic inflammation, where activated circulating leukocytes synthesise and release Gal-3 directly into the circulation (Fig. 8). This was tested by measuring gene expression of Gal-3 as well as MMP-9 and TNFα of circulating leukocytes in all four mouse models, with results indicating that only circulating leukocytes from I/R mice showed enhanced expression of all three genes. Known as a pro-inflammatory factor, Gal-3 upregulates the expression of inflammatory cytokines and activates macrophages, promoting regional and systemic inflammation commonly seen in settings of heart disease25. Unexpectedly, expression of the three genes in circulating leukocytes was reduced in β2-AR TG model. The reason for such suppressed expression remains unclear but this rules out the presence of a systemic inflammation in this model.
Activation of the sympatho-β-adrenergic system is a hallmark of heart disease and HF28. Following the findings of increased cardiac and circulating Gal-3 content in the β2-TG model, we studied the effect of the β-agonist ISO and observed a dose-dependent elevation of cardiac as well as circulating levels of Gal-3 by ISO treatment for 48 hours. Further, use of selective β-blockers lowered both cardiac and plasma levels of Gal-3, indicating involvement of both β1- and β2-AR activation in the changes of Gal-3 observed. Considering that Gal-3 is regarded as a clinical biomarker of heart disease and HF, future studies are warranted to explore the influence of β-blockers on dynamic changes of Gal-3 circulating levels in relation to therapeutic efficacy. Whereas high doses of ISO are known to induce cardiotoxicity including inflammatory infiltration, we reported a significant but moderate increase in inflammatory cells in ISO-treated mice (6 mg/kg/day for 48 hours). Thus, elevation of cardiac expression of Gal-3 is likely attributable to cardiac cells that are equipped in high density with β1-AR (e.g. cardiomyocytes) and β2-AR (e.g. fibroblasts, cardiomyocytes).
Recent studies have well established the renal mechanism responsible for elevated circulating level of Gal-3 by showing that Gal-3 concentrations are negatively correlated with eGFR but positively with renal biomarkers like cystatin-C13,21,23. In the present study, all four animal models had plasma levels of cystatin-C comparable to control levels. This excluded the renal factor in the interpretation of changes in blood Gal-3 levels. This is also indicated by our finding that Gal-3 levels only increased in DCM/ICM patients or obstructive HCM patients who exhibited renal dysfunction. Whereas a negative trans-renal Gal-3 gradient was noticed in HF patients, it is likely that insufficient renal clearance explains increased plasma Gal-3 levels in these patients. Our group size of HF patients and healthy subjects was too small to identify differences in renal clearance relative to the level of Gal-3 increment.
One limitation of the present study is that we did not obtain myocardial biopsies to measure cardiac Gal-3 expression in our patient cohorts. Nevertheless, Lopez et al. reported in patients with hypertensive heart disease, upregulation of Gal-3 at gene and protein levels endomyocardial biopsies20. Another limitation is that our clinical investigation did not include conditions associated with significant cardiac inflammation. Future studies should validate our pre-clinical findings in human patients with increased cardiac inflammatory cell infiltration (i.e. I/R or myocarditis). Finally, our patient cohorts were too small to identify the influence of the use of β-blockers on Gal-3 levels, and the presence of renal dysfunction as a confounding factor makes this attempt more difficult.
In conclusion, increase in circulating levels of Gal-3 in cardiomyopathy is aetiology-dependent and mediated by different mechanisms. In cardiomyopathy and HF patients, reduced renal clearance contributes to increased Gal-3 levels without evidence of cardiac release of Gal-3. However, findings from animal models (β2-TG cardiomyopathy, I/R or ISO treatment) demonstrate cardiac release of Gal-3 into the circulation as a key mechanism responsible for elevated circulating Gal-3 and highlight the significance of cardiac inflammation and enhanced β-AR stimulation in promoting cardiac Gal-3 release (Fig. 8). Our findings bear translational implications for interpretation of Gal-3 as a biomarker of heart disease and potential influence of Gal-3 levels by therapies like β-blockers.
Materials and Methods
Animal models of heart disease
All experimental procedures were approved by the Alfred Medical Research and Education Precinct’s Animal Ethics Committee and complied with the National Health and Medical Research Council of Australia Code for the Care and Use of Animals for Scientific Purposes (8th edition) and the ARRIVE guidelines. For surgical procedure or blood/tissue collection followed by euthanasia, mice were fully anaesthetized using the mixture of ketamine/xylazine/atropine (10/100/2 mg/kg, respectively).
All strains of mice were on the same C57Bl/6 genetic background. Male mice were used. Two transgenic (TG) mouse models of cardiomyopathy were due to cardiac-specific overexpression of mammalian sterile 20-like kinase-1 (Mst1-TG)41 or human β2-adrenoceptor (β2-TG)42. Our previous studies have shown phenotypes of DCM in the Mst1-TG model41,43 or an age-dependent development of fibrotic cardiomyopathy in β2-TG mice42,44. Age-matched non-TG (nTG) littermates were similarly studied.
I/R was induced in C57Bl/6 mice (aged 12∼14 weeks) by open-chest surgery for coronary artery occlusion-reperfusion (1 h ischemia followed by reperfusion) or sham-operated (SH), as previously described35. Isoproterenol (ISO, Sigma, USA) was administered to male 12∼14 week-old C57Bl/6 mice via subcutaneously implanted osmotic minipump (ALZET, USA). ISO was dissolved in saline containing 0.4 mM ascorbic acid and the doses were 2, 6 and 30 mg/kg/day, respectively for 2 days. In some experiments, animals received a combined treatment of ISO and selective β-AR antagonists atenolol (2 mg/kg/day, Sigma) or ICI-118551 (1 mg/kg/day, Sigma).
Sampling of heart and blood and isolation of circulating leukocytes from mice
Animals were anesthetised and blood was collected by left ventricular (LV) puncture using a 25 G needle. Blood was centrifuged and plasma was collected. Hearts were harvested and the LV was separated and used for biochemical assays or immunohistochemistry. In mice with I/R, areas of the ischemic and non-ischemic LV were identified and separated with the aid of a surgical microscope.
To determine the trans-cardiac gradient of circulating Gal-3, anesthetised animal was ventilated and the chest opened. Blood was then sampled (0.25 ml per site) by needle puncture from the right atrium (RA), LV and inferior vena cava (IVC), respectively, for determination of plasma concentrations of Gal-3.
Blood leukocytes were isolated from whole blood collected in ethylene-diamine-tetra-acetate (EDTA)-coated 10 ml centrifuge tubes. Blood was diluted 1:2 with PBS/2% FBS and centrifuged at 200 g for 10 min. Leukocytes forming a concentrated ‘buffy coat’ band were carefully collected and lysed with 1x red blood cell lysis buffer. Lysed samples were centrifuged at 800 g for 5 min and the supernatant was removed. RNA was then extracted from isolated leukocytes for RT-PCR.
Biochemical assays of mouse heart and blood samples
RNA from LV tissue or isolated leukocytes was extracted using Trizol® Reagent (Sigma) as previously described44. RNA (1 μg) was reverse-transcribed into cDNA and quantitative RT-PCR was performed using SYBR green reactions (Roche, Germany) and target primers (Sigma). The gene expression of Gal-3, matrix metalloproteinase-9 (MMP-9) and tumour necrosis factor-α (TNF-α) was measured, normalised to that of glyceraldehyle 3-phosphate dehydrogenase (GAPDH) or TATA-Box binding protein-associated factor 8 using the method of 2−ΔΔct, and presented relative to the control value.
Plasma levels of Gal-3 or cystatin-C (renal biomarker) were determined by ELISA (R&D Systems, USA). To measure cardiac content of gal-3 by using ELISA, LV tissues were homogenised using a lysis buffer with the addition of phenylmethylsulfonyl fluoride (1 mM), proteinase inhibitor cocktail (Sigma) and phosphatase inhibitor cocktail (Sigma).
Immunohistochemistry
Frozen-fixed LV sections (5 μm) were incubated with primary rat monoclonal antibodies against mouse CD45 (1:50, BD Pharmingen, USA) or mouse CD68 (1:200, AbD Serotec, USA), followed by incubation with secondary antibody Alexa Fluor 546-conjugated goat anti-rat IgG (1:500, Invitrogen, USA), as previously described35. Nuclei were stained with ProLong® Gold antifade reagent with 4′, 6-diamidino-2-phenylindole (Invitrogen). LV sections (8∼10 per heart) were acquired using Olympus BX61 fluorescence microscope and AnalySIS FIVE software (Olympus, Australia) at x20 magnification. The number of inflammatory cells was manually quantified, in a blinded fashion, using ImageJ (NIH, USA) and the average value was used.
Cohorts of patients with cardiomyopathy
Two patient cohorts were studied: advanced HF due to DCM or ICM, and patients with HCM. All patients and healthy volunteers were informed and consented to the study. The study was approved by the Alfred Hospital Research and Ethics Committee, which act in concordance with the guiding principles of the NHMRC of Australia.
Heart failure patients with dilated or ischemic cardiomyopathy
We studied 15 HF patients (NYHF class III∼IV) undergoing evaluation for potential heart transplantation. Hemodynamically unstable patients were excluded from the study. The aetiology of HF was non-ischemic DCM in 69% of patients whilst the rest was due to ICM. Patients continued their anti-HF medications at the time of study. Five healthy volunteers recruited from the general community were similarly studied. They were asymptomatic and were not treated for any form of cardiovascular disease, renal disease or diabetes.
An arterial line was placed into the radial or brachial artery under local anaesthesia for blood sampling. An introducer sheath was placed into the right internal jugular or median cubital vein under local anaesthesia. A sampling catheter was sequentially positioned in the coronary sinus, hepatic vein and renal vein under fluoroscopic guidance. The tip of the catheter was positioned at least 2 cm proximal to the orifice of each vessel. Echocardiography was performed to determine LVEF.
Patients with hypertrophic cardiomyopathy
HCM patients with confirmed HCM diagnosis by cardiac magnetic resonance (CMR) and non-HCM controls were studied. Patients were further divided into non-obstructive and obstructive HCM subgroups. All patients were in NYHF class I∼II. Peripheral venous blood samples were collected for assay of Gal-3 and creatinine. LV mass and LVEF were determined by CMR, as we previously described45.
Biochemical assays of human blood samples
Blood samples were collected in EDTA-coated tubes and were subsequently centrifuged with plasma stored at −70 °C until assay. The plasma concentrations of Gal-3 were measured by ELISA (R&D Systems). eGFR was calculated following a plasma creatinine test.
Statistical analyses
Animal data were presented as mean ± SEM and clinical data presented as mean ± SD unless otherwise stated. All results were analysed using Prism 7 Software or SigmaStat Software. Shapiro-Wilk’s and Levene’s tests were used to determine normality and equal variance, respectively. Statistical analyses included one- or two-way ANOVA followed by Bonferroni post-hoc test, unpaired student’s t-test (for datasets of only two groups) and paired student’s t-test (for paired data comparison). P < 0.05 was considered as statistically significant.
Data availability
All data generated or analysed during this study are included in this article.
Acknowledgements
This work was supported by grants from the National Health and Medical Research Council (NHMRC) of Australia (ID1081710 and ID1005329 to XJD, ID1036352 to AMD and DK) and the Victorian Government’s Operational Infrastructure Support Program. MNN is recipient of an Australian Government Research Training Program Scholarship. JRM, AMD, DK and XJD are NHMRC fellows (ID1078985, ID586656, ID1042495, ID1043026). WBZ is funded by China Scholarship Council.
Author Contributions
M.N.N. and X.J.D. conceived and coordinated the study. M.N.N., Y.S., D.V., L.F., A.H.E., W.B.Z., H.K., X.M.G., X.J.D. performed experiments; M.N.N., Y.S., L.F., A.H.E., W.B.Z., D.M.K. and X.J.D. analysed data; M.N.N. and X.J.D. prepared the figures and table, wrote the paper; M.N.N., L.F., H.K., J.S., A.J.T., J.R.M., A.M.D., D.M.K. and X.J.D. critically reviewed the paper. All authors reviewed the manuscript and approved the final version.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Rabinovich GA, Toscano MA. Turning ‘sweet’ on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. 2009;9:338–52. doi: 10.1038/nri2536. [DOI] [PubMed] [Google Scholar]
- 2.van der Velde AR, et al. Prognostic value of changes in galectin-3 levels over time in patients with heart failure: data from CORONA and COACH. Circ Heart Fail. 2013;6:219–226. doi: 10.1161/CIRCHEARTFAILURE.112.000129. [DOI] [PubMed] [Google Scholar]
- 3.Clementy N, et al. Serum Galectin-3 Levels Predict Recurrences after Ablation of Atrial Fibrillation. Sci Rep. 2016;6:34357. doi: 10.1038/srep34357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Francia P, et al. Osteopontin and galectin-3 predict the risk of ventricular tachycardia and fibrillation in heart failure patients with implantable defibrillators. J Cardiovasc Electrophysiol. 2014;25:609–616. doi: 10.1111/jce.12364. [DOI] [PubMed] [Google Scholar]
- 5.Hernandez-Romero D, et al. Galectin-3 as a marker of interstitial atrial remodelling involved in atrial fibrillation. Sci Rep. 2017;7:40378. doi: 10.1038/srep40378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Libhaber E, et al. Independent associations of circulating galectin-3 concentrations with aortic pulse wave velocity and wave reflection in a community sample. Hypertension. 2015;65:1356–1364. doi: 10.1161/HYPERTENSIONAHA.115.05159. [DOI] [PubMed] [Google Scholar]
- 7.Meijers WC, et al. Elevated plasma galectin-3 is associated with near-term rehospitalization in heart failure: a pooled analysis of 3 clinical trials. Am Heart J. 2014;167:853–860. doi: 10.1016/j.ahj.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 8.Wu CK, et al. Galectin-3 level and the severity of cardiac diastolic dysfunction using cellular and animal models and clinical indices. Sci Rep. 2015;5:17007. doi: 10.1038/srep17007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takemoto Y, et al. Galectin-3 Regulates Atrial Fibrillation Remodeling and Predicts Catheter Ablation Outcomes. JACC Basic Transl Sci. 2016;1:143–154. doi: 10.1016/j.jacbts.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Boer RA, et al. Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann Med. 2011;43:60–68. doi: 10.3109/07853890.2010.538080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maiolino G, et al. Galectin-3 predicts long-term cardiovascular death in high-risk patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2015;35:725–732. doi: 10.1161/ATVBAHA.114.304964. [DOI] [PubMed] [Google Scholar]
- 12.Stoltze Gaborit F, et al. Galectin 3: association to neurohumoral activity, echocardiographic parameters and renal function in outpatients with heart failure. BMC Cardiovasc Disord. 2016;16:117. doi: 10.1186/s12872-016-0290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.AbouEzzeddine OF, et al. Galectin-3 in heart failure with preserved ejection fraction. A RELAX trial substudy (Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Diastolic Heart Failure) JACC Heart Fail. 2015;3:245–252. doi: 10.1016/j.jchf.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jansen H, et al. Prognostic Utility of Galectin-3 for Recurrent Cardiovascular Events During Long-term Follow-up in Patients with Stable Coronary Heart Disease: Results of the KAROLA Study. Clin Chem. 2016;62:1372–1379. doi: 10.1373/clinchem.2016.257550. [DOI] [PubMed] [Google Scholar]
- 15.Singsaas EG, Manhenke CA, Dickstein K, Orn S. Circulating Galectin-3 Levels Are Increased in Patients with Ischemic Heart Disease, but Are Not Influenced by Acute Myocardial Infarction. Cardiology. 2016;134:398–405. doi: 10.1159/000445103. [DOI] [PubMed] [Google Scholar]
- 16.Besler C. et al. Plasma and Cardiac Galectin-3 in Patients With Heart Failure Reflects Both Inflammation and Fibrosis: Implications for Its Use as a Biomarker. Circ Heart Fail. 10 (2017). [DOI] [PubMed]
- 17.Lopez-Andres N, et al. Association of galectin-3 and fibrosis markers with long-term cardiovascular outcomes in patients with heart failure, left ventricular dysfunction, and dyssynchrony: insights from the CARE-HF (Cardiac Resynchronization in Heart Failure) trial. Eur J Heart Fail. 2012;14:74–81. doi: 10.1093/eurjhf/hfr151. [DOI] [PubMed] [Google Scholar]
- 18.Kornej J, et al. Galectin-3 in patients with atrial fibrillation undergoing radiofrequency catheter ablation. PLoS One. 2015;10:e0123574. doi: 10.1371/journal.pone.0123574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Truong QA, et al. Coronary sinus biomarker sampling compared to peripheral venous blood for predicting outcomes in patients with severe heart failure undergoing cardiac resynchronization therapy: the BIOCRT study. Heart Rhythm. 2014;11:2167–2175. doi: 10.1016/j.hrthm.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez B, et al. Galectin-3 and histological, molecular and biochemical aspects of myocardial fibrosis in heart failure of hypertensive origin. Eur J Heart Fail. 2015;17:385–392. doi: 10.1002/ejhf.246. [DOI] [PubMed] [Google Scholar]
- 21.Gopal DM, et al. Relationship of plasma galectin-3 to renal function in patients with heart failure: effects of clinical status, pathophysiology of heart failure, and presence or absence of heart failure. J Am Heart Assoc. 2012;1:e000760. doi: 10.1161/JAHA.112.000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meijers WC, et al. Renal handling of galectin-3 in the general population, chronic heart failure, and hemodialysis. J Am Heart Assoc. 2014;3:e000962. doi: 10.1161/JAHA.114.000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zamora E, et al. Renal function largely influences Galectin-3 prognostic value in heart failure. Int J Cardiol. 2014;177:171–177. doi: 10.1016/j.ijcard.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Drechsler C, et al. Galectin-3, Renal Function, and Clinical Outcomes: Results from the LURIC and 4D Studies. J Am Soc Nephrol. 2015;26:2213–2221. doi: 10.1681/ASN.2014010093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dick SA, Epelman S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ Res. 2016;119:159–176. doi: 10.1161/CIRCRESAHA.116.308030. [DOI] [PubMed] [Google Scholar]
- 26.Fang L, Du XJ, Gao XM, Dart AM. Activation of peripheral blood mononuclear cells and extracellular matrix and inflammatory gene profile in acute myocardial infarction. Clin Sci (Lond). 2010;119:175–183. doi: 10.1042/CS20100011. [DOI] [PubMed] [Google Scholar]
- 27.Peacock WF. How galectin-3 changes acute heart failure decision making in the emergency department. Clin Chem Lab Med. 2014;52:1409–1412. doi: 10.1515/cclm-2014-0221. [DOI] [PubMed] [Google Scholar]
- 28.Kaye DM, et al. Neurochemical evidence of cardiac sympathetic activation and increased central nervous system norepinephrine turnover in severe congestive heart failure. J Am Coll Cardiol. 1994;23:570–578. doi: 10.1016/0735-1097(94)90738-2. [DOI] [PubMed] [Google Scholar]
- 29.Calvier L, et al. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart Fail. 2015;3:59–67. doi: 10.1016/j.jchf.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Calvier L, et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler Thromb Vasc Biol. 2013;33:67–75. doi: 10.1161/ATVBAHA.112.300569. [DOI] [PubMed] [Google Scholar]
- 31.Frunza O, et al. Myocardial Galectin-3 Expression Is Associated with Remodeling of the Pressure-Overloaded Heart and May Delay the Hypertrophic Response without Affecting Survival, Dysfunction, and Cardiac Fibrosis. Am J Pathol. 2016;186:1114–1127. doi: 10.1016/j.ajpath.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Martinez E, et al. Galectin-3 blockade inhibits cardiac inflammation and fibrosis in experimental hyperaldosteronism and hypertension. Hypertension. 2015;66:767–775. doi: 10.1161/HYPERTENSIONAHA.115.05876. [DOI] [PubMed] [Google Scholar]
- 33.Sharma UC, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. 2004;110:3121–3128. doi: 10.1161/01.CIR.0000147181.65298.4D. [DOI] [PubMed] [Google Scholar]
- 34.Yu L, et al. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ Heart Fail. 2013;6:107–117. doi: 10.1161/CIRCHEARTFAILURE.112.971168. [DOI] [PubMed] [Google Scholar]
- 35.Gao XM, et al. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: a predominant role of anti-inflammation. J Mol Cell Cardiol. 2011;50:991–999. doi: 10.1016/j.yjmcc.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 36.Liu Y, et al. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arterioscler Thromb Vasc Biol. 2011;31:834–841. doi: 10.1161/ATVBAHA.110.220467. [DOI] [PubMed] [Google Scholar]
- 37.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Westman PC, et al. Inflammation as a Driver of Adverse Left Ventricular Remodeling After Acute Myocardial Infarction. J Am Coll Cardiol. 2016;67:2050–2060. doi: 10.1016/j.jacc.2016.01.073. [DOI] [PubMed] [Google Scholar]
- 39.Papaspyridonos M, et al. Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler Thromb Vasc Biol. 2008;28:433–440. doi: 10.1161/ATVBAHA.107.159160. [DOI] [PubMed] [Google Scholar]
- 40.van der Velde AR, et al. Galectin-3 and sST2 in prediction of left ventricular ejection fraction after myocardial infarction. Clin Chim Acta. 2016;452:50–57. doi: 10.1016/j.cca.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto S, et al. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest. 2003;111:1463–1474. doi: 10.1172/JCI17459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du XJ, et al. Age-dependent cardiomyopathy and heart failure phenotype in mice overexpressing β2-adrenergic receptors in the heart. Cardiovasc Res. 2000;48:448–454. doi: 10.1016/S0008-6363(00)00187-5. [DOI] [PubMed] [Google Scholar]
- 43.Pretorius L, et al. Reduced phosphoinositide 3-kinase (p110α) activation increases the susceptibility to atrial fibrillation. Am J Pathol. 2009;175:998–1009. doi: 10.2353/ajpath.2009.090126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen MN, et al. Spontaneous ventricular tachyarrhythmias in β2-adrenoceptor transgenic mice in relation to cardiac interstitial fibrosis. Am J Physiol Heart Circ Physiol. 2015;309:H946–H957. doi: 10.1152/ajpheart.00405.2015. [DOI] [PubMed] [Google Scholar]
- 45.Ellims AH, et al. Diffuse myocardial fibrosis in hypertrophic cardiomyopathy can be identified by cardiovascular magnetic resonance, and is associated with left ventricular diastolic dysfunction. J Cardiovasc Magn Reson. 2012;14:76. doi: 10.1186/1532-429X-14-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this article.