

ABSTRACT
The transcription factor GATA2 is required for expansion and differentiation of hematopoietic stem cells (HSCs). In mesenchymal stem cells (MSCs), GATA2 blocks adipogenesis, but its biological relevance and underlying genomic events are unknown. We report a dual function of GATA2 in bone homeostasis. GATA2 in MSCs binds near genes involved in skeletal system development and colocalizes with motifs for FOX and HOX transcription factors, known regulators of skeletal development. Ectopic GATA2 blocks osteoblastogenesis by interfering with SMAD1/5/8 activation. MSC-specific deletion of GATA2 in mice increases the numbers and differentiation capacity of bone-derived precursors, resulting in elevated bone formation. Surprisingly, MSC-specific GATA2 deficiency impairs the trabecularization and mechanical strength of bone, involving reduced MSC expression of the osteoclast inhibitor osteoprotegerin and increased osteoclast numbers. Thus, GATA2 affects bone turnover via MSC-autonomous and indirect effects. By regulating bone trabecularization, GATA2 expression in the osteogenic lineage may contribute to the anatomical and cellular microenvironment of the HSC niche required for hematopoiesis.
KEYWORDS: GATA2, bone, cell differentiation, mesenchymal stem cell, osteoblast, trabecularization
INTRODUCTION
GATA2 belongs to a family of six structurally related zinc finger transcription factors (GATA1 to -6) and plays a critical role in hematopoiesis. Mice that lack GATA2 die during embryonic development due to severe anemia upon impaired proliferation and survival of early hematopoietic stem cells (HSCs) (1). Expression of GATA2 in HSCs is required for the expansion of multipotent hematopoietic cells and the formation of mast cells but was found to be dispensable for the terminal differentiation of erythroid cells and macrophages (2). In humans, germ line mutations in GATA2 are associated with the GATA2 deficiency syndrome, which manifests as a complex array of hematologic, neoplastic, dermatologic, and pulmonary symptoms that can be accompanied by certain viral infections and congenital deafness (3). Acquired mutations have been linked to myelodysplastic syndrome, acute myeloid leukemia, and blast crisis transformation of chronic myeloid leukemia (4).
Besides its expression in hematopoietic precursors, GATA2 is expressed in mesenchymal stem/stromal cells (MSCs) (5), common precursors for, e.g., adipocytes, myocytes, osteocytes, and chondrocytes. In part owing to the embryonic lethality of GATA2-deficient mice, GATA2's function in MSCs is much less investigated, and most insights derive from studying its role during adipogenesis. GATA2 was shown to block differentiation of both white and brown precursor cells (5–7) by reducing peroxisome proliferator-activated receptor γ (PPARγ) promoter activity and by interfering with CCAAT/enhancer binding protein (C/EBP)-mediated transcription (8).
Here, we report the first genome-wide analysis of GATA2 binding sites in mesenchymal cells and implicate this transcription factor in bone homeostasis. GATA2 binds canonical motifs near genes involved in skeletal development and blocks osteoblastogenesis. Mice that lack GATA2 specifically in MSCs developed normally but, surprisingly, showed reduced bone trabecularization and bone strength involving lower osteoprotegerin (encoded by Opg) expression in MSCs and higher osteoclast numbers. Moreover, MSC-specific deletion of GATA2 reduced red blood cell counts, which may imply a much broader role for GATA2 in the control of hematopoiesis than previously thought.
RESULTS
GATA2 regulates and binds the Zfpm1 gene in mesenchymal 3T3-L1 cells.
GATA factors regulate gene expression via their interaction with friend of GATA (FOG)/zinc finger protein and FOG family member (ZFPM) cofactors (9). ZFPM1 is also a transcriptional target of GATA factors in hematopoietic cells, and binding sites near the Zfpm1 gene locus (kb +0.7 and +24.4 of the transcriptional start site [TSS]) have been previously identified in G1E-ER cells, an erythroid cell line (10). We focused on Zfpm1 in order to identify a functionally relevant binding site of GATA2 in 3T3-L1 cells, an adipocyte lineage-committed mesenchymal cell line (11). As previously reported (6, 12), GATA2 is downregulated during adipogenesis (Fig. 1A, before [day 0] and 14 days after the initiation of differentiation). Similarly, mRNA expression of Zfpm1 was reduced (Fig. 1B), in accordance with a recent study (13), suggesting that GATA2 regulates Zfpm1 expression in 3T3-L1 cells. Indeed, retroviral overexpression of GATA2 in preadipocytes upregulated ZFPM1 protein (Fig. 1C). We performed chromatin immunoprecipitation (ChIP) of endogenous GATA2 and found that binding was conserved at kb +0.7 but not kb +24.7 of the Zfpm1 TSS in 3T3-L1 cells and absent in adipocytes (Fig. 1D), consistent with the low expression of GATA2 after differentiation. An upstream site (kb −1.4) served as a negative control. GATA2 binding to kb +0.7 of Zfpm1 was used as a control/validation site for all further ChIP experiments. Input and GATA2-enriched chromatin of undifferentiated 3T3-L1 cells (>5-fold enriched at kb +0.7 of Zfpm1) was subjected to high-throughput sequencing (ChIP-seq).
FIG 1.

GATA2 binds at canonical WGATAR motifs in the genome of 3T3-L1 cells and is enriched near genes involved in skeletal development. (A) GATA2 protein expression in 3T3-L1 preadipocytes (day 0) and adipocytes (day 14 after the initiation of differentiation) determined by immunoblotting (IB). RAN protein served as a loading control. (B) Zfpm1 mRNA in 3T3-L1 preadipocytes and adipocytes was analyzed by qPCR. (C) GATA2 was retrovirally overexpressed in 3T3-L1 cells, and protein expression of GATA2 and ZFPM1 was determined by immunoblotting. RAN protein served as a loading control. (D) ChIP of endogenously expressed GATA2 in undifferentiated and differentiated 3T3-L1 cells revealed preadipocyte-specific binding of GATA2 to the Zfpm1 gene locus (kb +0.7). (E) Genomic localization of the 1,975 GATA2 binding sites in 3T3-L1 cells called by the MACS algorithm. UTR, untranslated region; prox., proximal. (F) Top 3 enriched de novo motif clusters identified by SeqPos. (G) (Top) Identification of E-box motifs in GATA2-bound regions by de novo motif analysis with SeqPos. (Bottom) Top four clusters of known transcription factor motifs enriched in GATA2 binding sites determined by SeqPos. (H) PhastCons evaluation of GATA2 binding sites for evolutionary sequence conservation. (I) GO analysis of nearest genes (70 kb 5′ or 3′ of binding sites; n = 2,230 genes) showing the term for skeletal system development genes (n = 56 genes; see Table S1 in the supplemental material) represented among the top-ranked clusters. The data are presented as means and SD; *, P < 0.05.
GATA2 binds genomic WGATAR motifs and is enriched at skeletal-development genes.
We identified a total of 1,975 peaks (false-discovery rate [FDR], 1%) (see Table S1 in the supplemental material), and more than 90% localized to intergenic and intronic regions. Only a small fraction mapped to proximal promoters (73 peaks within 3 kb 5′ of the TSS) (Fig. 1E). Binding to five randomly selected sites near the genes Prickle, Tinag, Cdk4, Tsen, and RetSat was validated and confirmed by ChIP-quantitative PCR (qPCR) (data not shown). De novo motif analysis of genome-wide GATA2 binding sites by SeqPos (14) revealed that GATA-containing sequences represented the top three motif clusters (Fig. 1F, plus and minus strands), matching the consensus WGATAR motif (W is T or A; R is G or A) (15–17) to a great extent. Of note, E-box motifs of CANNTG-WGATAR-containing composite elements, known to be important for GATA's cooperative function with other transcription factors (16, 18), were also enriched, although much less significantly (Fig. 1G, top). Interrogating known binding motifs in peak regions identified either GATA factors or transcription factors with binding motifs that contained GATA (Fig. 1G, bottom). The binding sites showed evolutionary conservation when assessed by PhastCons, which is based on a two-state phylogenetic hidden Markov model (19) (Fig. 1H). Next, nearby genes (located 70 kb 5′ or 3′ of GATA2 binding sites; n = 2,230 genes) (see Table S2 in the supplemental material) were analyzed by gene ontology (GO) analysis and were enriched in pathways involving transcription, nucleic acids, and nitrogen compound metabolic processes (Fig. 1I). Strikingly, one of the top-ranking pathways mapped to skeletal system development (n = 56 genes) (Fig. 1I; see Table S2 in the supplemental material), suggesting a role for GATA2 in osteoblast differentiation and bone homeostasis. Binding to six randomly selected sites near genes of this pathway (Foxc2, Cobl, Sfrp2, Igsf10, Man2a1, and Ptgs2) was validated and confirmed by ChIP-qPCR (data not shown).
HSC- versus MSC-specific binding of GATA2.
We then intersected the 3T3-L1 GATA2 cistrome with a published data set of GATA2 binding sites in FDCPmix cells (20), a bone marrow-derived cell line with HSC-like characteristics (21). The numbers of peaks/binding sites (see Table S1 in the supplemental material) and nearby genes (see Table S2 in the supplemental material) were much higher in these cells (FDR, 1%) and overlapped about 54% of the genes within 70 kb of GATA2 binding sites in 3T3-L1 cells (Fig. 2A). 3T3-L1-specific, overlapping, and mouse HSC (mHSC)-specific genomic GATA2 binding sites were mapped to H3K4me1, H3K27ac, and chromatin DNase I hypersensitivity sequencing (DHS) data of 3T3-L1 preadipocytes (Fig. 2B). We found strong enrichment of H3K4me1 and H3K27ac on both 3T3-L1-specific and overlapping GATA2 binding sites, whereas DHS was strongest on overlapping sites, followed by 3T3-L1- and mHSC-specific sites. These results support the notion that cell-type-specific chromatin accessibility determines GATA2 binding. Moreover, when analyzed separately for gene ontology, embryonic skeletal system morphogenesis and development were now the two top-ranking pathways of genes located near GATA2 sites that are specific to 3T3-L1 cells (n = 1,011 genes), whereas both overlapping genes (n = 1,219 genes) and genes near HSC-specific binding sites (n = 6,316) mapped to other, non-skeletal-system-related pathways (Fig. 2C, left; Table S2 in the supplemental material lists the corresponding genes). We then asked which motifs besides WGATAR enriched cell-type-specific peaks. Motifs for transcription factors of the FOX and HOX families, known regulators of neural crest cells and skeletal development (22, 23), were found near 3T3-L1-specific binding sites, whereas motifs for a variety of factors involved in hematopoiesis were identified near HSC-specific binding sites (Fig. 2C, right). Thus, GATA2 appears to colocalize with specific lineage-determining transcription factors that may prime and/or facilitate cell-type-selective binding. GATA2 binding sites near Zfpm1 and Ccna1 were conserved in C3H10T1/2 cells (Fig. 2D, left), which, in contrast to adipocyte lineage-committed 3T3-L1 cells, are mesenchyme-derived cells that exhibit multipotency (24). GATA2 ChIP-seq in these cells identified 1,517 binding sites (FDR, 1%) (see Table S1 in the supplemental material) and the GATA consensus motif as the most enriched sequence (Fig. 2D, top right). Of the 1,936 genes located near GATA2 binding sites in C3H10T1/2 cells (see Table S2 in the supplemental material), 989 overlapped those in 3T3-L1 (Fig. 2D, bottom right). As expected, heat map visualization showed a much greater overlap of GATA2 binding sites between the two mesenchymal cell lines than binding in HSCs (Fig. 2E).
FIG 2.

Cell-type-specific binding of GATA2 in mesenchymal and hematopoietic cells. (A) Intersection of nearest genes (70 kb 5′ or 3′ of binding sites) between mesenchymal 3T3-L1 and hematopoietic FDCPmix (mHSC) cells. (B) Average profile of the ChIP-seq signals of the indicated chromatin marks and DHS in 3T3-L1 preadipocytes around GATA2 binding sites. (C) (Left) GO analysis of cell-type-specific or overlapping nearest genes from panel A showing the top two ranked clusters. (Right) The corresponding cell-type-specific peaks were analyzed for known transcription factor binding motifs besides the GATA motif. (D) GATA2 binding in C3H10T1/2 cells near Zfpm1 and Ccna1 is conserved, and ChIP-seq identified 1,517 peaks and WGATAR as the most enriched motif (de novo); 51% of the nearest genes (70 kb 5′ or 3′ of binding sites) in C3H10T1/2 cells overlapped those in 3T3-L1 cells. The data are presented as means and SD; *, P < 0.05. (E) Heat map visualization of genome-wide GATA2 read coverage between 3T3-L1, C3H10T1/2, and FDCPmix cells.
GATA2 blocks osteoblastogenesis and impairs SMAD signaling.
To test the hypothesis that GATA2 regulates osteoblast differentiation, we retrovirally overexpressed GATA2 in C3H10T1/2 cells (Fig. 3A) and stimulated osteoblast conversion. Remarkably, ectopic GATA2 strongly suppressed osteoblastogenesis as assessed by alkaline phosphatase (ALPL) staining, alizarin staining of calcium deposition, and the expression of osteoblast marker genes after 8 days of differentiation (Fig. 3B and C). Since Wnt and SMAD signaling are pivotal in controlling osteoblast differentiation (25, 26), we tested whether ectopic GATA2 interfered with these pathways. GATA2 blocked the BMP2-induced activation of a SMAD1/5/8 luciferase reporter (Fig. 3D) but had no discernible effect on the basal or LiCl-mediated activation of a Wnt reporter system (Fig. 3E). We then isolated Lin− Sca1+ MSCs from subcutaneous white adipose tissue (sqWAT) and ectopically expressed GATA2 in these primary cells (Fig. 3F). Affymetrix gene expression profiling showed that ectopic expression of GATA2 in undifferentiated cells regulated 805 of the 2,230 genes with nearby GATA2 binding (see Table S3 in the supplemental material), 41 of 102 (q [FDR-adjusted P value] < 0.05) genes defined by the GO terms related to skeletal system development (Fig. 3G; see Table S2 in the supplemental material), and several SMADs and their respective target genes (Fig. 3H). Since the majority of these genes were downregulated (n = 29 versus 12), GATA2 seems to function primarily as a repressor of genes involved in skeletal development/osteoblast differentiation. Moreover, GATA2 also potently inhibited osteoblastogenesis in primary MSCs, as shown by reduced ALPL and alizarin staining and lower expression of osteoblast marker genes after 8 days of differentiation (Fig. 3I and J). Taking the data together, GATA2 inhibits osteoblastogenesis of MSCs, at least in part, by interfering with genes related to skeletal development, such as BMP2-driven SMAD signaling.
FIG 3.
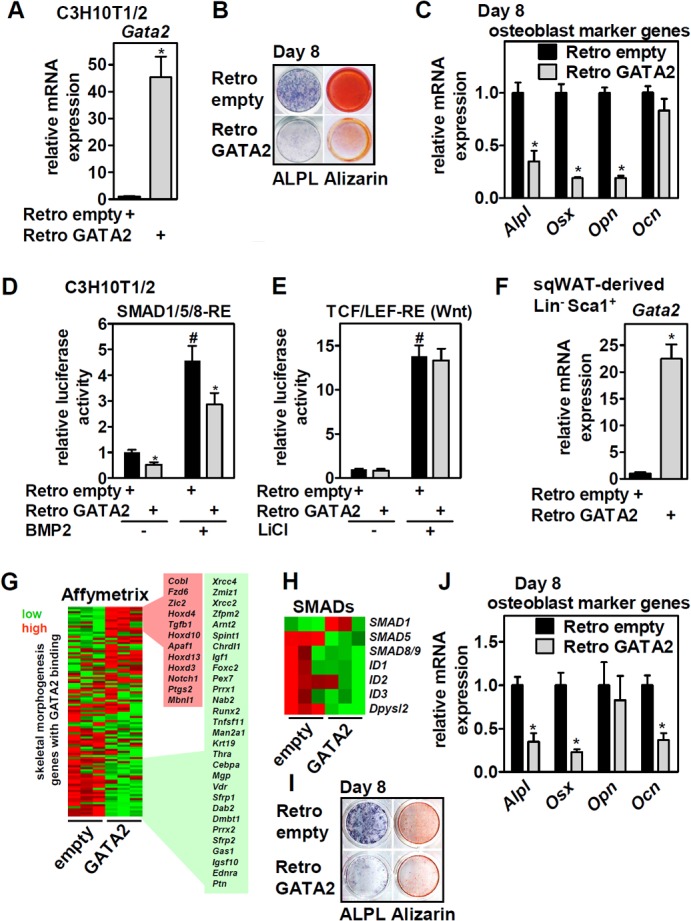
Ectopic expression of GATA2 in MSCs blocks osteoblast differentiation and interferes with BMP-driven SMAD activation. (A) C3H10T1/2 cells were infected with empty or GATA2-expressing retroviruses, and Gata2 mRNA levels were determined by qPCR; then, the cells were induced to differentiate into osteoblasts. (B and C) Eight days later, differentiation was assessed by ALPL staining and calcium precipitation with alizarin (B) and by the expression of osteoblast marker genes by qPCR (C). (D and E) Undifferentiated C3H10T1/2 cells shown in panel A were transfected with SMAD1/5/8-RE-driven (D) or TCF/LEF-RE-driven (E) reporter vectors and stimulated with 300 ng/ml BMP2 or 32 mM LiCl as indicated, and luciferase activity was determined. (F) sqWAT-derived Lin− Sca+ cells were infected as described for panel A, and Gata2 mRNA levels were determined. (G) Cells shown in panel F were analyzed by Affymetrix gene expression profiling. Expression of GATA2-bound genes (70 kb 5′ or 3′ of binding sites) mapped to GO skeletal morphogenesis- and development-related pathways (n = 102 genes) were visualized in a heat map. Gene symbols are given for genes up- or downregulated (n = 41; q < 0.05). (H) Heat map of selected genes involved in SMAD signaling. (I and J) Cells were induced to differentiate into osteoblasts, and 8 days later, differentiation was assessed by ALPL staining and calcium precipitation with alizarin (I) and the expression of osteoblast marker genes by qPCR (J). Data are presented as means and SD; *, P < 0.05 for Retro GATA2 versus Retro empty; #, P < 0.05 for BMP2 and LiCl treatment versus untreated Retro empty.
MSC-specific GATA2 deletion increases precursor cell numbers in bone and enhances in vitro osteoblastogenesis.
We then addressed the effects of GATA2 deficiency by crossing mice with floxed Gata2 alleles (27) with Prx1-Cre mice deleting GATA2 specifically in MSCs but not CD45+ hematopoietic or CD31+ endothelial cell populations (28, 29). This strategy allowed deletion in MSCs derived from bone and sqWAT but not in those from epididymal (eWAT) or brown adipose tissue (BAT) (Fig. 4A), consistent with previous reports on the Prx1-Cre line (30, 31). Notably, GATA2 deletion was not detectable when analyzing whole-tissue mRNA, presumably due to GATA2 expression in cell types that are not targeted by Prx1-Cre (data not shown). Loss of GATA2 in sqWAT MSCs was confirmed on the protein level (Fig. 4B) and did not induce an upregulation of other GATA family members as a compensatory response (Fig. 4C). We found that GATA2 deletion increased the number of bone-resident Lin− Sca1+ and Lin− Sca1− Pdgfra+ MSCs (Fig. 4D), precursor populations with high adipogenic and osteoblastogenic capacity, respectively (29). When assessed for in vitro osteoblast differentiation by alizarin staining and the expression of osteoblast marker genes, adipogenic Lin− Sca1+ cells lacking GATA2 showed striking enhancement of osteoblast differentiation (Fig. 4E and F), whereas there was no difference in the differentiation of Lin− Sca1− Pdgfra+ MSCs (Fig. 4G and H). Thus, deletion of GATA2 increases the numbers of bone-residing precursor cells and enhances the osteoblastic potential of Lin− Sca1+ precursors. Interestingly, and in contrast to bone, GATA2 deletion in sqWAT-resident MSCs did not affect Lin− Sca1+ or Lin− Sca1− cell numbers, sqWAT tissue mass, or in vitro adipogenesis of Lin− Sca1+ cells when assessed by Oil Red O lipid staining and the expression of adipocyte marker genes (data not shown).
FIG 4.
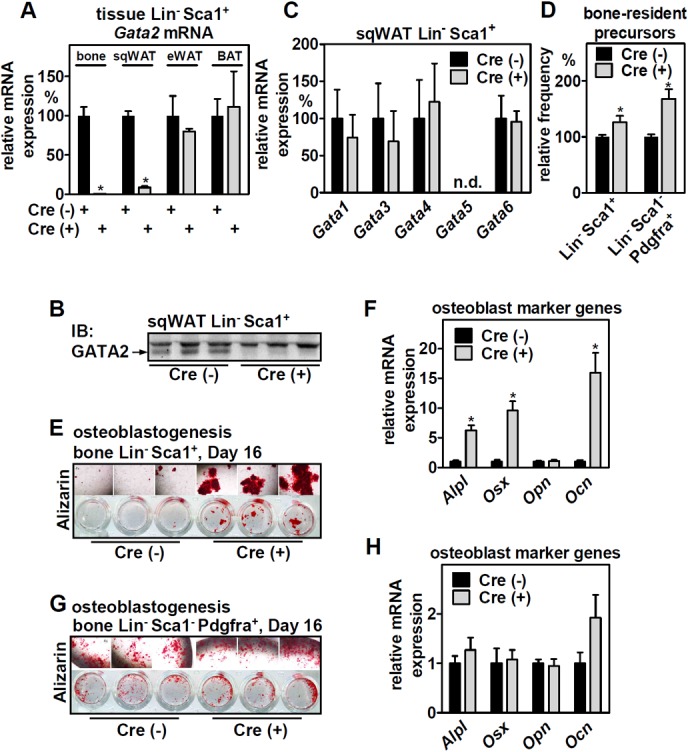
MSC-specific deletion of GATA2 in mice increases the number of bone-residing precursor cells and promotes their osteoblastogenic differentiation in vitro. (A) Lin− Sca1+ cells from various tissues of mesenchymal origin from Prx1-Cre-positive/negative mice with floxed Gata2 alleles were isolated and analyzed for expression of Gata2 by qPCR (n = 4 and 4). (B) Deletion of GATA2 protein in sqWAT-derived Lin− Sca1+ cells was validated by immunoblotting. (C) Lin− Sca1+ cells with or without Gata2 expression were analyzed for compensatory upregulation of other GATA factors by qPCR (n = 4 and 4). (D) Relative abundances of adipogenic (Lin− Sca1+) and osteoblastic (Lin− Sca1− Pdgfra+) precursors isolated from bones of Prx1-Cre-positive/negative mice with floxed Gata2 alleles (n = 14 and 11). (E and F) Ostoblastogenesis of Lin− Sca+ cells derived from bone was assessed by alizarin staining of calcium precipitation (E) and the expression of osteoblast marker genes by qPCR (F) (n = 3 and 3). (G and H) Ostoblastogenesis of Lin− Sca1+ Pdgfra+ cells derived from bone was assessed by alizarin staining of calcium precipitation (G) and the expression of osteoblast marker genes by qPCR (H) (n = 3 and 3). The data are presented as means and SD (means and SEM in panel D); *, P < 0.05 between Cre+ and Cre− cells or mice.
MSC-specific GATA2 deletion impairs trabecularization and mechanical strength of bone.
We next analyzed the bone structure of mice with MSC-specific GATA2 deletion and found no differences in the cartilage or bone morphogenesis of embryonic day 18.5 (E18.5) fetuses (Fig. 5A, top) or in growth plate morphology/height (Fig. 5A, bottom) and tibia length (17.6 ± 0.23 mm in Cre− versus 17.8 ± 0.14 mm in Cre+ mice; n = 4 and 4) at 3 months of age. However, micro–computed tomography (µCT) analyses revealed profound impairment of tibial trabecularization in the transitional section from the metaphysis to the diaphysis of 3-month-old mice (Fig. 5B), with bone surface and trabecula numbers strongly reduced (Fig. 5C). This observation was surprising and opposite to the phenotype we would have predicted from our findings regarding the increase in numbers and osteoblastic potential of bone-resident MSCs. Three-point bone bending, which depends predominantly on mid-diaphysis structure, showed that femora of aged mice with MSC-specific GATA2 deletion exhibited reduced cortical strength (Fig. 5D). To determine whether increased MSC numbers (Fig. 4D) could improve bone regeneration, we assessed the healing of a stabilized tibia fracture histomorphometrically but found no significant differences in mineralized and cartilaginous tissues between genotypes that would point to an altered healing process (data not shown). Hence, GATA2 expression in bone-resident MSCs is required for bone trabecularization and its cortical strength but is dispensable for the regeneration of fractured bone.
FIG 5.

MSC-specific deletion of GATA2 impairs trabecular bone structure and bone durability in aged mice. (A) (Top) E18.5 Prx1-Cre-positive/negative embryos with floxed Gata2 alleles were stained with alizarin red and alcian blue to assess mineralization and cartilage patterns. (Bottom) Alcian blue staining of longitudinal sections of proximal tibia growth plates and height quantifications from 3-month-old mice (n = 5 and 6). (B) μCT analyses of metaphysis (1), proximal diaphysis (2), and mid-diaphysis (3) of tibiae from 3-month-old mice. (C) Histomorphometric analysis of the medulary cavity of proximal diaphysis for bone surface and trabecula numbers in 3-month-old mice of the indicated genotypes (n = 6 and 6). (D) Three-point bending of tibiae from 3-month-old (n = 4 and 4) and 6-month-old (n = 7 and 4) mice of the indicated genotypes. The data are presented as means and SEM; *, P < 0.05 between Cre+ and Cre− mice. Scale bars: 2.5 mm (A, top), 100 μm (A, bottom), 1 mm (B, left), and 0.5 mm (B, right).
MSC-specific GATA2 deletion activates osteoclasts involving reduced osteoprotegerin and alters blood cell counts.
In order to identify the reason for impaired trabecularization in mice with MSC-specific GATA2 deletion, we first investigated bone anabolism and determined the mineral apposition rate (MAR) by calcein double staining. MSC-specific deletion of GATA2 increased the MAR in trabecular bone (Fig. 6A), whereas there was no effect in cortical bone (data not shown). This indicates that reduced trabecularization is unlikely to be due to reduced bone formation. We next assessed whether differentiation of GATA2-deficient bone-resident MSCs shifted toward adipocytes and found that in vitro adipogenesis of bone-derived Lin− Sca1+ cells was indeed enhanced (increased Oil Red staining and mRNA expression of the adipogenic marker genes Pparγ2 [18.7- ± 1.99-fold], Cebpa [6.8- ± 0.92-fold], and aP2 [33.5- ± 2.26-fold] in Cre+ compared to Cre− cells after the induction of differentiation). In contrast, hematoxylin and eosin (H&E) staining of bone sections and mRNA expression of adipocyte marker genes in bone failed to support a significant increase in bone marrow adipocytes (data not shown). Instead, bone mRNA expression of osteoclast marker genes and the number of osteoclasts determined to be tartrate-resistant acid phosphatase (TRAP)-positive, multinucleated cells was increased (Fig. 6B and C). This suggests that impaired trabecularization could be caused by an imbalance of catabolic over anabolic bone cells. Since GATA2 deletion in these mice is specific to the mesenchymal lineage, we analyzed known osteoblast-derived signals that affect osteoclast differentiation and activity. Expression levels of receptor activator of the nuclear factor kappa B ligand gene (Rankl) (32, 33) and the colony-stimulating factor 1 gene (Csf1) (34) were unchanged in bones of mice with GATA2 deleted, whereas that of Opg (35) was reduced by 50% (Fig. 6D). Since OPG is a decoy receptor for the osteoclast differentiation factor RANKL (36, 37), this gene expression pattern is consistent with the observed increase in osteoclasts. Thus, reduced trabecularization may be a consequence of lower expression of Opg and increased osteoclast differentiation. To investigate whether Opg is a direct transcriptional target of GATA2 in MSCs, we analyzed GATA2 binding near its genomic locus and found two binding sites at kb 13.0 and 75.4 upstream of its TSS in C3H10T1/2 cells but not in HSCs (Fig. 6E; validated by ChIP-qPCR [Fig. 6F]). Deleting GATA2 by adenoviral Cre expression reduced Opg expression in bone-derived Gata2 (flox/flox [fl/fl]) Lin− Sca1+ MSCs (Fig. 6G). A similar downregulation was observed in mouse embryonic fibroblasts (MEFs) and in Lin− Sca1+ MSCs derived from sqWAT in an adenoviral Cre dose- and time-dependent manner (data not shown), indicating that the regulation of Opg by GATA2 is cell autonomous and conserved between different mesenchymal cell types. To address whether altered morphology and metabolism of the trabecular bone of mice with MSC-specific GATA2 deletion affects hematopoiesis, we analyzed blood parameters. GATA2 deletion caused a slight reduction in red blood cell counts and hemoglobin levels, whereas there was no difference in the numbers of white blood cells and platelets (Fig. 6H).
FIG 6.

MSC-specific deletion of GATA2 affects bone turnover and blood cell counts. (A) Assessment of MAR by quantification of double-calcein-labeled trabecular sections of tibial bones of 3-month-old mice (n = 4 and 5). (B) Bone marrow-free femora of 6-month-old mice with floxed Gata2 alleles and the indicated genotypes were analyzed for osteoclast marker gene expression by qPCR (n = 3 and 3). (C) TRAP staining and quantification in proximal tibiae of 6-month-old mice of the indicated genotypes (n = 4 and 4). (D) mRNA expression of Rankl, Csf1, and Opg in the material shown in panel B was determined by qPCR. (E) GATA2 binding near the Tnfrsf11b (= Opg) gene locus in mesenchymal (C3H10T1/2) and hematopoietic (FDCPmix) cells shown in the UCSC Genome Browser. (F) ChIP-qPCR validation of GATA2 binding upstream (kb −13.0 and −75.4) of the Opg TSS in C3H10T1/2 cells. (G) Bone-derived Lin− Sca1+ precursor cells, isolated from mice with floxed Gata2 alleles, were infected with GFP- or Cre-expressing adenoviruses. Opg mRNA expression was determined 96 h later by qPCR. (H) Blood cell analysis of 6- to 9-month-old Prx1-Cre-positive/negative mice with floxed Gata2 alleles (n = 25 and 20). (A to D) The data are presented as means and SEM. (F and G) The data are presented as means and SD. *, P < 0.05. Scale bars: 25 μm (A) and 50 μm (C).
DISCUSSION
In this study, we provide the first genome-wide analysis of GATA2 binding sites in mesenchymal cells and identify a novel function of the transcription factor in establishing bone trabecularization. The most common sequences in genomic regions enriched by GATA2 resemble the canonical WGATAR motif, suggesting that GATA2 affects transcription in these cells predominantly via direct DNA binding rather than being tethered to other transcription factors, like C/EBPs (8), or as components of recently identified mega-transcription factor complexes (38). Binding sites in MSCs but not in HSCs were enriched near genes involved in skeletal system development and morphogenesis and colocalized with binding motifs of the FOX and HOX families of transcription factors. This suggests that certain lineage-determining factors pioneer MSC-specific GATA2 binding at short recognition sequences, such as the WGATAR motif, which occurs in the genome rather frequently. Since MSC-specific, but not HSC-specific, GATA2 binding sites associated strongly with H3K4me1 and H3K27ac in mesenchymal cells, chromatin accessibility could account for cell-type-specific binding.
Enrichment of binding sites near genes involved in skeletal system development and morphogenesis prompted us to explore GATA2's role during osteoblast differentiation. Indeed, we found that ectopic expression of GATA2 inhibited, whereas genetic deletion of GATA2 in Lin− Sca1+ precursor cells enhanced, osteoblast differentiation, at least in part by interfering with BMP-driven SMAD signaling. Besides osteoblast differentiation, MSC-specific GATA2 deletion also increased the number of mesenchymal precursor cells in bone and enhanced adipocyte differentiation of Lin− Sca1+ precursor cells in vitro. Thus, GATA2's function is to limit precursor cell numbers in bone and the osteoblastic/adipogenic differentiation of certain precursor populations. Intriguingly, deletion of GATA2 in sqWAT-residing precursor cells did not affect precursor cell numbers, their adipogenic differentiation, or sqWAT mass, suggesting that GATA2 is biologically more relevant to proliferation and differentiation of MSCs in bone, rather than those residing in WAT.
At first, we were surprised that MSC-specific deletion of GATA2, despite increased numbers of bone-residing precursor cells and enhanced osteoblast differentiation, led to reduced bone trabecularization and lower mechanical strength of bone. Bone formation within trabecular sections was increased but was accompanied by elevated numbers of osteoclasts, suggesting that anabolic, but also catabolic, processes were activated upon loss of GATA2 in MSCs. Our finding that GATA2 is required for full expression of the osteoclastogenesis inhibitor gene Opg in bone and MSCs, including bone-derived Lin− Sca1+ precursors, suggests that MSC-specific loss of GATA2 shifts the balance of anabolism versus catabolism within certain bone sections toward catabolism, thus interfering with trabecularization. This is further supported by the phenotype of Opg-deficient mice, which display similar but much more severe trabecular impairment and cortical bone porosity (39, 40). Since osteoclasts are derived from HSCs and GATA2-deficient osteoclast precursor cells exhibit reduced proliferation (41–43), GATA2 controls osteoclastogenesis, not only in a direct and cell-autonomous manner but also indirectly, via the regulation of Opg expression from cells of the mesenchymal lineage. Another interesting observation is that Opg deficiency in mice causes hearing loss (44), suggesting that deafness due to mutation of GATA2 in humans (3) could involve reduced OPG expression. Moreover, mutated OPG in humans can result in defective vestibular morphology (45), and malformations in similar structures have been found in GATA2-deficient mice (27). Notably, other signals of the osteoblast and osteoclast cross talk may be affected by loss of GATA2 in MSCs and thereby involved in the observed phenotype. Further research is needed to elucidate the contributions of these factors.
Our study is in accordance with some of the findings of Li et al. (46), who observed increased osteoblastogenesis of GATA2-deficient MSCs derived from bone. Mechanistically, this was attributed to increased Wnt/β-catenin signaling (46), a pathway whose activity we found not to be affected by ectopic GATA2 expression. Both Li et al. and Kamata et al. (5) observed reduced MSC proliferation upon loss of GATA2 or small interfering RNA (siRNA)-mediated depletion of GATA2 in human MSCs, respectively. In contrast, we found increased numbers of bone-residing precursor cells in mice with MSC-specific deletion of GATA2, suggesting that MSC proliferation may be influenced by the bone microenvironment. The most striking difference was in regard to reduced trabecularization, since Li et al. reported the opposite finding of greater bone mass and trabecularization upon the loss of GATA2 in MSCs, despite using the same mouse genetic model (46). The reason for this is currently unknown but may involve the spatial stratifications we applied to our μCT analyses. Moreover, Li et al. found increased bone marrow adiposity, whereas we and others (47) did not, which is another aspect that requires further studies to consolidate these contradictory results.
An intriguing notion concerns the expression of GATA2 in different developmental lineages and why GATA2, as one of the master regulators of HSC differentiation, controls mesenchymal precursors, especially in bone. A plausible hypothesis could derive from the greatly advanced understanding of the adult marrow HSC niche (48), where both hematopoietic and mesenchymal cells warrant HSC self-renewal, proliferation, and differentiation (49). GATA2 expression in HSCs is required for their expansion and the formation of mast cells, whereas GATA2 in MSCs may support development of the anatomical and cellular microenvironment of the niche. The slight reduction in red blood cell counts upon MSC-specific GATA2 deletion could be secondary to reduced bone trabecularization and an impaired HSC niche. Notably, accelerated osteoclastogenesis and osteoporosis in Opg-deficient mice were accompanied by reduced HSC mobilization and colony formation (50), supporting a link between higher osteoclast numbers/activity, the microenvironment of the niche, and blood cell formation. Support for a role of GATA2 comes from a study that identified compromised colony formation of hematopoietic progenitor cells from mice lacking GATA2 in MSCs and lower numbers of common myeloid progenitors after the transplantation of CD45+ cells into mice that lacked GATA2 in bone marrow (47). On the other hand, lower Opg expression upon GATA2 deletion in MSCs could directly hinder HSC expansion, since OPG was shown to enhance the expansion of hematopoietic progenitor cells in vitro (51). These observations warrant further research to complete our understanding of the interconnection of lineage-specific actions of GATA2.
In summary, we have identified genome-wide binding sites of GATA2 in mesenchymal cells and a novel function of GATA2 in bone trabecularization and mechanical strength, suggesting that GATA2 deficiency syndrome patients may be more vulnerable to bone fractures. Moreover, our study could imply a therapeutic potential for correcting an abnormal skeletal system and bone morphology to treat certain blood diseases.
MATERIALS AND METHODS
Mouse experiments.
All experimental-animal procedures were in accordance with institutional guidelines and were approved by the Landesamt für Gesundheit und Soziales in Berlin, Germany. Mice were housed under 12-h/12-h light/dark cycles and fed standard chow (diet R/M-H from ssniff, Germany). C57BL/6 females with floxed Gata2 alleles (27) were mated with male B6.Cg-Tg(Prrx1-cre)1Cjt/J mice (28) (Jackson Laboratory; stock no. 005584) for MSC deletion. Male mice were used for experiments.
Culture and differentiation of cell lines.
C3H10T1/2 and 3T3-L1 cells (ATCC) were cultured according to the provider's instructions. 3T3-L1 cells were differentiated to adipocytes by incubation with insulin, dexamethasone, and 3-isobutyl-1-methylxanthine, as previously described (52). C3H10T1/2 cells were differentiated to osteoblasts using 300 ng/ml BMP-2 (eBioscience), 10 mM β-glycerophosphate, and 50 μg/ml ascorbic acid (Sigma) for 8 days. Calcium phosphate and alkaline phosphatase staining was performed using specific reagents (Alizarin Red S [Sigma] and a BCIP [5-bromo-4-chloro-3-indolylphosphate]/nitroblue tetrazolium [NBT] kit [U.S. Biologicals]).
Isolation, FACS, culturing, and differentiation of primary cells.
Isolation and maintenance of sqWAT-derived MSCs were performed as described previously (53). In short, male C57Bl/6J mice aged 8 to 12 weeks were sacrificed, and sqWAT was isolated. The tissue was cut into small pieces, digested with collagenase type II (Sigma), and filtered through a 70-μm mesh to obtain sqWAT MSCs. The freshly isolated cells were seeded on 6-well plates and maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (all ThermoFisher), and 10 mM HEPES. The cells were cultivated for at least 2 passages before inducing differentiation. MSC identity was analyzed by fluorescence-activated cell sorting (FACS). For differentiation, cells were seeded in 24- or 48-well plates and grown to ∼80% confluence. Osteoblastogenesis was induced with 10 mM β-glycerophosphate, 50 μg/ml ascorbic acid, and 50 ng/ml BMP-2, and the culture medium was changed every other day. Bone-derived MSCs were isolated as previously described (29). In brief, soft-tissue-free bones (tibia/femur) were crushed and digested with collagenase type II (Cell Systems) for 1 h at 37°C with constant agitation. The digestion was stopped by adding sorting buffer (2% FBS–phosphate-buffered saline [PBS]). The suspension was filtered through a 70-μm mesh and centrifuged, and the pellet was resuspended in ammonium chloride-potassium lysis buffer to eliminate red blood cells. After an additional wash step, the cells were resuspended in sorting buffer and antibody labeled (see Table S4 in the supplemental material). Flow cytometry and cell sorting were conducted on a FACSAria III cell sorter (BD Biosciences) and analyzed using FlowJo software (Tree Star). Living cells were gated for lack of propidium iodide fluorescence and staining of calcein (Sigma). Compensation, fluorescence minus one control-based gating, and FACS isolation were conducted as described previously (54). The freshly isolated cells were maintained and differentiated to adipocytes or osteoblasts as described previously (29). Lipid accumulation was determined by Oil Red O staining (Sigma) and calcium deposition by 2% Alizarin Red S staining (Carl Roth). Mouse embryonic fibroblasts were isolated by removing the heads, limbs, and livers of E13.5 embryos. The remaining tissue was minced and homogenized using an 18 G syringe and seeded in culture flasks. Fibroblasts were grown and expanded in AlphaMEM (Sigma) supplemented with 10% FBS and 1% penicillin-streptomycin.
Retroviral and adenoviral infections.
The coding sequences of the murine GATA2 sequence were cloned downstream of a Kozak consensus sequence into a retroviral pMSCV vector containing a puromycin resistance cassette (TaKaRa Clontech) and verified by sequencing. EcoPack 2-293 cells (TaKaRa Clontech) were transfected with GATA2 or empty pMSCV vectors using Lipofectamine 2000 (ThermoFisher). The retroviral-particle-containing medium was harvested 48 h later. The supernatants were supplemented with 10 μg/ml Polybrene and used to infect preconfluent C3H10T1/2 cells or primary sqWAT-derived MSCs for 24 h; 48 h later, the cells were cultured in the presence of 1 μg/ml puromycin. Adenoviruses expressing green fluorescent protein (GFP) or Cre were prepared as described previously (55, 56), and equal titers (infectious units [IFU]) were used to infect MSCs (1.25E8 IFU/ml [Fig. 6G]) or MEFs (3.2E6 IFU/ml) overnight.
Transfections and luciferase reporter assays.
C3H10T1/2 cells were transfected with pGL4.49 (luc2P/TCF-LEF RE) (Promega) or pGL3-Id1-BRE (57) vectors using Lipofectamine 2000 (ThermoFisher). Sixteen to 20 h later, the cells were stimulated with LiCl (32 mM) or BMP-2 (300 ng/ml) for an additional 4 to 8 h, and luciferase activity was determined. Firefly luciferase activity was normalized to that of coexpressed renilla luciferase (Dual-Luciferase reporter assay; Promega).
ChIP.
ChIP analyses in 3T3-L1 and C3H10T1/2 cells were performed as described previously (56). Approximately 100 μg of sonicated chromatin extracts was used for each immunoprecipitation (IP) and incubated with 10 μg of a GATA2 antibody (sc-9008x; Santa Cruz) overnight. qPCRs were normalized to the amplification of a fragment of the insulin or 36B4 gene. The primers for ChIP-qPCR are listed in Table S5 in the supplemental material.
ChIP-seq and analyses.
Pooled DNA from three independent GATA2 ChIPs was used to generate libraries for deep sequencing on an Illumina HiSeq instrument. High-quality reads were removed if aligning to simple repeat regions, and reads were mapped to the mm10 mouse genome assembly. Peaks of 2 independent experiments were called using the MACS2 algorithm (58) with a minimum cutoff of 1% FDR and combined. Called peaks overlapping ENCODE blacklisted regions for mm10 (59) were removed, as well as peaks located in chromosome 10 (chr10) 106613366 to 107858706, chr10 116174799 to 118176364, or chrX 143482886 to 143483277, since these regions are amplified in 3T3-L1 cells. PhastCons-based average conservation profiles across peak regions (3,000 nucleotides [nt] around the peak center) were computed with the conservation_plot.py script from Tao Liu's open source libraries for bioinformatics (60). Peaks were mapped to neighboring genes within 70 kb 5′ or 3′ using the Bedtools windowBed function (61). Genome-wide localization of enriched peak regions was determined using the CEAS package (62). Motif searching was conducted using SeqPos (14). Gene ontology analysis of genes near the GATA2 binding site was performed using DAVID (63, 64). A previously published GATA2 ChIP-seq data set from murine hematopoietic progenitor cells (20) was analyzed similarly. Overlapping peaks were identified using intersectBed from Bedtools (65). DeepTools (66) was used to generate heat maps and signal profiles of peak regions (center ± 2 kb). H3K4me1 and H3K27ac ChIP-seq data were obtained from GEO (GSE95533) (67). DHS data were obtained from GEO (GSE27826) (68).
Blood analysis.
Complete blood cell counts were performed with peripheral blood samples obtained by facial vein or heart puncture from 6- to 9-month-old male mice, immediately stored in tubes containing K3-EDTA anticoagulant (Sarstedt), and analyzed on an XS-800i hematology analyzer (Sysmex) on the same day. A Grubbs' test-based outlier analysis was performed using online outlier calculator software (GraphPad Prism), and values with a significance level (α) of <0.05 were excluded.
Protein isolation and immunoblotting.
Cellular proteins were isolated in radioimmunoprecipitation assay (RIPA) buffer and separated by SDS-PAGE. After incubation with primary antibodies for GATA2 (sc-9008 [Santa Cruz] or 4595 [Cell Signaling]), ZFPM1 (sc-9361; Santa Cruz), or RAN (BD Biosciences), a secondary horseradish peroxidase-conjugated antibody was added, and a chemiluminescent substrate kit (Roche) was used for detection.
Isolation of RNA and qPCR.
RNA was purified using spin column kits (Qiagen or Macherey-Nagel). cDNA was generated using the Sprint Powerscript system (Clontech) or Moloney murine leukemia virus (MMLV) reverse transcriptase (RT) (Promega). qPCR was carried out using Sybr green PCR Mastermix (Eurogentec) and evaluated according to the standard-curve method. All RNA expression data were normalized to 36B4 (RPLP0).
Affymetrix microarray and heat maps.
Affymetrix microarray analysis (Mouse Gene 1.1ST and the GeneTitan system) and statistical evaluation were performed at the Nutrigenomics technology platform of the University of Wageningen, Wageningen, Netherlands. Row-normalized heat maps were generated with Heatmap Builder (69).
Whole-mount embryo staining and histology.
Staining was performed according to a published method (70) with minor modifications. Pregnant females were sacrificed at E18.5. Embryos were skinned and eviscerated, fixed in 100% ethanol (EtOH) for 6 h, stained in 150 mg/liter alcian blue 8 GX (Sigma) for up to 20 h, and incubated in 100% EtOH overnight. Initial clearing was conducted by incubating the embryos in 2% KOH for 6 h. Follow-up staining of calcified tissue was done in 50 mg/liter Alizarin Red S (Sigma) in 2% KOH for 3 h. The embryos were sequentially cleared in 2% to 0.2% KOH and stored in 100% glycerol. For histology, limbs from 10- to 12-week- or 6-month-old mice were fixed in 4% paraformaldehyde (PFA), decalcified with Osteosoft (Merck), and paraffinized. Sections (1.5 to 3 μm) were stained with alcian blue 8GX and nuclear fast red (Sigma) as a counterstain for growth plate analysis, or with Naphthol AS-MX phosphate (Sigma) and hemalum as a counterstain for TRAP. Osteoclast numbers were determined by counting multinucleated (n ≥ 3) TRAP-positive cells in the proximal tibia using a Keyence BZ-9000 microscope and ImageJ software. Growth plate height analysis was performed by calculating the mean height from 100 randomly assigned perpendicular distances between the resting and hypertrophic cartilage zones across the complete growth plate in ImageJ.
Bone metabolic rate analysis.
For dynamic histomorphometry analysis, 10-week-old male Gata2 (fl/fl) Prx1-Cre-negative/positive mice were injected intraperitoneally with 30 μg calcein/g mouse body weight at two time points (9 and 2 days before sacrifice). Tibiae were collected, cleared of soft tissue, and fixed in 4% PFA for 2 days. Samples were washed and dehydrated gradually in 70%, 80%, 90%, and 100% EtOH over a period of 12 days. A Technovit 9100 system (Kulzer) was used for infiltration and polymerization. Calcein double labeling was captured with a Keyence BZ-9000 fluorescence microscope and analyzed with an ImageJ macro (71). Exclusion criteria were predefined as lack of two distinguishable fluorescent calcein labels. The MAR was calculated according to international standards (72).
Bone μCT and analyses.
Bone μCT analysis was adapted from our previously used method (73). Dissected hind limbs of 10- to 12-week-old or 8-month-old male mice were pruned of soft tissue and scanned on a VivaCT 40 platform (Scanco Medical AG) using a voxel size of 10.5 μm, an exposure value of 70 kV, an intensity value of 114 μA, and an integration time of 381 ms. Histomorphometric analysis for metaphysis and proximal and middiaphysis regions was performed using Scanco Medical software and the BoneJ plugin (74) for ImageJ software.
Biomechanical bone testing.
Biomechanical whole-bone strength was studied in femora from 10- to 12-week- and 6-month-old male mice in destructive three-point bending experiments using an LM 1 ElectroForce Test Bench (Bose). Explanted femora were mounted anterior side up for bending tests at an 8.5-mm span width between the end supports. The load was applied to the anterior midshaft of the femur at a constant deflection rate of 0.1 mm/s. Load (50 lb/225 N load cell) and displacement data were sampled at 100 Hz. Stiffness, maximum load, load to failure, and deflection at failure at fracture were calculated from the force-deflection curve using a routine written in MATLAB (Mathworks, Inc.).
Bone fracture-healing model.
The fracture-healing model was performed as described previously (29). Briefly, 8-month-old male mice were given an analgesic (MediGel; ClearH2O) starting 2 days prior to surgery. On the day of surgery, the mice were anesthetized, and a steel pin (diameter, 0.35 mm) was inserted into the medullary cavity through a small cutaneous incision at the knee joint for stabilization, followed by fracture induction with scissors 0.5 cm distal from the knee. Fourteen days after fracture induction, tibiae were harvested for analysis. After removal of the pin, the extracted tibiae were fixed, and a μCT analysis was conducted. Subsequently, the tibiae were decalcified, followed by paraffin embedding and sectioning at 3 mm per slice. Samples were stained using SafraninO/Fast green and Movat Pentachrome. ImageJ software was used for computer-assisted histomorphometric analysis of fracture calluses. Six representative sections of each callus were analyzed for bone, fibrous, and cartilaginous tissue areas in a blinded manner.
Statistical analysis.
Significance was determined by 2-tailed Student's t test or analysis of variance (ANOVA), as appropriate, and P values of <0.05 were deemed significant. Representative results of at least three independent cell culture experiments are shown and are presented as means and standard deviations (SD). Mouse data are presented as means and standard errors of the mean [SEM].
Accession number(s).
GATA2 ChIP-seq data for 3T3-L1 and C3H10T1/2 cells and Affymetrix microarray data for primary cells overexpressing GATA2 are available in the GEO database under accession number GSE101592.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Einstein Foundation Berlin (grant no. A-2011-83 to M. Schupp and S. Sauer), by a Career Integration Grant from the European Commission (CIG 291867), by the German Research Foundation (DFG) (Emmy Noether grant SCHU 2546/1-1), by the Deutsche Diabetes Stiftung (grant no. 280-12-10 to M. Schupp), and by a DynAge Focus Area grant to M. Schupp and T. J. Schulz.
We thank Mathias Treier (Max-Delbrück Center, Berlin Buch) for sharing Prx1-Cre mice and Christian Freise (Charité) for sharing WNT reporter constructs. We thank Dag Wulsten and Britt Wildemann (Berlin-Brandenburger Centrum für Regenerative Therapien and Julius Wolff Institut, Charité Universitätsmedizin Berlin) for the biomechanical characterization of bone and helpful comments on bone histology, respectively. pGL3 BRE luciferase was a gift from Martine Roussel and Peter ten Dijke (Addgene plasmid 45126). We thank Jan Tuckermann and colleagues (University of Ulm) for methodological help on bone characterization. This study was initiated in the laboratory of Mitch Lazar (Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA), and we are deeply grateful for his support.
A. Tolkachov and M. Schupp conceived and designed experiments. A. Tolkachov, C. Fischer, T. H. Ambrosi, M. Bothe, C.-T. Han, M. Muenzner, S. Mathia, M. Thiele, S. H. Meijsing, S. Sauer, T. J. Schulz, and M. Schupp performed experiments and/or analyzed data. M. Salminen provided a genetic mouse model, and G. Seifert assisted with blood analyses. G. N. Duda assisted with bone characterization. A. Tolkachov, C. Fischer, T. H. Ambrosi, S. H. Meijsing, S. Sauer, T. J. Schulz, and M. Schupp wrote and edited the manuscript.
Charité-Universitätsmedizin Berlin is a corporate member of Freie Universität Berlin, Berlin, Germany.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00599-17.
REFERENCES
- 1.Tsai FY, Keller G, Kuo FC, Weiss M, Chen J, Rosenblatt M, Alt FW, Orkin SH. 1994. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 371:221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- 2.Tsai FY, Orkin SH. 1997. Transcription factor GATA-2 is required for proliferation/survival of early hematopoietic cells and mast cell formation, but not for erythroid and myeloid terminal differentiation. Blood 89:3636–3643. [PubMed] [Google Scholar]
- 3.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, Arthur DC, Gu W, Gould CM, Brewer CC, Cowen EW, Freeman AF, Olivier KN, Uzel G, Zelazny AM, Daub JR, Spalding CD, Claypool RJ, Giri NK, Alter BP, Mace EM, Orange JS, Cuellar-Rodriguez J, Hickstein DD, Holland SM. 2014. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood 123:809–821. doi: 10.1182/blood-2013-07-515528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crispino JD, Horwitz MS. 2017. GATA factor mutations in hematologic disease. Blood 129:2103–2110. doi: 10.1182/blood-2016-09-687889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamata M, Okitsu Y, Fujiwara T, Kanehira M, Nakajima S, Takahashi T, Inoue A, Fukuhara N, Onishi Y, Ishizawa K, Shimizu R, Yamamoto M, Harigae H. 2014. GATA2 regulates differentiation of bone marrow-derived mesenchymal stem cells. Haematologica 99:1686–1696. doi: 10.3324/haematol.2014.105692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tong Q, Dalgin G, Xu H, Ting CN, Leiden JM, Hotamisligil GS. 2000. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science 290:134–138. doi: 10.1126/science.290.5489.134. [DOI] [PubMed] [Google Scholar]
- 7.Tsai J, Tong Q, Tan G, Chang AN, Orkin SH, Hotamisligil GS. 2005. The transcription factor GATA2 regulates differentiation of brown adipocytes. EMBO Rep 6:879–884. doi: 10.1038/sj.embor.7400490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong Q, Tsai J, Tan G, Dalgin G, Hotamisligil GS. 2005. Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol Cell Biol 25:706–715. doi: 10.1128/MCB.25.2.706-715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantor AB, Orkin SH. 2005. Coregulation of GATA factors by the Friend of GATA (FOG) family of multitype zinc finger proteins. Semin Cell Dev Biol 16:117–128. doi: 10.1016/j.semcdb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, Blobel GA, Chodosh LA, Weiss MJ. 2004. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 104:3136–3147. doi: 10.1182/blood-2004-04-1603. [DOI] [PubMed] [Google Scholar]
- 11.Green H, Meuth M. 1974. An established pre-adipose cell line and its differentiation in culture. Cell 3:127–133. doi: 10.1016/0092-8674(74)90116-0. [DOI] [PubMed] [Google Scholar]
- 12.Schupp M, Cristancho AG, Lefterova MI, Hanniman EA, Briggs ER, Steger DJ, Qatanani M, Curtin JC, Schug J, Ochsner SA, McKenna NJ, Lazar MA. 2009. Re-expression of GATA2 cooperates with peroxisome proliferator-activated receptor-gamma depletion to revert the adipocyte phenotype. J Biol Chem 284:9458–9464. doi: 10.1074/jbc.M809498200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jack BH, Crossley M. 2010. GATA proteins work together with friend of GATA (FOG) and C-terminal binding protein (CTBP) co-regulators to control adipogenesis. J Biol Chem 285:32405–32414. doi: 10.1074/jbc.M110.141317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, Mieczkowski P, Lieb JD, Zhao K, Brown M, Liu XS. 2010. Nucleosome dynamics define transcriptional enhancers. Nat Genet 42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin DI, Orkin SH. 1990. Transcriptional activation and DNA binding by the erythroid factor GF-1/NF-E1/Eryf 1. Genes Dev 4:1886–1898. doi: 10.1101/gad.4.11.1886. [DOI] [PubMed] [Google Scholar]
- 16.Fujiwara T, O'Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA, Choi K, Farnham PJ, Bresnick EH. 2009. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol Cell 36:667–681. doi: 10.1016/j.molcel.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans T, Reitman M, Felsenfeld G. 1988. An erythrocyte-specific DNA-binding factor recognizes a regulatory sequence common to all chicken globin genes. Proc Natl Acad Sci U S A 85:5976–5980. doi: 10.1073/pnas.85.16.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wadman IA, Osada H, Grutz GG, Agulnick AD, Westphal H, Forster A, Rabbitts TH. 1997. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J 16:3145–3157. doi: 10.1093/emboj/16.11.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.May G, Soneji S, Tipping AJ, Teles J, McGowan SJ, Wu M, Guo Y, Fugazza C, Brown J, Karlsson G, Pina C, Olariu V, Taylor S, Tenen DG, Peterson C, Enver T. 2013. Dynamic analysis of gene expression and genome-wide transcription factor binding during lineage specification of multipotent progenitors. Cell Stem Cell 13:754–768. doi: 10.1016/j.stem.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spooncer E, Heyworth CM, Dunn A, Dexter TM. 1986. Self-renewal and differentiation of interleukin-3-dependent multipotent stem cells are modulated by stromal cells and serum factors. Differentiation 31:111–118. doi: 10.1111/j.1432-0436.1986.tb00391.x. [DOI] [PubMed] [Google Scholar]
- 22.Rux DR, Wellik DM. 2017. Hox genes in the adult skeleton: novel functions beyond embryonic development. Dev Dyn 246:310–317. doi: 10.1002/dvdy.24482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kam MK, Lui VC. 2015. Roles of Hoxb5 in the development of vagal and trunk neural crest cells. Dev Growth Differ 57:158–168. doi: 10.1111/dgd.12199. [DOI] [PubMed] [Google Scholar]
- 24.Reznikoff CA, Brankow DW, Heidelberger C. 1973. Establishment and characterization of a cloned line of C3H mouse embryo cells sensitive to postconfluence inhibition of division. Cancer Res 33:3231–3238. [PubMed] [Google Scholar]
- 25.Wu M, Chen G, Li YP. 2016. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 4:16009. doi: 10.1038/boneres.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karsenty G. 2008. Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet 9:183–196. doi: 10.1146/annurev.genom.9.081307.164437. [DOI] [PubMed] [Google Scholar]
- 27.Haugas M, Lillevali K, Hakanen J, Salminen M. 2010. Gata2 is required for the development of inner ear semicircular ducts and the surrounding perilymphatic space. Dev Dyn 239:2452–2469. doi: 10.1002/dvdy.22373. [DOI] [PubMed] [Google Scholar]
- 28.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. 2002. Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 29.Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank AM, Bocian C, Woelk L, Fan H, Logan DW, Schurmann A, Saraiva LR, Schulz TJ. 2017. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell 20:771–784. doi: 10.1016/j.stem.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez-Gurmaches J, Hsiao WY, Guertin DA. 2015. Highly selective in vivo labeling of subcutaneous white adipocyte precursors with Prx1-Cre. Stem Cell Reports 4:541–550. doi: 10.1016/j.stemcr.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krueger KC, Costa MJ, Du H, Feldman BJ. 2014. Characterization of Cre recombinase activity for in vivo targeting of adipocyte precursor cells. Stem Cell Reports 3:1147–1158. doi: 10.1016/j.stemcr.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. 1998. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A 95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM. 1999. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi N, Udagawa N, Akatsu T, Tanaka H, Shionome M, Suda T. 1991. Role of colony-stimulating factors in osteoclast development. J Bone Miner Res 6:977–985. doi: 10.1002/jbmr.5650060912. [DOI] [PubMed] [Google Scholar]
- 35.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. 1997. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319. doi: 10.1016/S0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 36.Wada T, Nakashima T, Hiroshi N, Penninger JM. 2006. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med 12:17–25. doi: 10.1016/j.molmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 37.Horowitz MC, Xi Y, Wilson K, Kacena MA. 2001. Control of osteoclastogenesis and bone resorption by members of the TNF family of receptors and ligands. Cytokine Growth Factor Rev 12:9–18. doi: 10.1016/S1359-6101(00)00030-7. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman MJ, Song X, Zhang F, Ma Q, Ohgi KA, Krones A, Rosenfeld MG. 2014. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 159:358–373. doi: 10.1016/j.cell.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS. 1998. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meijome TE, Hooker RA, Cheng YH, Walker W, Horowitz MC, Fuchs RK, Kacena MA. 2015. GATA-1 deficiency rescues trabecular but not cortical bone in OPG deficient mice. J Cell Physiol 230:783–790. doi: 10.1002/jcp.24803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamane T, Kunisada T, Yamazaki H, Nakano T, Orkin SH, Hayashi SI. 2000. Sequential requirements for SCL/tal-1, GATA-2, macrophage colony-stimulating factor, and osteoclast differentiation factor/osteoprotegerin ligand in osteoclast development. Exp Hematol 28:833–840. doi: 10.1016/S0301-472X(00)00175-2. [DOI] [PubMed] [Google Scholar]
- 42.Wei W, Zeve D, Wang X, Du Y, Tang W, Dechow PC, Graff JM, Wan Y. 2011. Osteoclast progenitors reside in the peroxisome proliferator-activated receptor gamma-expressing bone marrow cell population. Mol Cell Biol 31:4692–4705. doi: 10.1128/MCB.05979-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei W, Zeve D, Suh JM, Wang X, Du Y, Zerwekh JE, Dechow PC, Graff JM, Wan Y. 2011. Biphasic and dosage-dependent regulation of osteoclastogenesis by beta-catenin. Mol Cell Biol 31:4706–4719. doi: 10.1128/MCB.05980-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kao SY, Kempfle JS, Jensen JB, Perez-Fernandez D, Lysaght AC, Edge AS, Stankovic KM. 2013. Loss of osteoprotegerin expression in the inner ear causes degeneration of the cochlear nerve and sensorineural hearing loss. Neurobiol Dis 56:25–33. doi: 10.1016/j.nbd.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grasemann C, Unger N, Hovel M, Arweiler-Harbeck D, Herrmann R, Schundeln MM, Muller O, Schweiger B, Lausch E, Meissner T, Kiewert C, Hauffa BP, Shaw NJ. 2017. Loss of functional osteoprotegerin: more than a skeletal problem. J Clin Endocrinol Metab 102:210–219. doi: 10.1210/jc.2016-2905. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Huynh H, Zuo H, Salminen M, Wan Y. 2016. Gata2 is a rheostat for mesenchymal stem cell fate in male mice. Endocrinology 157:1021–1028. doi: 10.1210/en.2015-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasegawa S, Fujiwara T, Okitsu Y, Kato H, Sato Y, Fukuhara N, Onishi Y, Shimizu R, Yamamoto M, Harigae H. 2017. Effects of in vivo deletion of GATA2 in bone marrow stromal cells. Exp Hematol 56:31–45. doi: 10.1016/j.exphem.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 48.Boulais PE, Frenette PS. 2015. Making sense of hematopoietic stem cell niches. Blood 125:2621–2629. doi: 10.1182/blood-2014-09-570192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kacena MA, Gundberg CM, Horowitz MC. 2006. A reciprocal regulatory interaction between megakaryocytes, bone cells, and hematopoietic stem cells. Bone 39:978–984. doi: 10.1016/j.bone.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 50.Miyamoto K, Yoshida S, Kawasumi M, Hashimoto K, Kimura T, Sato Y, Kobayashi T, Miyauchi Y, Hoshi H, Iwasaki R, Miyamoto H, Hao W, Morioka H, Chiba K, Yasuda H, Penninger JM, Toyama Y, Suda T, Miyamoto T. 2011. Osteoclasts are dispensable for hematopoietic stem cell maintenance and mobilization. J Exp Med 208:2175–2181. doi: 10.1084/jem.20101890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schweikle E, Baessler T, Yildirim S, Kanz L, Mohle R, Weisel KC. 2012. Osteoprotegerin positively regulates hematopoietic progenitor cells. Curr Stem Cell Res Ther 7:72–77. doi: 10.2174/157488812798483458. [DOI] [PubMed] [Google Scholar]
- 52.Witte N, Muenzner M, Rietscher J, Knauer M, Heidenreich S, Nuotio-Antar AM, Graef FA, Fedders R, Tolkachov A, Goehring I, Schupp M. 2015. The glucose sensor ChREBP links de novo lipogenesis to PPARgamma activity and adipocyte differentiation. Endocrinology 156:4008–4019. doi: 10.1210/EN.2015-1209. [DOI] [PubMed] [Google Scholar]
- 53.Siersbaek MS, Loft A, Aagaard MM, Nielsen R, Schmidt SF, Petrovic N, Nedergaard J, Mandrup S. 2012. Genome-wide profiling of peroxisome proliferator-activated receptor gamma in primary epididymal, inguinal, and brown adipocytes reveals depot-selective binding correlated with gene expression. Mol Cell Biol 32:3452–3463. doi: 10.1128/MCB.00526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schulz TJ, Huang TL, Tran TT, Zhang H, Townsend KL, Shadrach JL, Cerletti M, McDougall LE, Giorgadze N, Tchkonia T, Schrier D, Falb D, Kirkland JL, Wagers AJ, Tseng YH. 2011. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc Natl Acad Sci U S A 108:143–148. doi: 10.1073/pnas.1010929108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muenzner M, Tuvia N, Deutschmann C, Witte N, Tolkachov A, Valai A, Henze A, Sander LE, Raila J, Schupp M. 2013. Retinol-binding protein 4 and its membrane receptor STRA6 control adipogenesis by regulating cellular retinoid homeostasis and retinoic acid receptor alpha activity. Mol Cell Biol 33:4068–4082. doi: 10.1128/MCB.00221-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schupp M, Lefterova MI, Janke J, Leitner K, Cristancho AG, Mullican SE, Qatanani M, Szwergold N, Steger DJ, Curtin JC, Kim RJ, Suh M, Albert MR, Engeli S, Gudas LJ, Lazar MA. 2009. Retinol saturase promotes adipogenesis and is downregulated in obesity. Proc Natl Acad Sci U S A 106:1105–1110. doi: 10.1073/pnas.0812065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Korchynskyi O, ten Dijke P. 2002. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem 277:4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shin H, Liu T, Duan X, Zhang Y, Liu XS. 2013. Computational methodology for ChIP-seq analysis. Quant Biol 1:54–70. doi: 10.1007/s40484-013-0006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shin H, Liu T, Manrai AK, Liu XS. 2009. CEAS: cis-regulatory element annotation system. Bioinformatics 25:2605–2606. doi: 10.1093/bioinformatics/btp479. [DOI] [PubMed] [Google Scholar]
- 63.Huang Da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 64.Huang Da W, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinlan AR. 2014. BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics 47:11.12.1–11.12.34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. 2014. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Siersbaek R, Madsen JGS, Javierre BM, Nielsen R, Bagge EK, Cairns J, Wingett SW, Traynor S, Spivakov M, Fraser P, Mandrup S. 2017. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Mol Cell 66:420–435. doi: 10.1016/j.molcel.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 68.Siersbaek R, Nielsen R, John S, Sung MH, Baek S, Loft A, Hager GL, Mandrup S. 2011. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J 30:1459–1472. doi: 10.1038/emboj.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.King JY, Ferrara R, Tabibiazar R, Spin JM, Chen MM, Kuchinsky A, Vailaya A, Kincaid R, Tsalenko A, Deng DX, Connolly A, Zhang P, Yang E, Watt C, Yakhini Z, Ben-Dor A, Adler A, Bruhn L, Tsao P, Quertermous T, Ashley EA. 2005. Pathway analysis of coronary atherosclerosis. Physiol Genomics 23:103–118. doi: 10.1152/physiolgenomics.00101.2005. [DOI] [PubMed] [Google Scholar]
- 70.Mallo M, Brandlin I. 1997. Segmental identity can change independently in the hindbrain and rhombencephalic neural crest. Dev Dyn 210:146–156. doi:. [DOI] [PubMed] [Google Scholar]
- 71.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM. 2013. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 28:2–17. doi: 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Willie BM, Birkhold AI, Razi H, Thiele T, Aido M, Kruck B, Schill A, Checa S, Main RP, Duda GN. 2013. Diminished response to in vivo mechanical loading in trabecular and not cortical bone in adulthood of female C57Bl/6 mice coincides with a reduction in deformation to load. Bone 55:335–346. doi: 10.1016/j.bone.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 74.Doube M, Klosowski MM, Arganda-Carreras I, Cordelieres FP, Dougherty RP, Jackson JS, Schmid B, Hutchinson JR, Shefelbine SJ. 2010. BoneJ: free and extensible bone image analysis in ImageJ. Bone 47:1076–1079. doi: 10.1016/j.bone.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.