

ABSTRACT
Viral hemorrhagic septicemia virus (VHSV), a rhabdovirus infecting teleost fish, has repeatedly crossed the boundary from marine fish species to freshwater cultured rainbow trout. These naturally replicated cross-species transmission events permit the study of general and repeatable evolutionary events occurring in connection with viral emergence in a novel host species. The purpose of the present study was to investigate the adaptive molecular evolution of the VHSV glycoprotein, one of the key virus proteins involved in viral emergence, following emergence from marine species into freshwater cultured rainbow trout. A comprehensive phylogenetic reconstruction of the complete coding region of the VHSV glycoprotein was conducted, and adaptive molecular evolution was investigated using a maximum likelihood approach to compare different codon substitution models allowing for heterogeneous substitution rate ratios among amino acid sites. Evidence of positive selection was detected at six amino acid sites of the VHSV glycoprotein, within the signal peptide, the confirmation-dependent major neutralizing epitope, and the intracellular tail. Evidence of positive selection was found exclusively in rainbow trout-adapted virus isolates, and amino acid combinations found at the six sites under positive selection pressure differentiated rainbow trout- from non-rainbow trout-adapted isolates. Furthermore, four adaptive sites revealed signs of recurring identical changes across phylogenetic groups of rainbow trout-adapted isolates, suggesting that repeated VHSV emergence in freshwater cultured rainbow trout was established through convergent routes of evolution that are associated with immune escape.
IMPORTANCE This study is the first to demonstrate that VHSV emergence from marine species into freshwater cultured rainbow trout has been accompanied by bursts of adaptive evolution in the VHSV glycoprotein. Furthermore, repeated detection of the same adaptive amino acid sites across phylogenetic groups of rainbow trout-adapted isolates indicates that adaptation to rainbow trout was established through parallel evolution. In addition, signals of convergent evolution toward the maintenance of genetic variation were detected in the conformation-dependent neutralizing epitope or in close proximity to disulfide bonds involved in the structural conformation of the neutralizing epitope, indicating adaptation to immune response-related genetic variation across freshwater cultured rainbow trout.
KEYWORDS: VHSV, viral hemorrhagic septicemia virus, adaptation, convergent evolution, host-pathogen interactions, negative-strand RNA virus, viral emergence
INTRODUCTION
RNA viruses are counted among the most rapidly evolving biological entities on earth. They have high replication rates and large population sizes (1, 2). Furthermore, they have exceptionally high mutation rates due to an error-prone RNA polymerase without proofreading ability (1, 3). Consequently, RNA viruses exhibit high genetic variation that provides the potential for rapid adaptation to environmental changes or new host species (4–6). Despite this, only a fraction of RNA viruses are able to emerge successfully in new host species. Even though viruses occasionally cross species barriers, they often fail to adapt to the new host and to establish a sustained transmission network (4–6).
In contrast, viral hemorrhagic septicemia virus (VHSV) has repeatedly crossed species barriers, adapted to new host species, and established sustained within-species transmission networks. VHSV thus represents a rare opportunity to investigate repeated viral emergence under natural conditions.
VHSV is a single-stranded RNA virus of negative polarity that belongs to the genus Novirhabdovirus within the family Rhabdoviridae (7, 8). It was initially isolated from freshwater cultured rainbow trout (Oncorhynchus mykiss) (9). Since then, it has been isolated from more than 80 marine and freshwater fish species (10), with conclusive evidence of susceptibility obtained for at least 44 of these species (10–13). Furthermore, it is assumed to be endemic among a wide range of marine and anadromous fish species in the Northern Hemisphere (10).
Phylogenetic studies have revealed four VHSV genotypes (I, II, III, and IV) (14–17), eight sublineages (Ia, Ib, Ic, Id, Ie, IVa, IVb, and IVc) (14, 18–20), and two clades (Ia-1 and Ia-2) (21). This classification of genotypes, sublineages, and clades is used henceforth.
In general, isolates adapted to cultured rainbow trout group into separate phylogenetic clusters, whereas isolates adapted to marine host species do not form host-specific clusters. Rainbow trout-adapted isolates generally belong to sublineages of genotype I (except Ib), whereas isolates adapted to marine host species are represented in all four genotypes. Within genotype I, rainbow trout-adapted isolates belong to sublineages Ia, Ic, and Id, whereas marine host-adapted isolates belong mainly to sublineages Ib and Ie.
Moreover, phylogenetic analyses have demonstrated that rainbow trout-adapted isolates have a marine ancestry, indicating that VHSV has successfully emerged from the multispecies marine reservoir into stocks of cultured rainbow trout (22). Recent transmission events reported for rainbow trout reared in marine and brackish waters in Finland (23, 24), Norway (25), and Sweden (26, 27) indicate that VHSV emergence from the marine environment into cultured rainbow trout might occur occasionally (22).
Mechanisms directly involved in VHSV emergence in cultured rainbow trout have not been identified. However, many viral and environmental factors have been proposed to facilitate and enhance VHSV emergence (22). In addition to the rearing conditions and close environmental contact described by Kurath and Winton (22), a broad marine multihost reservoir may provide the potential to establish and maintain a large and continuously changing spectrum of genetic variation that might facilitate VHSV emergence.
One key component of VHSV cross-species transmission and successful emergence is the viral glycoprotein (G protein), which is responsible for host receptor attachment and cell entry. It represents the only viral protein exposed on the viral surface and has been demonstrated to be targeted directly by the host immune system (28–31). The G protein is thus involved in at least three of the crucial steps of viral emergence.
Viral emergence in a new host species represents an environmental change that is likely to exert novel selective pressures on the virus. Hence, emergence is followed by genetic changes promoting adaptation to the new pressures. Such changes are often traceable in the genome by investigating the substitution rate ratio, ω (ω = dN/dS) (32). The normalized rate of nonsynonymous substitutions (dN) exceeds the rate of synonymous substitutions (dS) only in regions where certain alterations of amino acids offer selective advantages and thus become fixed at rates higher than those of (presumably mostly neutral) synonymous changes (33). The substitution rate ratio (ω = dN/dS) thus provides an indication of selection pressures at the protein level, with ω values of 1, <1, and >1 indicating the presence of neutral evolution and purifying and positive selection pressures, respectively.
The purposes of the present study were (i) to test for adaptive molecular evolution associated with VHSV emergence in freshwater cultured rainbow trout, (ii) to investigate whether particular amino acid sites undergo positive selection, and (iii) to explore whether VHSV emergence in freshwater cultured rainbow trout follows a repeatable pattern. Evidence for positive selection during the process of adaptation to freshwater cultured rainbow trout was detected, and several amino acid sites potentially involved in rainbow trout adaptation were identified by a maximum likelihood analysis using codon substitution models that allow for heterogeneous substitution rate ratios among amino acid sites. Among those sites, distinct amino acid combinations were identified, and they appeared to be related to the host origin.
RESULTS
Phylogeny.
A maximum likelihood phylogeny based on the complete coding region of the VHSV G protein was constructed to assess the genetic relationships of 198 unique VHSV G gene sequences. The majority of VHSV isolates clustered into previously defined genotypes, sublineages, and clades, with bootstrap support levels exceeding 75% (genotype I, 94%; genotype II, 99%; genotype III, 100%; genotype IV, 98%; sublineage Ia, 86%; sublineage Ib, 95%; sublineage Ic, 100%; sublineage Id, 78%; sublineage VIa, 100%; sublineage IVb, 100%; clade Ia-1, 89%; and clade Ia-2, 80%) (Fig. 1; Table 1).
FIG 1.

Maximum likelihood phylogeny showing the relationships of 198 unique full-length VHSV G gene sequences, illustrated by a radial unrooted tree (A) and a rooted phylogram (B). Phylogenies were reconstructed using the GTR+G+I substitution model with 1,000 bootstrap replicates and were rooted using snakehead rhabdovirus (SRHR) (GenBank accession no. AF147498) as a genetically distinct outgroup. Branch lengths in panel A and horizontal branch lengths in panel B reflect genetic distances, and the associated scale bars refer to the expected number of substitutions per site. Color coding of tree tips represents the host species (green, rainbow trout; dark blue, non-rainbow trout), and the tree tip shapes represent the host environment (circles, freshwater; triangles, marine/brackish water). Values at nodes represent percent support from bootstrap replicates, with a bootstrap cutoff of 75%. The color and font type of bootstrap values indicate genotype branching events (black and bold), sublineage branching events (colored and bold), and clade branching events (colored and italic). Vertical bars to the right of the tree indicate isolate associations with genotypes I, III, II, and IV (top to bottom; black labels in bold), sublineages Ia, If, Ib, Id, Ic, Ie, IVb, and IVa (top to bottom; colored labels in bold), and clades Ia-1, Ia-3, Ia-4, and Ia-2 (top to bottom; colored labels in italics).
TABLE 1.
Description of defined sub-data sets based on the phylogenetic reconstruction conducted in this study
| Data seta | Sample size | Bootstrap value (%) | Host speciesb | Geographic origin(s)c |
|---|---|---|---|---|
| Genotypes | ||||
| I | 160 | 94 | M, FW (mainly RT) | Europe (see sublineages and clades) |
| II | 13 | 99 | M | Europe (Baltic Sea) |
| III | 9 | 100 | M | Europe (Atlantic Sea, Skagerrak, and Kattegat) |
| IV | 16 | 98 | M, FW (no RT) | North America and Asia |
| Sublineages | ||||
| Ia | 124 | 86 | Freshwater cultured RT, 1 T, 1 G | Europe (see clades) |
| Ib | 18 | 95 | M | Baltic Sea, Skagerrak, Kattegat, North Sea, English Channel |
| Ic | 7 | 100 | Freshwater cultured RT (prior to 1997) | AT (1), DE (3), DK (3) |
| Id | 3 | 78 | Marine cultured RT | Gulf of Finland |
| Ie | 1 | RT | GE (1) | |
| Ifd | 5 | 99 | Freshwater cultured RT | DE (3), FR (2) |
| IVa | 14 | 100 | M | CA (5), JP (3), KR (3), US (3) |
| IVb | 2 | 100 | FW (no RT) | Great Lakes |
| Clades | ||||
| Ia-1 | 49 | 89 | Freshwater cultured RT, 1 T, 1 G | DK (40), DE (8), UK (1) |
| Ia-2 | 64 | 80 | Freshwater cultured RT | AT (5), CH (1), DE (51), DK (5), FR (1), SI (1) |
| Ia-3e | 5 | 99 | Freshwater cultured RT | DE (2), DK (3) |
| Ia-4e | 5 | 99 | Freshwater cultured RT | DE (1), DK (4) |
Sub-data sets in bold were subjected to molecular evolutionary analysis with CODEML. The remaining sub-data sets violated requirements for evolutionary analysis (bootstrap support of at least 75%, sample size of at least 7 isolates, no phylogenetic subdivision containing marine- and rainbow trout-adapted phylogenetic subgroups). Deviations in sample size within genotype I were caused by two isolates that were affiliated with genotype I but did not cluster into any of the sublineages or clades and one isolate that was affiliated with sublineage Ia but did not cluster into any clade.
M, marine species; FW, freshwater species; RT, rainbow trout; T, turbot; G, grayling.
Geographic origins of rainbow trout-adapted isolates are specified to the country level, and numbers in parentheses indicate the numbers of isolates originating from the corresponding countries. AT, Austria; CA, Canada; CH, Switzerland; DE, Germany; DK, Denmark; FI, Finland; FR, France; GE, Georgia; IE, Ireland; JP, Japan; KR, South Korea; NO, Norway; SE, Sweden; SI, Slovenia; UK, United Kingdom; US, United States.
Novel phylogenetic sublineage identified in this study.
Novel phylogenetic clade identified in this study.
In addition, a potential new sublineage within genotype I was identified and denoted If, while two potential new clades within sublineage Ia were identified and denoted Ia-3 and Ia-4. The new sublineage, If, had a bootstrap support level of 71%. It contained five isolates recovered from freshwater cultured rainbow trout, three of German and two of French origin. Clades Ia-3 and Ia-4 each had a bootstrap support level of 99%. Both clades contained European isolates of Danish or German origin from freshwater cultured rainbow trout. Detailed information on isolates can be found in Table 1 and in Table SA1 in the supplemental material.
Apart from one isolate (FA280208-VG; originating from marine cultured rainbow trout), isolates from rainbow trout belonged exclusively to genotype I, where they clustered in sublineages Ia, Ib, Ic, Id, Ie, and If. Sublineages Ia, Ic, and If represented isolates recovered from freshwater cultured rainbow trout. Sublineage Id represented isolates recovered from rainbow trout cultured in brackish water in the Gulf of Finland (24). Sublineage Ib contained two isolates recovered from sea-reared rainbow trout. The majority of Ib isolates included in this study, however, were recovered from marine host species. Sublineage Ie contained an isolate recovered from freshwater cultured rainbow trout in Georgia (GE-1.2; included in this study) after marine water had flooded the farm, while the majority of Ie isolates (not included in this study) were recovered from marine host species in the Black Sea (34). In contrast, isolates originating from marine host species were found across all four major genotypes.
The phylogenetic reconstruction confirmed a marine ancestry of rainbow trout-adapted isolates. However, it neither rejected nor confirmed the previous hypothesis about repeatedly occurring independent instances of VHSV emergence in cultured rainbow trout due to low bootstrap support of inner branching orders within genotype I. Despite this, the phylogenetic reconstruction suggested the occurrence of several cross-species transmission events from marine species to freshwater cultured rainbow trout (formation of sublineages Ia, Ic, and If) and from marine species to marine cultured rainbow trout (formation of sublineage Id and rainbow trout-adapted isolates in sublineages Ib and Ie and in genotype III) (Fig. 1).
Molecular evolutionary analysis. (i) Detection of adaptive evolution.
The presence of adaptive evolution in the VHSV G protein was investigated in separate parts of the phylogeny using CODEML, a maximum likelihood-based program implemented in the software package PAML 4.6 that applies different codon substitution models, allowing for heterogeneous substitution rate ratios among amino acid sites to determine site-specific selection pressures. The average ω value (averaged across all amino acid sites of the G protein) observed within each investigated data set did not exceed 0.36, indicating a predominance of purifying selection acting upon the G protein (Table SA3). Nevertheless, likelihood ratio tests conducted to compare codon substitution models (M1 versus M2 as well as M7 versus M8) demonstrated the presence of local positive selection in some data sets (sublineage-specific data sets Ia and Ic and clade-specific data sets Ia-1 and Ia-2) (Table 2). Evidence of positive selection was detected in all investigated data sets representing isolates recovered from freshwater cultured rainbow trout. In contrast, data sets representing isolates recovered from host species other than rainbow trout (mainly of marine origin) did not show any evidence of positive selection (genotype-specific data sets II, III, and IV and sublineage-specific data sets Ib and IVa). This was further supported by results established with BUSTED, where evidence of positive selection was detected only when isolates recovered from freshwater cultured rainbow trout were selected as a foreground branch (sublineage Ic versus the rest, clade Ia-2 versus the rest, and rainbow trout versus non-rainbow trout isolates) (Table SA4).
TABLE 2.
LRT statistics used to detect presence of positive selection pressurea
| Data setb | Environment | M1 vs M2 |
M7 vs M8 |
||
|---|---|---|---|---|---|
| LRT | P value | LRT | P value | ||
| Genotypes | |||||
| I | M, FW, wildlife, cultured | NA | NA | NA | NA |
| II | M, wildlife | 0.000 | NS | −0.001 | NS |
| III | M, wildlife | 1.578 | NS | 1.586 | NS |
| IV | M, FW, wildlife, cultured | 0.762 | NS | 1.083 | NS |
| Sublineages | |||||
| Ia | FW, cultured RT | 68.156 | <0.0001 | 68.957 | <0.0001 |
| Ib | M, wildlife, cultured | 1.599 | NS | 1.842 | NS |
| Ic | FW, cultured RT | 12.089 | 0.0024 | 12.282 | 0.0022 |
| Id | M, cultured RT | NA | NA | NA | NA |
| Ie | M, wildlife | NA | NA | NA | NA |
| If | FW, cultured RT | NA | NA | NA | NA |
| IVa | M, wildlife, cultured | 1.102 | NS | 1.107 | NS |
| IVb | FW, wildlife, cultured | NA | NA | NA | NA |
| Clades | |||||
| Ia-1 | FW, cultured RT | 31.102 | <0.0001 | 33.722 | <0.0001 |
| Ia-2 | FW, cultured RT | 21.049 | <0.0001 | 26.423 | <0.0001 |
| Ia-3 | FW, cultured RT | NA | NA | NA | NA |
| Ia-4 | FW, cultured RT | NA | NA | NA | NA |
LRTs were conducted for nested discrete models (M1 and M2) as well as nested continuous models (M7 and M8), comparing twice the log-likelihood difference to a χ2 distribution (df = 2). Significant LRT results demonstrate evidence of positive selection pressure. M, marine or brackish water; FW, freshwater; RT, rainbow trout; NS, not significant; NA not analyzed.
Data sets in bold were subjected to molecular evolutionary analysis with CODEML.
(ii) Detection of amino acid sites under adaptive evolution.
To identify amino acid sites under positive selection pressure per site, posterior probabilities were estimated using the Bayes empirical Bayes approach implemented in CODEML. Six amino acid sites were found to be under positive selection pressure, including amino acid 6 (aa6), aa212, aa258, aa259, aa505, and aa506. These amino acid sites were located within the N-terminal signal peptide of the G protein (aa6); the central region of the G protein (aa212, aa258, and aa259), corresponding to the region hosting the major neutralizing epitope (28); and the C-terminal and intracellular region of the G protein (aa505 and aa506). Of those, amino acid sites aa212, aa258, aa505, and aa506 were also detected using the FUBAR algorithm with a posterior probability threshold of 0.95 (Table SA5).
Three of the six adaptive amino acid sites (aa212, aa258, and aa259) were detected across phylogenetic groups. All three sites were shared between sublineage Ia, clade Ia-1, and clade Ia-2. The three remaining adaptive amino acid sites (aa6, aa505, and aa506) were data set specific, with aa6 detected in sublineage-specific data set Ic, aa505 detected in sublineage-specific data set Ia, and aa506 detected in clade-specific data set Ia-2 (Table 3). Relaxing the posterior probability threshold for adaptive amino acid sites from 0.95 to 0.7 increased the number of shared adaptive amino acid sites for data sets Ia, Ia-1, and Ia-2, with aa212, aa258, aa259, and aa506 found to be under positive selection in all three data sets and aa505 found to be under positive selection in data sets Ia and Ia-2. However, adaptive amino acids detected in data set Ic still remained data set specific (Table 3).
TABLE 3.
Amino acid sites detected under positive selection pressure in VHSV with associated posterior probability estimates and potential functional relevancea
| Amino acid | Probability of positive selectionb |
Location | |||
|---|---|---|---|---|---|
| Ia | Ia-1 | Ia-2 | Ic | ||
| 6c | 0.990 | Signal peptide | |||
| 8c | 0.790 | Signal peptide | |||
| 212 | 1.000 | 0.997 | 0.982 | Proximity of Cys215 | |
| 214 | 0.907 | Proximity of Cys215 | |||
| 229 | 0.823 | UNK | |||
| 258 | 1.000 | 1.000 | 0.999 | Conformation-dependent neutralizing epitope, proximity of Cys256 | |
| 259 | 1.000 | 0.966 | 0.993 | Conformation-dependent neutralizing epitope | |
| 284 | 0.838 | Conformation-dependent neutralizing epitope, proximity of Cys285 | |||
| 290 | 0.869 | UNK | |||
| 476 | 0.810 | UNK | |||
| 486 | 0.850 | Proximity of membrane, extracellular side | |||
| 492 | 0.911 | UNK | |||
| 505 | 0.979 | 0.887 | Proximity of membrane, intracellular side | ||
| 506 | 0.904 | 0.768 | 0.973 | Proximity of membrane, intracellular side | |
Amino acid sites under positive selection pressure were detected by employing substitution model M8. UNK, unknown.
Values in bold indicate posterior probabilities above 95%; missing values indicate posterior probabilities below 70%.
Amino acid belonging to the signal peptide, which is removed at the endoplasmic reticulum (ER) during protein synthesis.
(iii) Haplotypes at amino acid sites under positive selection.
For each phylogenetic group, the genetic variation at the six adaptive amino acid sites was recorded (Table 4; Table SA1), the amino acid combinations across those sites were identified, and the distributions of the positive selection site-restricted haplotypes were compared across phylogenetic groups. In total, 46 unique haplotypes were identified and were denoted H1 to H46. Only nine haplotypes were detected among the 55/198 G gene sequences representing VHSV isolates from species other than rainbow trout and collected worldwide, whereas 41 haplotypes were found among the remaining 143 G gene sequences representing rainbow trout-adapted VHSV isolates that were collected across Europe. Only four haplotypes (H28, H36, H37, and H38) were detected both among isolates from rainbow trout and among non-rainbow trout isolates. Haplotype H38 was found both in isolates SE-SVA-1033 and SE-SVA14, from sea-reared rainbow trout belonging to sublineage Ib, and in 27 isolates collected from marine species from the same geographic area. Haplotype H37 of isolate FR-L59X, from wild European eel (Anguilla anguilla) collected in brackish water (35), was also found in sublineages Ic and If, representing isolates from freshwater cultured rainbow trout from the same sampling period. Haplotype H36 of the GE-1.2 isolate, recovered from a rainbow trout farm flooded by marine water from the Black Sea, was furthermore found among marine genotype III isolates. Finally, haplotype H28 of four older rainbow trout isolates belonging to clade Ia-1 was also found in the turbot isolate Dsteinbutt, phylogenetically grouped into the same clade. The remaining 42 haplotypes were specific to either rainbow trout- or non-rainbow trout-adapted isolates. Four haplotypes (H33, H34, H35, and H40) were specific to VHSV isolates adapted to marine host species other than rainbow trout, two haplotypes (H9 and H32) were specific to VHSV isolates from freshwater host species other than rainbow trout, and 36 haplotypes were specific to rainbow trout-adapted isolates (33 for freshwater rainbow trout, 2 for marine rainbow trout, and 1 for marine and freshwater rainbow trout).
TABLE 4.
Amino acid combinations observed for amino acid sites under positive selection pressure (positive selection site-restricted haplotypes)a
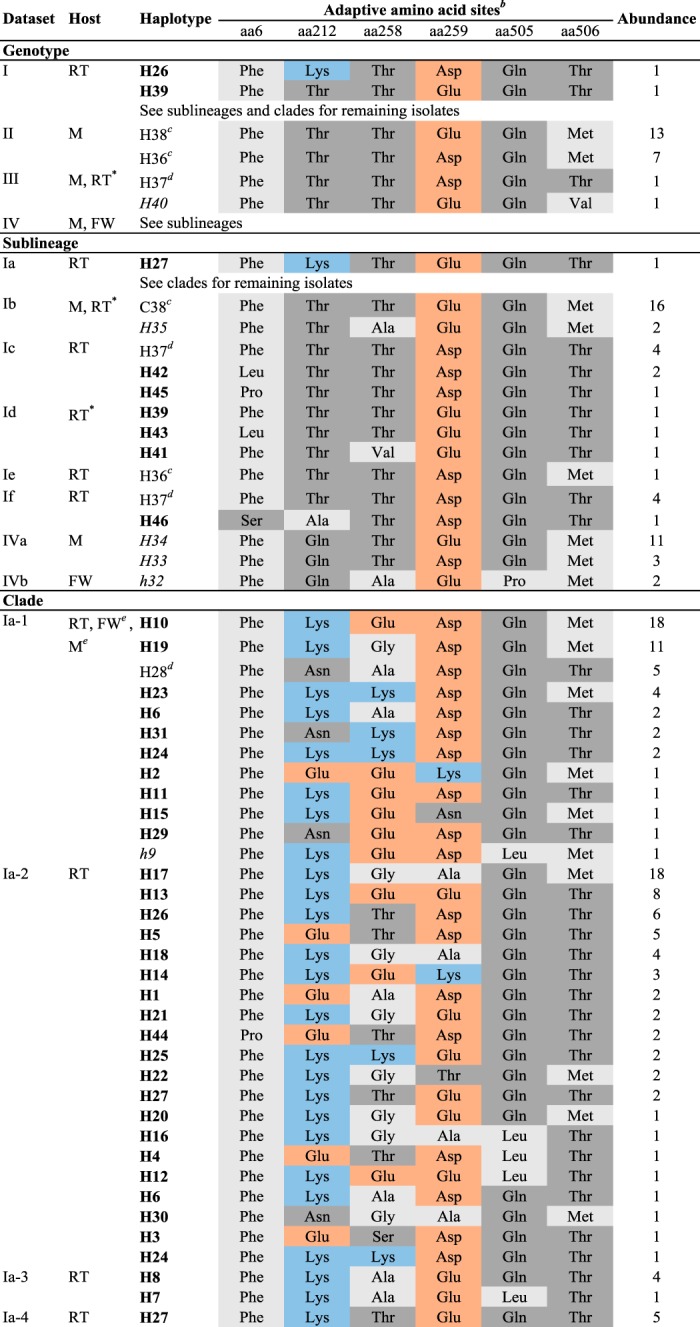
Haplotypes marked in bold are specific to rainbow trout-adapted isolates (n = 36). Haplotypes in italics are specific to non-rainbow trout-adapted isolates of marine origin (n = 4). Haplotypes in italics and indicated by a lowercase “h” are specific to non-rainbow trout-adapted isolates of freshwater origin (n = 2). Observed haplotypes and haplotype abundance are indicated for each phylogenetic group separately. Abbreviations: RT, rainbow trout; M, marine and brackish water species; FW, freshwater species other than rainbow trout. *, sea-reared rainbow trout.
bAmino acids are color coded according to chemical characteristics, as follows: light gray, neutral, nonpolar amino acids; dark gray, neutral, polar amino acids; orange, acidic amino acids; and blue, basic amino acids. Amino acid abbreviations are as follows: Asn, asparagine; Leu, leucine; Pro, proline; Asp, aspartate; Glu, glutamate; Gln, glutamine; Lys, lysine; Thr, threonine; Ala, alanine; Val, valine; Ser, serine; Phe, phenylalanine; Gly, glycine; and Met, methionine.
cHaplotypes detected in non-rainbow trout- and rainbow trout-adapted isolates, with an overrepresentation in non-rainbow trout-adapted isolates and rainbow trout-adapted isolates representing an exception.
dHaplotypes shared by non-rainbow trout- and rainbow trout-adapted isolates, with an overrepresentation in rainbow trout-adapted isolates and non-rainbow trout-adapted isolates representing an exception.
eOnly a single isolate, representing an exception for this phylogenetic group.
(iv) Patterns of adaptive evolution at amino acid sites under positive selection.
Substitution patterns detected at amino acid sites under positive selection revealed parallel evolution toward the maintenance of genetic variation or toward the same amino acid (convergent evolution). Evolution toward the maintenance of genetic variation was detected at amino acid sites aa212, aa258, and aa259 in sublineage Ia, especially within clades Ia-1 and Ia-2, indicating that genetic variation within the neutralizing epitope gives an evolutionary advantage in the majority of rainbow trout-adapted isolates. In contrast, rainbow trout-adapted isolates belonging to sublineages Ic, Id, Ie, and If were almost genetically fixed at aa212, aa258, and aa259, with amino acids resembling those found in marine isolates (Table 5).
TABLE 5.
Results for individually analyzed data sets and observed amino acid substitutions at amino acid sites identified to undergo positive selection pressureb
aAmino acids are color coded according to chemical characteristics, as follows: light gray, neutral, nonpolar amino acids; dark gray, neutral, polar amino acids; orange, acidic amino acids; and blue, basic amino acids. Amino acids are furthermore ranked according to abundance, starting with the most prevalent amino acid. Numbers in parentheses indicate abundances. Amino acid abbreviations are as follows: Asn, asparagine; Leu, leucine; Pro, proline; Asp, aspartate; Glu, glutamate; Gln, glutamine; Lys, lysine; Thr, threonine; Ala, alanine; Val, valine; Ser, serine; Phe, phenylalanine; Gly, glycine; and Met, methionine.
Abbreviations: RT, rainbow trout; M, marine and brackish water species; FW, freshwater species other than rainbow trout. *, sea-reared rainbow trout.
Moreover, substitution patterns observed for aa212, aa258, and 259 in clades Ia-1 and Ia-2 were not random but were limited to a specific set of polymorphic changes of identical or similar amino acids across phylogenetic groups (Table 5). Amino acids observed at those sites differed in chemical characteristics from amino acids observed at corresponding amino acid sites in marine isolates. Amino acid sites aa212 and aa258 of marine isolates were characterized by neutral-polar (threonine and glutamine) or neutral-nonpolar (alanine and valine) amino acids. In contrast, in the majority of rainbow trout-adapted isolates (sublineage Ia), aa212 showed an overrepresentation of basic (lysine) or acidic (asparagine and glutamic acid) amino acids, especially within clades Ia-1 and Ia-2, and aa258 was mainly characterized by basic (lysine), acidic (glutamic acid), and neutral (glycine, alanine, and threonine) amino acids (Table 5). The same but less-pronounced patterns were observed for aa259. At this site, however, acidic amino acids were present in marine isolates, and only the basic and neutral amino acids were specific to rainbow trout-adapted isolates.
Strictly convergent evolution was detected at amino acid site aa506, representing an amino acid change from methionine in marine isolates toward threonine in rainbow trout-adapted isolates. Threonine at aa506 was specific to rainbow trout-adapted isolates and represented the major amino acid variant in rainbow trout-adapted isolates (for threonine, n for rainbow trout [nRT]= 80 isolates; for methionine, nRT = 63 isolates) (Table 5). Furthermore, the amino acid change from methionine to threonine was not restricted to isolates adapted to freshwater cultured rainbow trout but was furthermore observed in isolates adapted to marine cultured rainbow trout.
Structural analysis.
Structural prediction of VHSV G proteins of 3 isolates was performed using the G protein of vesicular stomatitis virus (VSV) as a reference. The G gene sequence identity for VHSV and VSV was 20.5% for DK-3592b (sublineage Ia isolate of rainbow trout origin), 19.8% for DK-5e59 (sublineage Ib isolate of marine origin), and 21.3% for BC93372 (sublineage IVa isolate of marine origin). Predictions based on template and alignment information alone obtained GA341 scores below the recommended threshold of 0.6 (36). Protein structure predictions, including information on previously identified disulfide bond locations (Cys49—Cys359, Cys64—Cys315, Cys110—Cys152, Cys192—Cys197, Cys215—Cys285, and Cys251—Cys256) (37) in addition to template and alignment information, in contrast, achieved high reliability scores, exceeding 0.8 (for DK-3592b, 0.82; for DK-5e59, 0.98; and for BC93372, 0.99), and thus facilitated the investigation of amino acid sites under positive selection in a structural context.
Mapping of codon sites under positive selection onto the protein structure revealed that aa212, aa258, and aa259 are structurally located in close proximity to each other (Fig. 2A and B). Furthermore, all residues at codon sites experiencing positive selection were located on the protein surface, and all extracellular sites under positive selection pressure were located in the pleckstrin homology domain that holds the major neutralizing epitope (Fig. 2B and 3B and C) (38).
FIG 2.

Predicted three-dimensional (3D) protein structure of the extracellular region (aa21 to aa462) of the VHSV G protein (accession no. KC778774) from the rainbow trout-adapted freshwater isolate DK-3592b, a representative of sublineage Ia. The predicted protein structure was used to visualize the location (A), surface exposure (B), and disulfide bond proximity (C) of amino acid sites under positive selection pressure. Residues colored red (bold position numbers) and yellow (position numbers not in bold) represent sites under positive selection pressure, with probabilities exceeding 95% and 70%, respectively. The coordinate axes in the lower left corner of each protein presentation indicate the protein orientation. (A) NewCartoon representation of the predicted protein structure, providing a simplified representation of the protein based on secondary structure. Alpha-helices are drawn as coiled ribbons, beta-sheets as solid arrows, and all other structures as tubes. Amino acid sites under positive selection pressure are visualized for each corresponding phylogenetic group detected under adaptive evolution pressure. (B) QuickSurface representation providing a spatial representation of the predicted protein structure. Residues 212 to 284, corresponding to the region hosting the major neutralizing epitope, are shown in green. The 3D structure was rotated clockwise to visualize the surface exposure of adaptive amino acid sites in three different orientations. (C) NewCartoon representation of the predicted protein structure (details as described for panel A), showing locations of previously detected disulfide bonds and their proximity to adaptive amino acid sites. Disulfide bonds are indicated by color-coded cysteine residues, which are shown in VDM representation and color coded by atomic mass (Cys position numbers are shown in italics).
FIG 3.

Comparison of predicted 3D protein structures of the VHSV G protein across phylogenetic groups. Structural prediction of the extracellular region (aa21 to aa462) of the VHSV G protein in postfusion conformation was performed for isolate DK-3592b (Ia), isolate DK-5e59 (Ib), and isolate BC93372 (IVa). (A) Predicted proteins (DK-3592b, yellow; DK-5e59, light gray; and BC93372, dark gray) are shown in NewCartoon representation, providing a simplified representation of the postfusion protomers based on secondary structure. Alpha-helices are drawn as coiled ribbons, beta-sheets as solid arrows, and all other structures as tubes. All predicted protein structures are shown in identical protein orientations. The coordinate axes located in the lower left corner indicate the protein orientation. Residues colored red represent sites under positive selection pressure, with a probability exceeding 95%. Residues colored blue represent the conformation-dependent major neutralizing epitope (MNE) (aa212 to aa284). The N terminus of the predicted protein structure is colored cyan blue (aa21). The last residue of the extracellular region of the VHSV G protein structure (aa462) is colored magenta, indicating the location of the adjacent transmembrane region (TM), which was removed from the prediction. (B) QuickSurface representation of the predicted protein structures (DK-3592b, yellow; DK-5e59, light gray; and BC93372, dark gray), providing a spatial representation of the protein. Color coding of residues is as described for panel A. For ease in structural comparisons, structure trajectories of distinctive protein grooves detected in DK-3592b were traced by use of bold lines (red and blue) and subsequently transposed onto the other two protein predictions. (C) Predicted protein structure of DK-3592b, showing the following putative protein domains previously identified for the VSV G protein: DI, beta-sheet-rich lateral domain (red); DII, central domain involved in trimerization of G protomers (blue); DIII, pleckstrin homology domain holding the conformation-dependent major neutralizing epitope (orange); and DIV, fusion domain (yellow).
Visual comparison of the three predicted VHSV G protein structures suggested slight changes in the postfusion conformation, which affects the groove structure in close proximity to the major neutralizing epitope, and a slight bending of the fusion domain, especially in the sublineage IVa isolate (Fig. 3). In addition, the surface exposure of the sites under positive selection in the major neutralizing epitope differed slightly between isolates (Fig. 3).
DISCUSSION
Repeated emergence of VHSV from marine host species into freshwater cultured rainbow trout is accompanied by bursts of adaptive evolution in the G protein. Positive selection occurs in phylogenetic groups containing isolates adapted to freshwater cultured rainbow trout, whereas phylogenetic groups containing isolates adapted to marine host species do not show evidence of positive selection. Furthermore, we detected indications of parallel evolution across phylogenetic groups of rainbow trout-adapted isolates, suggesting VHSV adaptation to cultured rainbow trout through convergent routes of evolution.
Detection of adaptive evolution.
Evidence of adaptive evolution in the VHSV G protein was demonstrated through the detection of amino acid sites under positive selection pressure in separate parts of the phylogeny. The detection of amino acid sites under positive selection pressure was restricted to data sets representing phylogenetic groups of isolates originating from freshwater cultured rainbow trout. This indicates that emergence of VHSV in freshwater cultured rainbow trout is accompanied by a burst of adaptive evolution in the G protein following cross-species transmission. Adaptive evolution in external proteins of mammalian RNA viruses has been reported repeatedly, including for rabies virus (39–41), VSV (42–44), foot-and-mouth disease virus (45), influenza A virus (46, 47), dengue virus (48), and infectious hematopoietic necrosis virus (IHNV) (49). Those studies revealed that only a small percentage of amino acid sites showed evidence of positive selection pressure, whereas the most prevalent evolutionary force was purifying selection. This is in general agreement with the results obtained in this study. Evidence of positive selection pressure was detected at only 6 of 507 amino acid sites investigated across the VHSV G protein. The average ω value for the G protein observed within each individually investigated phylogenetic group did not exceed 0.36, indicating an action of purifying selection, which reflects a high level of structural constraints to maintain protein function. Residues undergoing positive selection were not detected among marine isolates. Whether this reflects a sampling bias of fewer marine isolates per host species than the value for included rainbow trout isolates or differences in substitution and replication rates in the different environments (14) that might reduce the power of detecting selection footprints needs further investigation. However, we speculate that VHSV isolates might have developed different adaptation strategies, i.e., a specialized strategy for high-density monocultured host species and a more general strategy that can infect multiple, less-dense host species in nature.
Sites detected under adaptive evolution.
Three of the six amino acid sites detected under positive selection pressure (aa212, aa258, and aa259) were located in the midregion of the G protein, within the conformation-dependent major neutralizing epitope, ranging from aa212 to aa288 (28). Positive selection within the conformation-dependent major neutralizing epitope was further supported by complementary detection approaches. In agreement with previous findings, the majority of adaptive amino acid sites detected in the G protein of IHNV, a novirhabdovirus closely related to VHSV, were located within the conformation-dependent neutralizing epitope (49–51). Furthermore, Huang and coworkers (50) demonstrated that changes in the midregion of the IHNV G protein enabled mutants to escape from neutralizing monoclonal antibodies.
Amino acid sites aa212, aa258, and aa259, moreover, were located in close proximity to two of the six disulfide bonds detected in the VHSV G protein (37), namely, Cys215—Cys285 and Cys251—Cys256 (Fig. 2C). Both disulfide bonds are involved in the structural conformation of the neutralizing epitope. The Cys215—Cys285 disulfide bond spans the neutralizing epitope, whereas the Cys251—Cys256 bond forms a loop-like structure within the neutralizing epitope. It was previously demonstrated that the Cys215—Cys285 bond is essential for correct folding of the conformation-dependent neutralizing epitope of the VHSV G protein (37). Amino acid changes in close proximity to either of the two disulfide bonds thus might affect bonding properties and potentially cause conformational changes of the neutralizing epitope. However, further research is needed to determine whether changes at positively selected amino acid sites aa212, aa258, and aa259 enable VHSV to either directly escape neutralizing antibodies or provide the mutants with a selective advantage by facilitating escape from other host immune responses.
In addition, evidence of positive selection was detected in the N-terminal region of the G protein, within the signal peptide (aa6), as well as in the C-terminal region, located on the intracellular side of the cell membrane (aa505 and aa506). Adaptive changes within the signal peptide might indicate differences in the host cell environment, as the viruses may require continuous adjustment of intrinsic signals that govern movement within the cell. However, positive selection at aa6 (within the signal peptide) was not confirmed by complementary detection approaches, and the actual biological impact is beyond the scope of this study and needs further investigation. In contrast, adaptive changes in the C-terminal region were confirmed by complementary detection approaches. These changes might indicate responses to species-specific differences in the host membrane. Alternatively, adaptive changes in the C-terminal region, located on the intracellular side of the cell membrane, might be a response to adaptive changes in the matrix (M) protein of VHSV. Efficient budding of VHSV particles is the result of a concerted action of both the M protein and cell membrane-associated G proteins (7, 52, 53), where M proteins mediate the transport of ribonucleocapsid molecules to the host cell membrane and subsequently initiate viral assembly and budding of new VHSV virions through interaction with the cytoplasmic tails of the membrane-bound G proteins (7, 53). Hence, adaptive changes observed at aa505 and aa506 might indicate coevolution with the M protein, representing selection pressures acting upon viral assembly and cell budding.
Convergent routes of VHSV evolution.
Four adaptive amino acid sites identified in this study (aa212, aa258, aa259, and aa506) revealed indications of convergent evolution following VHSV emergence in freshwater cultured rainbow trout. These findings are in general agreement with studies of foot-and-mouth disease virus (45), VSV (42–44), and bacteriophages (54) that demonstrated viral adaptation through highly convergent routes of evolution. Convergent evolution may furthermore be promoted by the small genome size and the strong functional constraints of RNA virus proteins. However, convergent evolution has been demonstrated mainly under laboratory conditions representing a highly homogenous environment. Adaptive evolution investigated under natural conditions, in rabies virus (41), influenza A virus (55, 56), and IHNV (49), in contrast, revealed that viral adaptation was established through alternative routes of evolution. Nonetheless, this study demonstrates the recurrence of identical changes in connection with independent instances of viral emergence in the same host species (i.e., the existence of convergent viral evolution) under natural conditions.
The strongest incidence of convergent evolution across phylogenetic groups was detected for amino acid site aa506. Convergent evolution at aa506 was characterized by a recurrent change from methionine to threonine. Although significant evidence of positive selection pressure at aa506 was detectable only for clade Ia-2, a comparison of substitution patterns at aa506 across phylogenetic groups revealed that an amino acid change toward threonine was common to all phylogenetic groups representing rainbow trout-adapted isolates. Isolates originating from host species other than rainbow trout all carried methionine at aa506, whereas the majority of rainbow trout-adapted isolates carried threonine at this position. Furthermore, the amino acid change from methionine to threonine was not restricted to isolates adapted to freshwater cultured rainbow trout but was also observed in isolates belonging to sublineage Id, a phylogenetic group representing isolates adapted to marine cultured rainbow trout. In contrast, isolates adapted to marine cultured rainbow trout clustering into sublineage Ib and genotype III, phylogenetic groups mainly representing marine non-rainbow trout-adapted isolates, carried methionine and thus resembled the group of isolates adapted to marine host species. Assuming that marine isolates represent the ancestral stage, adaptation to rainbow trout is likely supported by an amino acid change from methionine to threonine at aa506. It remains to be explored whether the observed convergent evolution at this site might indicate membrane differences across host species or coevolution of the M and G proteins.
Further signals of convergent evolution across phylogenetic groups were detected at amino acid sites aa212, aa258, and aa259 for clades Ia-1 and Ia-2. Convergent evolution at those sites was characterized by maintenance of genetic variation, indicating that those sites allow for some degree of genetic variation without impairing vital protein functions. Detection of maintained genetic variation might be the result of relaxed selection pressure, but all three sites were identified under positive selection pressure and did not resemble a random set of polymorphic changes but instead a specific set of polymorphic changes containing the same amino acids across phylogenetic groups. Moreover, maintenance of genetic variation was not randomly located. Structural prediction of the VHSV G protein revealed that all three sites were found adjacent to each other in the groove structure associated with the pleckstrin homology domain (domain DIII) that holds the major neutralizing epitope (Fig. 2B and 3B and C) and were in close proximity to disulfide bonds involved in the structural conformation of the neutralizing epitope (Cys215—Cys285 and Cys251—Cys256) (Fig. 2C). Visual comparison of predicted protein structures across phylogenetic groups (Ia, Ib, and IVa) revealed minor structural differences in this specific groove region (Fig. 3A and B). Although structural differences were minor, one must keep in mind that the protein predictions were based on the postfusion structure of a G protein protomer. G proteins associated with the viral membrane, however, form trimers and are in a prefusion conformation when potentially exposed to the host immune system. In the prefusion conformation, the beta-sheet-rich lateral domain (DI) and the central domain involved in trimerization of G protomers (DII) are flipped to the inside of the trimer, and the major neutralizing epitope located within the DIII domain is exposed at the top of the protein (38, 57). Thus, slight structural changes in the groove associated with the major neutralizing epitope might cause important changes in the prefusion conformation, where antibody interaction takes place.
However, signals of convergent evolution in clades Ia-1 and Ia-2 might not be associated with independently recurring viral emergence in cultured rainbow trout but rather with descent from a single host shift event. Nonetheless, following branching, the evolutionary paths of the two phylogenetic groups continue to be parallel for the three sites detected under positive selection pressure, supporting maintenance of genetic variation as an adaptive process associated with viral emergence in cultured rainbow trout.
Surprisingly, the two other clades of sublineage Ia—Ia-3 and Ia-4—did not show maintenance of genetic variation. The same was true for the remaining phylogenetic groups representing rainbow trout-adapted isolates and potentially originating from independently recurring transmission events (Ic, Id, Ie, and If). Whether this resembles a sample size bias or an intermediate state of rainbow trout adaptation cannot be clarified here. Alternatively, the lack of positive selection pressure at aa212, aa258, and aa259 and the absence of genetic variation in rainbow trout-representing phylogenetic groups other than Ia-1 and Ia-2 might indicate that VHSV possesses the potential to adapt to rainbow trout through alternative routes of evolution, as previously demonstrated for IHNV (49). Sublineage-specific patterns of positive selection detected for sublineage Ic might further support this hypothesis. In sublineage Ic, only one amino acid site was shown to be under positive selection (aa6). None of the other phylogenetic groups representing rainbow trout-adapted isolates showed evidence of positive selection pressure at aa6. Lowering the posterior probability threshold to 70% increased the number of amino acid sites detected under positive selection pressure in sublineage Ic, but the sites detected were still sublineage specific. The same was true for comparable detection approaches. Hence, the existence of alternative routes of VHSV adaptation to freshwater cultured rainbow trout cannot be excluded. However, sublineage Ic represents a group of old isolates that were recovered prior to 1997, and since then, no further VHSV isolates clustering into sublineage Ic have been recovered. Hence, observed differences in positive selection patterns in sublineage Ic may represent an earlier stage in rainbow trout adaptation that most likely has been outcompeted by other rainbow trout-adapted isolates or eliminated as a result of frequent eradication (stamping-out) programs in this period.
Haplotype patterns of adaptive evolution in VHSV.
Amino acid combinations observed at amino acid sites under positive selection pressure revealed a large number of positive selection site-restricted haplotypes. In total, 46 different haplotypes were identified. However, only four haplotypes (H33, H34, H35, and H40) were specific to VHSV isolates from marine host species other than rainbow trout, and two haplotypes (H9 and H32) were specific to isolates from freshwater host species other than rainbow trout. In contrast, 36 haplotypes were specific to rainbow trout-adapted VHSV isolates (33 for freshwater rainbow trout, 2 for marine rainbow trout, and 1 for marine and freshwater rainbow trout). The remaining four haplotypes (H28, H36, H37, and H38) were found in both non-rainbow trout- and rainbow trout-adapted isolates. However, H28 and H37 were found mainly in rainbow trout-adapted isolates, with the exception of one isolate characterized by H28 that was recovered from a turbot, representing the only non-rainbow trout-adapted isolate in clade Ia-1, and one characterized by H37 that was recovered from eel captured in brackish water. In contrast, H36 and H38 were found mainly in non-rainbow trout-adapted isolates, with the exception of one rainbow trout-adapted isolate characterized by H36 that was recovered from freshwater cultured rainbow trout in Georgia after marine water from the Black Sea had flooded the farm and two rainbow trout-adapted isolates characterized by H37 that were recovered from sea-reared rainbow trout. Accordingly, VHSV isolates included in this study could be separated into non-rainbow trout- and rainbow trout-adapted isolates based on haplotypes restricted to the six amino acid sites detected under positive selection pressure.
The non-rainbow trout-adapted haplotypes were characterized by small neutral polar and nonpolar side chains (threonine, glutamine, and alanine) at aa212 and aa258, in combination with acidic side chains (aspartic acid and glutamic acid) at aa259 and methionine at aa506. In contrast, sublineage Ia-specific haplotypes, including 31/36 rainbow trout-specific haplotypes, were characterized by an overrepresentation of large basic and acidic side chains at either of the three central amino acid sites (aa212, aa258, and aa259), and the majority of those included a combination of at least two amino acids containing charged side chains (lysine, aspartic acid, glutamic acid). Moreover, amino acids with large basic side chains (lysine) at sites under positive selection pressure were found only for sublineage Ia and were overrepresented at position aa212. These findings indicate significant steric and polarity changes in the neutralizing epitope for rainbow trout-adapted isolates, likely reducing the recognition by neutralizing antibodies and nicely supporting the other findings.
Hence, a continuous response to immune-mediated selection, resulting in continuous selection for protein variants with new antigenic configurations to escape existing immunity, might present one potential strategy of VHSV emergence in densely cultured rainbow trout, which has been demonstrated for mammalian RNA viruses (46). However, rainbow trout have a relatively short lifetime, and infection with VHSV often results in rapid mortality, limiting continuous immune-mediated selection. Nonetheless, recurrent infection in rainbow trout can occur (N. Lorenzen, personal communication) and thus may support immune-mediated selection, or at least maintenance of genetic variation in the population to facilitate antigenic changes. Adaptation to within-host species variation would be impossible in a multi-host-species environment but might become very advantageous in adapting to a single-host-species environment, which would support our speculations on VHSV specialist and generalist strategies in densely monocultured host species versus limited host access in nature.
Phylogenetic reconstruction.
The phylogenetic reconstruction of VHSV was in agreement with previous studies, confirming clustering into recently defined major genotypes (I to IV) (14–17), sublineages (Ia to Ie and IVa to IVc) (14, 15, 18–20), and clades (Ia-1 and Ia-2) (21). However, internal branching orders deviated slightly from those in previous studies. A potential new sublineage (If) and two potential new clades (Ia-3 and Ia-4) were identified within genotype I. Previous phylogenetic analyses (14, 15, 18–20, 58) were based on more limited data sets. Reduced numbers of isolates clustering into the newly detected sublineage If and clades Ia-3 and Ia-4 most likely affected the bootstrap support, and thus previous phylogenetic analyses might not have had the power to detect those phylogenetic groups. Furthermore, previous phylogenetic analyses often were conducted without a proper outgroup to resolve evolutionary directions, which likely explains the minor deviations observed for internal branching orders. As demonstrated by Einer-Jensen and coworkers (15), our phylogenetic analysis supports the assumption of a marine origin and subsequent viral emergence from the marine environment into freshwater cultured rainbow trout. However, our results neither reject nor confirm the possibility of repeated independent VHSV emergences from the marine environment into cultured rainbow trout, which was hypothesized previously (22). Nonetheless, the occurrence of VHSV isolates in marine cultured rainbow trout that genetically resemble marine VHSV isolates occurring in the geographic vicinity (23–27) and instances of VHSV infection following marine floods of aquaculture systems (34) suggest the occurrence of repeated and independent instances of VHSV emergence.
In conclusion, we provide evidence of positive selection pressure on six residues of the VHSV G protein, within the signal peptide, the putative major antibody neutralization site, and the intracellular tail. Evidence of positive selection was restricted to data sets representing isolates adapted to freshwater cultured rainbow trout, indicating that repeated emergences of VHSV in freshwater cultured rainbow trout were accompanied by bursts of adaptive evolution in the G protein. Amino acid combinations found at the six sites under positive selection could clearly differentiate between rainbow trout- and non-rainbow trout-adapted isolates. Furthermore, recurring identical changes across phylogenetic groups of rainbow trout-adapted isolates were identified, suggesting that potentially independent instances of VHSV emergence in freshwater cultured rainbow trout were established through convergent routes of evolution. The majority of adaptive sites identified were located either directly within the conformation-dependent neutralizing epitope or in close proximity to functional regions affecting the structural conformation of the neutralizing epitope, such as disulfide bonds, suggesting adaptation to immune response-related genetic variation across rainbow trout. This was further supported by the occurrence and maintenance of polymorphic amino acid spectra at adaptive sites located within the conformation-dependent neutralizing epitope.
MATERIALS AND METHODS
Sequences.
A total of 273 full-length G protein sequences were retrieved from NCBI GenBank (http://www.ncbi.nlm.nih.gov/nuccore/). Pairwise distances across all sequences were estimated using MEGA 6.06 (59), and sequences with P distances of <0.001 were treated as identical. All but one of the identical sequences was removed from the final data set, resulting in a panel of 198 unique sequences analyzed. Sequences covered all four genotypes (I to IV) and various geographic regions, sampling time points, and host species. The numbers of isolates from host species were as follows: 143 from rainbow trout, 23 from herring, 4 from Japanese flounder, 3 each from olive flounder and turbot, 2 each from Atlantic salmon, cod, coho salmon, eel, and sprat, and 1 each from blue whiting, dab, grayling, haddock, lamprey, muskellunge, Norway pout, Pacific sardine, rock gunnel, rockling, round goby, and whiting. Sequence metadata are summarized in Table SA1 in the supplemental material.
Sequence analysis. (i) Alignment.
Nucleotide sequences were aligned using CLC Genomic Workbench (GWB) V9.5.5 software by Qiagen, using the “very accurate (slow)” alignment parameter settings, with gap costs of 10 for opening a gap and 1 for gap extension. The nucleotide sequence length used for phylogenetic analysis was 1,524 bases, which corresponds to the entire length of the glycoprotein gene (G gene).
(ii) Phylogenetic analysis.
Phylogenetic relationships among isolates were analyzed using GWB V9.5.5 software. Briefly, a starting tree was constructed using the “create tree” tool, using the neighbor-joining approach in combination with the Jukes-Cantor nucleotide distance measure. The tree was subsequently used for substitution model comparison by using the “model testing” tool, based on the Akaike information criterion (AIC) and the Bayesian information criterion (BIC) estimated during substitution model comparison. AIC as well as BIC identified the general time-reversible substitution model with invariant sites and gamma-distributed rate heterogeneity (GTR+I+G) as the best-fitting substitution model. Alignment of the G gene sequences was finally used as a starting point for construction of the maximum likelihood tree by use of the GTR+I+G substitution model, along with 1,000 bootstrap replications. The root position was determined using snakehead rhabdovirus (SHRV) (GenBank accession no. AF147498) as a genetically related but distinct outgroup.
Molecular evolutionary analysis. (i) Detection of adaptive evolutionary processes.
To enable tracing of mechanisms of adaptive evolution occurring during VHSV emergence in cultured rainbow trout, sequences were divided into sub-data sets according to phylogenetic affiliation, giving genotype-specific data sets, sublineage-specific data sets, and clade-specific data sets (Table 1). The sub-data sets were subjected to molecular evolutionary analysis if their corresponding phylogenetic groups (genotypes, sublineages, or clades) had a bootstrap support level of at least 75% and included at least 7 isolates. For genotype-specific data sets that contained rainbow trout-adapted as well as marine-adapted sublineages (data set I, representing isolates assigned to genotype I), only the sublineage-specific data sets were subjected to molecular evolutionary analysis for tracing of adaptive events occurring during VHSV emergence in cultured rainbow trout. Accordingly, the following 9 data sets were tested for the presence of adaptive evolution: 3 genotype-specific data sets (II, III, and IV), 4 sublineage-specific data sets (Ia, Ib, Ic, and IVa), and 2 clade-specific data sets (Ia-1 and Ia-2). Four of the data sets (Ia, Ic, Ia-1, and Ia-2) represented rainbow trout-adapted isolates, and 5 data sets (II, III, IV, Ib, and IVa) represented marine-adapted isolates. Data sets are summarized in Table 1.
The presence of adaptive evolution was investigated using CODEML, implemented in the software package PAML 4.6 (60, 61). CODEML is a maximum likelihood-based program that applies different codon substitution models that allow for heterogeneous ω ratios among amino acid sites to determine site-specific selection pressures. CODEML requires a sequence alignment file and a corresponding phylogenetic tree file as input. Sequence alignment was conducted using CLUSTAL W (62), with the gap open penalty value set to 30 and default parameters otherwise. The alignment was inspected by eye, and the last three nucleotides, creating the terminal stop codon, were removed. A maximum likelihood tree was created in MEGA 6.06 (59), using the GTR+I+G substitution model. CODEML requires a single, unrooted tree, and thus the maximum likelihood tree was created without bootstrap resampling and without an outgroup. The sub-data-set-specific trees were subsequently unrooted using PHYLIP 3.695 (63).
All data sets were analyzed using the substitution models M0, M1, M2, M7, and M8, developed and described by Nielsen and Yang (64) and by Yang et al. (65) and implemented in CODEML. For all models, the following parameters were used for calculating ω values and maximum likelihood estimates: phylogenetic branch length, transition/transversion rate ratio (κ), and base frequency at the three codon positions. Model M0 assumes a uniform selection pressure, expressed by a single ω parameter shared across all amino acid sites (33). Model M1, representing evolutionary neutrality with purifying selection, assumes a proportion (p0) of conserved amino acid sites, with 0 < ω0 < 1, and a proportion (p1; p1 = 1 − p0) of neutral amino acid sites, where ω1 = 1. Model M2 is an extension of M1 which also allows for positive selection of a proportion p2 (p2 = 1 − p0 − p1), where ω2 > 1, estimated from the data. Models M7 and M8 represent continuous models in which the ω values follow a beta distribution, Β(p, q), which can assume different shapes on the interval (0, 1) (65). M7 represents a neutral selection regimen not allowing for positive selection, whereas M8 adds a discrete class of amino acid sites accounting for positive selection of proportion p1 (p1 = 1 − p0), where ω1 > 1, estimated from the data as a free parameter. All models are summarized in Table SA2 in the supplemental material. Because model M1 is nested within M2 and M7 is nested within M8, their relative fits were evaluated using a likelihood ratio test (LRT) comparing twice the log-likelihood ratio to a χ2 distribution (df = 2). Evidence of positive selection was indicated when the LRT statistically favored M2 over M1 or M8 over M7.
(ii) Detection of amino acid sites under adaptive evolution.
In cases where substitution models accounting for positive selection were statistically favored, per-site posterior probabilities were estimated using the Bayes empirical Bayes (BEB) approach, implemented in CODEML and described by Yang et al. (66). Accordingly, amino acid sites with ω values of >1 and a related high posterior probability represent amino acid sites under positive selection pressure and thus indicate adaptive evolution.
(iii) Validation of detection robustness.
Findings established with CODEML were confirmed using two complementary algorithms implemented in the HyPhy package (67): the branch site unrestricted statistical test for episodic diversification (BUSTED) algorithm (68) and the fast, unconstrained Bayesian approximation (FUBAR) algorithm (69). BUSTED was applied to infer gene-wide signs of positive selection, investigating whether the VHSV G gene experienced positive selection in at least one site on at least one branch of the entire phylogenetic tree. Accordingly, the nine sub-data sets generated for the CODEML analysis were each in turn defined as foreground branches in the entire data set, whereas the remaining phylogenetic tree was defined by background branches. For each phylogenetic partition (foreground or background branch), a codon model with three rate classes was fitted, constraining the dN/dS ratio to ω1 ≤ ω2 ≤ 1 ≤ ω3 in the alternative model, accounting for purifying, neutral, and positive selection. The model fit of the alternative model was compared to that of a null model with an ω3 value of 1 (disallowing positive selection) for the defined foreground branches but with an ω3 value of >1 for the background branch (68). The relative fits of the models were tested as described for the CODEML analysis. BUSTED analyses were conducted using the online version implemented at http://datamonkey.org (70, 71), using the universal code and default settings otherwise and running each foreground branch test in separate analyses.
FUBAR, a site-by-site approach, was applied to detect individual sites under positive selection pressure in sub-data sets of the foreground branches detected under positive selection pressure by either of the two detection approaches. As with CODEML, individual sites under positive selection pressure were identified through per-site posterior probabilities estimated using a Bayesian approach described by Murrell et al. (69). FUBAR analyses were conducted for sub-data sets separately by using the online version implemented at http://datamonkey.org (70, 71), with default settings as follows: 5 MCMC chains were generated, chain length was set to 2,000,000, burn-in was 1,000,000 samples, 100 samples were drawn from each chain, and the Dirichlet prior was set to 0.5.
Structural prediction and visualization of positions under adaptive evolution.
Efforts to crystallize the G protein of VHSV have so far been unsuccessful. However, the G protein structure of VSV, a rhabdovirus of the genus Vesiculovirus, was previously characterized (38). Furthermore, the majority of cysteine positions are comparable among rhabdoviruses and even conserved within rhabdovirus genera (37, 72), indicating conservation of the overall protein structure. The overall similarity among rhabdovirus G protein structures thus facilitated potential protein structure prediction of the VHSV G protein through homology modeling using known rhabdoviral G protein structures as the template. The G protein sequence of DK-3592b, a rainbow trout-adapted isolate (genotype I, sublineage Ia, clade Ia-3), was chosen for protein prediction analysis. The first 20 amino acids, representing the signal peptide of the G protein, are not part of the mature protein (37) and thus were removed from the G protein sequence of DK-3592b. However, the applied amino acid numbering nevertheless includes the signal peptide, i.e., the first residue in the mature protein corresponds to aa21. In addition, potential membrane-spanning regions (aa462 to aa488; potential transmembrane helix) and adjacent intercellular regions were predicted using TMPred software (http://www.ch.embnet.org/software/TMPRED_form.html) and were also removed from the G protein sequence of DK-3592b. The final protein sequence used for protein structure prediction had a length of 441 amino acids, spanning aa21 to aa462 of the encoded protein sequence. Based on homology modeling using CPHmodels software (73), the G protein of VSV in its postfusion conformation (Protein Data Bank [PDB] accession no. 2CMZ) was identified as a potential homolog template and subsequently used to predict the G protein structure of VHSV. Structural prediction of the G protein of DK-3592b was performed with Modeler 9.12 (74), using the G protein of VSV (accession no. 2CMZ) as the template. The following two predictions were conducted: a simple prediction based on template and alignment information and a constrained prediction that allowed restraining of previously identified disulfide bonds (37). For each prediction, five potential models were constructed and evaluated using the ModEval online server provided at http://modbase.compbio.ucsf.edu/evaluation (75–77). To assess model quality, ModEval estimates a GA341 score, which is based on target-template sequence identity, structural compactness, and potential energy of the predicted protein and was described in detail by John and Sali (36). Based on the GA341 score, the protein structure with the highest prediction probability was depicted and structural positions of amino acids under positive selection pressure were visualized using VMD 1.9.1 (78). To investigate potential structural differences between phylogenetic groups, protein structure prediction was repeated in a comparable manner for isolate DK-5e59 (genotype I, sublineage Ib; marine isolate) and isolate BC93372 (genotype IV, sublineage IVa; marine isolate).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from The Danish Agency for Science, Technology and Innovation (grants 09-066097/FTP, 09-065033/FTP, and 11-100968).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00436-18.
REFERENCES
- 1.Holmes EC. 2009. The evolution and emergence of RNA viruses. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 2.Manrubia SC. 2012. Modelling viral evolution and adaptation: challenges and rewards. Curr Opin Virol 2:531–537. doi: 10.1016/j.coviro.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Steinhauer DA, Domingo E, Holland JJ. 1992. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 122:281–288. doi: 10.1016/0378-1119(92)90216-C. [DOI] [PubMed] [Google Scholar]
- 4.Crill WD, Wichman HA, Bull JJ. 2000. Evolutionary reversals during viral adaptation to alternating hosts. Genetics 154:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duffy S, Shackelton LA, Holmes EC. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet 9:267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 6.Ferris MT, Joyce P, Burch CL. 2007. High frequency of mutations that expand the host range of an RNA virus. Genetics 176:1013–1022. doi: 10.1534/genetics.106.064634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flint SJ, Enquist LW, Racaniello VR, Skalka AM. 2009. Principles of virology, 3rd ed, vol 1 ASM Press, Washington, DC. [Google Scholar]
- 8.Wolf K. 1988. Fish viruses and fish viral diseases. Cornell University Press, Ithaca, NY. [Google Scholar]
- 9.Jensen MH. 1963. Preparation of fish tissue cultures for virus research. Bull Off Int Epizoot 59:131–134. [Google Scholar]
- 10.OIE. 2012. Manual of diagnostic tests for aquatic animals. http://www.oie.int/en/international-standard-setting/aquatic-manual/.
- 11.Al-Hussinee L, Huber P, Russell S, Lepage V, Reid A, Young KM, Nagy E, Stevenson RM, Lumsden JS. 2010. Viral haemorrhagic septicaemia virus IVb experimental infection of rainbow trout, Oncorhynchus mykiss (Walbaum), and fathead minnow, Pimphales promelas (Rafinesque). J Fish Dis 33:347–360. doi: 10.1111/j.1365-2761.2009.01128.x. [DOI] [PubMed] [Google Scholar]
- 12.Gadd T, Jakava-Viljanen M, Einer-Jensen K, Ariel E, Koski P, Sihvonen L. 2010. Viral haemorrhagic septicaemia virus (VHSV) genotype II isolated from European river lamprey Lampetra fluviatilis in Finland during surveillance from 1999 to 2008. Dis Aquat Organ 88:189–198. doi: 10.3354/dao02169. [DOI] [PubMed] [Google Scholar]
- 13.Skall HF, Olesen NJ, Mellergaard S. 2005. Viral haemorrhagic septicaemia virus in marine fish and its implications for fish farming—a review. J Fish Dis 28:509–529. doi: 10.1111/j.1365-2761.2005.00654.x. [DOI] [PubMed] [Google Scholar]
- 14.Einer-Jensen K, Ahrens P, Forsberg R, Lorenzen N. 2004. Evolution of the fish rhabdovirus viral haemorrhagic septicaemia virus. J Gen Virol 85:1167–1179. doi: 10.1099/vir.0.79820-0. [DOI] [PubMed] [Google Scholar]
- 15.Einer-Jensen K, Ahrens P, Lorenzen N. 2005. Parallel phylogenetic analyses using the N, G or Nv gene from a fixed group of VHSV isolates reveal the same overall genetic typing. Dis Aquat Organ 67:39–45. doi: 10.3354/dao067039. [DOI] [PubMed] [Google Scholar]
- 16.Snow M, Bain N, Black J, Taupin V, Cunningham CO, King JA, Skall HF, Raynard RS. 2004. Genetic population structure of marine viral haemorrhagic septicaemia virus (VHSV). Dis Aquat Organ 61:11–21. doi: 10.3354/dao061011. [DOI] [PubMed] [Google Scholar]
- 17.Snow M, Cunningham CO, Melvin WT, Kurath G. 1999. Analysis of the nucleoprotein gene identifies distinct lineages of viral haemorrhagic septicaemia virus within the European marine environment. Virus Res 63:35–44. doi: 10.1016/S0168-1702(99)00056-8. [DOI] [PubMed] [Google Scholar]
- 18.Benmansour A, Basurco B, Monnier AF, Vende P, Winton JR, de Kinkelin P. 1997. Sequence variation of the glycoprotein gene identifies three distinct lineages within field isolates of viral haemorrhagic septicaemia virus, a fish rhabdovirus. J Gen Virol 78:2837–2846. doi: 10.1099/0022-1317-78-11-2837. [DOI] [PubMed] [Google Scholar]
- 19.Lumsden JS, Morrison B, Yason C, Russell S, Young K, Yazdanpanah A, Huber P, Al-Hussinee L, Stone D, Way K. 2007. Mortality event in freshwater drum Aplodinotus grunniens from Lake Ontario, Canada, associated with viral haemorrhagic septicemia virus, type IV. Dis Aquat Organ 76:99–111. doi: 10.3354/dao076099. [DOI] [PubMed] [Google Scholar]
- 20.Nishizawa T, Iida H, Takano R, Isshiki T, Nakajima K, Muroga K. 2002. Genetic relatedness among Japanese, American and European isolates of viral hemorrhagic septicemia virus (VHSV) based on partial G and P genes. Dis Aquat Organ 48:143–148. doi: 10.3354/dao048143. [DOI] [PubMed] [Google Scholar]
- 21.Kahns S, Skall HF, Kaas RS, Korsholm H, Bang Jensen B, Jonstrup SP, Dodge MJ, Einer-Jensen K, Stone D, Olesen NJ. 2012. European freshwater VHSV genotype Ia isolates divide into two distinct subpopulations. Dis Aquat Organ 99:23–35. doi: 10.3354/dao02444. [DOI] [PubMed] [Google Scholar]
- 22.Kurath G, Winton J. 2011. Complex dynamics at the interface between wild and domestic viruses of finfish. Curr Opin Virol 1:73–80. doi: 10.1016/j.coviro.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Husu-Kallio J, Suokko M.. 2000. Two outbreaks of VHS (viral hemorrhagic septicaemia) in Finland. Standing Veterinary Committee, Veterinary and Food Department, Ministry of Agriculture and Forestry, Helsinki, Finland. [Google Scholar]
- 24.Raja-Halli M, Vehmas TK, Rimaila-Parnanen E, Sainmaa S, Skall HF, Olesen NJ, Tapiovaara H. 2006. Viral haemorrhagic septicaemia (VHS) outbreaks in Finnish rainbow trout farms. Dis Aquat Organ 72:201–211. doi: 10.3354/dao072201. [DOI] [PubMed] [Google Scholar]
- 25.Dale OB, Orpetveit I, Lyngstad TM, Kahns S, Skall HF, Olesen NJ, Dannevig BH. 2009. Outbreak of viral haemorrhagic septicaemia (VHS) in seawater-farmed rainbow trout in Norway caused by VHS virus genotype III. Dis Aquat Organ 85:93–103. doi: 10.3354/dao02065. [DOI] [PubMed] [Google Scholar]
- 26.Nordblom B. 1998. Report on an outbreak of viral haemorrhagic septicaemia in Sweden. Department for Animal Production and Health, Jönköping, Sweden. [Google Scholar]
- 27.Nordblom B, Norell AW. 2000. Report on an outbreak of VHS (viral hemorrhagic septicaemia) in farmed fish in Sweden. Swedish Board of Agriculture, Jönköping, Sweden. [Google Scholar]
- 28.Bearzotti M, Monnier AF, Vende P, Grosclaude J, de Kinkelin P, Benmansour A. 1995. The glycoprotein of viral hemorrhagic septicemia virus (VHSV): antigenicity and role in virulence. Vet Res 26:413–422. [PubMed] [Google Scholar]
- 29.Jørgensen PEV, Einer-Jensen K, Higman KH, Winton JR. 1995. Sequence comparison of the central region of the glycoprotein gene of neutralizable, non-neutralizable, and serially passed isolates of viral haemorrhagic septicaemia virus. Dis Aquat Organ 23:77–82. doi: 10.3354/dao023077. [DOI] [Google Scholar]
- 30.Lorenzen N, Lorenzen E, Einer-Jensen K, Heppell J, Davis HL. 1999. Genetic vaccination of rainbow trout against viral haemorrhagic septicaemia virus: small amounts of plasmid DNA protect against a heterologous serotype. Virus Res 63:19–25. doi: 10.1016/S0168-1702(99)00054-4. [DOI] [PubMed] [Google Scholar]
- 31.Lorenzen N, Olesen NJ, Jorgensen PE. 1990. Neutralization of Egtved virus pathogenicity to cell cultures and fish by monoclonal antibodies to the viral G protein. J Gen Virol 71:561–567. doi: 10.1099/0022-1317-71-3-561. [DOI] [PubMed] [Google Scholar]
- 32.Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426. [DOI] [PubMed] [Google Scholar]
- 33.Goldman N, Yang Z. 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11:725–736. [DOI] [PubMed] [Google Scholar]
- 34.Ogut H, Altuntas C. 2014. Survey of viral haemorrhagic septicaemia virus in wild fishes in the southeastern Black Sea. Dis Aquat Organ 109:99–106. doi: 10.3354/dao02728. [DOI] [PubMed] [Google Scholar]
- 35.Castric J, Jeffroy J, Bearzotti M, de Kinkelin P. 1992. Isolation of viral haemorrhagic septicaemia virus (VHSV) from wild elvers Anguilla anguilla. Bull Eur Assoc Fish Pathol 12:21–23. [Google Scholar]
- 36.John B, Sali A. 2003. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res 31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Einer-Jensen K, Krogh TN, Roepstorff P, Lorenzen N. 1998. Characterization of intramolecular disulfide bonds and secondary modifications of the glycoprotein from viral hemorrhagic septicemia virus, a fish rhabdovirus. J Virol 72:10189–10196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roche S, Bressanelli S, Rey FA, Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- 39.Badrane H, Tordo N. 2001. Host switching in Lyssavirus history from the Chiroptera to the Carnivora orders. J Virol 75:8096–8104. doi: 10.1128/JVI.75.17.8096-8104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmes EC, Woelk CH, Kassis R, Bourhy H. 2002. Genetic constraints and the adaptive evolution of rabies virus in nature. Virology 292:247–257. doi: 10.1006/viro.2001.1271. [DOI] [PubMed] [Google Scholar]
- 41.Streicker DG, Altizer SM, Velasco-Villa A, Rupprecht CE. 2012. Variable evolutionary routes to host establishment across repeated rabies virus host shifts among bats. Proc Natl Acad Sci U S A 109:19715–19720. doi: 10.1073/pnas.1203456109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuevas JM, Elena SF, Moya A. 2002. Molecular basis of adaptive convergence in experimental populations of RNA viruses. Genetics 162:533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Llewellyn ZN, Salman MD, Pauszek S, Rodriguez LL. 2002. Growth and molecular evolution of vesicular stomatitis serotype New Jersey in cells derived from its natural insect-host: evidence for natural adaptation. Virus Res 89:65–73. doi: 10.1016/S0168-1702(02)00113-2. [DOI] [PubMed] [Google Scholar]
- 44.Novella IS, Zarate S, Metzgar D, Ebendick-Corpus BE. 2004. Positive selection of synonymous mutations in vesicular stomatitis virus. J Mol Biol 342:1415–1421. doi: 10.1016/j.jmb.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 45.Fares MAA, Moya C, Escarmis E, Baranowski E, Domingo E, Barrio E. 2001. Evidence for positive selection in the capsid protein-coding region of the foot-and-mouth disease virus (FMDV) subjected to experimental passage regimens. Mol Biol Evol 18:10–21. doi: 10.1093/oxfordjournals.molbev.a003715. [DOI] [PubMed] [Google Scholar]
- 46.Bush RM, Fitch WM, Bender CA, Cox NJ. 1999. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol Biol Evol 16:1457–1465. doi: 10.1093/oxfordjournals.molbev.a026057. [DOI] [PubMed] [Google Scholar]
- 47.Forsberg R, Christiansen FB. 2003. A codon-based model of host-specific selection in parasites, with an application to the influenza A virus. Mol Biol Evol 20:1252–1259. doi: 10.1093/molbev/msg149. [DOI] [PubMed] [Google Scholar]
- 48.Twiddy SS, Woelk CH, Holmes EC. 2002. Phylogenetic evidence for adaptive evolution of dengue viruses in nature. J Gen Virol 83:1679–1689. doi: 10.1099/0022-1317-83-7-1679. [DOI] [PubMed] [Google Scholar]
- 49.LaPatra SE, Evilia C, Winston V. 2008. Positively selected sites on the surface glycoprotein (G) of infectious hematopoietic necrosis virus. J Gen Virol 89:703–708. doi: 10.1099/vir.0.83451-0. [DOI] [PubMed] [Google Scholar]
- 50.Huang C, Chien MS, Landolt M, Batts W, Winton J. 1996. Mapping the neutralizing epitopes on the glycoprotein of infectious haematopoietic necrosis virus, a fish rhabdovirus. J Gen Virol 77:3033–3040. doi: 10.1099/0022-1317-77-12-3033. [DOI] [PubMed] [Google Scholar]
- 51.Troyer RM, Kurath G. 2003. Molecular epidemiology of infectious hematopoietic necrosis virus reveals complex virus traffic and evolution within southern Idaho aquaculture. Dis Aquat Organ 55:175–185. doi: 10.3354/dao055175. [DOI] [PubMed] [Google Scholar]
- 52.Mebatsion T, König M, Conzelmann K. 1996. Budding of rabies virus particles in the absence of the spike glycoprotein. Cell 84:941–951. doi: 10.1016/S0092-8674(00)81072-7. [DOI] [PubMed] [Google Scholar]
- 53.Mebatsion T, Weiland F, Conzelmann KK. 1999. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J Virol 73:242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bull JJ, Badgett MR, Wichman HA, Huelsenbeck JP, Hillis DM, Gulati A, Ho C, Molineux IJ. 1997. Exceptional convergent evolution in a virus. Genetics 147:1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dunham EJ, Dugan VG, Kaser EK, Perkins SE, Brown IH, Holmes EC, Taubenberger JK. 2009. Different evolutionary trajectories of European avian-like and classical swine H1N1 influenza A viruses. J Virol 83:5485–5494. doi: 10.1128/JVI.02565-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2012. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336:1534–1541. doi: 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roche S, Albertini AA, Lepault J, Bressanelli S, Gaudin Y. 2008. Structures of vesicular stomatitis virus glycoprotein: membrane fusion revisited. Cell Mol Life Sci 65:1716–1728. doi: 10.1007/s00018-008-7534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Studer J, Janies DA. 2011. Global spread and evolution of viral haemorrhagic septicaemia virus. J Fish Dis 34:741–747. doi: 10.1111/j.1365-2761.2011.01290.x. [DOI] [PubMed] [Google Scholar]
- 59.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetic Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556. [DOI] [PubMed] [Google Scholar]
- 61.Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 62.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Felsenstein J. 2005. Using the quantitative genetic threshold model for inferences between and within species. Philos Trans R Soc Lond B Biol Sci 360:1427–1434. doi: 10.1098/rstb.2005.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nielsen R, Yang ZH. 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang Z, Nielsen R, Goldman N, Pedersen AM. 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang Z, Wong WS, Nielsen R. 2005. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 67.Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 68.Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, Eren K, Pollner T, Martin DP, Smith DM, Scheffler K, Kosakovsky Pond SL. 2015. Gene-wide identification of episodic selection. Mol Biol Evol 32:1365–1371. doi: 10.1093/molbev/msv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K. 2013. FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol Biol Evol 30:1196–1205. doi: 10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pond SL, Frost SD. 2005. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- 72.Walker PJ, Kongsuwan K. 1999. Deduced structural model for animal rhabdovirus glycoproteins. J Gen Virol 80:1211–1220. doi: 10.1099/0022-1317-80-5-1211. [DOI] [PubMed] [Google Scholar]
- 73.Nielsen M, Lundegaard C, Lund O, Petersen TN. 2010. CPHmodels-3.0—remote homology modeling using structure-guided sequence profiles. Nucleic Acids Res 38:W576–W581. doi: 10.1093/nar/gkq535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 75.Melo F, Sanchez R, Sali A. 2002. Statistical potentials for fold assessment. Protein Sci 11:430–448. doi: 10.1002/pro.110430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pieper U, Webb BM, Barkan DT, Schneidman-Duhovny D, Schlessinger A, Braberg H, Yang Z, Meng EC, Pettersen EF, Huang CC, Datta RS, Sampathkumar P, Madhusudhan MS, Sjolander K, Ferrin TE, Burley SK, Sali A. 2011. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res 39:D465–D474. doi: 10.1093/nar/gkq1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shen MY, Sali A. 2006. Statistical potential for assessment and prediction of protein structures. Protein Sci 15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.