

ABSTRACT
Herpesvirus membrane fusion depends on the core fusion machinery, comprised of glycoproteins B (gB) and gH/gL. Although gB structurally resembles autonomous class III fusion proteins, it strictly depends on gH/gL to drive membrane fusion. Whether the gH/gL complex needs to be membrane anchored to fulfill its function and which role the gH cytoplasmic (CD) and transmembrane domains (TMD) play in fusion is unclear. While the gH CD and TMD play an important role during infection, soluble gH/gL of herpes simplex virus 1 (HSV-1) seems to be sufficient to mediate cell-cell fusion in transient assays, arguing against an essential contribution of the CD and TMD. To shed more light on this apparent discrepancy, we investigated the role of the CD and TMD of the related alphaherpesvirus pseudorabies virus (PrV) gH. For this purpose, we expressed C-terminally truncated and soluble gH and replaced the TMD with a glycosylphosphatidylinositol (gpi) anchor. We also generated chimeras containing the TMD and/or CD of PrV gD or HSV-1 gH. Proteins were characterized in cell-based fusion assays and during virus infection. Although truncation of the CD resulted in decreased membrane fusion activity, the mutant proteins still supported replication of gH-negative PrV, indicating that the PrV gH CD is dispensable for viral replication. In contrast, PrV gH lacking the TMD, membrane-anchored via a lipid linker, or comprising the PrV gD TMD were nonfunctional, highlighting the essential role of the gH TMD for function. Interestingly, despite low sequence identity, the HSV-1 gH TMD could substitute for the PrV gH TMD, pointing to functional conservation.
IMPORTANCE Enveloped viruses depend on membrane fusion for virus entry. While this process can be mediated by only one or two proteins, herpesviruses depend on the concerted action of at least three different glycoproteins. Although gB has features of bona fide fusion proteins, it depends on gH and its complex partner, gL, for fusion. Whether gH/gL prevents premature fusion or actively triggers gB-mediated fusion is unclear, and there are contradictory results on whether gH/gL function requires stable membrane anchorage or whether the ectodomains alone are sufficient. Our results show that in pseudorabies virus gH, the transmembrane anchor plays an essential role for gB-mediated fusion while the cytoplasmic tail is not strictly required.
KEYWORDS: herpesvirus, pseudorabies virus, glycoproteins, gH/gL complex, gB, membrane fusion, virus entry
INTRODUCTION
Infectious entry of enveloped viruses into cells requires fusion of the viral envelope with a host cell membrane. To meet this challenge, enveloped viruses have evolved specialized surface glycoproteins enabling them to bind to a cellular receptor and to catalyze membrane merging. Many well-studied enveloped viruses, e.g., influenza viruses or rhabdoviruses, require only a single viral protein to mediate both functions. In contrast, herpesviruses use a more complex apparatus, which involves multiple viral glycoproteins. The core fusion machinery consists of glycoprotein B (gB) and the heterodimeric complex of gH and gL (gH/gL). This subset of glycoproteins is conserved across the Herpesviridae, while receptor binding is primarily mediated by nonconserved subfamily-specific glycoproteins (reviewed in references 1 and 2). The alphaherpesviruses pseudorabies virus (PrV; Suid alphaherpesvirus 1) and herpes simplex viruses 1 and 2 (HSV-1/2; Human alphaherpesvirus 1/2), for example, require gD and a gD-specific cellular receptor such as nectin-1 or herpesvirus entry mediator (HVEM) (3). Other herpesviruses lack a gD homolog, and receptor binding function is mediated by, e.g., varicella-zoster virus (VZV; human herpesvirus 3) glycoprotein E (gE) (4, 5) or gp42 of Epstein-Barr virus (EBV) (6–8).
The current model for herpesvirus membrane fusion assumes that the entry glycoproteins are activated in a cascade-like fashion to promote membrane fusion (9, 10). Activation of the Simplexvirus fusion machinery is initiated by binding of gD to one of its cellular receptors, which leads to a conformational change in the C-terminal region of the gD ectodomain, as shown for HSV-1 (11–14). This activated gD is thought to trigger gH/gL, which, in turn, is presumed to activate the bona fide fusion protein gB by direct interaction of their respective ectodomains (9, 15–18). A similar mechanism has been proposed for the Varicellovirus PrV (19). The related Varicellovirus VZV, on the other hand, has to initiate fusion via a fundamentally different mechanism, since it completely lacks a gD homolog (4, 5).
The crystal structures of the gB ectodomains resemble those of typical class III fusion proteins, including a trimeric fold, internal bipartite fusion loops, and a central alpha-helical coiled-coil (20–23). Despite its similarities to other class III fusion proteins, such as the G protein of vesicular stomatitis virus or baculovirus gp64 (24, 25), gB is not able to drive membrane fusion on its own but depends on the presence of the gH/gL complex (16, 18). However, the role of the gH/gL complex during fusion is still elusive.
Unlike gB, the crystal structures of EBV gH/gL (26), HSV-2 gH/gL (16), VZV gH/gL (27), and a core fragment of PrV gH (28) revealed no features typical for fusion proteins, and the experimental data point to a regulatory role (16, 21, 24, 26–28). While correct folding and transport of gH depends on the presence of gL in most herpesviruses (16, 29), it is not essential for gH virion incorporation in PrV (30). Moreover, infection of PrV can occur in the absence of gL, and/or the gL-binding domain in gH, when compensatory mutations in gD, gH, and/or gB are present (31–33), indicating that gL is not directly involved in the membrane fusion process, at least in PrV. In contrast, gL is required for fusion in HSV-1 and HSV-2, and no gL-negative infectious virus mutants have been reported in the simplexviruses.
For membrane fusion, a direct interaction between the ectodomains of gB and gH/gL has been proposed (9). Nevertheless, previous data demonstrated important functions for the transmembrane (TMD) and the cytoplasmic domains (CD) of gB and gH, for which structural information is currently lacking. However, the gB CD obviously restricts membrane fusion, since truncation of or point mutations within this region are reported to increase the fusogenic activity in several herpesviruses (17, 34–36). The 93-amino-acid (aa) CD of PrV gB contains two predicted alpha-helical domains. A C-terminally truncated PrV gB lacking one of these domains (gB008) (34), including an overlapping endocytosis motif, resulted in significantly enhanced cell surface expression as well as increased fusion activity in virus-free cell-cell fusion assays (32, 34, 37). PrV gB mutants lacking both alpha-helical domains (gB007), the complete CD (gB006), or the CD and the TMD (gB005) were unable to complement gB-negative PrV (34). Surprisingly, the defect in gB007, which still mediated direct cell-to-cell spread, was compensated for by two mutations in the gB ectodomain (38), showing a functional interplay between sequences in the CD and the ectodomain. In addition, the CDs of EBV and HSV-1 gB were reported to directly interact with lipid membranes (39, 40), indicating that the gB CD functions as a clamp, stabilizing the prefusion form of gB (41).
In contrast to gB, the gH CD is very short, with an only poorly conserved amino acid sequence even within the alphaherpesvirus subfamily (Fig. 1A). The CDs of HSV-1, VZV, and EBV gH had been linked to regulation of cell fusion (7, 42, 43), and a direct interaction between the CDs of EBV gH and gB was reported recently (7), pointing to an important role of the CDs in fusion. However, truncations of the cytoplasmic domains in HSV-1, VZV, and EBV gH had different consequences in terms of fusion activity. Stepwise truncations of the 14-aa HSV-1 gH CD resulted in a gradual reduction in fusion levels, whereas deletion of the complete HSV-1 gH CD completely abrogated cell-cell fusion activity, even in the presence of hyperfusogenic gB variants (41), as well as its ability to complement a gH-negative mutant (42). These studies indicate that the CD, or at least part of it, is essential for HSV-1 gH function. It had been hypothesized that it acts as a wedge to release the gB CD clamp, allowing gB to exert its fusion function (41). Surprisingly, a 5-aa insertion in the membrane-proximal part of the HSV-1 gH CD also prevented cell-cell fusion and complementation (44). Thus, either the length of the gH C tail is crucial or the membrane-proximal amino acids of the CDs, which are the most highly conserved, at least within the alphaherpesvirus gH homologs, comprising a basic amino acid (K or R, except in VZV gH) followed by a pair of neutral nonaromatic residues, execute specific functions.
FIG 1.
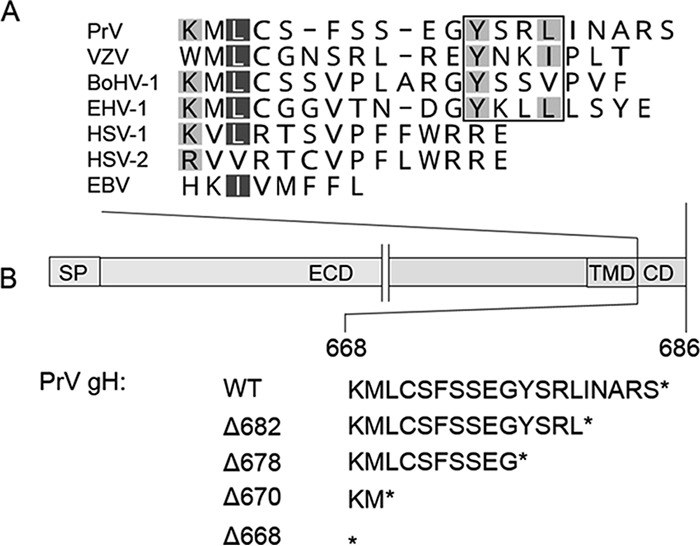
Sequence alignment of gH cytoplasmic domains of different herpesviruses and schematic representation of the generated gH mutants. (A) The alignment was generated using ClustalW (Geneious 10.3.2). Residues are colored according to their conservation (white letters on dark gray background, 80 to 100%; black letters on light gray, 60 to 80%). The YXXϕ endocytosis motif is boxed. Sequences of herpesvirus gH used for the alignment correspond to gH of pseudorabies virus (PrV; AAA47466), varicella-zoster virus (VZV; AH010537.2), bovine alphaherpesvirus 1 (BoHV-1; CAA41677), equid alphaherpesvirus 1 (EHV-1; AY464052), human alphaherpesvirus 1 (HSV-1; AJE60179), human alphaherpesvirus 2 (HSV-2; CAB06746), and Epstein-Barr virus (EBV; V01555.2). (B) The PrV gH open reading frame and the amino acid sequence of the cytoplasmic domains of the generated truncation mutants are shown. SP, signal peptide; ECD, ectodomain; TMD, transmembrane domain; CD, cytoplasmic domain.
In contrast to HSV-1, PrV, and EBV, full-length VZV gH is not functional in cell-cell fusion assays but requires truncation of the last 8 amino acids, comprising a predicted endocytosis motif, for function in these assays (43, 45, 46). In addition, it was shown that the length of the VZV gH CD is important for fusion regulation but not its amino acid composition. However, in these studies the membrane-proximal sequences were not altered, leaving their importance for fusion unclear (43). It was speculated that the gH CD acts as a gate keeper to control access to functional domains of neighboring proteins, e.g., allowing or preventing phosphorylation of the gB CD (43).
The EBV gH CD comprises only eight residues and was found to regulate fusion by altering gH binding to gp42 and epithelial cell attachment (7). Deletion of the last four amino acids had only marginal effects on cell-cell fusion. However, truncation to only two amino acids or complete deletion of the CD reduced fusion to background levels (7). Here, in contrast to VZV gH, the CD amino acid composition seems to be important, since replacement by foreign sequences resulted in significantly reduced fusion levels (7).
Besides the CD, the functional role of the gH TMD has also been discussed controversially. Mutations in the HSV-1 gH TMD, substitution of the gH TMD by analogous domains from other glycoproteins, or replacement by a glycosylphosphatidylinositol (gpi) anchor resulted in a nonfunctional protein (42, 47). These data indicate that HSV gH/gL needs to be anchored in the membrane by a specific TMD to fulfil its function. In contrast, recent studies suggested that soluble forms of HSV gH/gL are sufficient to induce gB-mediated fusion (9), while this was not observed for EBV (8, 48).
The poor sequence conservation of the gH CD and the divergent results in different herpesviruses highlight the need to study each gH homolog individually, especially in light of the different requirements to induce fusion. Thus, whereas in PrV gB and gH/gL are sufficient for the induction of syncytium formation by direct cell-cell fusion in transfection-based fusion assays (37), gD is required in HSV-1 (49). In VZV, which lacks a gD homolog, expression of gB and gH/gL is sufficient to induce membrane fusion in cell fusion assays, resembling the situation in PrV (5, 46). Moreover, EBV depends on the presence of receptor-binding gp42 for fusion of B cells (50, 51). Since the role of the CD and the TMD of PrV gH during membrane fusion had not been characterized, we investigated the functional relevance of the CD and the TMD of PrV gH by generating different C-terminal truncations and site-specific mutations, soluble gH, a gpi-anchored gH, as well as chimeras containing either only the TMD or TMD and CD of HSV-1 gH or PrV gD. These proteins were characterized for their ability to function in virus-free membrane fusion as well as virus infection.
RESULTS
Function of the PrV gH cytoplasmic domain.
To investigate the functional relevance of the 19-amino-acid (aa) cytoplasmic domain (CD) of PrV gH, different truncated proteins were generated lacking either the C-terminal 5, 9, or 17 aa or the complete CD (gHΔ682, gHΔ678, gHΔ670, and gHΔ668, respectively) (Fig. 1B). Correct protein expression was analyzed by Western blotting of rabbit kidney cells (RK13) after cotransfection with expression plasmids encoding the different gH variants and gL. Expression of gH was detected using the monospecific anti-gH serum (31). In Western blots, immature gH (gH imm) as well as several processed forms (gH mat) were detected for all C-terminally truncated mutants retaining at least the two most membrane-proximal amino acids (Fig. 2A). After complete deletion of the CD (gHΔ668), predominantly immature gH was detected, indicating that the amino acids immediately following the transmembrane domain (TMD) are involved in efficient protein trafficking and/or maturation (Fig. 2A). In contrast to HSV gH, which depends on the presence of gL for correct maturation (52), gL is dispensable for maturation of PrV gH (30, 53). Thus, absence of gL did not influence processing of any of the gH proteins (data not shown).
FIG 2.
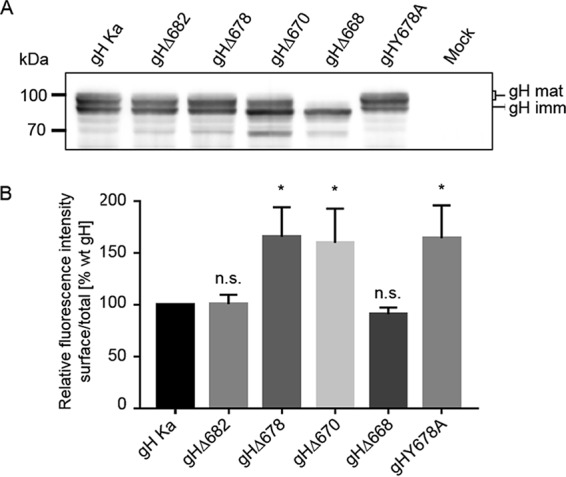
Expression and surface localization of gH truncation mutants. (A) Western blot analysis. Lysates prepared 48 h after transfection with expression plasmids for wild-type (gH Ka) or mutant gH in combination with gL or only the empty vector (mock) were separated by SDS-PAGE. Blots were probed for presence of mature (gH mat) and immature gH (gH imm). Molecular masses (kDa) of marker proteins are indicated on the left. (B) Flow cytometry. RK13 cells were transfected in parallel assays with expression plasmids for either wild-type or C-terminally truncated gH in combination with gL. After 24 h, cells were either permeabilized or not and then stained with a monoclonal antibody against gH and Alexa-Fluor 488-conjugated secondary antibodies, and fluorescence was measured. Bars represent the ratio between the mean fluorescence intensity (MFI) of cells expressing gH at the surface and MFI of total gH-positive cells. Results obtained for gH Ka were set as 100%. Mean values and standard deviations from three independent experiments are shown. Significance of differences from results obtained with wild-type gH is marked (*, P < 0.05). n.s., not significantly different from gH Ka.
Transport of the gH variants to the plasma membrane was investigated by flow cytometry of RK13 cells cotransfected with gH and gL expression plasmids. The analysis revealed that gHΔ682 and gHΔ668 were present on the surface at levels similar to those of wild-type (wt) gH (derived from PrV strain Kaplan [PrV-Ka]) (Fig. 2B), whereas deletion of the predicted tyrosine-based endocytosis motif YSRL, located at positions 679 to 682, as in gHΔ678 and gHΔ670 (Fig. 1), resulted in enhanced surface expression, indicating that the motif is functional (Fig. 2B). To further investigate this finding, tyrosine at position 678 was replaced by alanine (gHY678A). Flow cytometry of transfected cells revealed enhanced surface expression for gHY678A (Fig. 2B).
To analyze the role of PrV gH CD in membrane fusion, the different truncated mutants and gHY678A were tested in transient-transfection-based cell-cell fusion assays (Fig. 3A) (32, 54). Fusion activity was analyzed after cotransfection of RK13 cells with expression plasmids encoding either wild-type gH or the mutated PrV gH variants in combination with plasmids expressing wild-type gB and gL as well as enhanced green fluorescent protein (EGFP), which was used to facilitate evaluation by fluorescence microscopy (54). Assays with wild-type gH, gB, and gL served as positive controls, which were set as 100%, and assays with the gH expression plasmid replaced by the empty vector pcDNA3 served as negative controls (vector). Figure 3A shows that fusion activity obtained with gHΔ682, lacking the terminal 5 aa, was comparable to that of wild-type gH. gHΔ678, devoid of the last 9 aa, including the endocytosis motif, showed a slight increase in fusion activity, which was also evident in gHY678A-expressing cells, indicating that the enhanced cell surface localization promotes cell-cell fusion. However, despite efficient surface expression, expression of gHΔ670, with only two amino acids in the CD left, resulted in only approximately 50% fusion activity compared to that of the positive control. gHΔ668, lacking the complete CD, although showing impaired protein processing (Fig. 2A), was still able to mediate fusion to a limited extent, and fusion activity reached values of around 20%, while no fusion was observed in the absence of gH (Fig. 3A). These results indicate that the membrane-proximal part of the PrV gH CD contains sequences required for correct protein processing but is also involved in fusion regulation. However, the PrV gH CD was apparently not absolutely essential to mediate cell-cell fusion.
FIG 3.
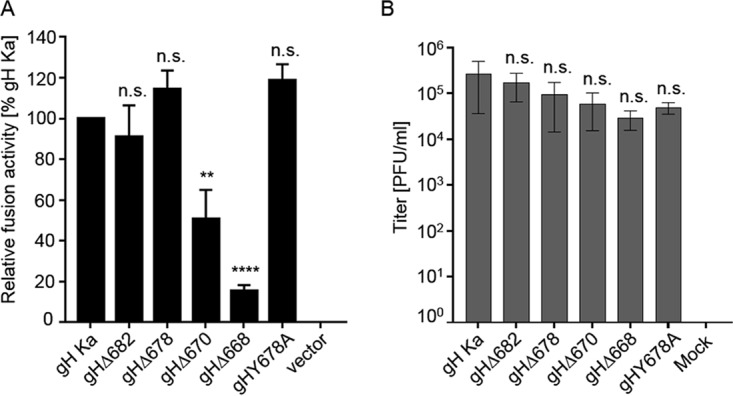
Cell-cell fusion activity of gH truncation mutants and transcomplementation of PrV-ΔgH. (A) RK13 cells were cotransfected with 200 ng of expression plasmids for EGFP in combination with full-length gB, gL, gH, or C-terminally truncated gH. Twenty-four hours posttransfection, the areas of green fluorescing syncytia were measured, and total fusion activity was determined by multiplication of the mean syncytium area with the number of syncytia in 10 fields of view. Fusion activities obtained with wild-type gH Ka, gB, and gL were set as 100%. Cells transfected with empty vector pcDNA3 served as a negative control (vector). Shown are mean relative values and standard deviations from four independent experiments with the corresponding standard deviations. Values significantly differing from those obtained with gH Ka are marked (**, P < 0.01; ****, P < 0.0001; all by unpaired t test with Welch correction). n.s., not significantly different from gH Ka. (B) gH Ka- or mutant gH-expressing RK13 cells were infected with PrV-ΔgH. Progeny virus titers were determined on wild-type PrV gH/gL-expressing cells and are given in PFU per ml. Cells transfected with the empty vector pcDNA3 served as a negative control. Shown are mean values for three independent assays with corresponding standard deviations. n.s., not significantly different from gH Ka.
The ability of the gH mutants to function in virus entry was determined by transcomplementation of a gH-negative PrV mutant (Fig. 3B) (55). To this end, RK13 cells were transfected with expression plasmids encoding wild-type gH or the different gH mutants and subsequently infected with PrV-ΔgH at a multiplicity of infection (MOI) of 3. Progeny virus titers were determined on RK13-gH/gL cells (56). As shown in Fig. 3B, all gH mutants were able to complement gH-negative virus, but titers were gradually reduced with decreasing length of the CD. Titers derived from cells expressing gH completely lacking the CD (gHΔ668), however, showed only an approximately 10-fold reduction (Fig. 3B) despite compromised protein processing (Fig. 2A). No infectious virus could be detected after infection of vector-transfected (mock) cells. These results show that the PrV gH CD also is not essential for gH function during virus replication.
Expression of soluble, gpi-anchored, and chimeric gH constructs.
To test for functional relevance of the transmembrane anchor, soluble gH (gHs) comprising only the PrV gH ectodomain (aa 1 to 646), as well as a glycosylphosphatidylinositol (gpi)-anchored version of gH (gH-gpi; gpi was added at position 646), were constructed (Fig. 4A). To investigate whether the gH TMD possesses specific features important for its function, gH chimeras were generated in which the TMD, or the TMD and CD of PrV gH, were replaced by the equivalent sequences from HSV-1 gH (gH-Hs-Hp and gH-Hs-Hs) or PrV gD (gH-D-H and gH-D-D) (Fig. 4A). Protein expression was analyzed by Western blotting and flow cytometry (Fig. 5). In contrast to wild-type gH, mature gHs was easily detectable in the supernatant of transfected cells, while immature forms of gHs were exclusively found in the cell lysate, demonstrating efficient secretion of anchorless gH (Fig. 5A). gH-gpi was also found in the supernatant after cleavage with phospholipase C (PLC), indicating correct processing (Fig. 5B). In Western blot experiments, expression patterns of all mutants were comparable to those of wild-type gH, indicating that the substitutions had no effect on protein processing and maturation (Fig. 5C).
FIG 4.
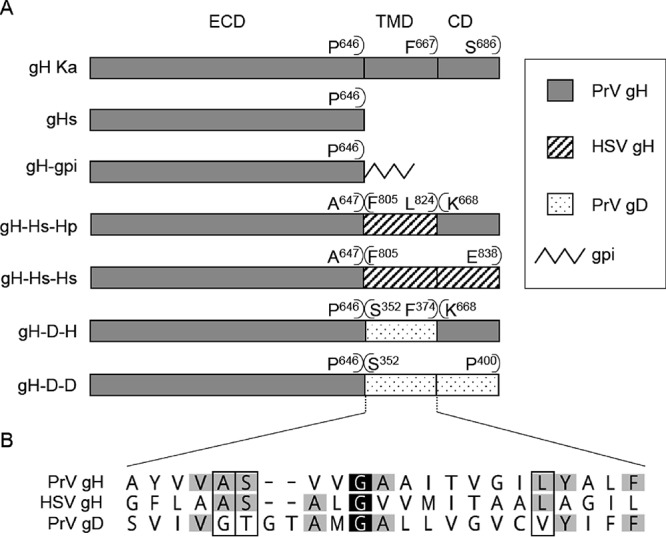
Schematic representation of the constructed gH mutants. (A) The PrV gH transmembrane domain (TMD) and/or cytoplasmic domain (CD) were deleted or replaced with a gpi anchor or the analogous region from HSV-1 or PrV gD. The constructs are illustrated, and each is named to indicate its composition with reference to the ectodomain (ECD), TMD, and CD. The first and last amino acids of the predicted (Geneious 10.3.2) components of the chimeric proteins are given above the respective bars. Amino acid positions refer to the sequences of PrV gH (GenBank accession number AAA47466), HSV-1 gH (GenBank accession number AJE60179), and PrV gD (GenBank accession number AFI70842.1). The proteins are not drawn to scale. (B) Sequence alignment of the predicted transmembrane domains of PrV and HSV-1 gH and PrV gD. The alignment was generated using ClustalW (Geneious 10.3.2). Residues are colored according to their conservation (white letters on black, 100%; black letters on light gray, 60 to 80%). Amino acids conserved in PrV and HSV gH but not in gD are boxed.
FIG 5.

Expression and surface localization of gHs, gH-gpi, and gH chimeras. Cell lysates (CL) or supernatants (SN) harvested 48 h after transfection with expression plasmids for wild-type gH Ka, gHs (A), gH-gpi (B), the different gH chimeras (C), or the empty vector (mock) were separated by SDS-PAGE. To test whether the predicted gH-gpi is membrane associated via the gpi anchor, wild-type gH and gH-gpi-expressing cells were treated with PLC. Supernatants and lysates of PLC-treated cells were separated by SDS-PAGE. Blots were incubated with a PrV gH-specific rabbit polyclonal antiserum. Molecular masses (kDa) of marker proteins are indicated. (D) Total and cell surface expression of wild-type gH and the gH mutants was analyzed by flow cytometry. Bars represent the ratio between the mean fluorescence intensity (MFI) of cells expressing gH at the surface and MFI of total gH-positive cells. Results obtained for gH Ka were set as 100%. Mean values and standard deviations for three independent experiments are shown. Significance of differences from results obtained with gH Ka is marked (*, P < 0.05; **, P < 0.01). n.s., not significantly different from gH Ka.
Total expression and surface localization of the gH proteins were analyzed by flow cytometry of RK13 cells cotransfected with expression plasmids for gH and gL (Fig. 5D). These analyses revealed that gH-gpi was efficiently expressed at the cell surface, while only a significantly smaller proportion of gHs could be detected on the surface (Fig. 5D). Surface expression of gH-Hs-Hp, gH-D-H, and gH-D-D was comparable to that of gH Ka, whereas a significant increase in surface expression compared to that of wild-type gH was observed for gH-Hs-Hs (Fig. 5D).
Functional analysis of soluble, gpi-anchored, and chimeric gH constructs.
The ability of the different gH mutants to function in cell-cell fusion and entry was investigated by fusion (Fig. 6A) and complementation assays (Fig. 6B). In contrast to the closely related HSV-1, in which soluble forms of gH/gL were able to trigger fusion, although at only low levels (9), soluble PrV gH/gL was completely unable to induce fusion (Fig. 6A), even in the presence of hyperfusogenic gB variants (data not shown). Despite localization on the cell surface, no fusion activity could be observed for gH-gpi, gH-D-H, or gH-D-D. However, chimeras containing the HSV-1 gH TMD were able to efficiently mediate membrane fusion to levels comparable to that of wild-type gH.
FIG 6.

Cell-cell fusion activity of gHs, gH-gpi, and gH chimeras and transcomplementation of PrV-ΔgH. (A) Results of cell-cell fusion assays are expressed as percent fusion activity of wild-type gH in combination with gB and gL set as 100%. (B) Viral titers obtained in the transcomplementation assay are shown as gray bars and correspond to the number of PFU per ml. Shown are mean values and corresponding standard deviations for four independent experiments. Values significantly differing from those obtained with wild-type gH are marked (****, P < 0.0001 by unpaired t test with Welch correction). n.s., not significantly different from gH Ka.
In line with these results, gHs, gH-gpi, gH-D-H, and gH-D-D were not able to complement gH-negative PrV (Fig. 6B). However, chimeras containing the HSV-1 gH TMD efficiently complemented PrV-ΔgH, with only marginally reduced viral titers (Fig. 6B). Thus, our data demonstrate that membrane anchorage of PrV gH via a TMD is crucial for its function in fusion. This TMD has to fulfil specific requirements not provided by the PrV gD TMD but by the HSV-1 gH TMD. In summary, we show that the PrV gH TMD is important for its function during the fusion process.
DISCUSSION
Infectious entry of enveloped viruses requires fusion of the viral envelope with cellular membranes. In herpesviruses, this involves the conserved fusion machinery consisting of gB and the heterodimeric gH/gL complex. However, the molecular details of this process remain to be uncovered. Although it is accepted that gB is the actual fusion protein, the exact role of the gH/gL complex remains elusive. Despite its presence in all herpesviruses, the amino acid sequence of gH is only poorly conserved, even between members from the same subfamily (28). Not only the poor amino acid sequence conservation of gH but also the different requirements to induce fusion in different herpesviruses highlight the need to study each protein individually to elucidate common features and differences for a better understanding of the fusion process.
In contrast to that of HSV (49), PrV gL is not necessary for correct transport, processing, or virion incorporation of gH and, moreover, the gL-binding domain in PrV gH is dispensable for the fusion process in the presence of compensatory mutations in gH and/or gB (31, 33). Reports on HSV-1 indicated that soluble gH/gL, lacking the membrane anchor, is sufficient to mediate low levels of membrane fusion in transient assays (9). This was surprising in light of earlier data ascribing an important function to the transmembrane and cytoplasmic domains of HSV-1 gH (42, 47). However, a soluble form of EBV gH/gL was not sufficient to trigger membrane fusion (8, 48). These fundamental differences in the requirement for gH in different herpesviruses prompted us to investigate whether the requirements for fusion activation differ for members of different subfamilies or even between the closely related alphaherpesviruses HSV-1 and PrV.
For this purpose, we first generated C-terminally truncated gH variants. While deletion of the 5 C-terminal amino acids had no effect on cell-cell fusion or complementation of PrV-ΔgH, deletion or site-specific mutagenesis of the predicted endocytosis motif (678YSRL681) resulted in an enhanced cell surface expression (Fig. 2B) concomitant with higher levels of fusion activity (Fig. 3A). However, gHΔ670, which retained only the two membrane-proximal amino acids despite enhanced surface expression (Fig. 2B), showed a 50% reduction in fusion activity (Fig. 3A), pointing to a regulatory role of the gH CD, particularly the N-terminal half.
Congruent with our results, approximately half of the gH CD (10 of 19 aa, as in PrV gHΔ678) appears to be sufficient for full function, as was shown for EBV gH (4 of 8 aa) (7) and HSV-1 (8 of 14 aa) (41). Further deletion, however, resulted in a significant reduction of fusion activity, again pointing to a regulatory role for the membrane-proximal part of the gH CD (Fig. 3A). In contrast, despite its similarity to the PrV gH CD in length, the C-terminal half of the VZV gH CD (8 of 18 aa) obviously impairs cell-cell fusion and had to be removed for function in transfection-based cell-cell fusion assays (43, 46). This enhanced fusion activity resulted in increased syncytium formation in infected cells concomitant with a significant reduction in virus titers (43). VZV is characterized by its syncytial phenotype in vivo, is highly cell associated, and lacks a gD homolog (57–59). It can be speculated that the VZV gH CD partly fulfills the regulatory role which is otherwise encoded in accessory proteins as, e.g., gD in HSV-1 or gp42 in EBV. PrV, which also belongs to the Varicellovirus genus, obviously exhibits an intermediate phenotype between HSV-1 and VZV regarding the importance of gD. Cell-cell fusion and direct cell-to-cell spread in PrV occur independently of gD (37, 60, 61) but without a significant syncytial phenotype in vitro or in vivo. Free virions even enter cells in the absence of gD when compensatory mutations in gB and gH are present (62). These data point to a well-balanced but virus-specific control of membrane fusion by a complex interplay between the receptor-binding factor and the components of the core fusion machinery which differ between the different herpesviruses.
The membrane-proximal gH CD residues of different alphaherpesviruses show a higher degree of conservation, comprised of a basic residue (K or R; the only exception is VZV) followed by two neutral nonaromatic residues (M, V, and L) (Fig. 1A). These residues, or at least the first two amino acids, appear to be important for efficient protein translocation, presumably by involvement in lipid interactions in the plasma membrane or the viral envelope (63). Correct trafficking is often influenced by the CD of type I transmembrane proteins. Since PrV gHΔ670 was efficiently processed and transported to the cell surface (Fig. 2B), the two remaining membrane-proximal residues seem to be sufficient for efficient transport through the secretory pathway. In contrast, gHΔ668, which completely lacks the CD, was mainly detected in its immature form (Fig. 2A). Nevertheless, surface expression of gHΔ668 was only slightly reduced compared to that of wt gH (Fig. 2B), and gHΔ668 still complemented PrV-ΔgH to only approximately 10-fold-reduced titers (Fig. 3B), indicating that the PrV gH CD, in contrast to the HSV-1 gH CD (41, 42), is not required for incorporation into the virus envelope to function during entry. In contrast to EBV gH (7), surface expression of HSV-1 gH lacking the CD seemed to be unaffected (41). However, the importance of the membrane-proximal residues might explain why a five-amino-acid insertion immediately after the TMD completely abrogated function (44, 64), as did complete deletion of the CD (41, 42).
The HSV-1 and VZV gH CDs were suggested to function as gatekeepers, utilizing their length to control access of functional domains within neighboring proteins (41, 43). Since PrV gH mutants gHΔ670 and gHΔ668 showed reduced fusion levels in combination with wild-type gB (Fig. 3A) or a more fusogenic variant (data not shown), it is possible that the PrV gH CD, although not essential, modulates fusion in a manner similar to that proposed for HSV. The HSV gH CD was suggested to act as a wedge to release the gB CD clamp, which enables activation of gB (41). Moreover, the conformation of the gH ectodomain may be altered by the truncations influencing interactions between the gB and gH ectodomains, although reactivity of truncated gH variants of HSV, VZV, and EBV with several conformation-dependent monoclonal antibodies was not affected. However, binding of the ectodomain of a truncated EBV gH to gp42 was shown to be altered (7, 41, 43). Because monoclonal antibodies for PrV gH recognizing conformational epitopes are lacking, a conformational alteration of the PrV gH ectodomain cannot be excluded.
Since the PrV gH CD was found to be dispensable for gH function during virus infection, we analyzed whether the membrane-spanning region is required. Thus, we expressed soluble gH (gHs; aa 1 to 646) lacking the CD and TMD (Fig. 4A). gHs was efficiently secreted into the supernatant of transiently transfected RK13 cells (Fig. 5A). However, no fusion activity or functional complementation was detectable (Fig. 6), even when gB variants with increased fusogenicity were coexpressed or when soluble forms of more fusogenic gH were used (data not shown). Thus, we conclude that membrane anchorage is essential for PrV gH function in fusion.
Replacement of the TMD (and CD) by a lipid anchor (gH-gpi), thereby anchoring the protein in only one leaflet of the lipid membrane, resulted in a nonfunctional protein (Fig. 6) despite efficient cell surface expression (Fig. 5D), again highlighting the importance of a membrane-spanning domain. This prompted us to investigate whether any transmembrane domain is sufficient for this purpose. To this end, we generated PrV gH chimeras in which the TMD (or TMD and CD) of PrV gH was replaced by the equivalent sequence from PrV gD or HSV-1 gH (Fig. 4A). All chimeras were efficiently expressed and processed (Fig. 5C). While gH-D-H and gH-Hs-Hp, both containing the CD of PrV gH, as well as gH-D-D, were present at levels on the cell surface similar to those of the wt gH, gH-Hs-Hs revealed an increase in cell surface localization (Fig. 5D). The HSV CD does not show a typical endocytosis motif (Fig. 1A), which could explain the enhanced gH-Hs-Hs cell surface presentation. The PrV gD CD, however, contains a functional endocytosis motif (YRLL) (65), which is apparently also active in gH-D-D. Despite efficient processing and surface expression, neither gH-D-H nor gH-D-D was functional in cell-cell fusion and transcomplementation assays (Fig. 6). In line with our data, the HSV-1 gH TMD could not be replaced by the TMD of HSV-1 gD or other type I membrane proteins, such as CD8 and influenza HA, pointing to a gH-specific feature within the TMD of herpesvirus gH (42). In contrast, the HSV-gH TMD efficiently substituted for the TMD in PrV gH in both assays. This is consistent with previous results showing that the complete HSV-1 gH C-terminal part comprising the conserved domain IV, as well as TMD and CD, could functionally substitute for the corresponding part in PrV gH, pointing to functional conservation (53).
Sequence comparison between the PrV gD, PrV gH, and HSV-1 gH TMD reveals only one strictly conserved amino acid, a centrally located glycine (G) residue at amino acid position 655 in PrV and 812 in HSV gH (Fig. 4B). Although a crucial role for gH function was attributed to this particular glycine, its presence in the gD TMD (aa 362) apparently is not sufficient for functional complementation in gH (42). Three further amino acids are conserved between PrV and HSV gH TMD that are not present in the gD TMD (Fig. 4B). Interestingly, in HSV-1, mutation of two of them, namely, alanine (A) 808 and serine (S) 809, which correspond to A651 and S652 in PrV gH, resulted in severely compromised cell-cell fusion (42). With respect to the observed functional conservation of the TMD of HSV and PrV, a similar effect on fusion seems likely for PrV. The third conserved amino acid absent from gD is leucine (L) at amino acid position 663 in PrV. In HSV and PrV gH, L663, together with A651, S652, and G655, are located on one face of an alpha-helical wheel plot (42). This suggests that the gH TMD has an intrinsic property to interact with lipids or other molecules within the membrane, involving those four amino acids. However, the 21-aa TMD of HSV-1 gH is close to the lower limit for a transmembrane-spanning helix (42). The PrV gD TMD, in contrast, is comprised of 23 residues (38, 65). Thus, we cannot completely exclude that the difference in length and not the amino acid composition is responsible for the nonfunctional phenotype.
Our data highlight the importance of the PrV gH TMD, with its specific features (length and amino acid composition) for function in fusion, whereas, in contrast to HSV-1 (41, 42), the PrV gH CD is apparently not an essential component of the fusion machinery. Moreover, as observed for EBV (48) and unlike HSV (9), soluble forms of PrV gH/gL are unable to promote cell-cell fusion, even in the presence of gB proteins with increased fusogenic potential. Interestingly, the gD TMD is not able to substitute functionally for the gH TMD, although localization at the plasma membrane, indicative of proper membrane anchorage, was observed. We hypothesize that the PrV gH TMD specifically interacts with membrane components such as lipids or other molecules as a prerequisite for triggering membrane fusion. These specific interactions could also be mediated by the HSV gH TMD but not by the TMD of gD.
In summary, we demonstrate here the relevance of the PrV gH TMD and CD in membrane fusion. Whereas a specific gH TMD is essential for triggering fusion, the gH CD modulates fusion efficiency. Given the importance of the TMD with its specific requirements, fusion induction by soluble HSV-1 gH/gL or in trans with gB and gH/gL present in different membranes, as shown for HSV-1 (9) and human cytomegalovirus (66), is difficult to reconcile. Either fusion can be triggered differently by the homologous gH/gL proteins, which seems improbable, or the influence of the TMD is indeed more quantitative than qualitative, with a higher dependence of PrV gH for a boost in fusion activity by the TMD. In either case the presence of a correct TMD is of high relevance for PrV gH function.
MATERIALS AND METHODS
Cells and viruses.
Rabbit kidney (RK13) and RK13-gH/gL cells (56) were grown in Dulbecco's modified Eagle's minimum essential medium (MEM) supplemented with 10% fetal calf serum (FCS) at 37°C and 5% CO2. The PrV mutant lacking gH (PrV-gHABF; here designated PrV-ΔgH) (55), which was derived from PrV strain Kaplan (PrV-Ka) (67), was propagated on RK13-gH/gL cells.
Expression plasmids.
Construction of expression plasmids for PrV-Ka and HSV-1 F gB, gH, and gL has been described previously (37, 53). Expression plasmids for the truncated versions of gH and soluble gH were generated by PCR, using pcDNA-gH as the template, DH-3 as forward primer, and the corresponding reverse primers gH Δ668-686, gH Δ670-686, gH Δ678-686, gH Δ682-686, and gH ECD R (Table 1). PCR products were cloned after cleavage with EcoRI and XhoI into correspondingly digested pcDNA3 (Invitrogen). The expression plasmid for gHY678A was generated by site-directed mutagenesis (QuikChange II XL kit; Agilent) with the complementary pair of oligonucleotide primers (Table 1) and pcDNA-gH as the template. For generation of pcDNA-gH-gpi, the 5′-terminal part of the PrV gH open reading frame (ORF) was amplified from pcDNA-gH Ka using primers gH Ka HindIII F and gH ECD EcoRV (Table 1) and cleaved with HindIII and EcoRV. The 110-bp sequence comprising a gpi addition signal was purified from pspGPIsig (kind gift from G. Keil, Insel Riems, Germany) by cleavage with SmaI and EcoRI. The two fragments were ligated and subsequently cloned into pcDNA3 via HindIII and EcoRI restriction sites.
TABLE 1.
Oligonucleotide primers for mutagenesis, PCR and sequence analyses
| Primer namea | Restriction site | Sequenceb (5′–3′) | Nucleotide positionc |
|---|---|---|---|
| Primers for construction of gH CD mutants | |||
| DH-3 | EcoRI | AGA ATT CAA AGT TTG CCG TGC CCG TC | 60748–60766 |
| gH_ECD R | XhoI | CACACTCGAGCTACGGGGACACGGGCGC | 62693–62707 (r) |
| gH Δ668-686 | XhoI | CACACTCGAGCTAGAATAGGGCGTACAGGATCCC | 62750–62770 (r) |
| gH Δ670-686 | XhoI | CACACTCGAGCTACATCTTGAATAGGGC | 62762–62776 (r) |
| gH Δ678-686 | XhoI | CACACTCGAGCTAGCCCTCGGAGGA | 62780–62809 (r) |
| gH Δ682-686 | XhoI | CACACTCGAGTAACCGAGAATAGCC | 62812–62798 (r) |
| Primers for construction of gH Y678A | |||
| gH Y679A F | CCTCCGAGGGCGCTTCTCGGTTAATAAACG | 62790–62819 | |
| gH Y679A R | CGTTTATTAACCGAGAAGCGCCCTCGGAGG | 62790-62819 (r) | |
| Primer for construction of gH-gpi | |||
| gH Ka HindIII F | HindIII | CACAAGCTTAAAGTTTGCCGTGCCCGTC | 60748–60766 |
| gH ECD EcoRV R | EcoRV | CACAGATATCCGGGGACACGGGCGCGAGCCG | 62687–62707 (r) |
| Primers for construction of gH chimeras | |||
| P CD H TMD F | TGGTTATGATTACCGCCGCCCTGGCTGGCATCCTA/AAGATGCTCTGCAGC | H, 43828–43862; P, 62771–62785 | |
| P ECD H TMD R | ATCATAACCACGCCCAGCGCAGAGGCGGCCAGAA/AGGCCGGGGACACGGG | H, 43853–43886; P, 62696–62711 (r) | |
| P ECD H TMD CD F | TCGCGCCCGTGTCCCCGGCC/TTTCTGGCCGCCTCTGCG | H, 43870–43887; P, 62691–62711 | |
| P ECD H TMD CD R | GCCAGCCAGGGCGGCGGTAATCATAACCACGCCCAGCGCAGAGGCGGCCAGAA/AGGCCGGGGACACGGGCGCGAGCC | H, 43834–42886; P, 62688–62711 (r) | |
| H TMD P CD | GCCGCCTCTGCGCTGGGCGTGGTTATGATTACCGCCGCCCTGGCTGGCATCCTA/ATGCTCTGCAGCTTCTCCTCCGAGG | H, 43828–43881; P, 62774–62798 | |
| gH Ka ECD gD TMD R | CGTGCCGACGATCACCGA/CGGGGACACGGGCGCGAGC | gD Ka, 122199–122215; gH Ka, 62689–62707 (r) | |
| gH Ka ECD gD TMD F | GCTCGCGCCCGTGTCCCCG/TCGGTGATCGTCGGCACG | gH Ka, 62689–62707; gD Ka, 122198–122215 | |
| gD TM gH CD F | ACCGCGATGGGCGCGCTCCTGGTGGGCGTGTGCGTCTACATCTTCTTC/AAGATGCTCTGCAGCTTC | gH Ka, 62771–62788; gD Ka, 122219–122266 | |
| gD TM gH CD R | AGGAGCGCGCCCATCGCGGTGCCCGTGCCGACGATCACCGAC/GGGGACACGGGCGCGAG | gH Ka, 62707–62690; gD Ka, 122198–122238 (r) | |
| HHV1gH-R | XhoI | CACACTCGAG TTA TTCGCGTCTCC | 43783–43796 (r) |
| US6-rev | EcoRI | CACAGAATTCC ATC GACGCCGGTACTGC | 121353–122370 (r) |
| DHrev1 | XbaI | CATCTAGACACGCGCACGCAGAGAGT | 62856–62874 (r) |
| Primer for sequencing | |||
| T7 | TAATACGACTCACTATAGGG | pcDNA3 | |
| SP6 | CTCTAGCATTTAGGTGACACTATAG | pcDNA3 (r) | |
| gH-WH3 | TGCACGAGAGCGACGACTACC | 61479–61499 | |
| gH-WH6 | GGACATCGCCAACCCGCTCG | 62356–62375 |
Designations and components of the OE primers specify the virus. P, PrV; H, HSV-1; TMD, transmembrane domain; CD, cytoplasmic domain; ECD, ectodomain; F, forward; R, reverse. Nucleotide positions in the PrV Ka genome refer to GenBank accession no. JQ809328 and for HSV-1 strain F to GenBank accession no. GU734771.
Nonmatching nucleotides are printed in boldface, and restriction sites for convenient cloning of PCR products are underlined. HSV-1 and PrV gD sequences are shown in italics.
Reverse-strand PCR primers are indicated (r).
Expression plasmids for gH chimeras gH-Hs-Hs, gH-Hs-Hp, gH-D-D, and gH-D-H were generated by overlap extension PCR (OE-PCR) (68). The 5′-terminal parts of the PrV gH ORF were amplified from pcDNA-gH using primer DH-3 and the corresponding reverse OE primers (Table 1). For constructs with the TMD from HSV-1 gH but comprising the CD from PrV gH, the 3′-terminal part of the PrV gH ORF was amplified from pcDNA-gH using the desired forward OE primers in combination with the reverse primer DHrev1 (Table 1). The 3′-terminal parts of the HSV-1 gH or PrV gD ORF were amplified from pcDNA-H1FgH (53) or pcDNA-gD (32), respectively, with the appropriate forward OE primers and the reverse primer HHV1 gH-R or US6-rev, respectively (Table 1). The PCR products were purified, and 10 ng of each of the matching products, which overlapped by ≥15 bp, was mixed and amplified in a final PCR using only the primers for the termini. PCR products were cloned via restriction sites which had been introduced by the primers into correspondingly digested pcDNA3 (Table 1). Correct mutagenesis of all constructs was verified by sequencing (data not shown). Geneious software, version 10.2.3 (Biomatters), was used for analysis and comparison of DNA and protein sequences.
Western blot analyses.
Protein expression was tested after transfection of the expression plasmids into RK13 cells. For this, approximately 1.8 × 105 RK13 cells per well were seeded onto 24-well cell culture plates. The following day, cells were transfected with 200 ng of the corresponding gH expression plasmid in 100 μl Opti-MEM using 1 μl Lipofectamine 2000 (Thermo Fisher Scientific). Empty vector (pcDNA3) served as a negative control. The mixture was incubated for 20 min at room temperature and subsequently added to the cells. After 3 h, cells were washed with phosphate-buffered saline (PBS) and incubated in MEM supplemented with 2% FCS at 37°C. Cells were lysed 48 h posttransfection, and samples were separated by discontinuous SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with the monospecific rabbit PrV gH antiserum (31), which was used at a dilution of 1:15,000. Binding of peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) was detected with Clarity Western ECL substrate (Bio-Rad) and recorded with a VersaDoc 4000 MP imager (Bio-Rad).
PLC treatment.
Correct membrane attachment of gH by a gpi anchor was tested by treatment with phospholipase C (PLC). For this, transfected cells were incubated with PBS containing 0.2 U PLC (Boehringer) per ml. After 1 h on a shaker at room temperature, the supernatant was collected, centrifuged for 2 min at 13,000 rpm, and subsequently used for Western blot or immunofluorescence analysis.
Flow cytometry.
RK13 cells were cotransfected with expression plasmids for gH and gL as described above and were detached after 24 h with 2 mM EDTA in PBS for 1 h at 4°C. Subsequently, cells were fixed with 4% paraformaldehyde (PFA) for 20 min at 4°C and washed with PBS supplemented with 2% bovine serum albumin (BSA) and 2 mM EDTA. Each sample was split into two fluorescence-activated cell sorting (FACS) tubes. While one aliquot was permeabilized with 0.5% saponin in PBS with 0.2% BSA for 30 min at 4°C to assay total gH expression, the second was incubated without saponin to test for plasma membrane localization of gH. After a washing step with 2 mM EDTA in PBS, cells were probed with a monoclonal gH antibody (31) at a dilution of 1:5 in PBS with 2% BSA for 1 h at 4°C. After another washing step with 2 mM EDTA in PBS, cells were incubated for 1 h at 4°C with a secondary antibody (Alexa Fluor 488, goat anti-mouse IgG; Invitrogen) at a dilution of 1:1,000 in PBS with 2% BSA. Samples were analyzed using a MACSQuant analyzer (Miltenyi Biotec). Surface expression of gH was quantified as the mean fluorescence intensity (MFI) of Alexa-488-positive, nonpermeabilized cells divided by the MFI of Alexa-488-positive, permeabilized cells. Results obtained for gH Ka were set as 100%. Mean values and standard deviations from three independent assays were determined.
Transient-transfection-based cell-cell fusion assays.
Fusion activity of the different gH mutants was determined after transient transfection of RK13 cells as described recently (54). Briefly, cells were transfected with 200 ng each of the expression plasmids for EGFP (pEGFP-N1; Clontech) and for PrV glycoproteins gB, gL, and gH or mutant gH in 100 μl Opti-MEM using 1 μl Lipofectamine 2000 (32, 37). Empty vector (pcDNA3) served as the negative control. Cells were fixed 24 h posttransfection with 3% PFA. Syncytium formation was analyzed using an Eclipse Ti-S fluorescence microscope and NIS-Elements Imaging Software (Nikon). Total fusion activity was determined by multiplication of the area of cells with three or more nuclei by the number of syncytia within 10 fields of view (5.5 mm2 each). Each experiment was repeated four times, and average percent values of positive-control transfections as well as standard deviations were calculated.
Transcomplementation assay.
The ability of the gH mutants to function in virus entry was determined by transcomplementation of PrV-ΔgH (55). Cells were transfected with the gH expression plasmids as described above. Twenty-four hours posttransfection cells were infected with PrV-ΔgH at a multiplicity of infection (MOI) of 3 and consecutively incubated on ice for 1 h and at 37°C for 1 h. Subsequently, the inoculum was removed, nonpenetrated virus was inactivated by low-pH treatment (69), and 1 ml fresh medium was added. After 24 h at 37°C, the cells and supernatant were harvested and lysed by freeze-thawing (−80°C and 37°C). Progeny virus titers were determined on RK13-gH/gL cells (56).
Statistical analyses.
The statistical significance of differences observed in flow cytometry, transient fusion assays, and transcomplementation assays was evaluated using unpaired t test with Welch correction provided by GraphPad Prism 7 software (GraphPad Software, Inc., San Diego, CA).
ACKNOWLEDGMENTS
These studies were supported by the Deutsche Forschungsgemeinschaft (DFG grant Me 854/11-2).
The technical assistance of Karla Günther is greatly appreciated. We thank Birke A. Tews, Michael Knittler, and Elke Rufer for their help with FACS analysis.
REFERENCES
- 1.Harrison SC. 2015. Viral membrane fusion. Virology 479-480:498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 65:1653–1668. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Q, Krogmann T, Ali MA, Tang WJ, Cohen JI. 2007. The amino terminus of varicella-zoster virus (VZV) glycoprotein E is required for binding to insulin-degrading enzyme, a VZV receptor. J Virol 81:8525–8532. doi: 10.1128/JVI.00286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davison AJ, Scott JE. 1986. The complete DNA sequence of varicella-zoster virus. J Gen Virol 67(Part 9):1759–1816. doi: 10.1099/0022-1317-67-9-1759. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Kenyon WJ, Li Q, Mullberg J, Hutt-Fletcher LM. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J Virol 72:5552–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Jardetzky TS, Longnecker R. 2016. The cytoplasmic tail domain of Epstein-Barr virus gH regulates membrane fusion activity through altering gH binding to gp42 and epithelial cell attachment. mBio 7:e01871-16. doi: 10.1128/mBio.01871-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowe CL, Connolly SA, Chen J, Jardetzky TS, Longnecker R. 2013. A soluble form of Epstein-Barr virus gH/gL inhibits EBV-induced membrane fusion and does not function in fusion. Virology 436:118–126. doi: 10.1016/j.virol.2012.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A 104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog 7:e1002277. doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazear E, Whitbeck JC, Zuo Y, Carfi A, Cohen GH, Eisenberg RJ, Krummenacher C. 2014. Induction of conformational changes at the N-terminus of herpes simplex virus glycoprotein D upon binding to HVEM and nectin-1. Virology 448:185–195. doi: 10.1016/j.virol.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J 24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connolly SA, Whitbeck JJ, Rux AH, Krummenacher C, van Drunen Littel-van den Hurk S, Cohen GH, Eisenberg RJ. 2001. Glycoprotein D homologs in herpes simplex virus type 1, pseudorabies virus, and bovine herpes virus type 1 bind directly to human HveC(nectin-1) with different affinities. Virology 280:7–18. doi: 10.1006/viro.2000.0747. [DOI] [PubMed] [Google Scholar]
- 15.Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J Biol Chem 284:17370–17382. doi: 10.1074/jbc.M109.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper RS, Heldwein EE. 2015. Herpesvirus gB: a finely tuned fusion machine. Viruses 7:6552–6569. doi: 10.3390/v7122957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84:3825–3834. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li A, Lu G, Qi J, Wu L, Tian K, Luo T, Shi Y, Yan J, Gao GF. 2017. Structural basis of nectin-1 recognition by pseudorabies virus glycoprotein D. PLoS Pathog 13:e1006314. doi: 10.1371/journal.ppat.1006314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 21.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A 106:2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burke HG, Heldwein EE. 2015. Crystal structure of the human cytomegalovirus glycoprotein B. PLoS Pathog 11:e1005227. doi: 10.1371/journal.ppat.1005227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandramouli S, Ciferri C, Nikitin PA, Calo S, Gerrein R, Balabanis K, Monroe J, Hebner C, Lilja AE, Settembre EC, Carfi A. 2015. Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat Commun 6:8176. doi: 10.1038/ncomms9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roche S, Bressanelli S, Rey FA, Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- 25.Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat Struct Mol Biol 15:1024–1030. doi: 10.1038/nsmb.1484. [DOI] [PubMed] [Google Scholar]
- 26.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xing Y, Oliver SL, Nguyen T, Ciferri C, Nandi A, Hickman J, Giovani C, Yang E, Palladino G, Grose C, Uematsu Y, Lilja AE, Arvin AM, Carfi A. 2015. A site of varicella-zoster virus vulnerability identified by structural studies of neutralizing antibodies bound to the glycoprotein complex gHgL. Proc Natl Acad Sci U S A 112:6056–6061. doi: 10.1073/pnas.1501176112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc Natl Acad Sci U S A 107:22635–22640. doi: 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roop C, Hutchinson L, Johnson DC. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J Virol 67:2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klupp BG, Fuchs W, Weiland E, Mettenleiter TC. 1997. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J Virol 71:7687–7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klupp BG, Mettenleiter TC. 1999. Glycoprotein gL-independent infectivity of pseudorabies virus is mediated by a gD-gH fusion protein. J Virol 73:3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schröter C, Vallbracht M, Altenschmidt J, Kargoll S, Fuchs W, Klupp BG, Mettenleiter TC. 2015. Mutations in pseudorabies virus glycoproteins gB, gD, and gH functionally compensate for the absence of gL. J Virol 90:2264–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vallbracht M, Rehwaldt S, Klupp BG, Mettenleiter TC, Fuchs W. 2017. Functional relevance of the N-terminal domain of pseudorabies virus envelope glycoprotein H and its interaction with glycoprotein L. J Virol 91:e00061-17. doi: 10.1128/JVI.00061-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nixdorf R, Klupp BG, Karger A, Mettenleiter TC. 2000. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J Virol 74:7137–7145. doi: 10.1128/JVI.74.15.7137-7145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weise K, Kaerner HC, Glorioso J, Schroder CH. 1987. Replacement of glycoprotein B gene sequences in herpes simplex virus type 1 strain ANG by corresponding sequences of the strain KOS causes changes of plaque morphology and neuropathogenicity. J Gen Virol 68(Part 7):1909–1919. doi: 10.1099/0022-1317-68-7-1909. [DOI] [PubMed] [Google Scholar]
- 36.Fan Z, Grantham ML, Smith MS, Anderson ES, Cardelli JA, Muggeridge MI. 2002. Truncation of herpes simplex virus type 2 glycoprotein B increases its cell surface expression and activity in cell-cell fusion, but these properties are unrelated. J Virol 76:9271–9283. doi: 10.1128/JVI.76.18.9271-9283.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klupp BG, Nixdorf R, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J Virol 74:6760–6768. doi: 10.1128/JVI.74.15.6760-6768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nixdorf R, Klupp BG, Mettenleiter TC. 2001. Role of the cytoplasmic tails of pseudorabies virus glycoproteins B, E and M in intracellular localization and virion incorporation. J Gen Virol 82:215–226. doi: 10.1099/0022-1317-82-1-215. [DOI] [PubMed] [Google Scholar]
- 39.Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J Virol 86:8171–8184. doi: 10.1128/JVI.00932-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia NJ, Chen J, Longnecker R. 2013. Modulation of Epstein-Barr virus glycoprotein B (gB) fusion activity by the gB cytoplasmic tail domain. mBio 4:e00571-12. doi: 10.1128/mBio.00571-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rogalin HB, Heldwein EE. 2015. Interplay between the herpes simplex virus 1 gB cytodomain and the gH cytotail during cell-cell fusion. J Virol 89:12262–12272. doi: 10.1128/JVI.02391-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harman A, Browne H, Minson T. 2002. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J Virol 76:10708–10716. doi: 10.1128/JVI.76.21.10708-10716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang E, Arvin AM, Oliver SL. 2014. The cytoplasmic domain of varicella-zoster virus glycoprotein H regulates syncytia formation and skin pathogenesis. PLoS Pathog 10:e1004173. doi: 10.1371/journal.ppat.1004173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson JO, Longnecker R. 2010. Reevaluating herpes simplex virus hemifusion. J Virol 84:11814–11821. doi: 10.1128/JVI.01615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pasieka TJ, Maresova L, Grose C. 2003. A functional YNKI motif in the short cytoplasmic tail of varicella-zoster virus glycoprotein gH mediates clathrin-dependent and antibody-independent endocytosis. J Virol 77:4191–4204. doi: 10.1128/JVI.77.7.4191-4204.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A 107:866–871. doi: 10.1073/pnas.0913351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones NA, Geraghty RJ. 2004. Fusion activity of lipid-anchored envelope glycoproteins of herpes simplex virus type 1. Virology 324:213–228. doi: 10.1016/j.virol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 48.Kirschner AN, Omerovic J, Popov B, Longnecker R, Jardetzky TS. 2006. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J Virol 80:9444–9454. doi: 10.1128/JVI.00572-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 50.Mullen MM, Haan KM, Longnecker R, Jardetzky TS. 2002. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol Cell 9:375–385. doi: 10.1016/S1097-2765(02)00465-3. [DOI] [PubMed] [Google Scholar]
- 51.Rowe CL, Chen J, Jardetzky TS, Longnecker R. 2015. Membrane anchoring of Epstein-Barr virus gp42 inhibits fusion with B cells even with increased flexibility allowed by engineered spacers. mBio 6:e02285-14. doi: 10.1128/mBio.02285-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J Virol 66:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Böhm SW, Backovic M, Klupp BG, Rey FA, Mettenleiter TC, Fuchs W. 2016. Functional characterization of glycoprotein h chimeras composed of conserved domains of the pseudorabies virus and herpes simplex virus 1 homologs. J Virol 90:421–432. doi: 10.1128/JVI.01985-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vallbracht M, Schröter C, Klupp BG, Mettenleiter TC. 2017. Transient transfection-based fusion assay for viral proteins. Bio-Protocol 7:e2162. doi: 10.21769/BioProtoc.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Böhm SW, Eckroth E, Backovic M, Klupp BG, Rey FA, Mettenleiter TC, Fuchs W. 2015. Structure-based functional analyses of domains II and III of pseudorabies virus glycoprotein H. J Virol 89:1364–1376. doi: 10.1128/JVI.02765-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klupp B, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J Virol 82:6299–6309. doi: 10.1128/JVI.00386-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Litwin V, Jackson W, Grose C. 1992. Receptor properties of two varicella-zoster virus glycoproteins, gpI and gpIV, homologous to herpes simplex virus gE and gI. J Virol 66:3643–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheatham WJ, Dolan TF Jr, Dower JC, Weller TH. 1956. Varicella: report of two fatal cases with necropsy, virus isolation, and serologic studies. Am J Pathol 32:1015–1035. [PMC free article] [PubMed] [Google Scholar]
- 59.Esiri MM, Tomlinson AH. 1972. Herpes zoster. Demonstration of virus in trigeminal nerve and ganglion by immunofluorescence and electron microscopy. J Neurol Sci 15:35–48. [DOI] [PubMed] [Google Scholar]
- 60.Rauh I, Mettenleiter TC. 1991. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J Virol 65:5348–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peeters B, de Wind N, Broer R, Gielkens A, Moormann R. 1992. Glycoprotein H of pseudorabies virus is essential for entry and cell-to-cell spread of the virus. J Virol 66:3888–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmidt J, Gerdts V, Beyer J, Klupp BG, Mettenleiter TC. 2001. Glycoprotein D-independent infectivity of pseudorabies virus results in an alteration of in vivo host range and correlates with mutations in glycoproteins B and H. J Virol 75:10054–10064. doi: 10.1128/JVI.75.21.10054-10064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilson DW, Davis-Poynter N, Minson AC. 1994. Mutations in the cytoplasmic tail of herpes simplex virus glycoprotein H suppress cell fusion by a syncytial strain. J Virol 68:6985–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jackson JO, Lin E, Spear PG, Longnecker R. 2010. Insertion mutations in herpes simplex virus 1 glycoprotein H reduce cell surface expression, slow the rate of cell fusion, or abrogate functions in cell fusion and viral entry. J Virol 84:2038–2046. doi: 10.1128/JVI.02215-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ficinska J, Van Minnebruggen G, Nauwynck HJ, Bienkowska-Szewczyk K, Favoreel HW. 2005. Pseudorabies virus glycoprotein gD contains a functional endocytosis motif that acts in concert with an endocytosis motif in gB to drive internalization of antibody-antigen complexes from the surface of infected monocytes. J Virol 79:7248–7254. doi: 10.1128/JVI.79.11.7248-7254.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wille PT, Wisner TW, Ryckman B, Johnson DC. 2013. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 4:e00332-13. doi: 10.1128/mBio.00332-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaplan AS, Vatter AE. 1959. A comparison of herpes simplex and pseudorabies viruses. Virology 7:394–407. doi: 10.1016/0042-6822(59)90068-6. [DOI] [PubMed] [Google Scholar]
- 68.Heckman KL, Pease LR. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc 2:924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 69.Mettenleiter TC. 1989. Glycoprotein gIII deletion mutants of pseudorabies virus are impaired in virus entry. Virology 171:623–625. doi: 10.1016/0042-6822(89)90635-1. [DOI] [PubMed] [Google Scholar]