

ABSTRACT
Several members of the APOBEC3 DNA cytosine deaminase family can potently inhibit Vif-deficient human immunodeficiency virus type 1 (HIV-1) by catalyzing cytosine deamination in viral cDNA and impeding reverse transcription. HIV-1 counteracts restriction with the virally encoded Vif protein, which targets relevant APOBEC3 proteins for proteasomal degradation. HIV-1 Vif is optimized for degrading the restrictive human APOBEC3 repertoire, and, in general, lentiviral Vif proteins specifically target the restricting APOBEC3 enzymes of each host species. However, simian immunodeficiency virus SIVmac239 Vif elicits a curiously wide range of APOBEC3 degradation capabilities that include degradation of several human APOBEC3s and even human APOBEC3B, a non-HIV-1-restricting APOBEC3 enzyme. To better understand the molecular determinants of the interaction between SIVmac239 Vif and human APOBEC3B, we analyzed an extensive series of mutants. We found that SIVmac239 Vif interacts with the N-terminal domain of human APOBEC3B and, interestingly, that this occurs within a structural region homologous to the HIV-1 Vif interaction surface of human APOBEC3G. An alanine scan of SIVmac239 Vif revealed several residues required for human APOBEC3B degradation activity. These residues overlap HIV-1 Vif surface residues that interact with human APOBEC3G and are distinct from those that engage APOBEC3F or APOBEC3H. Overall, these studies indicate that the molecular determinants of the functional interaction between human APOBEC3B and SIVmac239 Vif resemble those between human APOBEC3G and HIV-1 Vif. These studies contribute to the growing knowledge of the APOBEC-Vif interaction and may help guide future efforts to disrupt this interaction as an antiviral therapy or exploit the interaction as a novel strategy to inhibit APOBEC3B-dependent tumor evolution.
IMPORTANCE Primate APOBEC3 proteins provide innate immunity against retroviruses such as HIV and SIV. HIV-1, the primary cause of AIDS, utilizes its Vif protein to specifically counteract restrictive human APOBEC3 enzymes. SIVmac239 Vif exhibits a much wider range of anti-APOBEC3 activities that includes several rhesus macaque enzymes and extends to multiple proteins in the human APOBEC3 repertoire, including APOBEC3B. Understanding the molecular determinants of the interaction between SIVmac239 Vif and human APOBEC3B adds to existing knowledge on the APOBEC3-Vif interaction and has potential to shed light on what processes may have shaped Vif functionality over evolutionary time. An intimate understanding of this interaction may also lead to a novel cancer therapy because, for instance, creating a derivative of SIVmac239 Vif that specifically targets human APOBEC3B could be used to suppress tumor genomic DNA mutagenesis by this enzyme, slow ongoing tumor evolution, and help prevent poor clinical outcomes.
KEYWORDS: APOBEC3B, APOBEC3G, SIV, Vif, cancer mutation, host-pathogen interaction, innate immunity
INTRODUCTION
APOBEC3 (A3) enzymes are single-stranded DNA cytosine deaminases involved in innate immunity against a variety of pathogens, including retroviruses and retrotransposons (reviewed in references 1, 2, and 3). Up to seven family members, namely, A3A, A3B, A3C, A3D, A3F, A3G, and A3H, are expressed in primate cells. Extensive overexpression, separation-of-function, and gain-of-function studies using various model systems, including T cell lines and primary T lymphocytes, have demonstrated that human A3D, A3F, A3G, and A3H contribute to restricting the replication of Vif-deficient human immunodeficiency virus type 1 (HIV-1). These A3 enzymes package into virions and catalyze C-to-U deamination reactions in viral cDNA upon reverse transcription. This leads to G-to-A hypermutation of the viral genome, greatly affecting its coding capacity and therefore its ability to replicate and transmit. A3 enzymes also elicit various levels of deaminase-independent restriction activity, which is likely due to a strong affinity for RNA that provides a steric block to reverse transcription.
HIV-1 encodes the accessory protein Vif to counteract A3 enzymes and prevent restriction. Vif hijacks cellular core-binding factor beta (CBF-β) to form an E3 ubiquitin ligase complex with Cullin-5 (CUL5), Elongin B (ELOB), ELOC, and RING-box protein 2 (RBX2), which effectively targets restrictive A3 enzymes for polyubiquitination and proteasomal degradation (4–11). Vif utilizes several conserved residues and motifs to interact with different components of the complex. For instance, the SLQ motif is required for interacting with ELOC, and this is conserved among various strains of HIV-1, HIV-2, and simian immunodeficiency virus (SIV) (12). The highly conserved zinc-coordinating HCCH motif of Vif is required for the interaction with CUL5 (13). Moreover, the Vif–CBF-β interaction (also conserved between HIV and SIV) involves an extensive hydrophobic surface, largely provided by residues within the N-terminal half of Vif (14). Interestingly, despite these conserved and essential interactions, HIV-1 Vif is still capable of using different regions to target structurally similar A3 proteins (A3F, A3G, and A3H). For instance, the YRHHY motif of HIV-1 Vif is specifically required for A3G interaction and degradation, whereas the DRMR motif is specific for A3F (15, 16). Residue F39 is essential for the A3H interaction, and changing it to valine results in a separation-of-function protein that degrades A3G and A3F but not A3H (17, 18).
Cross-species comparisons have been instrumental in elucidating molecular determinants of the A3-Vif interaction. This is possible because A3 restriction factors are under positive selection and show considerable variation between species despite overall functional conservation. For instance, human and rhesus macaque A3Gs are 78% identical, and interspecies chimeras showed that D128 is required for the human enzyme to interact with HIV-1 Vif whereas K128 is required for the rhesus macaque enzyme to interact with SIVmac239 Vif (19–22). Similarly, E289 and E324 are required for the human A3F interaction with HIV-1 Vif, and K289 and K324 are required for the interaction with SIVmac239 Vif (23–25). These and additional cross-species comparisons have helped to define the A3 protein surfaces involved in these interactions and have led to robust structural models (26–30), although structures of complete A3-Vif ligase complexes have yet to be determined. In general, rapidly evolving A3 repertoires and far more rapidly evolving lentiviral Vif proteins, selected by strong adaptive immune pressure, have resulted in present-day host-pathogen interactions that are largely but not exclusively species specific.
One clear exception to this rule is SIVmac239 Vif, the Vif protein encoded by a SIV clone that infects rhesus macaques, which degrades all of the restrictive rhesus macaque A3 enzymes as well as multiple human enzymes, including A3B and A3G (31–33). A functional interaction between SIVmac239 Vif and human A3B is curious because neither the rhesus macaque nor the human enzyme restricts Vif-deficient HIV-1 replication in T cell model systems (SupT11 or CEM-SS) (33, 34). To shed light on the molecular determinants of this functional interaction, we performed mutagenesis studies on both human A3B and SIVmac239 Vif. Using 293T cells as a model system, we found that SIVmac239 Vif exclusively targets the N-terminal domain of human A3B and particularly residues within the loop 7 region. Based on the various A3-interacting surfaces of HIV-1 Vif, we mutated single amino acid residues of SIVmac239 Vif at corresponding regions. Functional tests of these mutants against human A3B revealed a putative physical interaction surface, which is largely similar to the A3G-interacting region on HIV-1 Vif but distinct from those that engage A3F and A3H.
RESULTS
SIVmac239 Vif mediates degradation of human APOBEC3 proteins.
Multiple groups have shown that SIVmac239 Vif efficiently degrades both human A3G and A3B, whereas HIV-1 Vif targets human A3G but not A3B (31, 33, 35). To confirm and extend these results, we performed immunoblotting of 293T cells expressing SIVmac239 Vif or HIV-1IIIB Vif together with A3G or A3B from human or rhesus macaque (Fig. 1A). As a positive control, both Vif proteins promoted the degradation of human A3G, and mutation of the conserved ELOC-binding motif (SLQ to AAA) abrogated most of this effect. As reported previously (33, 36), the Vif SLQ-to-AAA mutant still caused a partial reduction in A3G steady-state levels (e.g., compare lanes 4 and 20 in Fig. 1A). Only SIVmac239 Vif mediated the degradation of rhesus macaque A3G. In contrast, HIV-1IIIB Vif did not degrade human or rhesus macaque A3B, whereas SIVmac239 Vif efficiently, but not completely, promoted the degradation of both human and rhesus macaque A3B. The abilities of wild-type SIVmac239 Vif to degrade human A3B and A3G were further demonstrated through a titration experiment, in which SIVmac239 Vif was shown to degrade human A3B in a dose-dependent manner (Fig. 1B). Overall, these results confirmed that SIVmac239 Vif elicits a wider range of A3 degradation capabilities that, unlike those with HIV-1 Vif, are not limited to the A3 enzymes of its host species (Fig. 1C).
FIG 1.

SIVmac239 Vif degrades both human APOBEC3B and APOBEC3G. (A) Immunoblots of 293T cells expressing the indicated rhesus macaque (rh) or human (hu) A3 enzymes together with SIVmac239 Vif, HIV-1IIIB Vif, or SLQ-AAA mutant (m) derivatives. A3 enzymes were detected using an anti-HA antibody for a C-terminal HA epitope tag. Vif was detected using an anti-MYC antibody for a C-terminal MYC epitope tag, and anti-α-tubulin was used as a loading control. (B) Bar graphs for quantification (bottom) of immunoblots (top and not shown) of 293T cells expressing fixed amounts of the indicated human A3 enzymes together with an empty vector control (−) or various amounts of SIVmac239 Vif (WT) or a SLQ-AAA mutant (m) derivative (analyzed with the same antibodies as for panel A). A3 expression levels were determined by quantifying band intensities normalized to those of the corresponding Vif-null condition. Each error bar represents the standard deviation for three biological replicates. (C) Schematic summarizing the immunoblotting results from panels A and B. Open squares represent a functional Vif-A3 interaction evidenced by A3 degradation.
SIVmac239 Vif interacts with the N-terminal domain of human APOBEC3B.
Previous studies on the A3-Vif interaction revealed motifs and single amino acid residues that, when mutated, abrogate the degradation phenotype (19–25, 37–39). Most of these studies have focused on HIV-1 Vif and human A3 proteins, such as A3G, A3F, or A3H. For instance, the 128DPD130 motif of human A3G has a well-defined role in conferring sensitivity to HIV-1 Vif-mediated degradation, and the analogous regions of several nonhuman primate A3G proteins are also required for SIV Vif-mediated degradation (19–22, 39, 40). The human A3G 128DPD130 motif aligns with human A3B residues 128ERD130 (Fig. 2A). To begin to determine how SIVmac239 Vif interacts with human A3B, we asked if the A3B 128ERD130 motif is necessary by mutating the glutamic acid and aspartic acid to positively charged lysines. The resulting E128K, D130K, and double mutants were evaluated by immunoblotting of 293T cells expressing wild-type or mutant A3B together with increasing amount of SIVmac239 Vif or SLQ mutant (a representative immunoblot is shown in Fig. 2B and quantification in Fig. 2E). As a positive control, SIVmac239 Vif readily degrades wild-type human A3B, and this degradation phenotype is reduced by the SLQ-to-AAA mutation. Both single mutants and the E128K D130K double mutant of A3B are expressed at levels similar to those of wild-type A3B in the absence of Vif, and these two lysine substitutions alone or combined confer resistance to degradation by wild-type SIVmac239 Vif. This resistance phenotype is not altered by the SLQ-to-AAA change. These results demonstrate that human A3B and A3G use at least one partly similar surface for interacting with Vif.
FIG 2.

SIVmac239 Vif interacts with the N-terminal domain of human APOBEC3B. (A) Schematic of human A3B and A3G with N- and C-terminal halves shaded orange and green, respectively. N-terminal Vif-interacting amino acids and C-terminal catalytic domain residues are labeled. (B) Immunoblots of 293T cells expressing full-length human A3B, wild-type (WT) or the indicated mutants, together with SIVmac239 Vif or an SLQ-AAA derivative (SLQ). A3B was detected using an anti-HA antibody for a C-terminal HA epitope tag. Vif was detected using an anti-MYC antibody for a C-terminal MYC epitope tag, and anti-α-tubulin was used as a loading control. (C) Immunoblots of 293T cells expressing full-length human A3B (WT), the N-terminal half (NTD), or the C-terminal half (CTD) together with an empty vector (−), SIVmac239 Vif, or an SLQ-AAA derivative. Analysis was with the same antibodies as for panel B. (D) Immunoblots of 293T cells expressing the N-terminal half of human A3B (NTD) or the indicated single-amino-acid substitution mutants together with an empty vector (−), SIVmac239 Vif, or an SLQ-AAA derivative. Analysis was with the same antibodies as for panel B. (E, F, and G) Quantification of data in panels B, C, and D, respectively. The A3B level of each immunoblot lane was first normalized to tubulin, and then resistance-to-degradation values were calculated as the normalized A3B level in the presence of wild-type SIVmac239 Vif relative to the SIVmac239 Vif SLQ mutant. Each histogram bar reports the mean ± the difference between two independent reactions (E and G) or the mean ± standard deviation from three independent reactions (F).
E128 and D130 are located on the surface of the N-terminal domain of human A3B (41). HIV-1 Vif is known to interact exclusively with the N-terminal domain of human A3G (4, 25). To determine whether this is also the case for A3B, full-length, N-terminal domain, and C-terminal domain constructs were cotransfected into 293T cells with wild-type SIVmac239 Vif, the SLQ mutant, or empty vector (a representative immunoblot is shown in Fig. 2C and quantification in Fig. 2F). As described above, wild-type SIVmac239 Vif leads to decreased levels of full-length A3B, and the degradation phenotype is largely reversed by mutating the SLQ motif. The same trend was observed for the N-terminal domain of A3B, indicating that the N-terminal domain is sensitive to degradation by SIVmac239 Vif. However, the levels of A3B C-terminal domain are approximately the same when cells are cotransfected with wild-type versus mutant Vif, indicating that the C-terminal domain is resistant to SIVmac239 Vif. Therefore, analogous to the case for HIV-1 Vif and A3G, SIVmac239 Vif interacts functionally with the N-terminal domain of human A3B.
To identify additional interacting residues within the N-terminal domain of human A3B, we performed a focused alanine scan of the 121RLYYYW127 loop region upstream of the 128ERD130 motif. The sensitivities of these mutants to SIVmac239 Vif were determined using the cotransfection assay described above. Apart from Y124A and Y125A, all other alanine substitutions affected the steady-state levels of the A3B N-terminal domain to various degrees as demonstrated by the expression levels in the absence of Vif (a representative immunoblot is shown in Fig. 2D and quantification in Fig. 2G). Although several of these N-terminal domain mutants were expressed at lower-than-wild-type levels, immunoblots indicate that amino acid substitutions R122A, L123A, Y126A, and W127A confer resistance to degradation by wild-type SIVmac239 Vif, while Y124A and Y125A mutants remain sensitive (e.g., compare band intensities for reactions with wild-type and SLQ mutant Vif for each A3B N-terminal domain construct). Taken together, these data show that SIVmac239 Vif requires several residues in N-terminal domain loop 7 for degradation of human A3B.
Mutagenesis of SIVmac239 Vif delineates human APOBEC3B-interacting residues.
SIVmac239 Vif is more closely related to HIV-2 Vif than to HIV-1 Vif, sharing 70% and 25% amino acid identity, respectively (Fig. 3A). To determine critical regions of SIVmac239 Vif required for degradation of human A3B, we constructed single-amino-acid mutants targeting potential A3-interacting residues. Several studies have mapped residues in HIV-1 Vif required for interaction with human A3F (28, 29), and these can be grouped into three interacting regions (shaded yellow in Fig. 3B and C). In comparison to wild-type SIVmac239 Vif, all of the amino acid substitution derivatives informed by A3F-interacting regions 1 and 3 were similarly capable of promoting the degradation of human A3B, with the exception of R18A (a representative immunoblot is shown in Fig. 3D and quantification in Fig. 3G). In comparison, substitutions in the A3F-interacting region 2, W73A and an insertion of a single Gly after P77 (P77+G), compromised human A3B degradation activity. Moreover, the majority of mutants with amino acid substitutions of human A3H-interacting residues (17, 18), such as C40A, C40F, W49A, Q62Δ, and G64A, retained the ability to degrade human A3B, whereas a minority (W49Δ and W51A/Δ) were compromised (Fig. 3E and G). These two sets of partial loss-of-function mutants are discussed below in the context of a structural model.
FIG 3.

Human APOBEC3B degradation capabilities of SIVmac239 Vif and mutant derivatives. (A) Phylogenetic tree showing the relationship between SIVmac239 Vif, HIV-1IIIB Vif, and related lentiviral Vif proteins. (B) Schematic of HIV-1 Vif (blue) and CBF-β (purple) highlighting known A3F (yellow)-, A3G (red)-, and A3H (green)-interacting amino acid residues (Vif–CBF-β structure from PDB 4N9F). (C) Amino acid sequence alignment of HIV-1IIIB Vif, HIV-2ROD Vif, and SIVmac239 Vif. Established A3F-, A3G-, and A3H-interacting residues of HIV-1 Vif are highlighted with the same colors as in panel B. Asterisks and colons indicate identical and similar residues, respectively. Red-shaded and underlined SIVmac239 Vif residues were targeted by site-directed mutagenesis. (D to F) Immunoblots of 293T cells expressing full-length human A3B and an empty vector (−) or three different amounts of SIVmac239 Vif (WT) or the indicated mutant derivative. A3B was detected using an anti-HA antibody for a C-terminal HA epitope tag. Vif was detected using an anti-MYC antibody for a C-terminal MYC epitope tag, and anti-α-tubulin was used as a loading control. (G) Quantification of immunoblots in panels D to F and additional data not shown. The A3B level in each lane was normalized first using the loading control, tubulin, and then against the A3B levels in the presence of wild-type SIVmac239 Vif. Each histogram bar reports mean ± standard deviation from at least three independent reactions. The asterisks represent statistically significant differences from WT (P ≤ 0.05, two-sample t test). The red dashed line indicates normalized A3B levels of 1, in the presence of wild-type SIVmac239 Vif.
Interestingly, all SIVmac239 Vif amino acid substitution mutants, which represent A3G-interacting residues in HIV-1 Vif (15, 25, 37, 42–44), lost the capacity to degrade human A3B (Fig. 3F and G). The largest stretch of A3G-interacting amino acids in HIV-1 Vif is 40YRHHY44 (25). Substitutions at any of the analogous positions in SIVmac239 Vif compromised human A3B degradation activity (i.e., P43A, H44A, F45A, K46A, V47A, and G48A). An additional A3G-interacting residue in HIV-1 Vif is K26 (15, 42), and replacement of the analogous lysine in SIVmac239 Vif by alanine (K27A), as well as such replacement of a nearby positively charged residue (K30A), also abrogated human A3B degradation activity. Taken together, these mutagenesis results indicate that multiple residues in SIVmac239 Vif are required for degrading human A3B, and these are analogous to residues in HIV-1 Vif required for degrading human A3G.
SIVmac239 Vif mutants show differential abilities to counteract human A3B, A3G, A3F, and A3H.
To confirm the results from the cotransfection experiments described above and expand the scope of these studies to compare with other human APOBEC3 proteins, we selected three SIVmac239 Vif variants that showed compromised human A3B degradation capabilities and near-wild-type expression levels (K27A, K30A, and H44A) and further tested their abilities to counteract A3-mediated restriction in dose-responsive HIV-1 single-cycle infectivity assays. Although human A3B is not a restriction factor of HIV-1 produced in T lymphocytes, it potently restricts virus produced in 293T cells with or without HIV-1 Vif (33, 35). We therefore performed single-cycle infectivity assays in 293T cells with Vif-deficient HIV-1 complemented in trans with wild-type or a mutant SIVmac239 Vif and expressed together with human A3B, A3G, A3F, or A3H haplotype II (infectivity data are shown in Fig. 4A, representative immunoblots in Fig. 4B, and immunoblot quantification in Fig. 4C). Predetermined amounts of each A3 were used in order to reduce viral infectivity to >70% relative to that for the control reaction (i.e., no-A3 and no-Vif reactions) and enable proteins to be readily detectable by immunoblotting. In comparison to these reactions, wild-type SIVmac239 Vif partly degrades A3B and completely or nearly completely degrades A3G, A3F, and A3H. As expected, SIVmac239 Vif causes corresponding recoveries in viral infectivity, with partial recovery for reactions with A3B and complete (or near-complete) recovery for those with A3G, A3F, and A3H. In comparison, all three SIVmac239 Vif mutants show a compromised ability to degrade human A3B, resulting in increased A3B packaging and decreased infectivity compared to the case for wild-type SIVmac239 Vif. When tested against human A3G, the K27A mutant shows drastically compromised degradation and counteraction activities, indicating that this residue is also required for interaction with human A3G. The K30A and H44A substitutions also affect A3G degradation and counteraction, but the infectivity phenotypes are intermediate relative to those caused by wild-type SIVmac239 Vif and the K27A mutant. The three SIVmac239 Vif mutants retained partial A3F degradation activity but mediated nearly full recoveries in virus infectivity. In contrast, none of the three substitutions affected the ability of SIVmac239 Vif to degrade A3H, further showing that these mutants are fully active against one A3 enzyme and are likely to be true separation-of-function mutants.
FIG 4.

Abilities of SIVmac239 Vif and mutant derivatives to counteract restriction by human APOBEC3B, APOBEC3G, APOBEC3F, and APOBEC3H. (A) Bar graphs depicting relative infectivities of virus particles produced from 293T cells expressing the indicated A3 and Vif constructs. Infectivity is measured as the percentage of infected/fluorescent CEM-GFP reporter cells, and values are normalized to those under the A3-null condition (open bars, duplicated to position above the relevant lanes of the corresponding immunoblots). Each histogram bar represents the mean from two separate CEM-GFP infections, and the error bars represent the difference between these two values. The data on the left and right are from the same representative experiment and are separated only due to the necessity of requiring two separate immunoblots to analyze all of the reactions. (B) Immunoblots of virus particles and 293T cell lysates expressing the indicated constructs. A3B and A3G enzymes from virus particles and cell lysates were detected using an anti-HA antibody for a C-terminal HA epitope tag. A3F and A3H enzymes from virus particles and cell lysates were detected using specific anti-A3F and anti-A3H antibodies, respectively. Vif from cell lysates was detected using an anti-MYC antibody for a C-terminal MYC epitope tag. Anti-α-tubulin and anti-p24 (Gag) are loading controls for cell lysates and virus particles, respectively. (C) Quantification of the virus particle immunoblot data in panel B. The color scheme matches that in panel A, with A3B, A3G, A3F, and A3G represented by red, blue, orange, and purple, respectively. The A3 level in each lane was normalized first using the loading control, tubulin, and then against the A3 levels in the presence of wild-type SIVmac239 Vif. Each histogram bar reports the mean ± the difference between two independent reactions.
DISCUSSION
The Vif protein encoded by SIVmac239 has been shown previously to have cross-species degradation capabilities against human A3B and A3G (31–33). The degradation of human A3B by SIVmac239 Vif is particularly intriguing because human A3B is not an HIV-1 restriction factor in human T lymphocytes and is not targeted for degradation by HIV-1 Vif. Here we shed light on the molecular determinants of the functional interaction between SIVmac239 Vif and human A3B. We report key interacting amino acid residues on both Vif and A3B. Our data indicate that SIVmac239 Vif exclusively targets the N-terminal domain of human A3B. Amino acid substitutions E128K D130K within the 128ERD130 motif of human A3B, as well as single alanine substitutions of R122, L123, Y126, and W127 on the adjacent loop 7, confer resistance to SIVmac239 Vif-mediated degradation. Interestingly, these potential interacting amino acids reside within a region of human A3B that is highly similar to human A3G (121RLYYYWERD130 in A3B compared with 121RLYYFWDPD130 in A3G), and this conserved loop region of A3G has been implicated by mutagenesis and molecular modeling to interact directly with HIV-1 Vif (30, 45, 46). In addition, our mutagenesis studies targeting potential A3-interacting residues of SIVmac239 Vif revealed a panel of alterations that abrogated the human A3B degradation phenotype (i.e., R18A, K27A, K30A, P43A, H44A, F45A, K46A, V47A, G48A, W49Δ, W51A, W51Δ, W73A, and P77+G), suggesting that these residues contribute to the interaction with human A3B. Moreover, the majority of these human A3B-interacting residues map to or near known A3G-interacting regions of HIV-1 Vif (Fig. 5). Testing a subset of these mutants against a panel of human A3 enzymes in infectivity assays confirmed that these substitutions specifically compromise the abilities of SIVmac239 Vif to degrade and counteract human A3B and A3G, to a lesser extent A3F, and not at all A3H. Overall, these results indicate that interaction surfaces are shared between SIVmac239 Vif-human A3B and HIV-1 Vif-human A3G.
FIG 5.
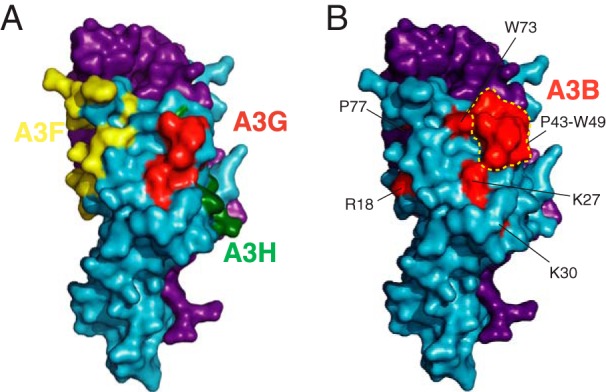
Structural depiction of the human APOBEC3B-interacting region of SIVmac239 Vif. (A) A larger representation of the schematic in Fig. 3B showing HIV-1 Vif (blue) and CBF-β (purple) and highlighting known A3F (yellow)-, A3G (red)-, and A3H-(green) interacting amino acid residues (PDB 4N9F). (B) Human A3B-interacting residues of SIVmac239 Vif highlighted upon the HIV-1 Vif (blue)–CBF-β (purple) X-ray crystal structure (PDB 4N9F). The red-shaded, A3B-interacting region (R18, K27, K30, P43-W49, W51, W73, and P77) largely overlaps with the red-shaded, A3G-interacting region in panel A.
Previous studies have demonstrated that the recruitment of CBF-β and other components of the E3 ligase complex, as well as the corresponding interacting residues within Vif, is highly conserved among various primate lentiviral Vif proteins (5, 10, 12, 13, 47, 48). Therefore, SIVmac239 Vif likely shares an overall similar structure with HIV-1 Vif due to these functional and structural constraints despite low similarity at the amino acid level (25%). This inference is further supported by data showing genetic requirements for A3 protein degradation for the SIVmac239 ELOC-interacting motif (SLQ), predicted CBF-β-interacting residues (W7, W13, and W22), and predicted zinc-coordinating residues (C116) (see, e.g., the SLQ-to-AAA data in Fig. 1 and 2; W7A, W13A, W22A, and C116A are quantified in Fig. 3G [additional data not shown]). Therefore, the human A3B-interacting residues of SIVmac239 Vif can be illustrated by highlighting the corresponding residues on the HIV-1 Vif crystal structure (Fig. 5A). The resulting image suggests that the A3B-interacting residues cluster within a surface that is similar to the human A3G-interacting surface of HIV-1 Vif and potentially even span a slightly larger surface area (compare Fig. 5A and B).
There are several possible explanations for why a functional interaction occurs between SIVmac239 Vif and human A3B in the absence of an apparent contemporary physiological context. One possibility is that the ability to degrade primate A3B arose as a necessary counterdefense in an ancestral simian lentivirus and that this activity has been maintained by a subset of present-day viruses, including SIVmac239 Vif. This idea is supported by reports showing human A3B degradation by HIV-2 Vif (49), by SIV stump-tailed macaque (SIVstm) Vif (31), and by several different SIV sooty mangabey (SIVsm) Vif proteins (31). This functional association is also consistent with the relatively recent origin of SIVmac, which is thought to have occurred in captivity by cross-species transmission from a sooty mangabey (SIVsm) (50). An alternative, albeit less likely, explanation is that the functional interaction between SIVmac239 Vif and human A3B may be due to molecular mimicry with the Vif-binding region of A3G, which based on cross-species comparisons has a long evolutionary history of lentivirus restriction (40). This alternative is supported by strong conservation of negative charge at amino acid position 130 (D in human A3B, human A3G, and almost all primate equivalents, including the sooty mangabey homologs). However, this alterative is refuted by divergence at amino acid position 128, which is variable among primate A3B and A3G enzymes. For instance, position 128 is a positively charged lysine in rhesus macaque A3G and a negatively charged aspartate in human A3G, whereas the corresponding position in rhesus macaque and human A3B is a negatively charged glutamate. Further studies, such as a full matrix of cross-species Vif/A3B comparisons, are needed to fully distinguish between these alternatives and additional possibilities such as Vif gain-of-function and/or Vif functional promiscuity within the SIVmac/sm clade.
Understanding the molecular interactions between SIVmac239 Vif and human A3B may also lead to the development of novel cancer therapies. Human A3B has been shown to cause mutations in multiple types of cancer and to contribute to the development of drug resistance in mouse models (51–53). A3B is therefore likely to have a major role in driving tumor evolution and has become both a diagnostic and a therapeutic target. We previously proposed using SIVmac239 Vif as a molecular probe to interrogate A3B function in vivo (31), but a direct application of this idea is complicated by promiscuous A3 degradation activity. Therefore, a better understanding of the interactions between SIVmac239 Vif and human A3B may help to engineer the requisite specificity for such an application. Indeed, our data show that the human A3B-interacting surface on SIVmac239 Vif is likely quite distinct and separable from the human A3F- or A3H-interacting surface. However, the large overlap between the human A3B- and A3G-interacting surfaces on SIVmac239 Vif is still a formidable hurdle in engineering perfect specificity. Nevertheless, the fact that three Vif mutants did not counter A3B and A3G identically suggests that such an outcome may be achieved with additional studies and particularly with comparative structural studies of multiple different Vif-A3 complexes.
MATERIALS AND METHODS
APOBEC3 expression constructs.
The constructs expressing huA3G (NM021822), huA3B (NM004900), rhA3G (AY331716), and rhA3B (JF714485) with C-terminal 3× hemagglutinin (3×HA) tags in the pcDNA3.1(+) vector (Invitrogen) have been reported previously (33). The pcDNA3.1(+) constructs expressing the C-terminally 3×HA-tagged N-terminal domain and C-terminal domain of huA3B have also been described previously (54). Mutants of human A3B (R122A, L123A, Y124A, Y125A, Y126A, W127A, E128K, D130K, and E128K D130K) were created by site-directed mutagenesis using forward primers 5′-ACC ATC TCT GCC GCC GCC CTC TAC TAC TAC TG-3′, 5′-TCT CTG CCG CCC GCG CCT ACT ACT ACT GGG-3′, 5′-TCT GCC GCC CGC CTC GCC TAC TAC TGG GAA AG-3′, 5′-GCC GCC CGC CTC TAC GCC TAC TGG GAA AGA GA-3′, 5′-CCG CCC GCC TCT ACT ACG CCT GGG AAA GAG ATT ACC-3′, 5′-GCC CGC CTC TAC TAC TAC GCG GAA AGA GAT TAC CGA AG-3′, 5′-GCC TCT ACT ACT ACT GGA AAA GAG ATT ACC GAA GG-3′, 5′-CTA CTA CTA CTG GGA AAG AAA GTA CCG AAG GGC GCT CTG C-3′, and 5′-CGC CTC TAC TAC TAC TGG AAA AGA AAG TAC CGA AGG GCG CTC TGC-3′ and corresponding reverse primers, respectively, verified by DNA sequencing, and used for transient expression in 293T cells. The constructs expressing untagged huA3F (NM145298) and huA3H haplotype II (FJ376614.1) in the pcDNA3.1(+) vector have been reported previously (36, 55).
Vif expression constructs.
The lentiviral Vif proteins from HIV-1IIIB (EU541617) and SIVmac239 (AY588946) were codon optimized (GenScript Corporation) and expressed with a C-terminal MYC tag from pVR1012 (36). Mutants of SIVmac239 Vif (W7A, W13A, P16A, E17A, R18A, R21A, W22A, K27A, K30A, K38A, C40A, C40F, P43A, H44A, F45A, K46A, V47A, G48A, W49A, W49Δ, W51A, W51Δ, Q62Δ, F64A, W73A, P77+G, C116A, R173A, D174A, N175A, R176A, R177A, and G178A) were created by site-directed mutagenesis using forward primers 5′-GAG GAA GAG AAG CGG GCG ATC GCA GTG CCT AC-3′, 5′-ATC GCA GTG CCT ACC GCG CGC ATT CCA GAG AG-3′, 5′-CTA CCT GGC GCA TTG CAG AGA GGC TCG A-3′, 5′-TGG CGC ATT CCA GCG AGG CTC GAG AGG-3′, 5′-GGC GCA TTC CAG AGG CGC TCG AGA GGT GGC-3′, 5′-CCA GAG AGG CTC GAG GCG TGG CAT AGC CTC AT-3′, 5′-AGA GAG GCT CGA GAG GGC GCA TAG CCT CAT CAA G-3′, 5′-GAG AGG TGG CAT AGC CTC ATC GCG TAC CTT AAA TAT AAG ACC AA-3′, 5′-GCA TAG CCT CAT CAA GTA CCT TGC ATA TAA GAC CAA AGA CCT GCA G-3′, 5′-AGA CCA AAG ACC TGC AGG CAG TGT GCT ATG TGC CCC-3′, 5′-CAA AGA CCT GCA GAA AGT GGC CTA TGT GCC CCA TTT TAA G-3′, 5′-AAA GAC CTG CAG AAA GTG TTC TAT GTG CCC CAT TTT AAG-3′, 5′-CAG AAA GTG TGC TAT GTG GCC CAT TTT AAG GTG G-3′, 5′-AAA GTG TGC TAT GTG CCC GCT TTT AAG GTG GGA TGG GC-3′, 5′-GTG CTA TGT GCC CCA TGC TAA GGT GGG ATG GGC C-3′, 5′-AAG TGT GCT ATG TGC CCC ATT TTG CGG TGG GAT GGG C-3′, 5′-GTG CCC CAT TTT AAG GCG GGA TGG GCC TGG-3′, 5′-CCC ATT TTA AGG TGG CAT GGG CCT GGT GGA C-3′, 5′-GCC CCA TTT TAA GGT GGG AGC GGC CTG GTG G-3′, 5′-GCC CCA TTT TAA GGT GGG AGC CTG GTG GA-3′, 5′-GGT GGG ATG GGC CGC GTG GAC CTG TTC C-3′, 5′-GGT GGG ATG GGC CTG GAC CTG TTC CA-3′, 5′-GTG ATT TTT CCC TTG GAG GGC TCC CAC CTG-3′, 5′-CCT TGC AGG AGG CCT CCC ACC TGG A-3′, 5′-GGA AGT TCA GGG ATA CGC GCA CCT GAC TCC TGA G-3′, 5′-TGG CAC CTG ACT CCT GGA GAG AAA GGC TGG CTG-3′, 5′-CTC CAC ATA CTT CCC CGC TTT CAC CGC CGG TGA G-3′, 5′-GAA GCA GTG GCG CGC CGA TAA CCG GCG C-3′, 5′-GTG GCG CCG CGC TAA CCG GCG CG-3′, 5′-AGC AGT GGC GCC GCG ATG CCC GGC GCG G-3′, 5′-GGC GCC GCG ATA ACG CGC GCG GGC TG-3′, 5′-CCG CGA TAA CCG GGC CGG GCT GAG GAT G-3′, and 5′-ATA ACC GGC GCG CGC TGA GGA TGG C-3′ and the corresponding reverse primers, respectively, verified by DNA sequencing, and used for transient expression in 293T cells.
Cell lines.
293T cells were maintained in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) and 0.5% penicillin-streptomycin (P-S). CEM-green fluorescent protein (GFP) cells were maintained in RPMI medium with 15% FBS and 0.5% P-S.
Vif degradation experiments.
293T cells were transfected with pcDNA3.1(+)-A3-HA or empty vector, along with pVR1012-Vif-MYC or empty vector, using polyethyleneimine (PEI) (Polysciences, Inc.) The following amounts of A3 and Vif expression constructs were transfected: 200 ng A3 and 200 ng Vif for Fig. 1A; 100 ng A3B, 200 ng A3G, and 25, 50, or 100 ng Vif for Fig. 1B; 200 ng A3 and 200 or 400 ng Vif for Fig. 2B and D; and 200 ng A3 and 200, 300, or 400 ng Vif for Fig. 2C and 3. After 48 h, cells were harvested for immunoblot analysis.
Immunoblotting experiments.
Cell and virus particle lysates were prepared by resuspension of washed cell pellets directly in 2.5× Laemmli sample buffer. Proteins were separated using discontinuous sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). HA-tagged A3 proteins were detected using monoclonal mouse anti-HA (BioLegend). Human A3F was detected using a polyclonal rabbit anti-A3F serum 675 created by contracted immunization of animals (Covance) with a peptide corresponding to the last 30 residues of the protein. A3H proteins were detected using previously generated monoclonal mouse anti-A3H (P1D8) (38). MYC-tagged Vif proteins were detected using polyclonal rabbit anti-MYC (Sigma-Aldrich). Tubulin was detected using a monoclonal mouse anti-α-tubulin antibody (Covance). HIV-1 Gag was detected using monoclonal mouse anti-HIV-1 p24 (NIH AIDS Reagent Program) (56). Immunoblots were quantified using ImageJ.
HIV single-cycle infection with replication-proficient virus.
The single-cycle infection assay was carried out as described previously (33). 293T cells were transfected (TransIT-LT1; Mirus) with 1 μg of a Vif-deficient HIV proviral expression construct along with 0, 25, or 50 ng of a Vif expression construct and 25 ng of an A3 expression construct. After 48 h, CEM-GFP HIV-1 reporter cells were infected with virus-containing supernatants, and cell and virus particle lysates were prepared for immunoblotting.
Flow cytometry.
HIV-infected CEM-GFP cells were prepared for flow cytometry by fixation using 4% paraformaldehyde in phosphate-buffered saline (PBS). GFP fluorescence was measured on a BD FACSCanto II flow cytometer. Data were analyzed using FlowJo flow cytometry analysis software (version 8.8.6). The fraction of GFP-positive cells among all live cells was quantified for each sample.
Sequence alignments and structural images.
All amino acid sequences were aligned using Clustal Omega. The amino acid sequences of human A3B, human A3G, HIV-1IIIB Vif, HIV-2ROD Vif, and SIVmac239 Vif correspond to GenBank accession numbers NM004900, NM021822, EU541617, P04595, and AY588946, respectively. The secondary structure elements of human A3B N-terminal domain, human A3B C-terminal domain, and human A3G C-terminal domain were annotated based on reported crystal structures (41, 57, 58). The secondary structural elements of human A3G N-terminal domain were predicted based on a crystal structure of the homologous rhesus macaque A3G N-terminal domain (59). Structural images were generated with PyMOL (pymol.org).
ACKNOWLEDGMENTS
We thank D. Salamango for comments on the manuscript and B. Wei and J. Lalli for assistance with plasmid constructions.
This work was supported by NIAID R37 AI064046 and NCI R21 CA206309. J.W. received partial salary support from an Interdisciplinary Doctoral Fellowship from the University of Minnesota Graduate School. R.S.H. is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished McKnight University Professor, and an Investigator of the Howard Hughes Medical Institute.
REFERENCES
- 1.Desimmie BA, Delviks-Frankenberrry KA, Burdick RC, Qi DF, Izumi T, Pathak VK. 2014. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J Mol Biol 426:1220–1245. doi: 10.1016/j.jmb.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris RS, Dudley JP. 2015. APOBECs and virus restriction. Virology 479:131–145. doi: 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakano Y, Aso H, Soper A, Yamada E, Moriwaki M, Juarez-Fernandez G, Koyanagi Y, Sato K. 2017. A conflict of interest: the evolutionary arms race between mammalian APOBEC3 and lentiviral Vif. Retrovirology 14:31. doi: 10.1186/s12977-017-0355-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conticello SG, Harris RS, Neuberger MS. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol 13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 5.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 6.Marin M, Rose KM, Kozak SL, Kabat D. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med 9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 7.Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 8.Stopak K, de Noronha C, Yonemoto W, Greene WC. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell 12:591–601. doi: 10.1016/S1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 9.Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21–31. doi: 10.1016/S0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- 10.Jäger S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. 2011. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481:371–375. doi: 10.1038/nature10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Du J, Evans SL, Yu Y, Yu XF. 2011. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481:376–379. doi: 10.1038/nature10718. [DOI] [PubMed] [Google Scholar]
- 12.Yu YK, Xiao ZX, Ehrlich ES, Yu XH, Yu XF. 2004. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev 18:2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo K, Xiao ZX, Ehrlich E, Yu YK, Liu BD, Zheng S, Yu XF. 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci U S A 102:11444–11449. doi: 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo Y, Dong L, Qiu X, Wang Y, Zhang B, Liu H, Yu Y, Zang Y, Yang M, Huang Z. 2014. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature 505:229–233. doi: 10.1038/nature12884. [DOI] [PubMed] [Google Scholar]
- 15.Chen G, He Z, Wang T, Xu R, Yu XF. 2009. A patch of positively charged amino acids surrounding the HIV-1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J Virol 83:8674–8682. doi: 10.1128/JVI.00653-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell RA, Smith J, Barr R, Bhattacharyya D, Pathak VK. 2009. Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of HIV-1 Vif. J Virol 83:1992–2003. doi: 10.1128/JVI.01621-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Binka M, Ooms M, Steward M, Simon V. 2012. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J Virol 86:49–59. doi: 10.1128/JVI.06082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Refsland EW, Hultquist JF, Luengas EM, Ikeda T, Shaban NM, Law EK, Brown WL, Reilly C, Emerman M, Harris RS. 2014. Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLoS Genet 10:e1004761. doi: 10.1371/journal.pgen.1004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bogerd HP, Doehle BP, Wiegand HL, Cullen BR. 2004. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc Natl Acad Sci U S A 101:3770–3774. doi: 10.1073/pnas.0307713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, Pathak VK. 2004. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci U S A 101:5652–5657. doi: 10.1073/pnas.0400830101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schrofelbauer B, Chen D, Landau NR. 2004. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc Natl Acad Sci U S A 101:3927–3932. doi: 10.1073/pnas.0307132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mangeat B, Turelli P, Liao S, Trono D. 2004. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J Biol Chem 279:14481–14483. doi: 10.1074/jbc.C400060200. [DOI] [PubMed] [Google Scholar]
- 23.Land AM, Shaban NM, Evans L, Hultquist JF, Albin JS, Harris RS. 2014. APOBEC3F determinants of HIV-1 Vif sensitivity. J Virol 88:12923–12927. doi: 10.1128/JVI.02362-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albin JS, LaRue RS, Weaver JA, Brown WL, Shindo K, Harjes E, Matsuo H, Harris RS. 2010. A single amino acid in human APOBEC3F alters susceptibility to HIV-1 Vif. J Biol Chem 285:40785–40792. doi: 10.1074/jbc.M110.173161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell RA, Pathak VK. 2007. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol 81:8201–8210. doi: 10.1128/JVI.00395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ooms M, Letko M, Simon V. 2017. The structural interface between HIV-1 Vif and human APOBEC3H. J Virol 91:e02289-. doi: 10.1128/JVI.02289-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakashima M, Tsuzuki S, Awazu H, Hamano A, Okada A, Ode H, Maejima M, Hachiya A, Yokomaku Y, Watanabe N, Akari H, Iwatani Y. 2017. Mapping region of human restriction factor APOBEC3H critical for interaction with HIV-1 Vif. J Mol Biol 429:1262–1276. doi: 10.1016/j.jmb.2017.03.019. [DOI] [PubMed] [Google Scholar]
- 28.Nakashima M, Ode H, Kawamura T, Kitamura S, Naganawa Y, Awazu H, Tsuzuki S, Matsuoka K, Nemoto M, Hachiya A, Sugiura W, Yokomaku Y, Watanabe N, Iwatani Y. 2016. Structural insights into HIV-1 Vif-APOBEC3F interaction. J Virol 90:1034–1047. doi: 10.1128/JVI.02369-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richards C, Albin JS, Demir O, Shaban NM, Luengas EM, Land AM, Anderson BD, Holten JR, Anderson JS, Harki DA, Amaro RE, Harris RS. 2015. The binding interface between human APOBEC3F and HIV-1 Vif elucidated by genetic and computational approaches. Cell Rep 13:1781–1788. doi: 10.1016/j.celrep.2015.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Letko M, Booiman T, Kootstra N, Simon V, Ooms M. 2015. Identification of the HIV-1 Vif and human APOBEC3G protein interface. Cell Rep 13:1789–1799. doi: 10.1016/j.celrep.2015.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Land AM, Wang J, Law EK, Aberle R, Kirmaier A, Krupp A, Johnson WE, Harris RS. 2015. Degradation of the cancer genomic DNA deaminase APOBEC3B by SIV Vif. Oncotarget 6:39969–39979. doi: 10.18632/oncotarget.5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaur R, Strebel K. 2012. Insights into the dual activity of SIVmac239 Vif against human and African green monkey APOBEC3G. PLoS One 7:e48850. doi: 10.1371/journal.pone.0048850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol 85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haché G, Shindo K, Albin JS, Harris RS. 2008. Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC3G. Curr Biol 18:819–824. doi: 10.1016/j.cub.2008.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doehle BP, Schafer A, Cullen BR. 2005. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 36.LaRue RS, Lengyel J, Jonsson SR, Andresdottir V, Harris RS. 2010. Lentiviral Vif degrades the APOBEC3Z3/APOBEC3H protein of its mammalian host and is capable of cross-species activity. J Virol 84:8193–8201. doi: 10.1128/JVI.00685-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith JL, Pathak VK. 2010. Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3DE and African green monkey APOBEC3F that interact with HIV-1 Vif. J Virol 84:12599–12608. doi: 10.1128/JVI.01437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li MM, Wu LI, Emerman M. 2010. The range of human APOBEC3H sensitivity to lentiviral Vif proteins. J Virol 84:88–95. doi: 10.1128/JVI.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huthoff H, Malim MH. 2007. Identification of amino acid residues in APOBEC3G required for regulation by HIV-1 Vif and virion encapsidation. J Virol 81:3807–3815. doi: 10.1128/JVI.02795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Compton AA, Emerman M. 2013. Convergence and divergence in the evolution of the APOBEC3G-Vif interaction reveal ancient origins of simian immunodeficiency viruses. PLoS Pathog 9:e1003135. doi: 10.1371/journal.ppat.1003135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao X, Yang H, Arutiunian V, Fang Y, Besse G, Morimoto C, Zirkle B, Chen XS. 2017. Structural determinants of APOBEC3B non-catalytic domain for molecular assembly and catalytic regulation. Nucleic Acids Res 45:7494–7506. doi: 10.1093/nar/gkx362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dang Y, Wang XJ, Zhou T, York IA, Zheng YH. 2009. Identification of a novel WxSLVK motif in the N terminus of HIV and SIV Vif that is critical for APOBEC3G and APOBEC3F neutralization. J Virol 83:8544–8552. doi: 10.1128/JVI.00651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehle A, Wilson H, Zhang C, Brazier AJ, McPike M, Pery E, Gabuzda D. 2007. Identification of an APOBEC3G binding site in HIV-1 Vif and inhibitors of Vif-APOBEC3G binding. J Virol 81:13235–13241. doi: 10.1128/JVI.00204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. 2005. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog 1:e6. doi: 10.1371/journal.ppat.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lavens D, Peelman F, Van der Heyden J, Uyttendaele I, Catteeuw D, Verhee A, Van Schoubroeck B, Kurth J, Hallenberger S, Clayton R, Tavernier J. 2010. Definition of the interacting interfaces of APOBEC3G and HIV-1 Vif using MAPPIT mutagenesis analysis. Nucleic Acids Res 38:1902–1912. doi: 10.1093/nar/gkp1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhai C, Ma L, Zhang Z, Ding J, Wang J, Zhang Y, Li X, Guo F, Yu L, Zhou J, Cen S. 2017. Identification and characterization of loop7 motif and its role in regulating biological function of human APOBEC3G through molecular modeling and biological assay. Acta Pharm Sin B 7:571–582. doi: 10.1016/j.apsb.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han X, Liang W, Hua D, Zhou X, Du J, Evans SL, Gao Q, Wang H, Viqueira R, Wei W, Zhang W, Yu XF. 2014. Evolutionarily conserved requirement for core binding factor beta in the assembly of the human immunodeficiency virus/simian immunodeficiency virus Vif-cullin 5-RING E3 ubiquitin ligase. J Virol 88:3320–3328. doi: 10.1128/JVI.03833-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hultquist JF, Binka M, LaRue RS, Simon V, Harris RS. 2012. Vif proteins of human and simian immunodeficiency viruses require cellular CBFbeta to degrade APOBEC3 restriction factors. J Virol 86:2874–2877. doi: 10.1128/JVI.06950-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith JL, Izumi T, Borbet TC, Hagedorn AN, Pathak VK. 2014. HIV-1 and HIV-2 Vif interact with human APOBEC3 proteins using completely different determinants. J Virol 88:9893–9908. doi: 10.1128/JVI.01318-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Apetrei C, Kaur A, Lerche NW, Metzger M, Pandrea I, Hardcastle J, Falkenstein S, Bohm R, Koehler J, Traina-Dorge V, Williams T, Staprans S, Plauche G, Veazey RS, McClure H, Lackner AA, Gormus B, Robertson DL, Marx PA. 2005. Molecular epidemiology of simian immunodeficiency virus SIVsm in U.S. primate centers unravels the origin of SIVmac and SIVstm. J Virol 79:8991–9005. doi: 10.1128/JVI.79.14.8991-9005.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS. 2013. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burns MB, Temiz NA, Harris RS. 2013. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet 45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Law EK, Sieuwerts AM, LaPara K, Leonard B, Starrett GJ, Molan AM, Temiz NA, Vogel RI, Meijer-van Gelder ME, Sweep FC, Span PN, Foekens JA, Martens JW, Yee D, Harris RS. 2016. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci Adv 2:e1601737. doi: 10.1126/sciadv.1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stenglein MD, Matsuo H, Harris RS. 2008. Two regions within the amino-terminal half of APOBEC3G cooperate to determine cytoplasmic localization. J Virol 82:9591–9599. doi: 10.1128/JVI.02471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liddament MT, Brown WL, Schumacher AJ, Harris RS. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol 14:1385–1391. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 56.Chesebro B, Wehrly K, Nishio J, Perryman S. 1992. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol 66:6547–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shandilya SM, Nalam MN, Nalivaika EA, Gross PJ, Valesano JC, Shindo K, Li M, Munson M, Royer WE, Harjes E, Kono T, Matsuo H, Harris RS, Somasundaran M, Schiffer CA. 2010. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure 18:28–38. doi: 10.1016/j.str.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi K, Carpenter MA, Kurahashi K, Harris RS, Aihara H. 2015. Crystal structure of the DNA deaminase APOBEC3B catalytic domain. J Biol Chem 290:28120–28130. doi: 10.1074/jbc.M115.679951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao X, Li SX, Yang H, Chen XS. 2016. Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat Commun 7:12193. doi: 10.1038/ncomms12193. [DOI] [PMC free article] [PubMed] [Google Scholar]