

ABSTRACT
The RNA-binding motif 4 (RBM4) protein participates in cell differentiation via its role in regulating the expression of tissue-specific or developmentally regulated mRNA splice isoforms. RBM4 is expressed in embryonic brain during development; it is initially enriched in the ventricular zone/subventricular zone and subsequently distributed throughout the cerebral cortex. Rbm4a knockout brain exhibited delayed migration of late-born neurons. Using in utero electroporation, we confirmed that knockdown of RBM4 impaired cortical neuronal migration. RNA immunoprecipitation with high-throughput sequencing identified Disabled-1 (Dab1), which encodes a critical reelin signaling adaptor, as a potential target of RBM4. Rbm4a knockout embryonic brain showed altered Dab1 isoform ratios. Overexpression of RBM4 promoted the inclusion of Dab1 exons 7 and 8 (7/8), whereas its antagonist polypyrimidine tract-binding protein 1 (PTBP1) acted in an opposite manner. RBM4 directly counteracted the effect of PTBP1 on exon 7/8 selection. Finally, we showed that the full-length Dab1, but not exon 7/8-truncated Dab1, rescued neuronal migration defects in RBM4-depleted neurons, indicating that RBM4 plays a role in neuronal migration via modulating the expression of Dab1 splice isoforms. Our findings imply that RBM4 is necessary during brain development and that its deficiency may lead to developmental brain abnormality.
KEYWORDS: alternative splicing, cortex development, neuronal migration, Dab1
INTRODUCTION
Alternative splicing of precursor mRNAs (pre-mRNAs) generates multiple transcripts from a single gene and thereby greatly increases protein diversity. Alternative splicing can produce functionally distinct or cell-type-specific protein isoforms and can also modulate gene expression at the posttranscriptional level. Alternative splicing is prevalent in the mammalian brain and contributes significantly to brain development and functional complexity of the nervous system (1–3). Aberrant splicing of certain pre-mRNAs can impair neural development and cause neurological diseases. A number of RNA-binding proteins, e.g., Nova, Rbfox, nSR100/Srrm4, and polypyrimidine tract-binding proteins (PTBPs), are involved in splicing regulation during brain development (4). For example, nSR100/Srrm4 is important for development of the central and peripheral nervous systems by promoting cortical layer formation and neurite outgrowth (5). Rbfox2 is required for proper radial migration of Purkinje cells during cerebellar development as well as for the functional maturation of neurons (6, 7).
The protein RNA-binding motif 4 (RBM4) is a splicing regulator with binding preference for CU-rich sequences (8, 9, 10, 11). RBM4 regulates alternative splicing programs that impact differentiation of various cell types (8, 9, 12). RBM4 is expressed in most human adult tissues, and it has also been detected in the cortical plate of the mouse embryonic brain (13). RBM4 level is reduced in patients affected with fetal Down syndrome (14). We previously reported that RBM4 modulates alternative splicing of the Notch signaling inhibitor Numb and that RBM4-induced Numb isoforms potentiate the expression of the proneurogenesis gene Mash1 and hence promotes differentiation of mouse neuronal progenitor cells (15). In addition, RBM4 modulates the splice isoform switch of pyruvate kinase M (PKM) and thereby elevates mitochondrial oxidative phosphorylation and facilitates neuronal differentiation of mesenchymal stem cells (10). Mammalian genomes have two adjacent copies of Rbm4 (Rbm4a and Rbm4b) (8). Here, by using Rbm4a knockout (KO) mice, we observed aberrant distribution of certain cortical layer markers in mouse embryonic brain (see below). This finding prompted us to characterize the mechanism by which RBM4 modulates neuronal migration.
The mammalian neocortex comprises six layers, or laminae, during brain development. A remarkable feature of the neocortex is the birth date-dependent “inside-out” arrangement of projection neurons (16). Reelin secreted from Cajal-Retzius cells binds the cell surface receptors apolipoprotein E receptor 2 (ApoER2) and very-low-density lipoprotein receptor (VLDLR) and rapidly induces phosphorylation of Disabled-1 (Dab1) at multiple tyrosine residues in radially migrating neurons (17). Phosphorylated Dab1 subsequently modulates cell adhesion and cytoskeletal rearrangement that direct neuronal migration in the cortex (17). Intriguingly, proteasomal degradation of phosphorylated Dab1 is essential for termination of reelin-mediated signaling, indicating the importance of the dynamic control of Dab1 phosphorylation during cortical lamination. Thus, the reelin-Dab1 signaling pathway plays a vital role in neuronal positioning during brain development (18, 19). Reelin or Dab1 mutant mice exhibit defects in cortical layer formation (1, 20); dysregulated reelin expression is linked to brain disorders such as schizophrenia, lissencephaly, and epilepsy as well as neurodegenerative diseases (21).
Multiple splice isoforms of Dab1 are expressed during brain development. Notably, alternatively spliced exons 7 and 8 (7/8) of Dab1 encode several tyrosine residues that are subject to differential phosphorylation by reelin-induced signaling kinases. Exogenous expression of Dab1 isoforms lacking exon 7/8 results in neuronal migration defects with attenuated Dab1 tyrosine phosphorylation (22). Previous reports have shown that Nova2 deficiency results in aberrant inclusion of Dab1 exons 9b/c, which may also modulate the activity of tyrosine motifs, and hence causes neuronal migration defects (22). Nevertheless, the mechanism underlying the alternative selection of exons 7/8 remains unclear.
Here, we describe the role of RBM4 in neuronal migration. To explore the mechanism, we searched for RBM4 target transcripts in the mouse embryonic brain. The observed association of RBM4 with Dab1 transcripts prompted us to investigate how RBM4 regulates Dab1 pre-mRNA splicing and whether RBM4 contributes to neuronal migration via Dab1.
RESULTS
Expression of RBM4 in the developing cortex.
To characterize RBM4 involvement in cortical development, we carried out immunostaining for RBM4 in mouse coronal embryonic brain sections. The result revealed ubiquitous expression of RBM4 in the neocortex with enrichment in the ventricular zone (VZ) and subventricular zone (SVZ) at embryonic day 12.5 (E12.5) (Fig. 1A). At E18.5, RBM4 was more diffusely distributed in the cortical plate (Fig. 1A). The levels of RBM4 were not drastically changed between E12.5 and E18.5 although a slight increase was observed at E14.5 (for the details, see Fig. 4). Nevertheless, the expression and redistribution of RBM4 from VZ/SVZ to the cortical plate suggest its functional role in the developing brain.
FIG 1.

RBM4 deficiency affects the radial migration of neurons during cortical development. (A) RBM4 expression in the developing mouse cerebral cortex. Immunostaining was performed on coronal sections of each mouse E12.5 and E18.5 brain using anti-RBM4 (green). Merged images include immunostaining with anti-RBM4 (green) and anti-Tuj1 (red) and Hoechst staining (blue). MZ, marginal zone; CP, cortical plate; VZ, ventricular zone; SVZ, subventricular zone; IZ, intermediate zone. Scale bar, 100 μm. (B) The bar graph shows data for NeuN-positive cells in the coronal sections of mouse E18.5 brain neocortex. (C and D). Coronal sections of E18.5 Rbm4a knockout and wild-type neocortex were stained with the neuronal layer-specific markers FoxP2 (layers V and VI) (C) and SATB2 (layers II to V) (D) and with Hoechst, as indicated. Scatter plot vertical graphs, respectively, represent percentages of FoxP2- and SATB2-positive neurons in cortical layers. Cortical layers are essentially labeled as described in panel A. UCP and LCP represent upper and lower cortical plates, respectively. (E) Summary of the effect of RBM4 deficiency in neuronal migration. (F) Schemes show the strategy for in utero electroporation (IUE) using the shRBM4-containing or empty (control) EGFP expression vector; expression was driven by the U6 promoter (U6-pro; for shRNA) and synaptophysin promoter (SYP-pro; for EGFP). (G) The knockdown efficiency of RBM4 was examined by transfection of the control or shRBM4 vector into HEK293 cells; immunoblotting was performed using antibodies against RBM4 and GAPDH. (H) The vectors were each injected into the lateral ventricle of each E13.5 brain, followed by electroporation. Brains were harvested at E18.5 for analysis. Representative images of IUE show EGFP (green) and Hoechst stain (blue). The bar graph shows the ratios of GFP-positive cells to total cells in three arbitrarily dissected cortical layers. *, P < 0.05; **, P < 0.01; ***, P < 0.001. WT, wild type.
FIG 4.

RBM4 regulates alternative exon selection of Dab1. (A) The diagram illustrates the mouse Dab1 minigene and primers used for nested RT-PCR. HEK293 cells were cotransfected with either the empty vector (lanes 1 to 3) or Dab1 minigene (lanes 4 and 5) and either the empty or FLAG-RBM4 expression vector. RT-PCR and immunoblotting were performed to detect the indicated mRNAs (Dab1 and Gapdh) and proteins (RBM4 and GAPDH). Relative exon 7/8 inclusion (i.e., +E7/8 versus total; E12.5 was set as 1) was obtained from three independent experiments. CMV, cytomegalovirus; BGH, bovine growth hormone. (B) The diagram illustrates the mutant Dab1 minigenes that contained a mutated 5′ splice site (m5′SS; pink arrowheads) or lacked each of the indicated CU-rich sequences (green arrowheads); the 5′ SS and CU-rich sequences are indicated below the diagram. The positions of the CU-rich sequences are indicated (−, upstream of exon 6 or 7; +, downstream of exon 8). HEK293 cells were transfected with the wild-type or each of the mutant minigenes; Dab1 minigene splicing products and Gapdh were analyzed by RT-PCR. *, either +E7 or −E7 (C) The 5′ splice site sequences of introns 7/8 of mammalian, Gallus gallus (introns 6/7), and Danio rerio Dab1.
A potential role for RBM4 in neuronal migration during corticogenesis.
We examined the cerebral cortex of Rbm4a knockout embryos and their wild-type littermates at E18.5. Rbm4a knockout did not significantly affect cortical thickness at E18.5 (data not shown). Immunofluorescence of the neuron-specific marker NeuN revealed no drastic reduction in the number of NeuN-positive cells in Rbm4a knockout mice compared with the wild-type levels at both E18.5 (Fig. 1B) and postnatal day 10 (data not shown), indicating that deletion of one copy of Rbm4 did not significantly impair neurogenesis. We also performed immunostaining for layer-specific markers; although the distribution of forkhead box protein P2 (FoxP2)-positive neurons was apparently normal (Fig. 1C), neurons positive for transcription factor special AT-rich sequence-binding protein 2 (SATB2) were aberrantly distributed, i.e., significantly increased in the lower cortical layer and intermediate zone (Fig. 1D and E). This result suggested that Rbm4a knockout particularly impaired the migration of late-born neurons, indicating a role for RBM4 in cortical layer formation.
To confirm the role of RBM4 in neuronal migration, we attempted to knock down RBM4 in E13.5 cortical progenitor neurons by using in utero electroporation (IUE) of a vector expressing both an RBM4-targeted short-hairpin RNA (shRNA) and green fluorescent protein (GFP) (Fig. 1F). This RBM4 shRNA efficiently downregulated the expression of RBM4 in HEK293 cells (Fig. 1G). IUE followed by immunofluorescence of E18.5 cortical sections revealed that RBM4 depletion partially impaired the radial migration of newborn neurons (Fig. 1H). A similar result was observed in Rbm4a knockout embryonic brain (see Fig. 6 below). Therefore, RBM4 might play a role in neuronal migration.
FIG 6.

RBM4 modulates neuronal migration via Dab1. (A) The scheme illustrates the CAG-Dab1-mNES-DsRed and CAG-Dab1Δ78-mNES-DsRed vectors. mNES represents mutated NES. (B) The vectors shown in panel A were each transfected into HEK293 cells; immunoblotting was performed with antibodies against Dab1 and GAPDH. (C and D) IUE was performed as described in the legend to Fig. 1. In brief, for panel C, each wild-type E13.5 brain was injected with the GFP-containing control or shRBM4 expression vector alone or together with the Dab1-mNES or Dab1Δ78-mNES expression vector. For panel D, each Rbm4a KO E13.5 brain was injected with a dsRED-containing control or Dab1-mNES-DsRed expression vector (scale bar, 100 μm). In representative images, Hoechst stain (blue) indicates cell nuclei, and EGFP (green) and DsRed (Red) represent the expression of shRNA and recombinant Dab1, respectively. The bar graphs in panels C and D are presented as described in the legend to Fig. 1H. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Identification of RBM4-associated transcripts in the developing mouse brain.
To determine how RBM4 might regulate neuronal migration, we searched for its associated transcripts in the mouse embryonic brain. RBM4 ribonucleoprotein immunoprecipitation (RIP) was performed using E13.5 whole-brain lysates. Immunoblotting with anti-RBM4 confirmed efficient precipitation of RBM4 (similar to the results for RBM4 shown in Fig. 2E; also data not shown). Coprecipitated RNA was subjected to high-throughput RNA sequencing (RNA-Seq) using an Illumina HiSeq platform. Sequencing revealed a quality score of 30, indicating 99.9% base call accuracy. RIP-Seq was performed in duplicate; each experiment yielded total reads of 4.5 × 107. Compared with IgA control precipitates, the duplicate assessments identified 4,111 and 5,388 genes, respectively, as potential RBM4 targets (Fig. 2A). Analysis of the top 500 transcripts having a >3-fold increase in each RBM4-RIP revealed 68 genes common between the two RIPs (Fig. 2A; see also Table S1 in the supplemental material). Gene ontology analysis revealed that ∼80% of these 68 RBM4 target transcripts encode cellular signaling factors such as SH2 domain-binding proteins and nucleotide-binding proteins and kinases, and the remaining was a set of RNA-processing factors (Fig. 2B). Among the 68 targets, we determined that 11 genes are implicated in neuronal migration (Fig. 2C). Most notably, the reelin signaling regulator, Dab1, exhibits multiple splice isoforms during brain development (17). Dab1 was in the top 183 and 59 hits in the two RIP-Seq experiments (Fig. 2D). The result of immunoprecipitation and reverse transcription-PCR (RT-PCR) showed that RBM4 efficiently bound the transcripts of Dab1 but not those of Gapdh (Fig. 2E). The binding of RBM4 to the Atn1 and Csk transcripts was weak but reproducibly detected, whereas its binding of U6 appeared to be nonspecific (Fig. 2E).
FIG 2.

RBM4-associated brain transcripts include Dab1. (A) Flow diagram for the strategy used to identify RBM4-associated transcripts in embryonic mouse brain. Two independent immunoprecipitation experiments of E13.5 brain lysates were performed using control (IgA) or anti-RBM4. Immunoprecipitated RNA was subjected to RNA-Seq. A total of 68 genes were common between the top 500 of each individual batch obtained through similar transcripts and event identifications (ID) (TSS represents alternative 5′ first exon and transcription start site). (B) Gene ontology analysis of the aforementioned 68 RBM4-associated transcripts with respect to molecular function categories. (C) A list of 11 potential RBM4 targets that are involved in neuronal migration. (D) The graph shows the top 500 genes plotted by the ratio of RBM4/IgA (log10) versus rank in two experiments. Dab1 is indicated. (E) As in panel A, each E13.5 brain cell lysate was subjected to immunoprecipitation (IP) using anti-RBM4 or anti-IgA. RT-PCR and immunoblotting (IB) were performed to detect the indicated RNAs and proteins. Lane 1, 2% input.
RBM4 deficiency impairs Dab1 isoform expression in the developing brain.
The association of RBM4 with Dab1 transcripts prompted us to examine whether RBM4 is involved in the expression of Dab1 splice isoforms (Fig. 3A). We examined the expression of Dab1 splice isoforms in the Rbm4a knockout mouse brain. RT-PCR revealed that exon 7-containing Dab1 (Dab1+E7) was dominant in the E13.5 wild-type brain (Fig. 3B, WT). In Rbm4a knockout mice, both the full-length (+E7/8) and +E7 isoforms were reduced with a concomitant increase in the exon 7/8-skipped isoform (−E7/8) (Fig. 3B, KO). RBM4 deficiency reduced the expression of full-length Dab1 by ∼20% on average, suggesting that RBM4 contributes to exon 7/8 inclusion (Fig. 3B). A similar result was observed in Rbm4a or Rbm4b knockout mouse embryonic fibroblasts (data not shown). However, exon 9b/c usage was not significantly altered in the Rbm4a knockout brain (Fig. 3C). Therefore, RBM4 may function differently from Nova2 in regulating Dab1 isoform expression (22). Mouse embryonal carcinoma P19 cells can undergo neural differentiation upon exposure to retinoic acid. By using such an in vitro model for neural development, we previously showed that transient induction of RBM4 correlates with alternative splicing isoform switches in its target mRNAs during neuronal differentiation (15). Therefore, we examined Dab1 isoform expression in P19 cells. P19 cells initially form embryonic bodies upon treatment with retinoic acid and subsequently undergo neuronal differentiation in neurobasal medium. We observed an induction in RBM4 protein and the neuronal markers Map2 mRNA and Tuj1 protein, as previously reported (15), as well as the splicing switch of Dab1 from the −E7/8 to +E7/8 isoform (Fig. 3D). This observation further correlated RBM4 expression with exon 7/8 inclusion. Next, we overexpressed FLAG-RBM4 in undifferentiated P19 cells and observed a slight increase in the +E7/8 isoform (Fig. 3E). These data reiterated that RBM4 may regulate alternative exon selection of Dab1 and possibly contribute to exon 7/8 inclusion.
FIG 3.

RBM4-deficient brain exhibits aberrant expression of Dab1 exons 7/8 splice isoforms. (A) Diagram showing alternative splicing of mouse Dab1 exons 6 to 11 and primers used for RT-PCR. (B) Total RNA was isolated from each E13.5 Rbm4a knockout and wild-type (WT) mouse brain. RT-PCR was performed to analyze Dab1, Rbm4a, Rbm4b, and Gapdh mRNAs. The bar graph represents Dab1 splice forms. *, P > 0.05. (C) RT-PCR of E13.5 brain RNA was performed to analyze E9b/c-lacking or containing Dab1 and Gapdh mRNAs. (D) RT-PCR and immunoblotting (IB) were performed to examine Dab1, Map2, and Gapdh mRNAs and RBM4, Tuj1, and GAPDH proteins, respectively, throughout neuronal differentiation of P19 cells. UD, undifferentiated cells; EB, retinoic acid-induced embryonic body; D2, differentiation for 2 days. (E) Total RNA and protein from control-transfected or FLAG-RBM4 vector-transfected P19 cells were analyzed by RT-PCR (Dab1 and Gapdh) and immunoblotting (RBM4 and GAPDH). For panels B, D, and E, the percentage of exon 7/8 inclusion was measured as +E7/8 versus total, including +E7/8, +E7, and −E7/8; average and standard deviation were obtained from at least three independent experiments or samples.
RBM4 directly regulates alternative splicing of Dab1 pre-mRNA.
To confirm the role of RBM4 in splicing regulation of Dab1, we generated a Dab1 minigene reporter spanning exons 6 to 9 of mouse Dab1 (see Materials and Methods) (Fig. 4A). Overexpression of FLAG-RBM4 significantly promoted exon 7/8 inclusion of this reporter in HEK293 cells (Fig. 4A). To explore the cis elements that govern exon 7/8 selection, we further took advantage of this reporter. First, we suspected that the divergent 5′ splice sites (SS) of both introns 7 and 8 account for exon 7/8 skipping (Fig. 4B); therefore, these 5′ splice sites were individually mutated to conform to the consensus. In either mutant, exons 7/8 were completely included (Fig. 4B, lanes 6 and 7; m5'SS denotes the mutant 5′ splice site). Second, we suspected that the CU-rich sequences in introns 6 and 7 serve as the regulatory elements for exon 7/8 selection. We thus generated four different deletion mutants. Deletion of −26 to −12 of intron 6 or of −62 to −31 (mutant 7-1) or −30 to −17 (mutant 7-2) of intron 7 resulted in efficient inclusion of exons 7/8 (Fig. 4B, lanes 2 to 4). However, deletion of a short CU-rich sequence within intron 8 had no effect (Fig. 4B, lane 5). Minimal levels of both single-exon-containing products (+E7 and +E8) were detected in the mutant reporters (Fig. 4B), suggesting that this minigene was able to recapitulate in vivo conditions. Notably, the features of divergent 5′ splice sites (Fig. 4C) and long CU-rich sequences (data not shown) are conserved in vertebrate Dab1 genes, suggesting their advantage for functional selection of alternative exons. Together, both trans-acting factors (such as RBM4) and intronic cis elements (5′ SS and regulatory sequences) can dictate the alternative selection of Dab1 exons 7/8.
RBM4 antagonizes PTBP1 function in regulating alternative splicing of Dab1.
RBM4 regulates mRNA splicing essentially through binding to CU-rich motifs (8, 9, 10, 11). The above data (Fig. 4B) suggested that the CU-rich elements of introns 6/7 are recognized by splicing suppressors that counteract the effect of RBM4 in splicing regulation. PTBP1 and RBM4 compete for CU-rich sequences in target pre-mRNAs (11, 12). We thus assessed the expression of the RBM4, PTBP1, and PTBP2 (neuronal PTB) in the mouse embryonic brain. RBM4 was continuously expressed from E12.5 to E18.5, with a slight increase at E14.5. PTBP1 was detected only at E12.5, whereas PTBP2 was expressed throughout the period examined and particularly was upregulated at E14.5 to E16.5 (Fig. 5A). We performed RT-PCR and immunoblotting to examine Dab1 expression. The level of the full-length Dab1 transcript was increased at E14.5, likely via alternative splicing. Immunoblotting revealed that Dab1Δ78 (lacking exons 7/8) was dominant at E12.5 to E14.5 and began to switch to the full-length protein at E14.5. The splicing change of Dab1 was coincident with the minimal increase of RBM4 and disappearance of PTBP1, suggesting that PTBP1 and RBM4 regulate Dab1 isoform expression roughly in a sequential manner (Fig. 5A).
FIG 5.

RBM4 and PTBP1 oppositely modulate alternative splicing of Dab1. (A) Immunoblotting (IB) was performed to detect the expression of Dab1, PTBP1, PTBP2, RBM4, and GAPDH proteins in mouse brain cell lysates at the indicated embryonic days. FL, full-length Dab1. For panels B to E, HEK293 cells were cotransfected with the wild-type Dab1 minigene and shRNA (shLuc as a control, or shPTBP1) (B), the FLAG-PTBP1 (left panel) or HA-PTBP2 (right panel) or their respective empty vectors (C), the empty or FLAG-RBM4 vector alone or with different amounts of the FLAG-PTBP1 vector (D), or the empty or FLAG-PTBP1 vector alone or with different amounts of the FLAG-RBM4 vector (E). In panels D and E, FLAG-RBM4 (*) and FLAG-PTBP1 (**) are indicated. Dab1 minigene expression was examined by RT-PCR as described for panel A. Whole-cell lysates were subjected to immunoblotting to detect endogenous RBM4, PTBP1, and GAPDH and overexpressed PTBP1 (FLAG) and PTBP2 (HA). For panels A to E, relative percentages of exon 7/8 inclusion were measured as described in the legend of Fig. 4A. (F) The diagram illustrates the intronic CU-rich regions (thick lines) used for the probes in panels G to I. The positions of each probe are indicated (−, upstream of exon 7; +, exon 7); the shaded box indicates exon 7 sequence. (G and H) EMSA was performed using purified recombinant MBP, MBP-RBM4, or nontagged PTBP1 and each 32P-labeled probe (panel G, intron 6, 7 and 8; panel H, subregions 7-0, 7-1, and 7-2/3). The reaction products were fractionated on a 6% nondenaturing polyacrylamide gel. (I) For cross-linking experiments, MBP-RBM4 (lane 1), PTBP1 (lane 2), or PTBP1 and increasing amounts of MBP-RBM4 (lanes 3 to 6) were incubated with 32P-labeled probe 7 followed by UV cross-linking and digestion with RNase. The reaction products were fractionated by SDS-PAGE. Relative intensity of each protein-RNA cross-link is indicated below. (J) The model shows that RBM4 and PTBP1 might compete for the binding sites in Dab1 intron 7 and that their antagonistic action modulates exon 7/8 usage.
To assess the above possibility, we depleted or overexpressed PTBP1 in HEK293 cells. Figure 5B shows that an ∼50% reduction of PTBP1 by shRNA increased the level of Dab1+E7/8 by 40%. Overexpression of FLAG-PTBP1 significantly induced exon 7/8 skipping, i.e., reduced exon 7/8 inclusion by >60% (Fig. 5C, lane 2), whereas PTBP2 had only a minimal activity (lane 4). Next, we examined whether PTBP1 competes with RBM4 in Dab1 alternative splicing. Overexpression of FLAG-PTBP1 efficiently suppressed the effect of coexpressed RBM4 in exon 7/8 inclusion (Fig. 5D, lanes 3 and 4). FLAG-RBM4 was also able to reverse the effect of PTBP1 in Dab1+E7/8 isoform expression, albeit with less efficiency than PTBP1 in antagonizing RBM4 (Fig. 5E, lane 4). These results suggested competition between RBM4 and PTBP1.
Next, we investigated how RBM4 and PTBP1 regulate Dab1 exon selection. For electrophoretic mobility shift assays (EMSA), we purified a recombinant maltose-binding protein (MBP)-RBM4 fusion and a nontagged PTBP1 from bacterial lysates and used 32P-labeled probes derived from introns 6, 7, and 8 (Fig. 5F). EMSA revealed that both MBP-RBM4 and PTBP1, but not MBP, specifically bound to intron 7 (Fig. 5G). To further investigate the binding region, we divided the intron 7 probe into three subregions, namely, 7-0, 7-1, and 7-2/3. Using EMSA, we observed that MBP-RBM4 bound to all three subregions but exhibited highest affinity for region 7-1, whereas PTBP1 bound only to 7-1 (Fig. 5H). In fact, PTBP1 had a stronger affinity to region 7-1 than RBM4 (data not shown), consistent with the notion that this fragment contains four copies of the PTBP1-binding motif UCUU (Fig. 5F) (23). Finally, we titrated MBP-RBM4 in the binding reaction mixture containing PTBP1 and 32P-labeled 7-1 probe and observed that increasing amounts of MBP-RBM4 gradually reduced PTBP1 binding to the probe (Fig. 5I). The results so far revealed that PTBP1 had a higher affinity for the middle UCUU repeat-containing region, whereas RBM4 bound to a broader CU-rich region of intron 7, by which these two proteins may antagonistically regulate Dab1 exon 7/8 selection (Fig. 5J).
RBM4 regulates neuronal migration via Dab1.
The results shown in Fig. 1 showed that RBM4 deficiency impaired cortical neuronal migration and positioning. The above result indicated that RBM4 likely contributes to the expression of full-length Dab1. To test whether Dab1 could restore the migration defects caused by RBM4 depletion, we generated a vector that expressed Dab1 and the red fluorescent protein DsRed. However, this Dab1 fusion appeared to exert a dominant negative effect in neuronal migration due to its cytoplasmic accumulation (data not shown), as reported previously (24). To attenuate such a deleterious effect, we disrupted the nuclear export signals (NESs) of Dab1 (24) and generated NES-mutant Dab1 and its exon 7/8-lacking version (Fig. 6A, Dab1-mNES and Dab1Δ78-mNES, respectively); their expression was evaluated in HEK293 cells (Fig. 6B). IUE revealed that Dab1-mNES could almost fully rescue the neuronal migration defect caused by RBM4 depletion (Fig. 6C, Dab1-mNES). Furthermore, we evaluated the effect of Dab1-mNES in Rbm4a knockout embryonic cortices, which exhibited migration defects of developing neurons (Fig. 6D, vector). Overexpression of Dab1-mNES could also rescue the migration defect of Rbm4a knockout (Fig. 6D, Dab1-mNES). However, the exon 7/8-truncated, mutant NES-bearing Dab1 dominantly inactivated neuronal migration (Fig. 6C, DabΔ78-mNES). This result indicated that RBM4 participates in neuronal migration, likely via modulating the selection of Dab1 exons 7/8. Moreover, RBM4-promoted full-length Dab1 expression is likely important for neuronal migration during brain development.
DISCUSSION
RBM4 participates in differentiation of a variety of cell types, including muscle cells, pancreas cells, adipocytes, and neurons, via regulating alternative splicing (8–11, 15). RBM4 directs the expression of splice isoforms that promote neuronal differentiation from mouse embryonic carcinoma P19 cells, neural progenitor cells, and human mesenchymal cells (10, 15). The role of RBM4 in neuronal differentiation echoes its abundance in the VZ/SVZ in the early embryonic brain (Fig. 1). Although Rbm4a knockout only minimally reduced neuron numbers in E18.5 brains, it did not significantly affect neocortical thickness (data not shown). Nevertheless, the observation that RBM4 deficiency impaired neuronal migration and disorganized late-born neurons (Fig. 1) prompted us to focus on the role of RBM4 in neuronal migration in this study. Because Rbm4b knockout appears to cause infertility, we are presently unable to address whether complete loss of RBM4 may lead to severe neurogenesis or any other neuronal migration defects.
In this study, we identified and confirmed Dab1 transcripts as targets of RBM4 in the mouse embryonic brain (Fig. 2) and demonstrated that RBM4 promotes exon 7/8 selection of Dab1 (Fig. 4). Exons 7/8 contain several critical tyrosine (Y) residues that are differentially phosphorylated in response to reelin signaling (25). Upon reelin activation, phosphorylation of Y185/Y198 (on exons 6 and 7, respectively) by Src kinases promotes neuron migration, whereas Abl-induced phosphorylation of Y220/Y232 (on exons 8 and 9, respectively) leads to Dab1 degradation and subsequent termination of reelin signaling (18, 26). Therefore, the selection of alternative exons 7/8 finely modulates the reelin-induced cellular response and coordinates neuronal migration. The Dab1Δ78 isoform appears early during brain development; the single exon-containing isoforms (i.e., exon 7 or 8) are also transiently expressed, and their expression levels may be determined in response to subtle changes in the ratio of RBM4 to PTBP1/2. At later stages, inclusion of both exons 7 and 8 likely contributes to full activation of Dab1 as well as termination of neuronal migration. Therefore, ordered selection of exons 7/8 in Dab1 during cortical developmental is critical for proper reelin response. Our results define the regulatory mechanism for exon 7/8 selection, which will be important for further understanding of how splicing regulation integrates into reelin signaling.
Using the minigene reporter, we observed that overexpression of RBM4 promoted inclusion of exons 7/8 (Fig. 4). Moreover, our investigation into the cis elements of Dab1 indicated that weak 5′ splice sites and long CU-rich elements in the surrounding introns modulate alternative exon 7/8 usage. This feature is evolutionarily conserved (Fig. 4), suggesting a common regulatory mechanism across species. We deduced that RBM4 binding to CU-rich elements in intron 7 promotes spliceosome assembly on intron 7, which bears a poor 5′ splice site, and that exon 7 inclusion may obligatorily trigger exon 8 inclusion, resulting in the expression of full-length Dab1. In addition, a CU-rich sequence in intron 6 also serves as a negative element (Fig. 4), but which factors recognize this element needs further investigation. Moreover, alternative selection of exons 9b/c has been observed during brain development. Inappropriate inclusion of exons 9b/c also results in mislocalization of late-generated cortical neurons (22). Nevertheless, Rbm4a knockout did not affect this alternative splicing event (Fig. 3). We reasoned that this was attributable to the lack of sufficiently long CU-rich sequences within intron 9.
This study reveals that RBM4 and PTBP1 have opposite effects on exon 7/8 usage of Dab1 by competing for binding to the overlapping CU-rich sequences in intron 7 (Fig. 5). The level of PTBP1 protein, but not its mRNA, was sharply reduced at E14.5 (Fig. 5), suggesting that PTBP1 protein downregulation results from microRNA-mediated translational suppression of PTBP1 mRNA and/or PTBP1 protein instability. We deduced that PTBP1 maintains Dab1Δ78 expression until E12.5 and meanwhile/subsequently RBM4 competes off PTBP1 to promote exon 7 inclusion until PTBP1 is turned completely off. The level of PTBP2 increased between E14.5 and E16.5, but PTBP2 had much weaker activity in suppressing exon 7/8 usage than PTBP1 (Fig. 5 and illustration in Fig. 7). Therefore, the full-length Dab1 appears to predominate during the late stage of brain development. Moreover, the proper temporal control of splicing factor expression, particularly the shutoff of PTBP1, is likely critical for cortical lamination. In addition, whether RBM4 also acts in concert with Nova proteins and other splicing regulators to regulate the expression of a full spectrum of Dab1 isoforms remains to be investigated.
FIG 7.
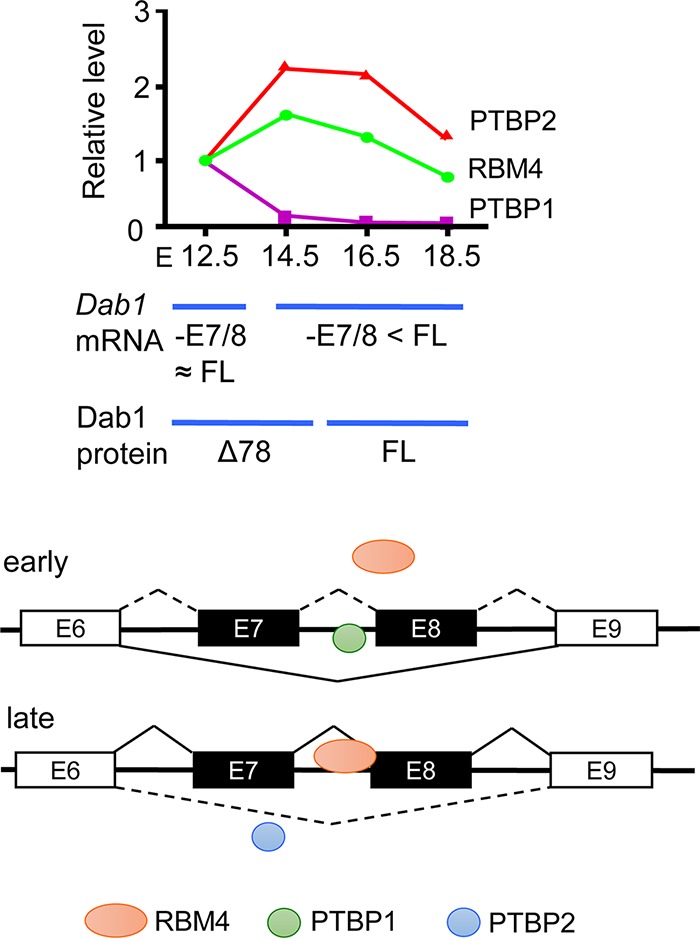
RBM4 functions in cortical lamination by modulating alternative splicing of Dab1. (Top) The diagram depicts the result of the experiment represented in Fig. 6A, showing the relative expression of RBM4, PTBP1, and PTBP2 throughout brain development (E12.5 was set to 1) and Dab1 mRNA and protein isoforms expressed at different stages. FL, full-length; 78 Dab1Δ78. (Bottom) The scheme illustrates the regulation of Dab1 exon 7/8 selection by the interplay between RBM4 and PTBP1/2 in the early and late stages of brain development; single exon (E7 or E8) selection is not depicted.
Down syndrome, also known as trisomy for human chromosome 21, is a genetic disorder commonly associated with intellectual disability. Down syndrome patients and model mice exhibit delayed or disorganized cortical lamination and a reduced number of dendrites and reduced synapse formation (27). Reduced RBM4 expression has been linked to fetal Down syndrome (14). Our study reveals that RBM4 deficiency causes aberrant distribution of cortical neurons in the embryonic brain (Fig. 1) and reduced neurite outgrowth in cultured neurons (15), providing a hint as to how RBM4 deficiency might cause abnormal brain developmental defects associated with Down syndrome. Thus, genetic defects or aberrant expression of RBM4 may cause developmental brain disorders.
MATERIALS AND METHODS
Plasmids.
The expression vectors encoding FLAG-tagged RBM4 (human RBM4A gene) and PTBP1 have been described previously (11), as has the expression vector pCEP-HA-nPTB (where HA is hemagglutinin) (28). The cDNAs for mouse Dab1 and exon 7/8-truncated Dab1 (Dab1Δ78) were obtained by RT-PCR using the transcripts of mouse E13.5 brain as the template. These two cDNAs were placed in frame at the N terminus of DsRED of a CAG promoter-based vector, resulting in pCAG-Dab1-DsRed and pCAG-Dab1Δ78-DsRed. The NES sequences for Dab1 were subsequently mutated using PCR-based mutagenesis according to Honda and Nakajima (29). The resulting vectors were in pCAG-Dab1mNES-DsRed and pCAG-Dab1Δ78mNES-DsRed. The pCAG-Cre-IRES-H2BmCherry and pLL 3.7-U6-Syp promoter-GFP vectors were obtained from T. K. Tang and Y.-S. Huang (Academia Sinica, Taipei, Taiwan), respectively. The shRNA expression vectors (pLKO.1-shRBM4 and pLKO.1-shluc-puro) have been previously described (15). The pLKO.1-shPTB-puro vector was purchased from the National RNAi Core Facility, Academia Sinica. For IUE, the RBM4-targeted shRNA oligonucleotides were inserted into the pLL3.7-U6-Syp-GFP vector using the Xma1 and SphI sites. To construct the minigene containing Dab1 exons 6 to 9, the PCR-amplified fragments from mouse genomic DNA were ligated and inserted into pcDNA 3.1 (GE Healthcare), including nucleotides 104679107 to 104680425 (1,319 bp), nucleotides 104681264 to 104682008 (745 bp), and nucleotides 104688005 to 104688732 (728 bp) of chromosome 4 (genome assembly GRCm38.p3; GenBank accession number NC_000070.6). To construct the plasmids for RNA probes, PCR-amplified intronic fragments of Dab1, i.e., 109 bp of intron 6 (nucleotides 104679977 to 104680085), 110 bp of intron 7 (nucleotides 104681501 to 104681610), and 106 bp of intron 8 (nucleotides 104681732 to 104681837), were each cloned into pGEM-T (Promega). Mutants were generated by PCR-based mutagenesis.
Splicing assay.
HEK293T cells were grown to 60 to 70% confluence in six-well plates before transfection. Subsequently, 1 to 4 μg of pcDNA-FLAG-RBM4 with or without pcDNA-FLAG-PTBP1 or pCEP4-HA-nPTB was cotransfected with 1 μg of the pcDNA3.1-Dab1 minigene vector using Lipofectamine 2000 (Life Technologies) or Hyperfectin (Omics Bio). Cells were harvested at 48 h posttransfection. For knockdown, cells were transfected with 1 μg of each pLKO.1-based shRNA vector (see above). Transfected cells were harvested at 48 and 72 h posttransfection. To collect RNA, cells were washed twice with ice-cold phosphate-buffered saline (PBS) followed by extraction with TRIzol reagent (Life Technologies). RT-PCR was performed using the primers listed in Table S2 in the supplemental material.
Animals and genotyping.
All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee (IACUC) 13-04-547 of Academia Sinica and compliant with the Ministry of Science and Technology, Taiwan. To produce the Rbm4a KO, male and female mice carrying Rbm4a+/−, b+/+ were used. Primers were used as earlier described by Lin et al. (8).
Culture and neuronal differentiation of P19 cells.
Culture and neuronal differentiation of P19 cells were performed according to the method of Tarn et al. (15). To detect endogenous Dab1 splicing, 1 µg of pcDNA-FLAG-RBM4 or control plasmid was transfected into P19 cells grown 60% confluence in 6-well plates by using Lipofectamine 2000 (Life Technologies).
RNA extraction and immunoprecipitation from mouse embryonic brain.
Timed pregnant mouse embryos from Rbm4a knockout (8) and wild-type littermates were collected. Total RNA was extracted using TRIzol reagent. RNA samples were treated with DNase I (Roche) at 37°C for 30 min to eliminate genomic DNA contaminants. To isolate RBM4-associated transcripts, E13.5 brains were collected and lysed in radioimmunoprecipitation assay (RIPA) lysis buffer containing 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 10% glycerol, protease inhibitor cocktail (Roche), and 20 U of RNase inhibitor (Enzymatics). The lysate was centrifuged at 12,000 × g for 15 min. The supernatant was subjected to precleaning with IgG beads for 2 h in a cold room and subsequently incubated with anti-RBM4-bound or control IgG-bound beads as described previously (8). RNA was converted to cDNA using a High-Capacity cDNA reverse transcription kit (Applied Biosystems), followed by PCR analysis (see below).
cDNA library preparation for RNA sequencing.
For RNA sequencing, immunoprecipitated RBM4 ribonucleoprotein complex was treated with TRIzol reagent followed by phenol-chloroform extraction. The cDNA library was constructed and sequenced by the High-Throughput Genomic Core at the Biodiversity Research Center, Academia Sinica, using an Illumina Hiseq 2500 instrument with a 2- by 150-bp paired-end configuration. The raw reads were processed by Genewiz (NJ). Alternative splicing (AS) events from RNA-Seq data were analyzed by using the Asprofile program, version 1.0.
RT-PCR.
In general, 1 μg of total RNA was treated with RQ1 RNase-free DNase (Promega). cDNA was generated using oligo(dT) and a SuperScript III first strand synthesis system kit (Thermo Fisher Scientific). PCR primers used are listed in Table S2 in the supplemental material. The products were fractionated by electrophoresis through 2 or 3% agarose gels or 6% polyacrylamide gels for Dab1 splicing analysis. Band intensity was quantified by using ImageJ, version 1.47.
Immunoblotting.
Immunoblotting was performed as described previously (11). Primary antibodies included affinity-purified polyclonal anti-RBM4 (11), anti-Dab1 (Santa Cruz Biotechnology), anti-FLAG (Sigma), anti-PTBP2 (Proteintech), and monoclonal antibodies against PTBP1 (Abcam), HA (gift from S.-C. Cheng, Academia Sinica), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Proteintech), and α-tubulin (GeneScript). Secondary antibody was horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG.
Purification of recombinant proteins.
The pET-29b-based vectors encoding MBP, MBP-RBM4, and nontagged PTBP1 were each transformed into Escherichia coli strain BL21(DE3) (Promega). Expression of recombinant proteins was induced by 0.2 mM isopropyl-β-d-1-thiogalactopyranoside at 22°C (MBP and MBP-RBM4) or 18°C (PTBP1) overnight. MBP-RBM4 and PTBP1 were purified using the pMAL protein fusion and purification system (New England BioLabs) and heparin-Sepharose CL-6B beads (GE Healthcare Bio-Sciences AB), respectively.
EMSA.
EMSA was performed according to Rio (30), with minor modification. RNA probes were in vitro transcribed using T7 RNA polymerase; the templates were NotI-digested linearized pGEM-T vectors containing a Dab1 intronic fragment corresponding to intron 6 (I6; GCUGGCUCUCUACUGUGCUGUAACAUCUGAAGAUAAUCUCACUUCUCUUCCUUUGCAUCCUUUGCUACUCCCUCCCUUCCCUGGCUGGGAUAGACCAUUUUGGAAGAGG), intron 7 (I7; CUGUGAUUUGCAUCUAAAUUGGAAAUACUUUUCUCGUCUUUCUUUCUUUCUUUUUUUUUUCCUUCUGCCUUUUCCUCUCUGGCUGCUCCUGCAAUAGUACAUUGUGUUUG), or intron 8 (I8; GCTGGCTCTCTACTGTGCTGTAACATCTGAAGATAATCTCACTTCTCTTCCTTTGCATCCTTTGCTACTCCCTCCCTTCCCTGGCTGGGATAGACCATTTTGGAAGAGG). For EMSA, 5 × 104 cpm (∼75 fmol) of 32P-labeled RNA probe was incubated with ∼20 pmol of each purified recombinant protein (MBP, MBP-RBM4, or PTBP1) in 30-μl reaction mixtures at 30°C for 30 min, with or without UV-cross-linking (UV Stratalinker 1800), followed by RNase digestion on ice for 10 min. The reaction products (1/6 volume) were then analyzed with electrophoresis on 8% SDS-PAGE or nondenaturing 6% polyacrylamide gel in 0.5× TBE buffer (90 mM Tris, 64.6 mM boric acid, 2.5 mM EDTA, pH 8.3). Dried gels were exposed to high-performance autoradiography films (GE Healthcare).
IUE.
The detailed procedure for IUE has been described in Tabata and Nakajima (31). In brief, we injected the shRNA-encoding vectors and/or recombinant Dab1 expression vectors into the lateral ventricle of each E13.5 mouse brain and collected samples at E18.5. After injection, electronic pulses of 30 V were given five times at 950-ms intervals using a pulse generator (CUY21Vivo-SQ; BEX Company). Throughout the experiments, mice were anesthetized with isoflurane.
Histology, immunohistochemistry, and quantification.
For histology, mice were transcardially perfused with PBS and subsequently with 4% paraformaldehyde in PBS (pH 7.4). Each isolated postnatal and/or E18.5 brain was fixed with 4% paraformaldehyde for 24 h, followed by cryoprotection with increasing concentrations of sucrose in PBS, i.e., 10%, 20%, and 30% (24 h). Brain sections (16 μm or 30 μm) were prepared with a Leica CM3050S cryostat and stored at −20°C until analysis. Sections were subjected to antigen retrieval using 0.001% sodium citrate (pH 7.4) at 90°C for 30 min, followed by permeabilization with 0.2% Triton X-100 for 10 min and blocking with 5% bovine serum albumin in PBS for 1 h at room temperature. Mouse brain coronal sections were incubated with the antibodies given below overnight at 4°C. Primary antibodies used were as follows: rabbit polyclonal anti-GFP, mouse monoclonal anti-GFP (3E6) (Life Technologies), rabbit polyclonal anti-mCherry and anti-NeuN (Abcam), goat polyclonal anti-DsRed (L-18), goat polyclonal anti-FoxP2 (N-16) (as above, from Santa Cruz Biotechnology), and mouse monoclonal anti-SATB2 (Abcam). Subsequently, hybridization using species-specific secondary antibodies was performed for 1 h, followed by Hoechst staining for 10 min at room temperature; antibodies used included donkey anti-goat IgG (Alexa Fluor 568), donkey anti-goat IgG (Alexa Fluor 647) (as above, from Abcam), goat anti-mouse IgG, goat anti-rabbit IgG (Alexa Fluor 488), and goat anti-rabbit IgG–rhodamine (as above, from Thermo Fisher Scientific).
Statistical analysis.
The Student t test was performed to determine the significance between the treatment groups. For all data, we performed at least three independent experiments or used at least three pairs of brain samples to obtain means and standard deviations (SD).
Supplementary Material
ACKNOWLEDGMENTS
We thank Tang K. Tang, Yi-Shiuan Huang, Guey-Shin Wang, and Soo-Chen Cheng for expression vectors and antibodies and/or experimental suggestions and Shen-Ju Chou for experimental materials and critical reading of the manuscript. We also thank the Core Facility of the Institute of Biomedical Sciences and Ching-Shu Suen for technical assistance and the NGS Core Facility for transcriptome sequencing.
This work was supported by Ministry of Science and Technology grant 106-2311-B-001-015.
D.D. performed all experiments and contributed to manuscript preparation. K.-Y.H. maintained the Rbm4a knockout mouse. W.-Y.T. oversaw the project, designed the experiments, and wrote the manuscript.
We declare that we have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00007-18.
REFERENCES
- 1.D'Arcangelo G, Miao GG, Chen SC, Scares HD, Morgan JI, Curran T. 1995. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 2.Johnson MB, Kawasawa YI, Mason CE, Krsnik Z, Coppola G, Bogdanovic D, Geschwind DH, Mane SM, State MW, Sestan N. 2009. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF, Black DL, Kharchenko PV, Sharp PA, Walsh CA. 2016. Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell 166:1147–1162. doi: 10.1016/j.cell.2016.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng S, Black DL. 2013. Alternative pre-mRNA splicing in neurons: growing up and extending its reach. Trends Genet 29:442–448. doi: 10.1016/j.tig.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quesnel-Vallieres M, Lrimia M, Cordes SP, Blencowe BJ. 2015. Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev 29:746–759. doi: 10.1101/gad.256115.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gehman LT, Meera P, Stoilov P, Shilu L, O'Brien JE, Meisler MHMA Jr, Otis TS, Black DL. 2012. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev 26:445–460. doi: 10.1101/gad.182477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vuong CK, Black DL, Zheng S. 2016. The neurogenetics of alternative splicing. Nat Rev Neurosci 17:265–281. doi: 10.1038/nrn.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin JC, Yan YT, Hsieh WK, Peng PJ, Su CH, Tarn WY. 2013. RBM4 promotes pancreas cell differentiation and insulin expression. Mol Cell Biol 33:319–327. doi: 10.1128/MCB.01266-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin JC, Tarn WY, Hsieh WK. 2014. Emerging role for RNA binding motif protein 4 in the development of brown adipocytes. Biochim Biophys Acta 1843:769–779. doi: 10.1016/j.bbamcr.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 10.Su CH, Hung KY, Hung SC, Tarn WY. 2017. RBM4 regulates neuronal differentiation of mesenchymal stem cells by modulating alternative splicing of pyruvate kinase M. Mol Cell Biol 37:00466–16. doi: 10.1128/MCB.00466-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin JC, Tarn WY. 2011. RBM4 down-regulates PTB and antagonizes its activity in muscle cell-specific alternative splicing. J Cell Biol 193:509–520. doi: 10.1083/jcb.201007131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin JC, Tarn WY. 2005. Exon selection in α–tropomyosin mRNA is regulated by the antagonistic action of RBM4 and PTB. Mol Cell Biol 25:10111–10121. doi: 10.1128/MCB.25.22.10111-10121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooks YS, Wang G, Yang Z, Smith KK, Bieberich E, Ko L. 2009. Functional pre-mRNA trans-splicing of coactivator CoAA and corepressor RBM4 during stem/progenitor cell differentiation. J Biol Chem 284:18033–18046. doi: 10.1074/jbc.M109.006999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernert G, Fountoulakis M, Lubec G. 2002. Manifold decreased protein levels of matrin 3, reduced motor protein HMP and hlark in fetal Down's syndrome brain. Proteomics 2:1752–1757. doi:. [DOI] [PubMed] [Google Scholar]
- 15.Tarn WY, Kuo HC, Ya HI, Lin SW, Tseng CT, Dhananjaya D, Hung KY, Tu CC, Chang SH, Huang GJ, Chiu IM. 2016. RBM4 promotes neuronal differentiation and neurite outgrowth by modulating Numb isoform expression. Mol Biol Cell 27:1676–1683. doi: 10.1091/mbc.E15-11-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper JA. 2008. A mechanism for inside-out lamination in the neocortex. Trends Neurosci 31:113–119. doi: 10.1016/j.tins.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Rice DS, Curran T. 2001. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci 24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- 18.Gao Z, Godbout R. 2013. Reelin-disabled-1 signaling in neuronal migration: splicing takes the stage. Cell Mol Life Sci 70:2319–2329. doi: 10.1007/s00018-012-1171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howell BW, Hawkes R, Soriano P, Cooper JA. 1997. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 20.Bock HH, May P. 2016. Canonical and non-canonical Reelin signaling. Front Cell Neurosci 10:166. doi: 10.3389/fncel.2016.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang B, Wang W, Zhang Z, Hu Y, Meng F, Wang F, Lou H, Zhu L, Godbout R, Duan Gao SZ. 2017. Alternative splicing of disabled-1 controls multipolar-to-bipolar transition of migrating neurons in the neocortex. Cereb Cortex 2017:1–11. doi: 10.1093/cercor/bhx212. [DOI] [PubMed] [Google Scholar]
- 22.Yano M, Hayakawa-Yano Y, Mele A, Darnell RB. 2010. Nova2 regulates neuronal migration through an RNA switch in disabled-1 signaling. Neuron 66:848–858. doi: 10.1016/j.neuron.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen M, David CJ, Manley JL. 2012. Concentration-dependent control of pyruvate kinase M mutually exclusive splicing by hnRNP proteins. Nat Struct Mol Biol 19:346–354. doi: 10.1038/nsmb.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Honda T, Nakajima K. 2016. Proper level of cytosolic disabled-1, which is regulated by dual nuclear translocation pathways, is important for cortical neuronal migration. Cereb Cortex 26:3219–3236. doi: 10.1093/cercor/bhv162. [DOI] [PubMed] [Google Scholar]
- 25.Gao Z, Poon HY, Li L, Li X, Palmesino E, Glubrecht DD, Colwill K, Dutta I, Kania A, Pawson T, Godbout R. 2012. Splice-mediated motif switching regulates disabled-1 phosphorylation and SH2 domain interactions. Mol Cell Biol 32:2794–2808. doi: 10.1128/MCB.00570-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sekine K, Kubo K, Nakajima K. 2014. How does reelin control neuronal migration and layer formation in the developing mammalian neocortex? Neurosci Res 86:50–58. doi: 10.1016/j.neures.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Haydar TF, Reeves RH. 2012. Trisomy 21 and early brain development. Trends Neurosci 35:81–91. doi: 10.1016/j.tins.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KM, Tarn WY. 2010. TRAP150 activates pre-mRNA splicing and promotes nuclear mRNA degradation. Nucleic Acids Res 38:3340–3350. doi: 10.1093/nar/gkq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honda T, Nakajima K. 2006. Mouse disabled 1 (DAB1) is a nucleocytoplasmic shuttling protein. J Biol Chem 281:38951–38965. doi: 10.1074/jbc.M609061200. [DOI] [PubMed] [Google Scholar]
- 30.Rio DC. 2014. Electrophoretic mobility shift assay for RNA-protein complexes. Cold Spring Harb Protoc 2014:435–440. doi: 10.1101/pdb.prot080721. [DOI] [PubMed] [Google Scholar]
- 31.Tabata H, Nakajima K. 2001. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience 103:865–872. doi: 10.1016/S0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.