

Abstract
The host cytokine IL-6 plays an important role in host defence and prevention of lung injury from various pathogens, making IL-6 an important mediator in the host’s susceptibility to respiratory infections. The cellular response to IL-6 is mediated through a Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) signal transduction pathway. Human metapneumovirus (hMPV) is an important causative agent of viral respiratory infections known to inhibit the IFN-mediated activation of STAT1. However, little is known about the interactions between this virus and other STAT signalling cascades. Herein, we showed that hMPV can attenuate the IL-6-mediated JAK/STAT3 signalling cascade in lung epithelial cells. HMPV inhibited a key event in this pathway by impeding the phosphorylation and nuclear translocation of STAT3 in A549 cells and in primary normal human bronchial epithelial cells. Further studies established that hMPV interrupted the IL-6-induced JAK/STAT pathway early in the signal transduction pathway by blocking the phosphorylation of JAK2. By antagonizing the IL-6-mediated JAK/STAT3 pathway, hMPV perturbed the expression of IL-6-inducible genes important for apoptosis, cell differentiation and growth. Infection with hMPV also differentially regulated the effects of IL-6 on apoptosis. Thus, hMPV regulation of these genes could usurp the protective roles of IL-6, and these data provide insight into an important element of viral pathogenesis.
Introduction
Human metapneumovirus (hMPV) is an important causative agent of acute viral respiratory tract infections in children, the elderly and immunocompromised patients. Infection with hMPV can result in bronchiolitis, bronchitis or pneumonia, and can exacerbate chronic obstructive pulmonary disease (Edwards et al., 2013; Greensill et al., 2003; Martinello et al., 2006; Williams et al., 2004). Seroprevalence studies have demonstrated that virtually all children have antibodies to hMPV by 10 years of age (Ebihara et al., 2003; Leung et al., 2005; van den Hoogen et al., 2001). Despite the high infection rate in children, there is a lifelong cycle of infection and reinfection with hMPV, suggesting that the initial infection does not provide complete long-term protective immunity (Ebihara et al., 2004). Thus, the ability of hMPV to evade the host’s innate immune system is an important factor in the success of the viral infection. hMPV is known to antagonize the type I IFN response, a known antiviral response (Dinwiddie & Harrod, 2008); however, little is known about how hMPV regulates other innate immune signalling pathways important during respiratory infections.
IL-6 is a pleiotropic cytokine produced in response to respiratory infections with various pathogens. In vivo studies have demonstrated that viral and bacterial pathogens that are innocuous in WT mice can cause disease in IL-6-deficient mice (Jones et al., 2006; Ladel et al., 1997; Murphy et al., 2008; van der Poll et al., 1997). Infections with respiratory syncytial virus (RSV), a virus closely related to hMPV, demonstrated a positive correlation between high IL-6 levels and disease severity (Welliver, 2008). Moreover, hMPV induced a more severe disease in mice than RSV, and these results were, in part, associated with higher levels of host IL-6 (Huck et al., 2007). These data demonstrate that IL-6 is a key determinant in host susceptibility to respiratory infection and could play an important role in hMPV pathogenicity (Guerrero-Plata et al., 2005; Huck et al., 2007; Laham et al., 2004).
IL-6 induces cellular responses to disease through various signalling cascades, including a Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) signal transduction pathway (reviewed by Kamimura, 2003). Similar to other JAK/STAT pathways, binding of IL-6 to the receptor complex activates a member of the JAK family (JAK1, JAK2 and Tyk2), which leads to tyrosine phosphorylation and dimerization of STAT3. The activated STAT3 translocates to the nucleus where it binds to DNA and modulates gene expression. Whilst genes stimulated by IL-6 are predominantly involved in the inflammatory response, they are also important in apoptosis, cell differentiation, cell proliferation, cell recruitment and the acute phase response. The disruption of IL-6 signal transduction pathways will modify the expression of several genes needed to maintain lung homeostasis, thereby increasing the host’s susceptibility to injury (Matsuzaki et al., 2006; Quinton et al., 2008).
hMPV inhibits STAT1 activation, thereby antagonizing the IFN-induced JAK/STAT signal transduction cascade, a known antiviral pathway (Dinwiddie & Harrod, 2008). However, the ability of this virus to regulate other JAK/STAT signalling pathways important for host defence is not well understood. Here, we report that hMPV infection inhibited IL-6-induced STAT3 signalling in lung epithelial cells and that this inhibition occurred early in the signal transduction cascade. By usurping the IL-6-induced JAK/STAT signalling cascade, hMPV altered the epithelial transcriptional response important for maintaining the mucosal tissue microenvironment. These findings provide new information on mechanisms by which hMPV affects lung pathogenesis following infection.
Results
Human metapneumovirus inhibits the expression of a STAT3 reporter gene
With the exception of the IFN-inducible JAK/STAT pathway, little is known about how hMPV regulates cytokine-induced JAK/STAT signalling cascades important in controlling virus infections of the lung. IL-6 is an important regulator of the host’s defence against pathogens and signals through a JAK/STAT3 signalling pathway (Jones et al., 2006; Ladel et al., 1997; Murphy et al., 2008; van der Poll, 1997; Zhang et al., 2011). To determine whether hMPV could regulate the IL-6-induced JAK/STAT3 signalling cascade, expression of a luciferase reporter gene under the control of STAT3 was examined. A549 cells were transfected with a STAT3 reporter construct, a negative-control reporter construct or a control reporter construct that constitutively expressed luciferase (positive control) 24 h prior to infection with hMPV. At 24 h post-infection (p.i.), cells were treated with IL-6 for 8 h and luciferase expression was then measured. Mock-infected cells transfected with the STAT3 reporter construct demonstrated a fold increase of 3.6±0.732 (mean±sd) in luciferase expression following induction with IL-6. In contrast, hMPV-infected cells had a fold increase of 1.28±0.629 following treatment with IL-6. Compared with mock-infected cells, hMPV infection significantly attenuated the fold increase of the STAT3-driven luciferase expression following IL-6 treatment (P<0.0001, two-way ANOVA; Fig. 1a). Confirmation of transfection and background reporter activity was determined using positive- and negative-control reporter constructs, respectively. Infection and/or treatment with IL-6 did not alter the luciferase expression of the control plasmids, suggesting that the results of the STAT3-driven luciferase expression were specific to the effects of hMPV infection and IL-6 treatment.
Fig. 1.
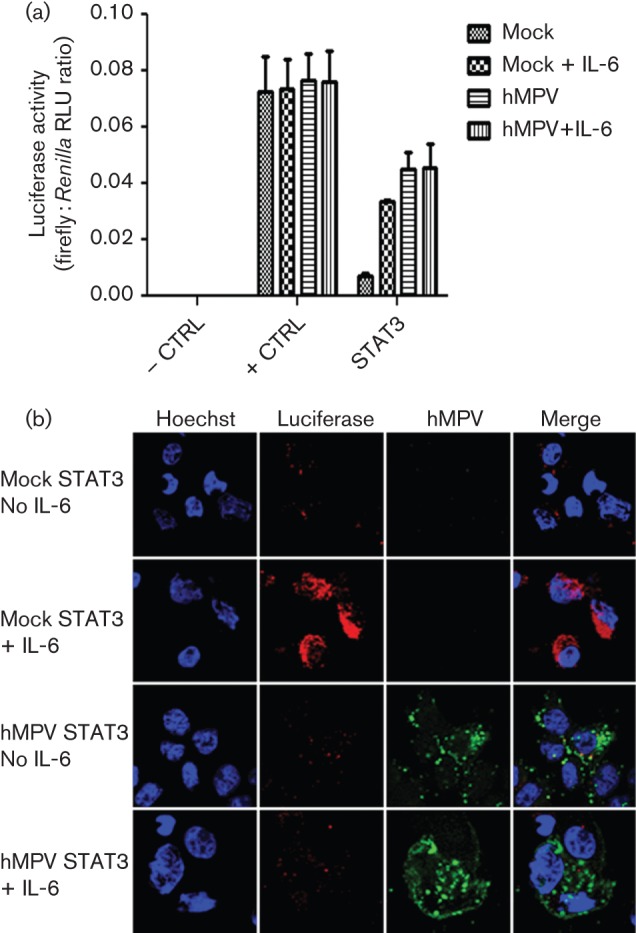
hMPV attenuates STAT3-inducible luciferase reporter gene expression. A549 cells were transfected with a plasmid that constitutively expresses firefly luciferase (luc; + CTRL), a negative control (– CTRL) or a STAT3–luc reporter plasmid. The cells were infected with hMPV at an m.o.i. of 4 and then treated with IL-6 (20 ng ml−1) for 8 h. (a) Luciferase expression is shown as the firefly : Renilla relative light units (RLU) ratio (means±sem for three independent experiments). (b) Infected cells transfected with the STAT3 reporter plasmid and treated with IL-6 were fixed in 4 % paraformaldehyde. Cells were stained for hMPV antigen (green) and firefly luciferase (red). Nuclei were counterstained with Hoechst (blue). Results are representative of three independent experiments.
hMPV-infected cells had higher luciferase activity than mock-infected cells. This could be due to IL-6 production by hMPV infection (data not shown) and the early induction of STAT3 reporter in uninfected cells. To further assess the effects of hMPV infection on the IL-6 response, immunofluorescence was utilized to demonstrate the inhibition of luciferase expression in virus-infected cells. A549 cells transfected with the STAT3 reporter construct were co-stained with an antibody against hMPV and an antibody against firefly luciferase (Fig. 1b). Firefly luciferase was detected in the cytoplasm of mock-infected cells treated with IL-6 but not in untreated cells. Conversely, the amount of luciferase detected in the cytoplasm of IL-6-treated and untreated hMPV-infected cells remained at background levels. Together, these data suggested that hMPV is capable of impeding the IL-6-stimulated JAK/STAT3 signalling cascade at the level of STAT3-mediated gene expression.
HMPV inhibits translocation of STAT3 to the nucleus
Transcription of STAT3-responsive genes requires nuclear translocation and DNA binding of STAT3. To determine the nuclear localization of STAT3 during hMPV infection, immunofluorescent detection of nuclear STAT3 was assessed in hMPV-infected lung epithelial cells following IL-6 stimulation (Fig. 2). Upon IL-6 stimulation of mock-infected cells, the cytoplasm : nucleus ratio of STAT3 decreased from 4.704 to 0.089 (Table S1, available in JGV Online), thereby revealing the nuclear localization of STAT3 following IL-6 induction. However, in hMPV-infected cells, the cytoplasm : nucleus ratio of STAT3 remained similar at 5.183 (untreated) and 3.635 (IL-6 treated) following IL-6 stimulation, indicating that hMPV infection prevented nuclear translocation of STAT3 in lung epithelial cells.
Fig. 2.
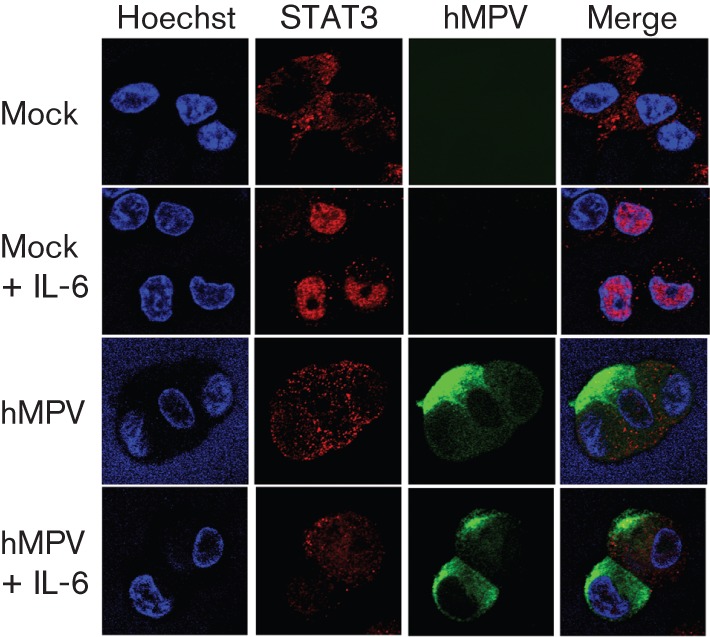
hMPV inhibits the nuclear localization of STAT3. A549 cells were infected with hMPV at an m.o.i. of 0.5. At 48 h p.i., cells were treated with IL-6 (20 ng ml−1) for 30 min and then fixed with 4 % paraformaldehyde. Cells were stained for hMPV antigen (green) and STAT3 (red). The nuclei were counterstained with Hoechst (blue). Results are representative of three independent experiments.
Phosphorylation of STAT3 is a key event preceding translocation to the nucleus. For this reason, the ability of hMPV to inhibit STAT3 phosphorylation was examined by Western blot analysis (Fig. 3). A549 cells were infected for 48 h with medium only, UV-inactivated virus or hMPV and then treated with IL-6 for 30 min. STAT3 was readily phosphorylated (p-STAT3) in uninfected cells (STAT3 : p-STAT3 ratio = 3.21) and in cells infected with UV-inactivated virus (STAT3 : p-STAT3 ratio = 3.50), but only background levels of p-STAT3 (STAT3 : p-STAT3 ratio = 14.67) were detected in hMPV-infected cells (Fig. 3a and Table S2A). The differences in p-STAT3 levels identified when comparing cells infected with hMPV and UV-inactivated virus suggested that the inhibition of p-STAT3 was not due to cellular factors produced during virus propagation. In addition, A549 cells were infected with an increasing m.o.i. and treated with IL-6 at 6, 12, 24 and 48 h p.i. to examine STAT3 phosphorylation over the course of infection (Fig. 3b). The appearance of p-STAT3 was evident in the mock-infected cells and in cells infected with the lowest m.o.i. tested (m.o.i. = 0.1) throughout the course of infection. The levels of p-STAT3 start to decrease at 12 h p.i. in cells infected with the highest m.o.i. tested (m.o.i. = 1), and a noticeable decrease in p-STAT3 occurred in cells infected with an m.o.i. of 0.5 and 1 at 24 and 48 h p.i. The results of densitometric analysis of the Western blot are shown in Table S2(B). Thus, the expression of pSTAT-3 decreased over time in cells infected at an m.o.i. of 0.5 and 1. Together, these data suggested that replicating virus is required for the inhibition of STAT3 phosphorylation.
Fig. 3.

hMPV replication is a potent inhibitor of STAT3 phosphorylation. To examine STAT3 phosphorylation, Western blot analysis was performed on cell lysates using antibodies toward phospho-STAT3 (p-STAT3), STAT3 and actin. (a) A549 cells were infected with medium only (mock), UV-inactivated virus (UVI) or hMPV at an m.o.i. of 0.5 and treated with IL-6 (20 ng ml−1) for 30 min. (b) A549 cells were infected with an increasing m.o.i. of hMPV and treated with medium only or with medium containing IL-6 (20 ng ml−1). (c) Cells were infected with medium only or with hMPV at an m.o.i. of 0.5 and treated with increasing concentrations of IL-6 at 24 (upper panel) and 48 (lower panel) h p.i. (d) At 48 h p.i., mock- or hMPV-infected cells were treated with medium only, IL-6 (20 ng ml−1) or IL-22 (20 ng ml−1). All images are representative of three independent experiments. No TXT, no cytokine (IL-6 or IL-22) treatment. In all experiments, actin was used as a loading control.
To examine the potency of hMPV on the inhibition of STAT3 phosphorylation, A549 cells were infected at an m.o.i. of 0.5 and treated with increasing concentrations of IL-6 at 24 and 48 h p.i. (Fig. 3c). p-STAT3 was identified by Western blot analysis in mock-infected cells treated with IL-6 at a concentration as low as 2 ng ml−1. However, p-STAT3 was not detected in hMPV-infected cells, even in cells treated at the highest concentration of IL-6 (200 ng ml−1), at either time point. The concentration of 200 ng ml−1 is approximately 2–3 logs higher than what has been reported for bronchoalveolar lavage fluid from infected mice (Guerrero-Plata et al., 2005; Huck et al., 2007) and in infected A549 cells (data not shown), demonstrating hMPV is a powerful inhibitor of the IL-6-mediated JAK/STAT3 signalling pathway at physiological conditions in lung epithelium.
STAT3 is activated by several cytokines (e.g. IL-22, IL-10 and IL-6) and thus a hMPV-induced reduction in STAT3 phosphorylation may have global implications beyond IL-6 signalling. To determine whether hMPV has a widespread effect on STAT3 phosphorylation, infected cells were treated with IL-6 or IL-22, as both are important for mediating lung host defence (Aujla & Kolls, 2009; Whittington et al., 2004). p-STAT3 was detected in mock-infected cells treated with IL-6 and IL-22, demonstrating that both cytokines induced STAT3 phosphorylation in A549 cells (Fig. 3d). Consistent with the previous findings, STAT3 phosphorylation was inhibited in hMPV-infected cells treated with IL-6. Conversely, similar levels of p-STAT3 were detected in mock-infected and hMPV-infected cells treated with IL-22. Quantification of the Western blots confirmed these results. The STAT : p-STAT3 ratio for hMPV-infected cells increased from 1.97 to 5.91 following induction with IL-6 but remained the same following IL-22 induction (Table S2C). These data demonstrated that hMPV inhibition of STAT3 phosphorylation is cytokine specific.
HMPV antagonism of the IL-6-induced JAK/STAT pathway in primary normal bronchial epithelial (NHBE) cells
To assess further the antagonistic ability of hMPV on the IL-6-induced JAK/STAT pathway, STAT3 phosphorylation was examined in primary NHBEs by Western blot analysis (Fig. 4a). P-STAT3 was identified in mock-infected NHBE cells treated with IL-6. However, when compared with the mock-infected lysate (STAT : p-STAT3 ratio = 5.81), the amount of p-STAT3 was markedly diminished in hMPV-infected lysate (STAT : p-STAT3 ratio = 14.36) following induction with IL-6 (Table S3A). Similar to A549 cells, total STAT3 levels did not decrease after infection, confirming that hMPV attenuation of p-STAT3 is not due to a decrease in total STAT3 levels.
Fig. 4.
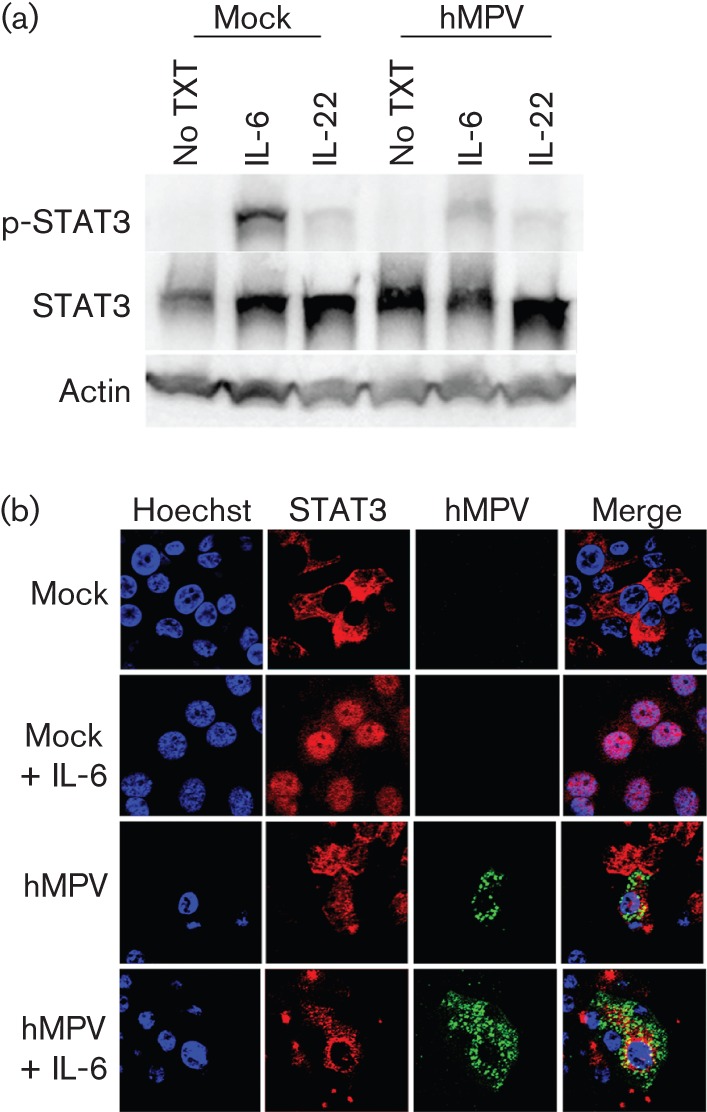
Inhibition of STAT3 phosphorylation and translocation in hMPV-infected NHBE cells. NHBE cells were infected at an m.o.i. of 0.5. At 48 h p.i., cells were treated with 20 ng IL-6 ml −1 for 30 min. (a) Cell lysates were analysed by Western blot analysis with antibodies against p-STAT3, STAT3 or actin. (b) Cells were fixed with 4 % paraformaldehyde and stained for viral antigen (green) and STAT3 (red). Nuclei were counterstained with Hoechst (blue). Results are representative of two independent experiments. No TXT, no cytokine (IL-6 or IL-22) treatment.
The increased amount of residual p-STAT3 recovered in hMPV-infected NHBE cells compared with A549 cells may be due to the heterogeneous cell population and decreased infection rate identified in the primary cell line (Dinwiddie & Harrod, 2008; Kuiken et al., 2004). Immunofluorescence microscopy was performed to verify the hMPV inhibition of STAT3 translocation to the nucleus of infected NHBE cells (Fig. 4b and Table S3B). Whilst nuclear localization of STAT3 appeared following induction with IL-6 in mock-infected cells (cytoplasm : nucleus ratio = 0.127), STAT3 remained predominantly in the cytoplasm of hMPV-infected cells following IL-6 stimulation (cytoplasm : nucleus ratio = 2.85). These results were consistent with the findings in A549 cells and provided further evidence of hMPV regulation of IL-6-induced JAK/STAT3 signalling in lung epithelial cells.
Inhibition of JAK phosphorylation by hMPV
JAK1, JAK2 and Tyk2 have been implicated in the IL-6-induced JAK/STAT signal transduction pathway (Heinrich et al., 1998). Inhibition of this step could suppress downstream events such as STAT3 phosphorylation, as phosphorylation of these kinases is required to initiate or amplify the signal. To identify the JAK involved in the IL-6-mediated JAK/STAT cascade in A549 cells, immunoprecipitation of JAK1 (Best et al., 2005) and Western blot analysis of Tyk2 and JAK2 was performed. As type I IFNs are known to signal through JAK1 and Tyk2, mock-infected cell lysates treated with 1000 U IFN-α ml−1 for 15 min were used as a positive control. As expected, JAK1 phosphorylation was observed in immunoprecipitates from mock-infected cells treated with IFN-α. In contrast, phosphorylated JAK1 was not detected in immunoprecipitates from mock-infected A549 cells treated with IL-6 (Fig. 5a). Similar results were obtained for Tyk2. Phosphorylated Tyk2 (p-Tyk2) was observed in mock-infected lysates treated with IFN-α but not in mock-infected lysates treated with IL-6 (Fig. 5b and Table S4A). This suggested that JAK1 and Tyk2 are not the primary kinases in the IL-6-induced JAK/STAT signalling pathway in A549 lung epithelial cells. IFN-γ utilizes JAK2 in mediating STAT1 phosphorylation. Therefore, mock-infected cells treated with IFN-γ (10 ng ml−1) for 15 min were used as a positive control to examine the involvement of JAK2 in the IL-6-induced signalling cascade. p-JAK2 was identified in lysates from mock-infected cells treated with IFN-γ (JAK2 : p-JAK2 ratio = 9.23) and in lysates from mock-infected cells treated with IL-6 (JAK2 : p-JAK2 ratio = 3.5), suggesting that JAK2 is involved in the IL-6-mediated STAT3 pathway in A549 cells (Fig. 5c and Table S4B). The involvement of JAK2 in the IL-6-induced STAT3 pathway has been reported previously in A549 cells (Huang et al., 2010; Zhang et al., 2009). Notably, JAK2 phosphorylation was not observed in hMPV-infected lysates treated with IL-6. By inhibiting phosphorylation of JAK2, hMPV antagonizes IL-6 early in the STAT3 signal transduction pathway.
Fig. 5.
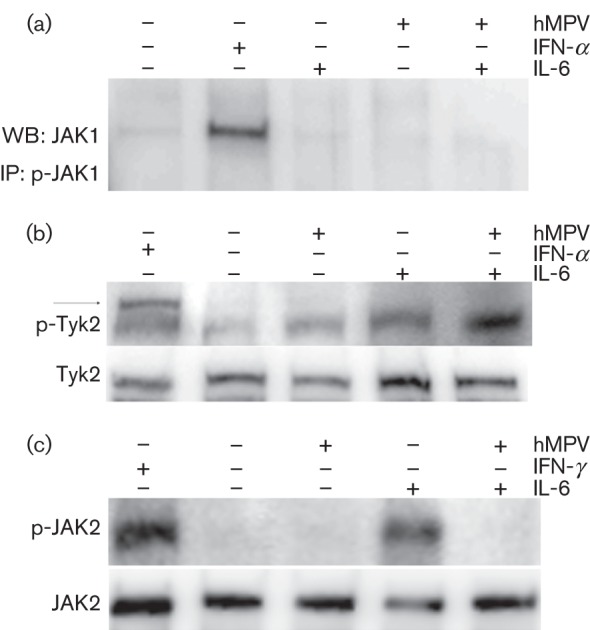
hMPV inhibits phosphorylation of JAK2. MPV- or mock-infected cells were treated with IL-6 (20 ng ml−1) and either INF-α (1000 U ml−1) or IFN-γ (10 ng ml−1). (a) Proteins were immunoprecipitated using phospho-JAK1 (p-JAK1)-specific antibody. The immunoprecipitates were examined by Western blot analysis utilizing an antibody against JAK1. (b, c) Total cell lysates were separated by SDS-PAGE and the membranes were probed with antibodies recognizing Tyk2 and phospho-Tyk2 (p-Tyk2; b) or JAK2 and p-JAK2 (c). Panels (a) and (b) are representative of three independent experiments, whilst (c) is representative of two independent experiments.
HMPV regulation of IL-6-induced JAK/STAT-dependent gene expression
The above data demonstrated that hMPV infection antagonizes the IL-6-induced JAK/STAT signal transduction pathway in the A549 lung epithelial cell line. By impeding this pathway, hMPV probably alters the expression of IL-6-inducible STAT3 target genes. A quantitative reverse transcription (RT)-PCR array was used to analyse differential expression of genes induced by IL-6 in mock- and hMPV-infected cells. This array was focused on a panel of genes relevant to JAK/STAT pathways. In mock-infected cells, IL-6 regulated the expression of 43 of the 84 genes (51 %) tested (Fig. 6a). These include genes important for the acute phase response (CRP and A2M), which are known to be upregulated through the IL-6-inducible STAT3 signalling cascade. Regulation of additional genes correlated with the different functions of IL-6, such as cell differentiation and growth (CCND1, OSM, INSR and IL2RG), apoptosis (CDKN1A, NFKB1 and PRLR) and the inflammatory and immune response (FCER1A, SIT1, SMAD1 and F2). Of the genes regulated by IL-6, nine genes (19 %) were regulated more than twofold by hMPV infection. Utilizing the david bioinformatics database (http://david.abcc.ncifcrf.gov/), the genes were classified into different functional groups (Table 1). Following IL-6 treatment, infection with hMPV altered the expression of genes important for transcription regulation by increasing the RNA levels of YY1 and SMAD2 compared with mock-infected cells. Genes important for cytokine production (IFNAR1 and SOCS3) were also regulated by hMPV. In addition, several genes regulated by hMPV following IL-6 induction were important for cell differentiation, proliferation and apoptosis. Infection with hMPV increased the expression of genes that function in cell differentiation and proliferation (SMAD2 and SOCS3) compared with mock-infected cells (Allen & Unterman, 2007; Cacalano et al., 2001; Kleinsteuber et al., 2012; Rossa et al., 2012; Ungefroren et al., 2011). hMPV-infected cells also differentially regulated the expression of IL-6-inducible genes involved in apoptosis/cell survival. hMPV infection increased the expression of IL-6-inducible anti-apoptotic genes (SOCS3, SRC and IL-4) and downregulated IL-6-inducible genes that promote apoptosis/cell killing (NOS2 and SMAD4) compared with mock-infected cells (Abu-Ghazaleh et al., 2001; Akbar et al., 1996; Karni et al., 1999; Ke et al., 2008; Li et al., 2005; Lu et al., 2006; Madonna et al., 2012; Renauld et al., 1995; Schmidt et al., 2004; Todaro et al., 2008; Zhang, 2007). In addition, JAK2, which functions as a pro- or anti-apoptotic gene and in cell differentiation, was downregulated at the RNA level by hMPV. Together, these data demonstrated that hMPV inhibition of the IL-6-induced JAK/STAT3 pathway regulates the RNA expression of a number of IL-6 inducible genes, especially those important in cell differentiation, cell proliferation and apoptosis.
Fig. 6.

hMPV differentially regulates expression of genes important for cell proliferation and apoptosis. (a) Scatterplot comparing gene expression levels between mock-infected cells and mock-infected cells treated with IL-6. Total RNA was isolated and an RT-PCR array was performed. Data were analysed utilizing the Bioscience web-based RT2 profiler PCR array data analysis program. Symbols outside the grey diagonal lines indicate genes whose levels were regulated more than threefold. Red circles indicate genes that were upregulated and green symbols indicate genes that were downregulated. (b, c) To examine the effects of hMPV on the role of IL-6 in apoptosis, A549 cells were infected at an m.o.i. of 1 for 24 h. Cells were (continued) Fig. 6. (continued) then treated for 18 h with IL-6 (20 ng ml−1) followed by a 6 h treatment with cycloheximide (CHX) and TNF-α. (b) Annexin V staining and flow cytometry were performed to determine number of apoptotic cells. The graph represents the mean of two experiments. An asterisk represents a significant change (P<0.05). (c) A Cell Death Detection ELISA kit (Roche) was utilized for the in vitro determination of DNA fragmentation, measured as changes in absorbance at 405 nm (A450). The graph represents the mean of two experiments.
Table 1. hMPV regulation of IL-6-inducible genes.
| Gene name | IL-6 regulation* | hMPV regulation† | Function |
| IFNAR1 | ↓ | ↑ | Cytokine production |
| IL-4 | ↓ | ↑ | Apoptosis |
| JAK2 | ↑ | ↓ | Cell differentiation and growth, apoptosis |
| NOS2 | ↑ | ↓ | Apoptosis |
| SMAD2 | ↓ | ↑ | Cell differentiation and growth, transcription |
| SMAD4 | ↑ | ↓ | Cell differentiation and growth, transcription, apoptosis |
| SOCS3 | ↓ | ↑ | Cytokine production |
| SRC | ↓ | ↑ | Apoptosis |
| YY1 | ↓ | ↑ | Transcription |
IL-6 regulation of genes in mock-infected cells.
hMPV regulation of IL-6 regulated genes (hMPV-infected cells+IL-6 vs mock-infected cells+IL-6).
To examine the effects of IL-6 on apoptosis and whether hMPV infection modified this effect, A549 cells were infected at an m.o.i. of 1 for 24 h. Apoptosis was induced chemically following IL-6 stimulation. Apoptotic cells were detected using annexin V staining and flow cytometry (Fig. 6b). IL-6 alone had no effect on apoptosis in mock- or hMPV-infected cells. However, when apoptosis was stimulated with cycloheximide and TNF-α, there was a significant increase in the number of apoptotic cells in mock-infected cells pre-treated with IL-6 compared with untreated mock-infected cells (P<0.05, two-way ANOVA). Conversely, in hMPV-infected cells, the number of apoptotic cells was decreased significantly in cells treated with IL-6 compared with untreated hMPV-infected cells (P<0.05, two-way ANOVA). Similar results were obtained for DNA fragmentation analysis (Fig. 6c). In apoptotic-induced cells, hMPV infection decreased the amount of DNA fragmentation following IL-6 induction, whilst mock-infected cells demonstrated an increase in DNA fragmentation. Although infection itself sensitized the cells to DNA fragmentation and apoptosis following treatment with cycloheximide and TNF-α, the effects of IL-6 on apoptosis were differentially regulated in hMPV-infected cells compared with mock-infected cells. Thus, these functional assays supported the RT-PCR array data demonstrating that hMPV inhibition of the IL-6 pathway antagonizes the pro-apoptotic role of IL-6 in A549 cells. By regulating these genes, hMPV could alter the role of IL-6 in maintaining lung homeostasis during infection and play an important role in pathogenesis.
Discussion
Many viruses have developed methods to evade the host IFN response, but little is known about interactions between viruses and other innate signalling cascades important in viral pathogenesis and lung injury. One such signal cascade is that induced by IL-6. IL-6 is a pleiotropic cytokine known to protect the lung from injury, and thus is an important determinant in the host’s susceptibility to respiratory infections (Kida et al., 2005; Ladel et al., 1997; Murphy et al., 2008; van der Poll et al., 1997; Ward et al., 2000; Yu et al., 2002). The studies presented in this paper examined the interaction between hMPV, an important causative agent of respiratory infections, and the IL-6-induced JAK/STAT signal transduction pathway. hMPV attenuated STAT3-inducible luciferase expression following induction with IL-6 through inhibition of JAK2 phosphorylation, thereby averting the downstream activation and nuclear translocation of STAT3. Further analysis demonstrated that hMPV could differentially regulate the expression of IL-6-inducible JAK/STAT-mediated genes important for apoptosis, cell proliferation and differentiation. The ability of hMPV to thwart the IL-6-induced JAK/STAT signal cascade altered the cytokine’s role in apoptosis.
Several respiratory viruses, including hMPV, cause the host to release IL-6 in response to infection. In the lung, IL-6 regulates inflammation, cell growth and apoptosis (DiCosmo et al., 1994; Moodley et al., 2003; Waxman et al., 2003). These responses are important for protection and maintenance of lung homeostasis and occur, in part, through a STAT3-dependent signal cascade. Although a role for STAT3 has been suggested during viral respiratory infections (Ghoshal et al., 2001; Liu et al., 2004), little is known about the ability of viruses to silence the effects of STAT3. Two paramyxoviruses, mumps virus and measles virus, inhibit cytokine-induced STAT3 signalling (Palosaari et al., 2003; Ulane et al., 2003). For both mumps and measles virus, the V protein is implicated in the inhibition of STAT3 activation (reviewed by Horvath, 2004). Mumps virus targets STAT1 and STAT3 for proteasomal degradation, whereas measles virus forms a high-molecular-mass complex with STAT1, STAT2 and STAT3, preventing nuclear translocation of STATs. The overall levels of STAT3 do not decrease during hMPV infection, and thus the mechanism for hMPV inhibition of STAT3 signalling is not similar to that of mumps virus. Currently, it is not known whether hMPV forms high-molecular-mass complexes similar to measles virus, thereby sequestering STATs in the cytoplasm, or whether hMPV inhibits JAK/STAT signalling by a unique mechanism. Notably, the V protein of measles and mumps virus also targets the IFN-induced JAK/STAT signalling cascade, demonstrating that a single virus protein can target multiple innate signalling pathways (Horvath, 2004). Although it is known that hMPV inhibits the IFN-inducible JAK/STAT signalling cascade, the viral determinant for this antagonism is unknown (Dinwiddie & Harrod, 2008). hMPV does not contain any sequence homologous to the V protein of measles or mumps virus, nor to the NS proteins of RSV known to modulate JAK/STAT pathways (Dinwiddie & Harrod, 2008). Thus, the immunomodulatory effects of hMPV on JAK/STAT signalling could be due to a different protein or to a novel protein function not used by the other paramyxoviruses.
The data in this paper clearly demonstrate that hMPV obstructs the IL-6-inducible JAK/STAT signalling cascade. However, the identity of the cellular target for hMPV inhibition of the IL-6 signalling pathway remains unknown. There is evidence suggesting that the target is likely to be the IL-6 receptor complex. hMPV-infected cells were treated with two different cytokines (IL-6 and IL-22). Both of these cytokines are important in lung host defence and signal through STAT3 (Aujla & Kolls, 2009; Jones et al., 2006; Murphy et al., 2008; Whittington et al., 2004). hMPV was able to inhibit STAT3 activation in cells induced with IL-6 but not in cells stimulated with IL-22, demonstrating that STAT3 is not a direct target of inhibition. In addition, as JAK2 was not phosphorylated during hMPV infection, inhibition is likely to occur prior to STAT3 activation. This would implicate either JAK or the receptor as a target. However, if JAK2 was targeted directly, other pathways that signal through JAK2 would also be inhibited. Previously, Western blot analysis demonstrated that hMPV does not inhibit STAT phosphorylation following induction with IFN-γ, a cytokine known to signal through JAK2 (Dinwiddie & Harrod, 2008). Thus, the inhibition of JAK2 activation identified in the IL-6 pathway is not due to a global inhibition of JAK2 phosphorylation by hMPV: IL-6 and IFN-γ signal through different receptors. IFN-γ signals through IFN-γR1 and IFN-γR2, whereas IL-6 signals through a receptor complex consisting of IL-6Ra and gp130, further implicating the receptor as the target for inhibition.
By usurping the IL-6-inducible JAK/STAT3 signalling cascade, hMPV modulated the role of IL-6 in maintaining homeostasis. Specifically, both the RT-PCR array data and the apoptotic functional assay demonstrated that hMPV infection promoted cell survival by decreasing the pro-apoptotic role of IL-6. Although the role of apoptosis in hMPV is not currently well described, current data, including a minimal cytopathic effect, suggest that apoptosis is not induced during hMPV infection. However, in this paper, hMPV sensitized the cells to cycloheximide- and TNF-α-induced apoptosis. This is similar to data obtained for RSV, a closely related virus. RSV sensitized cells to apoptosis following induction with exogenous TNF-related apoptosis-inducing ligand (Kotelkin et al., 2003). Without this stimulus, apoptosis was delayed until very late in RSV infection. This delay/inhibition of apoptosis in infected cells could be associated with the upregulation of anti-apoptotic factors by RSV (Kotelkin et al., 2003). Studies have suggested that this delay/inhibition of apoptosis by RSV is consistent with the minimal cytopathic effect observed during infection (Bitko et al., 2007; Groskreutz, 2007; Kotelkin et al., 2003). As viruses inhibit apoptosis as a method of ensuring virus replication (Itoh et al., 1998; Teodoro & Branton, 1997), it is probable that hMPV upregulation of anti-apoptotic genes and genes important for cell growth will support virus survival, replication and persistence (Alvarez et al., 2004; Liu et al., 2009). Further insight into the motive of hMPV in regulating the IL-6-inducible genes important for cell proliferation and apoptosis could reveal insightful information about hMPV pathogenesis.
Infection with hMPV does not provide long-term protective immunity and an individual can have lifelong, repeated infections, demonstrating the importance of the innate immune response during both the primary and subsequent infections. Understanding how these viruses evade both the canonical antiviral pathways, such as the IFN-induced cascade, and other innate signalling pathways important in host defence will provide more insight into viral pathogenesis and immunity. With the addition of these studies, hMPV is now known to interrupt two innate JAK/STAT signalling pathways important in host defence against respiratory viral infection. Uncovering the molecular mechanisms of these interactions will identify differences and congruities of hMPV antagonism of the various innate signalling pathways and could identify possible targets for the development of new vaccines or therapeutics.
Methods
Cells.
Human lung epithelial cells (A549) and monkey kidney cells, LLC-MK2 (Viromed) were maintained in Dulbecco’s minimal essential media (DMEM; Invitrogen) containing 10 % FBS, 4 mM l-glutamine, 3.7 g sodium bicarbonate l−1, 100 U penicillin ml−1, 100 µg streptomycin ml−1 and 0.25 µg amphotericin B ml−1 at 37 °C in 5 % CO2.
NHBE cells isolated from human trachea and primary bronchi were plated onto collagen-coated 100 mm dishes (Biocoat; Becton Dickinson) and grown in BEGM medium (Lonza) to 80 % confluency. The cells were passaged onto collagen-coated Transwell inserts (Corning) and maintained in BEGM : DMEM (1 : 1 ratio) supplemented with 50 nM retinoic acid, 1.5 µg BSA ml−1 (Sigma) and BEGM SingleQuots (Lonza). Once confluent, the NHBE cells were grown under air–liquid interface conditions until differentiation (21–28 days).
Reagents.
The following primary antibodies were used for the experiments in this paper. STAT3, p-STAT3 (Y705), p-Tyk2 (Y1054/1055) and p-JAK2 (Y1007/1008) antibodies were obtained from Cell Signalling. The antibodies generated against Tyk2 (clone EP1127Y) and firefly luciferase were acquired from Abcam. JAK1 (clone HR-785) and JAK2 (clone C20) were purchased from Santa Cruz Biotechnology. Antibodies to hMPV, pJAK1 and β-actin were obtained from Millipore, Invitrogen and Sigma, respectively. The HRP-conjugated secondary antibodies [goat anti-rabbit IgG(H+L) and goat anti-mouse IgG(H+L)] were purchased from Thermo Scientific and the Alexa Fluor-conjugated secondary antibodies (Alexa Fluor 594-conjugated goat anti-rabbit and Alexa Fluor 488-conjugated goat anti-mouse) were obtained from Invitrogen. All antibodies were used at the manufacturer’s recommended concentrations. The cytokines human IFN-α, human IFN-γ, human IL-6 and human IL-22 were acquired from PBL Interferon Source, Millipore, Invitrogen and Cell Signalling, respectively.
Virus propagation, titration and infections.
hMPV strain CAN97-83, a gift from Guy Boivin (Laval University, Québec City, Québec, Canada), was propagated in LLC-MK2 cells and titrated by a focus-forming assay as described previously (Dinwiddie & Harrod, 2008).
For experiments designed to examine virus interactions with JAK/STAT signalling, cells were infected with hMPV at an m.o.i. of 0.5 unless otherwise noted. Following a 1 h adsorption period, the inocula were removed and the cells washed with PBS. Infected cells were maintained in serum-free Opti-MEM containing 0.1 µg TPCK-trypsin ml−1 (Worthington Biochemical Corp.). Cells were treated with IL-6 for 30 min or 8 h.
Western blot analysis.
Cell were lysed in buffer containing 10 mM Tris/HCl (pH 7.5), 15 mM NaCl, 0.5 % NP-40, 1 % Triton X-100, 1 mM EDTA, 1 mM EGTA (pH 8.0), 0.2 mM sodium ortho-vanadate and 0.4 mM PMSF, and total protein was quantified with a bicinchoninic acid protein assay kit. Equal amounts of protein were separated by SDS-PAGE and transferred to PVDF. The membrane was incubated with the primary antibody overnight at 4 °C with rocking. Following three washes, the membranes were incubated with a HRP-conjugated secondary antibody at room temperature for 1 h. The membranes were developed using Western Lightening Plus-ECL (Perkin Elmer), the proteins visualized using an LAS4000 Luminescent Image Analyser (Fujifilm) and densitometry was performed using MultiGauge v3.0 (Fujifilm).
Immunofluorescence.
Following infection and cytokine treatment, A549 cells were fixed in 4 % paraformaldehyde. The cells were permeabilized for 5 min in 0.2 % Triton X-100. Cells were double stained with anti-hMPV and with either anti-STAT3 or anti-firefly luciferase antibody for 1 h at room temperature. Alexa Fluor-conjugated secondary antibodies were applied to the cells for 1 h at room temperature. Nuclei were counterstained with Hoechst (10 µg ml−1) and coverslips mounted using Mowiol. Fluorescence was visualized using a Zeiss LSM 510 META confocal microscope. Fluorescence was quantified using ImageJ software (National Institutes of Health).
Luciferase reporter assay.
To quantify activation of the STAT3 signal transduction pathway, a dual luciferase reporter assay (Cignal) was utilized. For transient expression of a reporter gene, A549 cells were transfected with the Cignal STAT3-luc construct, a Cignal positive-control construct that constitutively expresses firefly luciferase (positive control) or a Cignal negative-control construct (SABiosciences) using SureFECT. At 24 h post-transfection, cells were infected with hMPV at an m.o.i. of 4. The infection continued for 24 h. IL-6 (20 ng ml−1) was added to the cells and the cells were incubated for a further 8 h at 37 °C. Luciferase expression was detected using a dual luciferase reporter assay system and an AutoLumat Plus LB 953 (Berthold Technologies).
Analysis of host gene expression.
At 48 h p.i., A549 cells were treated with IL-6 (20 ng ml−1) or with medium only and incubated for 8 h at 37 °C. Total RNA was isolated using an RNeasy Mini kit (Qiagen), which included a 10 min on-column DNase treatment. A JAK/STAT RT2 Profiler PCR array (SABiosciences) was used to analyse host gene expression. The Bioscience web-based RT2 profiler PCR array data analysis program was used to calculate fold regulation.
Staining of apoptotic cells and DNA fragmentation analysis.
A549 cells were infected with hMPV at an m.o.i. of 1. At 24 h p.i., the cells were maintained in Opti-MEM supplemented with 2 % FBS and IL-6 (20 ng ml−1) for 20 h. To induce apoptosis, cycloheximide (5 µg ml−1) and TNF-α (10 ng ml−1) were added to the cells and the cells were incubated at 37 °C for 6 h. Annexin V and 7-aminoactinomycin D (eBioscience) was used to stain apoptotic cells. Annexin V was used at half the recommended dose and was added simultaneously with 7-aminoactinomycin D. The cells were stained whilst incubating on ice, and flow cytometry on a FACSCanto machine (BD Biosciences) was performed immediately after staining. A Cell Death Detection ELISA (Roche) was used, following the manufacturer’s instructions, for quantitative analysis of cytoplasmic histone-associated DNA fragments.
Data analysis.
Results were compared using two-way ANOVA with Bonferroni post-test findings and were consider statistically significant at P<0.05.
Acknowledgements
The authors thanks Dr Guy Boivin for providing hMPV strain CAN97-83, Jesse vanWestrienen and the Lovelace qRT-PCR core facility for providing Taqman reagents.
Supplementary Data
Supplementary Data
Footnotes
Four supplementary tables are available with the online version of this paper.
References
- Abu-Ghazaleh R., Kabir J., Jia H., Lobo M., Zachary I. (2001). Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. Biochem J 360, 255–264. 10.1042/0264-6021:3600255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar A. N., Borthwick N. J., Wickremasinghe R. G., Panayoitidis P., Pilling D., Bofill M., Krajewski S., Reed J. C., Salmon M. (1996). Interleukin-2 receptor common gamma-chain signaling cytokines regulate activated T cell apoptosis in response to growth factor withdrawal: selective induction of anti-apoptotic (bcl-2, bcl-xL) but not pro-apoptotic (bax, bcl-xS) gene expression. Eur J Immunol 26, 294–299. 10.1002/eji.1830260204 [DOI] [PubMed] [Google Scholar]
- Allen D. L., Unterman T. G. (2007). Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol 292, C188–C199. 10.1152/ajpcell.00542.2005 [DOI] [PubMed] [Google Scholar]
- Alvarez R., Harrod K. S., Shieh W. J., Zaki S., Tripp R. A. (2004). Human metapneumovirus persists in BALB/c mice despite the presence of neutralizing antibodies. J Virol 78, 14003–14011. 10.1128/JVI.78.24.14003-14011.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aujla S. J., Kolls J. K. (2009). IL-22: a critical mediator in mucosal host defense. J Mol Med (Berl) 87, 451–454. 10.1007/s00109-009-0448-1 [DOI] [PubMed] [Google Scholar]
- Best S. M., Morris K. L., Shannon J. G., Robertson S. J., Mitzel D. N., Park G. S., Boer E., Wolfinbarger J. B., Bloom M. E. (2005). Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol 79, 12828–12839. 10.1128/JVI.79.20.12828-12839.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitko V., Shulyayeva O., Mazumder B., Musiyenko A., Ramaswamy M., Look D. C., Barik S. (2007). Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-κB-dependent, interferon-independent mechanism and facilitate virus growth. J Virol 81, 1786–1795. 10.1128/JVI.01420-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacalano N. A., Sanden D., Johnston J. A. (2001). Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol 3, 460–465. 10.1038/35074525 [DOI] [PubMed] [Google Scholar]
- DiCosmo B. F., Geba G. P., Picarella D., Elias J. A., Rankin J. A., Stripp B. R., Whitsett J. A., Flavell R. A. (1994). Airway epithelial cell expression of interleukin-6 in transgenic mice. Uncoupling of airway inflammation and bronchial hyperreactivity. J Clin Invest 94, 2028–2035. 10.1172/JCI117556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinwiddie D. L., Harrod K. S. (2008). Human metapneumovirus inhibits IFN-α signaling through inhibition of STAT1 phosphorylation. Am J Respir Cell Mol Biol 38, 661–670. 10.1165/rcmb.2007-0285OC [DOI] [PubMed] [Google Scholar]
- Ebihara T., Endo R., Kikuta H., Ishiguro N., Yoshioka M., Ma X., Kobayashi K. (2003). Seroprevalence of human metapneumovirus in Japan. J Med Virol 70, 281–283. 10.1002/jmv.10391 [DOI] [PubMed] [Google Scholar]
- Ebihara T., Endo R., Kikuta H., Ishiguro N., Ishiko H., Hara M., Takahashi Y., Kobayashi K. (2004). Human metapneumovirus infection in Japanese children. J Clin Microbiol 42, 126–132. 10.1128/JCM.42.1.126-132.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards K. M., Zhu Y., Griffin M. R., Weinberg G. A., Hall C. B., Szilagyi P. G., Staat M. A., Iwane M., Prill M. M., Williams J. V., New Vaccine Surveillance Network (2013). Burden of human metapneumovirus infection in young children. N Engl J Med 368, 633–643. 10.1056/NEJMoa1204630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal K., Majumder S., Zhu Q., Hunzeker J., Datta J., Shah M., Sheridan J. F., Jacob S. T. (2001). Influenza virus infection induces metallothionein gene expression in the mouse liver and lung by overlapping but distinct molecular mechanisms. Mol Cell Biol 21, 8301–8317. 10.1128/MCB.21.24.8301-8317.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greensill J., McNamara P. S., Dove W., Flanagan B., Smyth R. L., Hart C. A. (2003). Human metapneumovirus in severe respiratory syncytial virus bronchiolitis. Emerg Infect Dis 9, 372–375. 10.3201/eid0903.020289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groskreutz D. J., Monick M. M., Yarovinsky T. O., Powers L. S., Quelle D. E., Varga S. M., Look D. C., Hunninghake G. W. (2007). Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J Immunol 179, 2741–2747. 10.1128/JVI.79.23.14992-14997.2005 [DOI] [PubMed] [Google Scholar]
- Guerrero-Plata A., Casola A., Garofalo R. P. (2005). Human metapneumovirus induces a profile of lung cytokines distinct from that of respiratory syncytial virus. J Virol 79, 14992–14997. 10.1128/JVI.79.23.14992-14997.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich P. C., Behrmann I., Müller-Newen G., Schaper F., Graeve L. (1998). Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334, 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath C. M. (2004). Silencing STATs: lessons from paramyxovirus interferon evasion. Cytokine Growth Factor Rev 15, 117–127. 10.1016/j.cytogfr.2004.02.003 [DOI] [PubMed] [Google Scholar]
- Huang W. L., Yeh H. H., Lin C. C., Lai W. W., Chang J. Y., Chang W. T., Su W. C. (2010). Signal transducer and activator of transcription 3 activation up-regulates interleukin-6 autocrine production: a biochemical and genetic study of established cancer cell lines and clinical isolated human cancer cells. Mol Cancer 9, 309. 10.1186/1476-4598-9-309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huck B., Neumann-Haefelin D., Schmitt-Graeff A., Weckmann M., Mattes J., Ehl S., Falcone V. (2007). Human metapneumovirus induces more severe disease and stronger innate immune response in BALB/c mice as compared with respiratory syncytial virus. Respir Res 8, 6. 10.1186/1465-9921-8-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M., Hotta H., Homma M. (1998). Increased induction of apoptosis by a Sendai virus mutant is associated with attenuation of mouse pathogenicity. J Virol 72, 2927–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. R., Quinton L. J., Simms B. T., Lupa M. M., Kogan M. S., Mizgerd J. P. (2006). Roles of interleukin-6 in activation of STAT proteins and recruitment of neutrophils during Escherichia coli pneumonia. J Infect Dis 193, 360–369. 10.1086/499312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura D., Ishihara K., Hirano T. (2003). IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol 149, 1–38. 10.1038/sj.onc.1202835 [DOI] [PubMed] [Google Scholar]
- Karni R., Jove R., Levitzki A. (1999). Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 18, 4654–4662. 10.1038/sj.onc.1202835 [DOI] [PubMed] [Google Scholar]
- Ke Z., Zhang X., Ma L., Wang L. (2008). Deleted in pancreatic carcinoma locus 4/Smad4 participates in the regulation of apoptosis by affecting the Bcl-2/Bax balance in non-small cell lung cancer. Hum Pathol 39, 1438–1445. 10.1016/j.humpath.2008.03.006 [DOI] [PubMed] [Google Scholar]
- Kida H., Yoshida M., Hoshino S., Inoue K., Yano Y., Yanagita M., Kumagai T., Osaki T., Tachibana I. & other authors (2005). Protective effect of IL-6 on alveolar epithelial cell death induced by hydrogen peroxide. Am J Physiol Lung Cell Mol Physiol 288, L342–L349. 10.1152/ajplung.00016.2004 [DOI] [PubMed] [Google Scholar]
- Kleinsteuber K., Heesch K., Schattling S., Sander-Juelch C., Mock U., Riecken K., Fehse B., Fleischer B., Jacobsen M. (2012). SOCS3 promotes interleukin-17 expression of human T cells. Blood 120, 4374–4382. 10.1182/blood-2011-11-392738 [DOI] [PubMed] [Google Scholar]
- Kotelkin A., Prikhod’ko E. A., Cohen J. I., Collins P. L., Bukreyev A. (2003). Respiratory syncytial virus infection sensitizes cells to apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J Virol 77, 9156–9172. 10.1128/JVI.77.17.9156-9172.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiken T., van den Hoogen B. G., van Riel D. A., Laman J. D., van Amerongen G., Sprong L., Fouchier R. A., Osterhaus A. D. (2004). Experimental human metapneumovirus infection of cynomolgus macaques (Macaca fascicularis) results in virus replication in ciliated epithelial cells and pneumocytes with associated lesions throughout the respiratory tract. Am J Pathol 164, 1893–1900. 10.1016/S0002-9440(10)63750-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladel C. H., Blum C., Dreher A., Reifenberg K., Kopf M., Kaufmann S. H. (1997). Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect Immun 65, 4843–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laham F. R., Israele V., Casellas J. M., Garcia A. M., Lac Prugent C. M., Hoffman S. J., Hauer D., Thumar B., Name M. I. & other authors (2004). Differential production of inflammatory cytokines in primary infection with human metapneumovirus and with other common respiratory viruses of infancy. J Infect Dis 189, 2047–2056. 10.1086/383350 [DOI] [PubMed] [Google Scholar]
- Leung J., Esper F., Weibel C., Kahn J. S. (2005). Seroepidemiology of human metapneumovirus (hMPV) on the basis of a novel enzyme-linked immunosorbent assay utilizing hMPV fusion protein expressed in recombinant vesicular stomatitis virus. J Clin Microbiol 43, 1213–1219. 10.1128/JCM.43.3.1213-1219.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Wu L., Oelschlager D. K., Wan M., Stockard C. R., Grizzle W. E., Wang N., Chen H., Sun Y., Cao X. (2005). Smad4 inhibits tumor growth by inducing apoptosis in estrogen receptor-α-positive breast cancer cells. J Biol Chem 280, 27022–27028. 10.1074/jbc.M505071200 [DOI] [PubMed] [Google Scholar]
- Liu T., Castro S., Brasier A. R., Jamaluddin M., Garofalo R. P., Casola A. (2004). Reactive oxygen species mediate virus-induced STAT activation: role of tyrosine phosphatases. J Biol Chem 279, 2461–2469. 10.1074/jbc.M307251200 [DOI] [PubMed] [Google Scholar]
- Liu Y., Haas D. L., Poore S., Isakovic S., Gahan M., Mahalingam S., Fu Z. F., Tripp R. A. (2009). Human metapneumovirus establishes persistent infection in the lungs of mice and is reactivated by glucocorticoid treatment. J Virol 83, 6837–6848. 10.1128/JVI.00379-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Fukuyama S., Yoshida R., Kobayashi T., Saeki K., Shiraishi H., Yoshimura A., Takaesu G. (2006). Loss of SOCS3 gene expression converts STAT3 function from anti-apoptotic to pro-apoptotic. J Biol Chem 281, 36683–36690. 10.1074/jbc.M607374200 [DOI] [PubMed] [Google Scholar]
- Madonna S., Scarponi C., Pallotta S., Cavani A., Albanesi C. (2012). Anti-apoptotic effects of suppressor of cytokine signaling 3 and 1 in psoriasis. Cell Death Dis 3, e334. 10.1038/cddis.2012.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinello R. A., Esper F., Weibel C., Ferguson D., Landry M. L., Kahn J. S. (2006). Human metapneumovirus and exacerbations of chronic obstructive pulmonary disease. J Infect 53, 248–254. 10.1016/j.jinf.2005.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki Y., Xu Y., Ikegami M., Besnard V., Park K. S., Hull W. M., Wert S. E., Whitsett J. A. (2006). Stat3 is required for cytoprotection of the respiratory epithelium during adenoviral infection. J Immunol 177, 527–537. [DOI] [PubMed] [Google Scholar]
- Moodley Y. P., Misso N. L., Scaffidi A. K., Fogel-Petrovic M., McAnulty R. J., Laurent G. J., Thompson P. J., Knight D. A. (2003). Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol Biol 29, 490–498. 10.1165/rcmb.2002-0262OC [DOI] [PubMed] [Google Scholar]
- Murphy E. A., Davis J. M., Brown A. S., Carmichael M. D., Ghaffar A., Mayer E. P. (2008). Effect of IL-6 deficiency on susceptibility to HSV-1 respiratory infection and intrinsic macrophage antiviral resistance. J Interferon Cytokine Res 28, 589–595. 10.1089/jir.2007.0103 [DOI] [PubMed] [Google Scholar]
- Palosaari H., Parisien J. P., Rodriguez J. J., Ulane C. M., Horvath C. M. (2003). STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol 77, 7635–7644. 10.1128/JVI.77.13.7635-7644.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinton L. J., Jones M. R., Robson B. E., Simms B. T., Whitsett J. A., Mizgerd J. P. (2008). Alveolar epithelial STAT3, IL-6 family cytokines, and host defense during Escherichia coli pneumonia. Am J Respir Cell Mol Biol 38, 699–706. 10.1165/rcmb.2007-0365OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renauld J. C., Vink A., Louahed J., Van Snick J. (1995). Interleukin-9 is a major anti-apoptotic factor for thymic lymphomas. Blood 85, 1300–1305. [PubMed] [Google Scholar]
- Rossa C., Jr, Sommer G., Spolidorio L. C., Rosenzweig S. A., Watson D. K., Kirkwood K. L. (2012). Loss of expression and function of SOCS3 is an early event in HNSCC: altered subcellular localization as a possible mechanism involved in proliferation, migration and invasion. PLoS ONE 7, e45197. 10.1371/journal.pone.0045197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S., Rainer J., Ploner C., Presul E., Riml S., Kofler R. (2004). Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death Differ 11 (Suppl 1), S45–S55. 10.1038/sj.cdd.4401456 [DOI] [PubMed] [Google Scholar]
- Teodoro J. G., Branton P. E. (1997). Regulation of apoptosis by viral gene products. J Virol 71, 1739–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro M., Lombardo Y., Francipane M. G., Alea M. P., Cammareri P., Iovino F., Di Stefano A. B., Di Bernardo C., Agrusa A. & other authors (2008). Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived interleukin-4. Cell Death Differ 15, 762–772. 10.1038/sj.cdd.4402305 [DOI] [PubMed] [Google Scholar]
- Ulane C. M., Rodriguez J. J., Parisien J. P., Horvath C. M. (2003). STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J Virol 77, 6385–6393. 10.1128/JVI.77.11.6385-6393.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungefroren H., Groth S., Sebens S., Lehnert H., Gieseler F., Fändrich F. (2011). Differential roles of Smad2 and Smad3 in the regulation of TGF-β1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: control by Rac1. Mol Cancer 10, 67. 10.1186/1476-4598-10-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoogen B. G., de Jong J. C., Groen J., Kuiken T., de Groot R., Fouchier R. A., Osterhaus A. D. (2001). A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med 7, 719–724. 10.1038/89098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Poll T., Keogh C. V., Guirao X., Buurman W. A., Kopf M., Lowry S. F. (1997). Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis 176, 439–444. 10.1086/514062 [DOI] [PubMed] [Google Scholar]
- Ward N. S., Waxman A. B., Homer R. J., Mantell L. L., Einarsson O., Du Y., Elias J. A. (2000). Interleukin-6-induced protection in hyperoxic acute lung injury. Am J Respir Cell Mol Biol 22, 535–542. 10.1165/ajrcmb.22.5.3808 [DOI] [PubMed] [Google Scholar]
- Waxman A. B., Mahboubi K., Knickelbein R. G., Mantell L. L., Manzo N., Pober J. S., Elias J. A. (2003). Interleukin-11 and interleukin-6 protect cultured human endothelial cells from H2O2-induced cell death. Am J Respir Cell Mol Biol 29, 513–522. 10.1165/rcmb.2002-0044OC [DOI] [PubMed] [Google Scholar]
- Welliver R. C., Sr (2008). The immune response to respiratory syncytial virus infection: friend or foe? Clin Rev Allergy Immunol 34, 163–173. 10.1007/s12016-007-8033-2 [DOI] [PubMed] [Google Scholar]
- Whittington H. A., Armstrong L., Uppington K. M., Millar A. B. (2004). Interleukin-22: a potential immunomodulatory molecule in the lung. Am J Respir Cell Mol Biol 31, 220–226. 10.1165/rcmb.2003-0285OC [DOI] [PubMed] [Google Scholar]
- Williams J. V., Harris P. A., Tollefson S. J., Halburnt-Rush L. L., Pingsterhaus J. M., Edwards K. M., Wright P. F., Crowe J. E., Jr (2004). Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N Engl J Med 350, 443–450. 10.1056/NEJMoa025472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Zheng X., Witschi H., Pinkerton K. E. (2002). The role of interleukin-6 in pulmonary inflammation and injury induced by exposure to environmental air pollutants. Toxicol Sci 68, 488–497. 10.1093/toxsci/68.2.488 [DOI] [PubMed] [Google Scholar]
- Zhang J., Kalyankrishna S., Wislez M., Thilaganathan N., Saigal B., Wei B., Ma L., Wistuba II, Johnson F. M. & other authors (2007). SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am J Pathol 170, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Yin P., Di D., Luo G., Zheng L., Wei J., Zhang J., Shi Y., Zhang J., Xu N. (2009). IL-6 regulates MMP-10 expression via JAK2/STAT3 signaling pathway in a human lung adenocarcinoma cell line. Anticancer Res 29, 4497–4501. [PubMed] [Google Scholar]
- Zhang X., Goel T., Goodfield L. L., Muse S. J., Harvill E. T. (2011). Decreased leukocyte accumulation and delayed Bordetella pertussis clearance in IL-6-/- mice. J Immunol 186, 4895–4904. 10.4049/jimmunol.1000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data