

ABSTRACT
Signal peptidase complex subunit 1 (SPCS1) is a newly identified host factor that regulates flavivirus replication, but the molecular mechanism is not fully understood. Here, using Japanese encephalitis virus (JEV) as a model, we investigated the mechanism through which the host factor SPCS1 regulates the replication of flaviviruses. We first validated the regulatory function of SPCS1 in JEV propagation by knocking down and knocking out endogenous SPCS1. The loss of SPCS1 function markedly reduced intracellular virion assembly and the production of infectious JEV particles but did not affect cell entry, RNA replication, or translation of the virus. SPCS1 was found to interact with nonstructural protein 2B (NS2B), which is involved in posttranslational protein processing and virus assembly. Serial deletion mutation of the JEV NS2B protein revealed that two transmembrane domains, NS2B(1–49) and NS2B(84–131), interact with SPCS1. Further mutagenesis analysis of conserved flavivirus residues in two SPCS1 interaction domains of NS2B demonstrated that G12A, G37A, and G47A in NS2B(1–49) and P112A in NS2B(84–131) weakened the interaction with SPCS1. Deletion mutation of SPCS1 revealed that SPCS1(91–169), which contains two transmembrane domains, was involved in interactions with both NS2B(1–49) and NS2B(84–131). Taken together, these results demonstrate that SPCS1 affects viral replication by interacting with NS2B, thereby influencing the posttranslational processing of JEV proteins and the assembly of virions.
IMPORTANCE Understanding virus-host interactions is important for elucidating the molecular mechanisms of virus propagation and identifying potential antiviral targets. Previous reports demonstrated that SPCS1 is involved in the flavivirus life cycle, but the mechanism remains unknown. In this study, we confirmed that SPCS1 participates in the posttranslational protein processing and viral assembly stages of the JEV life cycle but not in the cell entry, genome RNA replication, or translation stages. Furthermore, we found that SPCS1 interacts with two independent transmembrane domains of the flavivirus NS2B protein. NS2B also interacts with NS2A, which is proposed to mediate virus assembly. Therefore, we propose a protein-protein interaction model showing how SPCS1 participates in the assembly of JEV particles. These findings expand our understanding of how host factors participate in the flavivirus replication life cycle and identify potential antiviral targets for combating flavivirus infection.
KEYWORDS: assembly, flavivirus, host factor, protein-protein interactions, viral replication
INTRODUCTION
Japanese encephalitis virus (JEV) is a zoonotic mosquito-transmitted arbovirus belonging to the genus Flavivirus in the family Flaviviridae. In addition to JEV, many other flaviviruses are important human pathogens, such as West Nile virus (WNV), dengue virus (DENV), yellow fever virus (YFV), and Zika virus (ZIKV). These flaviviruses have similar replication life cycles and genomic structures. The genome consists of a single-stranded positive-sense RNA with a length of ∼11 kb. The single open reading frame (ORF) encodes a large polyprotein, which is processed by cellular and viral proteases into three structural proteins (C, prM/M, and E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (1). The C protein and genomic RNA form the nucleocapsid, which is enveloped by membrane anchored with prM/M and E proteins (2). Nonstructural proteins contribute to viral RNA replication, protein processing, and virion assembly (3–5).
Among all nonstructural proteins, NS2B acts as a cofactor for NS3 protein function, promoting protease activity to process the genome-encoded polyprotein (6). JEV NS2B is an integral membrane protein of 131 amino acids that contains three main domains: an N-terminal hydrophobic transmembrane domain (TMD) (containing two transmembrane regions, TM1 and TM2), a C-terminal hydrophobic transmembrane domain (TM3), and a central hydrophilic outer membrane domain required for the activation of the NS3 protease function (7–11). Other than its protease cofactor activity, little is known about other functions of NS2B, especially the transmembrane domains. A previous report showed that WNV NS2B interacts with itself and other membrane proteins, including NS2A, NS4A, and NS4B (12), and Li et al. (13) recently confirmed these interactions in JEV. Furthermore, through systematic mutagenesis of conserved flavivirus residues within the transmembrane domains of NS2B, these features of NS2B were found to be important for virion assembly. However, the molecular mechanism underpinning the participation of NS2B in virion assembly remains unknown.
Signal peptidase complex subunit 1 (SPCS1) is a component of the microsomal signal peptidase complex that is responsible for the cleavage of signal peptides of many secreted and membrane-associated proteins. Using genome-wide inhibition of WNV-induced cell death with a clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 knockout system, Zhang et al. (14) revealed that SPCS1 is needed for the proper cleavage of flavivirus structural proteins and the secretion of viral particles. The loss of SPCS1 expression resulted in markedly reduced yields of WNV, DENV, ZIKV, YFV, JEV, and hepatitis C virus (HCV) particles. For HCV, SPCS1 was identified as a host factor participating in virus assembly through interactions with E2 and NS2 (15). However, exactly how SPCS1 regulates the life cycle of flaviviruses remains to be elucidated.
In this study, we validated the regulatory effect of SPCS1 on JEV propagation by knocking down and knocking out endogenous SPCS1. SPCS1 was important for posttranslational protein processing and virus assembly but not cell entry, RNA replication, or translation. Using a bimolecular fluorescence complementation (BiFC) system, SPCS1 was shown to interact with NS2B transmembrane domains. Together with knowledge of the roles of NS2A and NS2B in virion assembly, our results demonstrate for the first time that the host factor SPCS1 participates in the virion assembly of flaviviruses through interactions with NS2B.
RESULTS
Loss of host factor SPCS1 function impairs JEV propagation.
To verify the role of endogenous SPCS1 in the propagation of JEV, three SPCS1-specific small interfering RNAs (siRNAs) or an uncorrelated control siRNA was transfected into HEK-293 cells, followed by infection with JEV. At 48 h postinfection (hpi), cells were fixed to detect JEV E antigen by an indirect immunofluorescence assay (IFA) with E protein-specific monoclonal antibody (MAb) 5E7. As indicated in Fig. 1A, among the three SPCS1-specific siRNAs, the greatest reduction in infectivity was observed with siRNA 2 (P < 0.001), and siRNA 3 also showed a significant reduction in infectivity (P < 0.05). Only siRNA 1 failed to show a significant reduction in infectivity. Furthermore, siRNA transfection had no significant effect on cell viability (Fig. 1B).
FIG 1.

Effect of SPCS1 knockdown on propagation of JEV. (A) HEK-293 cells were transfected with three different siRNAs targeted against SPCS1, or a control siRNA, at a final concentration of 15 nM. At 48 hpi, cells were infected with JEV at an MOI of 0.5. Two days after infection, JEV antigen-positive cells were identified by indirect immunofluorescence assays using JEV E protein-specific monoclonal antibodies. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Cell infectivity was examined by using an HCS system. The results are the averages of data from three independent experiments performed in triplicate. (B) Cell viability following siRNA transfection, determined by using 3-(4,5-dimethylthiazol-2-yl)-(2,5-diphenyltetrazolium bromide)-tetrazolium (MTT) cell viability assays. The data are pooled from three experiments in duplicate. Statistical significance was determined by analysis of variance with a multiple-comparison correction (**, P < 0.01; ***, P < 0.001).
To further investigate the effect of the loss of SPCS1 function on JEV propagation, we established an SPCS1 knockout (KO) cell line by the transfection of HEK-293 cells with the CRISPR-Cas9 system (Fig. 2A). Wild-type (WT) HEK-293 and SPCS1 KO cells were infected with JEV, and we compared the efficiencies of JEV replication. The level of infectivity of JEV toward SPCS1 KO cells was significantly lower than that toward WT cells (Fig. 2B and C). In SPCS1 KO cells, the cytopathic effects caused by JEV infection were almost completely eliminated when visualized by light microscopy (Fig. 2D) or by staining with crystal violet (Fig. 2E). Viral protein expression levels in SPCS1 KO cells were lower than those in WT cells (Fig. 2F), and the viral titer in the culture supernatant of SPCS1 KO HEK-293 cells was significantly lower than that in WT HEK-293 cells at 24, 48, and 72 hpi (Fig. 2G).
FIG 2.
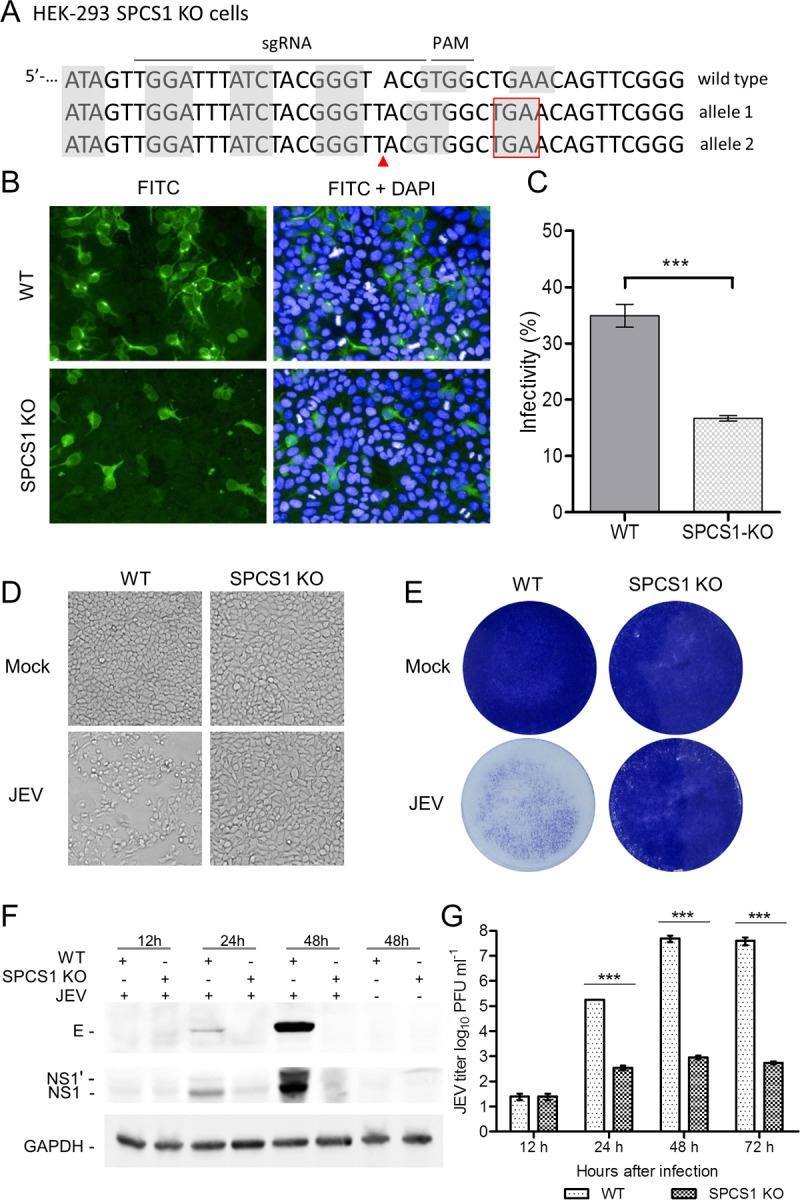
Effect of the loss of SPCS1 function on propagation of JEV particles. (A) Sequencing of SPCS1 alleles in gene-edited HEK-293 cells after limiting-dilution cloning. The subgenomic RNA targeting site and protospacer adjacent motif (PAM) sequences are highlighted above the WT gene, and the sequences of edited alleles are indicated. Nucleotide triplet codons are indicated by shaded boxes. Gene editing resulting in insertions of the T nucleotide is indicated with a red arrow, and a nucleotide insertion resulting in a stop codon is indicated with a red box. (B) WT and SPCS1 KO HEK-293 cells were infected with JEV at an MOI of 0.5. At 48 hpi, cells were fixed and probed with JEV E protein-specific MAb by an immunofluorescence assay. Data from one experiment of three are shown. FITC, fluorescein isothiocyanate. (C) Cell infectivity examined with an HCS system. The data are the averages of results from three independent experiments performed in triplicate. (D and E) Cell cytopathic effects were observed at 72 hpi by microscopy (D) or crystal violet staining (E). Data from one experiment of three are shown. (F) Expression of the E and NS1 proteins in infected cells, analyzed by Western blotting with E protein- and NS1 protein-specific MAbs. Data from one experiment of two are shown. (G) Comparison of viral titers in the supernatants of WT and SPCS1 KO cells infected with JEV at an MOI of 0.01. At 12, 24, 48, and 72 hpi, the titer of infectious JEV was determined by plaque-forming assays on BHK-21 cells. The data are pooled from three experiments in duplicate. Statistical significance was determined by Student's t test (***, P < 0.001).
We further evaluated the effect of the transcomplementation of SPCS1 on JEV propagation in SPCS1 KO cells. For this purpose, SPCS1 KO cells were transfected with a plasmid either expressing or not expressing SPCS1. In SPCS1 KO cells transfected with the SPCS1-expressing plasmid, a larger number of cells displayed positive fluorescence (Fig. 3A), and the ratio of positive cells was significantly higher (Fig. 3B) than for SPCS1 KO cells. Compensation of SPCS1 in SPCS1 KO cells also reversed the cytopathic effects of JEV infection (Fig. 3C). These results demonstrated that the impairment of JEV propagation by the knockout of SPCS1 was specific.
FIG 3.

Effects of JEV propagation in SPCS1 KO HEK-293 cells following complementation with a plasmid expressing SPCS1. (A) WT HEK-293 cells, SPCS1 KO HEK-293 cells, and SPCS1 KO HEK-293 cells cotransfected with a plasmid expressing SPCS1 (or a control plasmid) were infected with JEV at an MOI of 0.5. At 48 hpi, cells were fixed and probed with E protein-specific monoclonal antibodies by an IFA. Data from one experiment of three are shown. (B) Infectivity analyzed by using the HCS system. The data are pooled from three experiments in duplicate. Statistical significance was determined by Student's t test (****, P < 0.0001). (C) Cell cytopathic effects in infected cells observed at 72 hpi with the crystal violet staining method. Data from one experiment of three are shown.
SPCS1 does not regulate cell entry or RNA replication during JEV infection.
To examine whether SPCS1 affected the cell entry step during JEV infection, we examined the infectivity of single-round infectious reporter replicon particles (RRPs) toward WT and SPCS1 KO HEK-293 cells, for which we employed a WNV subgenomic replicon containing a green fluorescent protein (GFP) reporter gene (16) packaged with the JEV structural protein C-prM-E (Fig. 4). RRPs are unable to produce progeny particles due to a lack of structural protein-encoding genes in the replicon. This single-round infectious RRP system allows us to evaluate virus entry and replication without the influence of reinfection. The results showed that both WT and SPCS1 KO HEK-293 cells could be infected with RRPs, and GFP expression was evident (Fig. 5A). GFP positivity was analyzed by using a high-content screening (HCS) system, and there were no significant differences in GFP positivity between WT and SPCS1 KO HEK-293 cells (Fig. 5B). Furthermore, to investigate whether SPCS1 affects JEV genome RNA replication, we examined JEV genome RNA replication during the early stages of infection, between 4 and 12 hpi, using real-time PCR assays. The results demonstrated that there were no significant differences between SPCS1 KO cells and WT cells at 4, 6, 8, 10, and 12 hpi (Fig. 5C), suggesting that SPCS1 is not involved in virus entry into cells or viral genome replication.
FIG 4.

Generation of a cell line stably expressing the JEV C-prM-E protein and preparation of RRPs. (A) Schematic representation of the generation of a stable cell line expressing the JEV C-prM-E protein and the preparation of RRPs. The BJEV-CME cell line stably expressing the C-prM-E protein was generated by transfecting BHK-21 cells with the pCAG-opti-JEV-CME plasmid, followed by cloning and selection with G418. To produce RRPs, BJEV-CME cells were transfected with a DNA-based WNV replicon under the control of the cytomegalovirus (CMV) promoter. The replicon genome lacks the major coding sequence of the structural protein C-prM-E, and the corresponding sequence was replaced with a GFP-coding sequence following the foot-and-mouth disease virus (FMDV) 2A coding sequence. The replicon plasmid was transcribed by the cytomegalovirus promoter into the replicon RNA expressing GFP and the nonstructural replicase protein. BJEV-CME cells express the C-prM-E polyprotein, which is cleaved into the C, prM, and E proteins by replicon RNA-encoded nonstructural protease and endogenous cellular signal peptidase (SP). The replicon RNA amplifies itself again, and the three structural proteins package the replicon RNA into RRPs, which are secreted into the culture medium. When RRPs infect other JEV-susceptible cells, such as BHK-21 cells, the replicon RNA expresses GFP and nonstructural proteins, which amplifies more RNA. However, no structural proteins or additional RRPs are produced in RRP-infected cells, thereby preventing further replication. When BJEV-CME cells are infected with RRPs, the structural proteins expressed by the cells package the replicon RNA into progeny RRPs, and the infection spreads in rounds similar to those for the wild-type virus. UTR, untranslated region. (B) Cloned and selected stable cell lines were identified by an IFA with JEV E protein-specific MAb 5E7. (C) Production of RRPs from the WNV replicon plasmid by transfection of BJEV-CME cell lines. Green florescence was visualized when replicon plasmid pWNVrepdCME-GFP was transfected into BJEV-CME cells (first infection) and BHK-21 cells (second infection). Transfected-cell supernatants were passaged onto fresh cell cultures, as indicated by the arrows, and green florescence was observed only in BJEV-CME cells. At 3 days postinoculation, supernatants of cells infected first were used to inoculate cells, as indicated by the arrows, for a second infection, and green florescence was analyzed at 72 h postinfection. HRr, hammerhead ribozyme; HDVr, hepatitis delta virus ribozyme.
FIG 5.

Effects of the loss of SPCS1 function on virus entry into cells, genome RNA replication, and protein processing during JEV infection. (A) WT and SPCS1 KO HEK-293 cells were infected with JEV single-round infectious RRPs at an MOI of 0.5. At 48 hpi, cell nuclei were stained with Hoechst reagent. Cells positive for GFP fluorescence indicated infection with RRPs. Data from one experiment of three are shown. (B) Infectivity analyzed by using the HCS system. The data are pooled from three experiments in duplicate. Statistical significance was determined by Student's t test (*, P < 0.05). (C) Real-time qRT-PCR analysis of JEV during the early stages of infection. The data are pooled from three experiments in duplicate. Statistical significance was determined by Student's t test (*, P < 0.05). (D to F) WT and SPCS1 KO HEK-293 cells were infected with JEV at an MOI of 1.0. At 4, 6, 8, 10, and 12 hpi, infected cells were harvested, and JEV genome RNAs were analyzed by qRT-PCR. To examine the expression of the JEV E, prM, and NS1 proteins in infected cells, WT and SPCS1 KO HEK-293 cells were infected with JEV at an MOI of 20. At 48 hpi, cell lysates were blotted with anti-JEV E protein (D), anti-JEV prM protein (E), or anti-JEV NS1 protein (F) monoclonal antibodies. Higher-molecular-mass bands (Ehi, Mhi, and NS1hi) reacting with the respective monoclonal antibodies are indicated. Data from one experiment of two are shown.
SPCS1 regulates posttranslational protein processing and virus assembly.
To determine whether SPCS1 is required for virus translation, WT and SPCS1 KO HEK-293 cells were infected with a high dose of JEV at a multiplicity of infection (MOI) of 20, and the expression of the E, prM/M, and NS1 proteins was measured at 48 hpi by Western blotting. The results showed that E, prM/M, and NS1 were all expressed in both WT and SPCS1 KO HEK-293 cells. However, all three proteins were expressed at lower levels in SPCS1 KO cells. In SPCS1 KO cells, higher-molecular-mass bands reacted with anti-E, anti-prM/M, and anti-NS1 antibodies (Fig. 5D to F). These steady-state expression levels are consistent with previous findings by Zhang et al. (14), where translation was not impacted in SPCS1 knockout cells. We further investigated the effects of SPCS1 knockout on virus particle assembly in SPCS1 KO cells by negative-staining electron microscopy. In WT cells infected with JEV, virus particles were observed in the endoplasmic reticulum (ER) (Fig. 6A and B), but we did not observe virus particles in the ER of SPCS1 KO cells after JEV infection (Fig. 6C and D). These results suggest that the SPCS1 knockout did not affect viral protein translation but instead influenced posttranslational viral protein processing and virus assembly.
FIG 6.
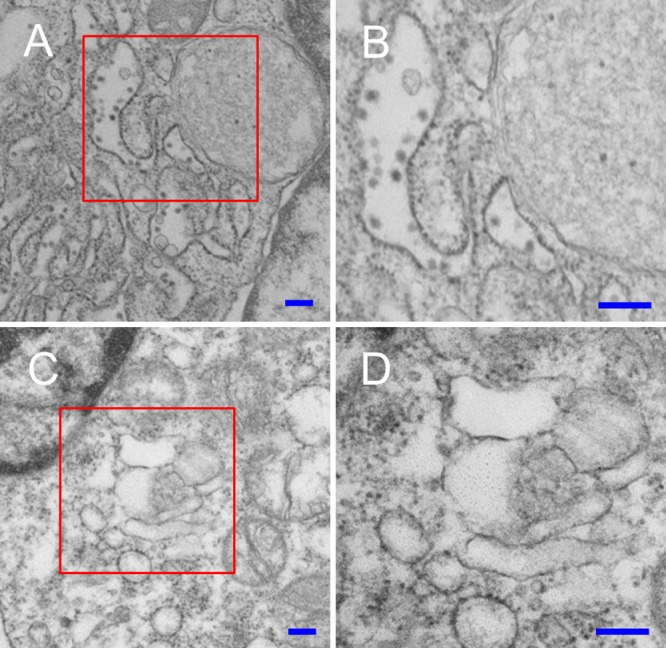
Electron micrographs of WT and SPCS1 KO HEK-293 cells infected with JEV. (A) Low-magnification image of JEV-infected WT HEK-293 cells. (B) Enlargement of the area boxed in red in panel A. Virus particles in the ER are indicated by red arrowheads. (C) Low-magnification image of JEV-infected SPCS1 KO HEK-293 cells. (D) Enlargement of the area boxed in red in panel C. Bars = 200 nm.
To assess whether SPCS1 is required for virus assembly or the release of infectious particles, we constructed a single-round infectious chimeric JEV reporter replicon particle packaging system (Fig. 7A). In WT HEK-293 cells, in addition to sporadic fluorescence, we observed GFP fluorescence foci, indicating that infectious virus particles were produced. The expression of GFP in infected Vero cells demonstrates the presence of infectious virions in the cell supernatant. However, in SPCS1 KO HEK-293 cells, we observed only sporadic GFP fluorescence but no GFP foci and no GFP expression in Vero cells infected with the supernatant. Infectious viruses were recovered when SPCS1 was complemented by the transfection of plasmid pCAG-SPCS1 (Fig. 7B). These results suggest that SPCS1 is involved in the assembly of infectious JEV particles.
FIG 7.

Effects of the loss of SPCS1 function on virus assembly. (A) Schematic diagram of the experimental workflow. SPCS1 KO HEK-293 cells were transfected with plasmid pCAG-J-CME expressing JEV structural proteins with or without plasmid pCAG-SPCS1 expressing SPCS1. WT HEK-293 cells were transfected with pCAG-J-CME as positive controls. Transfected cells were infected with WNV-GFP RRPs (21) at 24 hpi. At 48 hpi, supernatants were harvested and used to infect Vero cells. GFP expression in cells was observed by fluorescence microscopy at 48 hpi. (B) Visualization of GFP expression following transfection with plasmids and infection with WNV-GFP RRPs (top row) and in supernatants of infected Vero cells (bottom row). Progeny viruses represented by the presence of GFP foci are indicated with red arrows.
SPCS1 interacts with the JEV NS2B protein.
To further investigate the molecular mechanisms by which SPCS1 regulates JEV replication and assembly, we examined the interaction of SPCS1 with JEV genome-encoded proteins using a Venus-based BiFC system (Fig. 8A). SPCS1 was fused in frame with the N-terminal amino acids (amino acids 1 to 173) of the Venus protein (VN), and JEV genome-encoded proteins were individually fused with the C-terminal residues (amino acids 174 to 239) of the Venus protein (VC). After screening of all 10 JEV genome-encoded proteins, it was found that SPCS1 strongly interacted with the NS2B protein (Fig. 8B). To verify whether the interaction of SPCS1 with NS2B is prevalent in flaviviruses, we further examined the interaction of SPCS1 with NS2B proteins from WNV and ZIKV. The results demonstrated that SPCS1 also strongly interacts with WNV NS2B and ZIKV NS2B. These interactions were also verified by reverse-BiFC analysis, in which SPCS1 was fused with VC and NB2Bs were fused with VN (Fig. 8B).
FIG 8.

Interaction of SPCS1 with the JEV NS2B protein in mammalian cells. (A) Schematic diagram of BiFC analysis. Blue A's and purple B's represent a pair of proteins analyzed by BiFC. VN and VC represent the N-terminal fragment (residues 1 to 173) and the C-terminal fragment (residues 174 to 239) of the Venus protein, respectively. hpt, hours posttransfection. (B) Detection of the interaction of SPCS1-VN with VC fused to the NS2B proteins of JEV, WNV, and ZIKV and the corresponding reverse interactions of SPCS1-VC with VN fused to the NS2B proteins of JEV, WNV, and ZIKV. Data from one experiment of three are shown. (C) HEK-293T cells were cotransfected with separate plasmids expressing SPCS1-myc and FLAG-tagged NS2B. Cell lysates of transfected cells were immunoprecipitated with anti-myc antibody. The resulting precipitates and whole-cell lysates used for immunoprecipitation were examined by immunoblotting using anti-myc and anti-FLAG antibodies. Data from one experiment of two are shown. CoIP, coimmunoprecipitation. (D) HEK-293T cells were transfected with a plasmid expressing SPCS1-myc. Cells were infected with JEV at an MOI of 0.5 at 24 h posttransfection. At 48 hpi, cell lysates of infected cells were immunoprecipitated with anti-myc antibody. The resulting precipitates and whole-cell lysates used for immunoprecipitation were examined by immunoblotting using anti-myc and anti-NS2B antibodies. Data from one experiment of two are shown.
To further confirm the interaction between SPCS1 and the JEV NS2B protein, myc-tagged SPCS1 was coexpressed in HEK-293T cells with FLAG-tagged JEV NS2B (J-NS2B), followed by coimmunoprecipitation and immunoblotting. NS2B was shown to be coimmunoprecipitated with SPCS1 (Fig. 8C), which demonstrates that NS2B is associated with SPCS1. Additionally, the endogenous NS2B protein expressed after JEV infection also coimmunoprecipitated with transfected SPCS1 (Fig. 8D). These results provide further evidence that SPCS1 interacts with the flavivirus NS2B protein.
SPCS1 interacts with NS2B transmembrane domains.
The flavivirus NS2B protein has multiple transmembrane domains. According to TMpred (17), there are three transmembrane regions, TM1 (residues 4 to 22), TM2 (residues 31 to 47), and TM3 (residues 102 to 122), and one major domain outside the transmembrane region (residues 48 to 101) (Fig. 9A). To further determine the region(s) of NS2B responsible for the interaction with SPCS1, deletion mutants of JEV NS2B were constructed according to the predicted topology (Fig. 9A). Amino acid sequence alignment of NS2B proteins from different flaviviruses revealed five conserved residues (E6, G12, P32, G37, and G47) in the TM1-TM2 region and two conserved residues (P112 and I115) in the TM3 region (Fig. 9B).
FIG 9.

Schematic diagram of JEV NS2B deletion mutant constructs and sequence alignment of the transmembrane domains of flavivirus NS2B proteins. (A) JEV NS2B deletion mutant constructs tagged with the C-terminal fragment of the Venus protein or FLAG. Transmembrane segments are indicated in orange boxes. The main outer membrane domains (OMD) are indicated in blue boxes. The locations of NS2B deletion mutants are indicated on the left, with amino acid positions in superscript type. (B) Alignment of flavivirus NS2B transmembrane domain sequences. Conserved residues between groups are shaded in red. Five conserved residues in the TM1 and TM2 regions, and two conserved residues in the TM3 region, were selected for site-directed mutagenesis. For point mutations, all selected amino acid residues were replaced with alanine. MVEV, Murray Valley encephalitis virus; SLEV, Saint Louis encephalitis virus; DV1 to DV4, dengue virus 1 to 4.
BiFC assay results for SPCS1-VN and VC-fused NS2B deletion mutants coexpressed in HEK-293T cells revealed almost no fluorescence with the pair SPCS1-VN and NS2B(29–101), NS2B(29–83), NS2B(29–65), or NS2B(48–101). Meanwhile, weak fluorescence was observed with the pair SPCS1-VN and NS2B(1–30) or NS2B(102–131), and strong fluorescence was observed with the pair SPCS1-VN and NS2B(1–101), NS2B(1–47), NS2B(23–131), NS2B(48–131), NS2B(66–131), or NS2B(84–131) (Fig. 10A). Ratios of Venus fluorescence-positive cells were calculated by using the HCS system (Fig. 10B), and the results suggested that two separate regions interact with SPCS1: the NS2B N-terminal region containing TM1 and TM2 and the C-terminal region containing TM3. The two minimal regions possessing the ability to interact with SPCS1 in an unimpaired manner in full-length NS2B are NS2B(1–49) and NS2B(84–131). The expression of VC-tagged NS2B deletion mutants was verified by Western blotting (Fig. 10C). To further investigate the interaction of SPCS1 with NS2B in cells, we first verified the expression of FLAG-tagged NS2B deletion mutants in HEK-293T cells (Fig. 10D) and then coexpressed myc-tagged SPCS1 with the indicated FLAG-tagged NS2B deletion mutants in HEK-293T cells, followed by coimmunoprecipitation and immunoblotting. NS2B(84–131) coimmunoprecipitated with SPCS1 (Fig. 10E), but NS2B(1–49) did not coimmunoprecipitate with SPCS1 in this assay, possibly due to the poor expression of NS2B(1–49). These results demonstrated that SPCS1 interacts with NS2B(1–49) and NS2B(84–131), the two transmembrane domains of the JEV NS2B protein.
FIG 10.

Interaction of SPCS1 with JEV NS2B deletion mutants. (A) BiFC analysis of the interaction between SPCS1-VN and VC fused to JEV NS2B or its deletion mutants. Images were taken at 12 hpi by using the HCS system. Data from one experiment of three are shown. (B) The ratio of positive cells was determined by using the HCS system. The data are pooled from three experiments in duplicate. Statistical significance was determined by analysis of variance with a multiple-comparison correction (***, P < 0.001). (C) Expression of BiFC constructs. HEK-293T cells were transfected with NS2B deletion mutants tagged with VC. Cell lysates were examined by immunoblotting using anti-GFP or anti-GAPDH antibodies. Data from one experiment of two are shown. (D) Expression of FLAG-tagged JEV NS2B deletion mutants. HEK-293T cells were transfected with the indicated plasmids, and cell lysates were examined by immunoblotting using anti-FLAG or anti-GAPDH antibodies. Data from one experiment of two are shown. (E) Cells were cotransfected with the indicated NS2B deletion mutant plasmids, and lysates of transfected cells were immunoprecipitated with anti-myc antibody. The resulting precipitates and whole-cell lysates used for immunoprecipitation were examined by immunoblotting using anti-FLAG or anti-myc antibodies. Data from one experiment of two are shown.
Mutations G12A and P112A weaken interactions between NS2B transmembrane domains and SPCS1.
To further characterize the interactions between SPCS1 and the transmembrane domains of NS2B(1–49) and NS2B(84–131), conserved flavivirus amino acid residues were mutated, as indicated in Fig. 9B. We then used the BiFC assay to examine the effects of the mutations on interactions with SPCS1. After cotransfection, we found that all mutants of NS2B(1–49) (Fig. 11A) and NS2B(84–131) (Fig. 11B) retained the ability to interact with SPCS1. By using the high-content screening system, the ratio of positive cells was determined and was found to be lower with the G12A mutant of NS2B(1–49) than with WT NS2B(1–49) (Fig. 11C). Mutation P112A in NS2B(84–131) also significantly reduced the positive cell ratio compared with WT NS2B(84–131) (Fig. 11D). These results suggest that the conserved amino acid residues in the transmembrane domains are important for the interaction with SPCS1.
FIG 11.

Interaction of SPCS1 with point mutants of JEV NS2B(1–49) and NS2B(84–131). (A and B) Visualization of the interaction between SPCS1-VN and VC fused with point mutants of JEV NS2B(1–49) (A) and NS2B(84–131) (B) using the BiFC system. Images were taken at 12 hpi by using the HCS system. Data from one of two independent experiments performed in triplicate are shown. (C and D) The ratios of positive cells were determined. Shown are data from analyses of the statistical significance of differences between point mutants and the corresponding WT NS2B(1–49) (C) and NS2B(84–131) (D). Data are the averages of results from two independent experiments performed in triplicate. Statistical significance was determined by analysis of variance with a multiple-comparison correction (**, P < 0.01; ***, P < 0.001).
Characterization of the interaction between SPCS1 and JEV NS2B.
To determine the region of SPCS1 responsible for the interaction with NS2B, deletion mutants of VN-tagged SPCS1 were constructed (Fig. 12A) and coexpressed with VC-tagged full-length NS2B(1–131), the NS2B(1–49) N-terminal region, and the NS2B(84–131) C-terminal region. The expression of VN-tagged SPCS1 deletion mutants in HEK-293T cells was verified by Western blotting (Fig. 12B). The C-terminal region of SPCS1, SPCS1(91–169), which contains the two transmembrane regions, showed a strong interaction with full-length NS2B, NS2B(1–49), and NS2B(84–131) (Fig. 12C). However, there was almost no Venus fluorescence signal in cells coexpressing SPCS1(1–111) with NS2B, NS2B(1–49), or NS2B(84–131) and only a weak fluorescence signal (Fig. 12C) and a lower positive cell ratio (Fig. 12D to 12F) in cells coexpressing the SPCS1 deletion mutant SPCS1(1–132), SPCS1(1–92), SPCS1(112–169), or SPCS1(121–169) with NS2B, NS2B(1–49), or NS2B(84–131). These results suggest that residues 91 to 169 of SPCS1 are involved in the interaction with both NS2B(1–49) and NS2B(84–131). Coimmunoprecipitation assays indicated interactions only between NS2B(84–131) and SPCS1 deletion mutants (Fig. 12G and H) but not with NS2B(1–49) (data not shown), possibly due to the low expression level of NS2B(1–49).
FIG 12.

Interaction of SPCS1 deletion mutants with JEV NS2B, NS2B(1–49), and NS2B(84–131). (A) Schematic diagram of SPCS1 deletion mutant constructs tagged with the N-terminal fragment of the Venus protein or myc. Transmembrane segments are indicated in gray boxes. The locations of SPCS1 deletion mutants are indicated on the left, along with amino acid positions. (B) Expression of VN-tagged SPCS1 deletion mutants. The membrane was probed with mouse monoclonal antibodies against GFP. Data from one experiment of two are shown. (C) Plasmids were cotransfected into HEK-293T cells, images were captured at 12 hpi by using the HCS system, and the ratio of positive cells was determined. Data from one experiment of three are shown. (D) Positive cell ratios for the interaction between SPCS1 deletion mutants and JEV NS2B. (E) Positive cell ratios for the interaction between SPCS1 deletion mutants and JEV NS2B(1–49). (F) Positive cell ratios for the interaction between SPCS1 deletion mutants and JEV NS2B(84–131). The data are pooled from three experiments in duplicate. Statistical significance was determined by analysis of variance with a multiple-comparison correction (***, P < 0.001). (G) Expression of myc-tagged SPCS1 deletion mutants. HEK-293T cells were transfected with the indicated plasmids, and cell lysates were examined by immunoblotting using anti-myc or anti-GAPDH antibodies. Data from one experiment of two are shown. (H) Cells were cotransfected with JEV NS2B(84–131) and the indicated SPCS1 deletion mutants, and lysates of transfected cells were immunoprecipitated with anti-myc antibody. The resulting precipitates and whole-cell lysates used for immunoprecipitation were examined by immunoblotting using anti-FLAG or anti-myc antibodies. Data from one experiment of two are shown.
DISCUSSION
SPCS1 is a component of the signal peptidase complex that processes secreted or membrane-associated proteins in cells. The mammalian signal peptidase complex consists of five subunits, SPCS1, SPCS2 SPCS3, SEC11A, and SEC11C (18). Analysis of the topology of SPCS1 demonstrated that it contains two hydrophobic segments close to each other, and very few amino acids are exposed to the lumen of the ER. Thus, SPCS1 is not likely to function as a catalytic subunit (19). Suzuki et al. (15) reported that SPCS1 participates in the assembly of hepatitis C virus through interactions with E2 and NS2. Silencing of endogenous SPCS1 markedly reduced the production of infectious HCV, but the propagation of JEV was not affected by the knockdown of SPCS1. Using a genome-wide CRISPR-Cas9-based screen of host genes, Zhang et al. (14) found that a subset of ER-associated SPCS proteins is necessary for the proper cleavage of flavivirus structural proteins and the production of infectious virus particles. In that study, the loss of SPCS1 function markedly reduced the propagation of all tested members of the genus Flavivirus, including JEV, WNV, DENV, ZIKV, and YFV.
In this study, we first verified that SPCS1 is necessary for JEV replication, with results that are consistent with those reported by Zhang et al. (14), and we then evaluated the effect of the loss of SPCS1 function on different steps of the JEV replication cycle. We initially assessed the cell entry of JEV using single-round infectious GFP-expressing reporter replicon particles, and SPCS1 knockout cells showed infectivity similar to that for WT cells. This demonstrated that SPCS1 did not affect JEV entry into cells. Quantitative comparison of JEV genome copy numbers in SPCS1 knockout and WT cells during the early stages of infection revealed no significant differences. This finding was also consistent with data from a previous report (14). When cells were infected with JEV at a high MOI, structural (prM and E) and nonstructural (NS1) proteins were detected in cell lysates of both WT and SPCS1 KO cells. However, in SPCS1 KO cells, structural and nonstructural proteins were detected mainly as high-molecular-mass bands, which is consistent with data reported previously by Zhang et al. (14). Furthermore, by using reporter flavivirus replicon transfection experiments and 35S pulse-chase studies, Zhang et al. (14) revealed that SPCS1 did not affect viral protein translation but instead influenced posttranslational viral protein processing. Using a single-round infectious chimeric JEV reporter replicon particle packaging system, we revealed that SPCS1 knockout impaired the assembly of infectious virus particles (Fig. 7). Taken together, these results suggest that SPCS1 is most likely involved in the assembly of infectious viruses rather than cell entry, RNA replication, or viral protein expression during the JEV life cycle.
To dissect the molecular mechanisms by which SPCS1 modulates virus assembly, we employed the Venus-based BiFC system, found that the nonstructural JEV protein NS2B interacts with the host factor SPCS1, and confirmed interactions between SPCS1 and NS2B proteins from WNV and ZIKV, suggesting that the interaction is conserved throughout the flavivirus genus. The BiFC system was convenient for studying protein-protein interactions, but there are also limitations. The overexpression and the inherent capacity of fluorescent fragments to correlate without depending on the interactions between the proteins that are fused to the fragment may cause a false-positive signal. Thus, we verified the interaction results for SPCS1 and NS2B by coimmunoprecipitation. The flavivirus NS2B protein is known to function as a cofactor for viral NS3 protease (6). For the activation of NS3 protease, the central hydrophilic region of NS2B is both necessary and sufficient (7–11). However, the function(s) of the hydrophobic TMDs of N2SB remains unclear. Recently, Li et al. (13) found that NS2B plays a critical role in JEV virion assembly by employing systematic mutagenesis of conserved residues in the TMDs of NS2B. Interestingly, our systematic deletion mutation analysis revealed that both the N-terminal TMDs (TM1 and TM2) and the C-terminal TMD (TM3) interact with SPCS1. Furthermore, mutational analysis of SPCS1 revealed that residues 91 to 169 of SPCS1 mainly contribute to these interactions.
The flavivirus nonstructural protein NS2A is a nonenzymatic, integral membrane protein (20). In addition to playing an important role in viral RNA replication (21), NS2A also performs an essential role in virus assembly (3, 22–24). Previous studies revealed that NS2A interacts with NS2B in mammalian cells (12, 13, 24). We also surveyed the interaction of NS2B with NS2A using BiFC assays (data not shown). Consistent with data from previous reports (12, 13), we found that NS2B could interact with NS2A, and NS2B could interact with itself. The NS2B(1–49) and NS2B(84–131) regions are largely responsible for the interactions of NS2B with NS2A and with itself. Based on data in the existing literature (3–5, 13, 22–24) and the findings of this study, we propose a model of the protein-protein interactions of NS2B with SPCS1 and NS2A (Fig. 13). To date, some aspects of virus replication and assembly in the flavivirus life cycle have been reported (3–5, 13, 22–25). The viral polyprotein is co- and posttranslationally cleaved into 10 proteins, of which the structural proteins prM and E and the ER membrane-associated nonstructural proteins NS2A, NS2B, NS4A, and NS4B are anchored in the ER membrane through their transmembrane domains. ER membrane rearrangement under the induction of NS4A (4, 26) and NS4B (27) forms membrane vesicles, with the prM and E proteins being arranged on the surface of the vesicles, and the nascent viral genome and capsid protein are wrapped within vesicles (28). Through protein-protein interactions and possible hydrophobic interactions, NS2B, together with NS2A, NS4A, and NS4B, accumulates at the vesicle neck, which is open to the cytoplasm (29, 30). Interactions between host factors such as SPCS1 and viral proteins such as NS2B are involved in virus assembly, budding, and release into the ER lumen. Virus assembly is complicated and involves host factors and viral components. To fully understand the details of this virus assembly process, more studies are needed.
FIG 13.
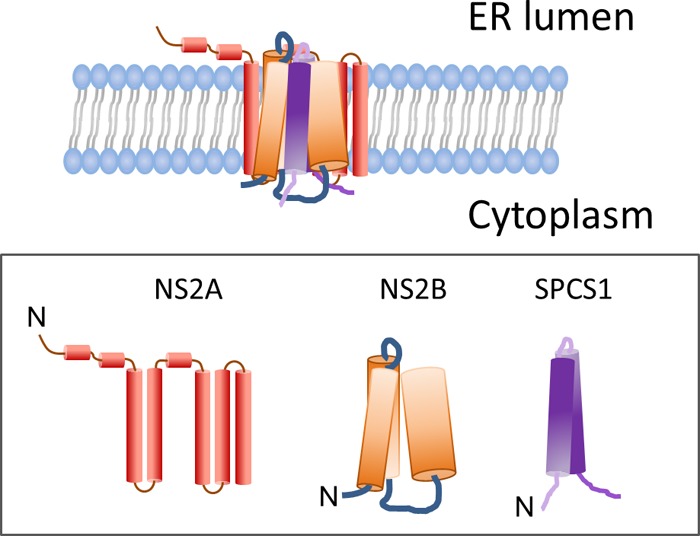
Proposed model for the interaction of NS2B with SPCS1 and NS2A.
In summary, our study provides the first evidence that the host factor SPCS1 regulates flavivirus assembly through interactions with hydrophobic transmembrane domains of the virus protein NS2B. Our findings provide clues for understanding the molecular mechanism of the assembly of infectious JEV and other flaviviruses.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
Human embryo kidney HEK-293T cells (ATCC CRL-3216), HEK-293 cells (ATCC CRL-1573), SPCS1 KO HEK-293 cells (generated from HEK-293 cells as parental cells in this study), and BHK-21 cells (ATCC CCL-10) were grown in Dulbecco's modified Eagle's medium (DMEM) (catalog number SH30021.01; HyClone) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (catalog number 10099-141; Gibco), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in 5% CO2. For HEK-293, SPCS1 KO, and HEK-293T cell cultures, cell culture plates were covered with poly-d-lysine hydrobromide (catalog number P7886; Sigma) at 50 μg/ml. The BJEV-CME cell line (generated from BHK-21 cells as parental cells in this study), which stably expresses the JEV C-prM-E protein, was maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 mg/ml G418 at 37°C in 5% CO2. The JEV SA14 strain was propagated and titrated onto BHK-21 cells. Mouse monoclonal antibodies against the JEV prM (31, 32), E (33), and NS1 (34, 35) proteins were generated previously. Mouse monoclonal antibody against JEV NS2B was a gift from Shengbo Cao (Huazhong Agricultural University, Wuhan, China). Mouse monoclonal antibodies against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from Proteintech (catalog number 10494-1-AP). Rabbit polyclonal antibodies against myc (catalog number MA1-980) and mouse monoclonal antibodies against FLAG (catalog number ma1-91878) were obtained from Thermo Fisher. Mouse monoclonal antibodies against hemagglutinin (HA) (catalog number H9658) were obtained from Sigma-Aldrich. Rabbit polyclonal antibodies (catalog number A01388-40) and mouse monoclonal antibodies (catalog number A00185-40) against GFP were obtained from GenScript. Alexa Fluor 680-conjugated donkey anti-mouse IgG antibodies were obtained from Thermo Fisher (catalog number A10038). IRDye 800CW-conjugated donkey anti-rabbit IgG antibodies were obtained from Li-Cor (catalog number 926-32213).
Plasmids.
WNV subgenomic replicon plasmid pWNVrepdCME-GFP was described previously (16). Plasmid pCAG-J-CME expressing the JEV C-prM-E protein was constructed by cloning the optimized C-prM-E-encoding sequence into the SacI and XhoI restriction sites of pCAGneo (33).
To generate plasmid pCAG-SPCS1-myc expressing SPCS1, the cDNA encoding SPCS1 with spacer and myc tag sequences (GPGEQKLISEEDL) at the C terminus was synthesized and inserted into pCAGneo between the SacI and XhoI restriction sites.
For the expression of all 10 JEV proteins (C, prM, E, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) with FLAG tags, cDNA sequences were amplified by PCR and inserted into pCAGneo. To generate plasmids for JEV NS2B deletion mutants, parts of JEV NS2B-encoding genes with a FLAG tag at the C terminus were amplified by PCR and inserted into pCAGneo.
For the construction of BiFC vectors, the two separate parts of the Venus-encoding sequence, the Venus N terminus (VN) (residues 1 to 173) and the Venus C terminus (VC) (residues 174 to 239) were synthesized with a GGGSGGGS linker sequence (13) at the 5′ end and inserted into pCAGneo between the XhoI and BglII restriction sites. The resulting VN and VC expression plasmids were named pCAG-VN and pCAG-VC, respectively. SPCS1, the 10 JEV genome-encoding proteins and the WNV NS2B, ZIKV NS2B, and JEV NS2B deletion mutants cDNA sequences were PCR amplified and inserted into vectors pCAG-VN and pCAG-VC between the SacI and XhoI restriction sites. cDNA sequences of NS2B(1–49) mutants and NS2B(84–131) mutants were synthesized and inserted into vector pCAG-VC between the SacI and XhoI restriction sites.
DNA transfection.
HEK-293T cells (90% confluent) were transfected with plasmid DNA using Lipofectamine LTX and Plus reagent (catalog number 15338-100; Invitrogen). HEK-293 cells (80% confluent) were transfected with plasmid DNA by using TransIT-293 transfection reagent (catalog number Mir 2705; Mirus).
Immunofluorescence assay.
HEK-293 cells and SPCS1 KO HEK-293 cells were seeded onto CellCarrier-96 microplates (catalog number 6005550; PerkinElmer) and infected with JEV at an MOI of 0.5. Twenty-four hours after infection, cells were fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) at 4°C for 10 min, and incubated with 4% bovine albumin V in PBS at 37°C for 30 min. JEV was stained with JEV E-specific mouse MAb 5E7 (33) at 37°C for 1 h or at 4°C overnight and then incubated with fluorescein-conjugated AffiniPure goat anti-mouse IgG (catalog number ZF-0312; ZSGB-Bio) and Hoechst 33342 reagent (catalog number 62249; ThermoFisher). After three washes with PBS, cells were visualized by using a high-content screening microplate imaging reader (Operetta; PerkinElmer). The infection ratio of cells was calculated by using Acapella high-content imaging and analysis software.
PFU assay.
Approximately 2 × 105 BHK-21 cells/well in 0.5 ml complete medium were seeded into 24-well plates, and 24 h later, the monolayer of BHK-21 cells was inoculated with 100 μl of the serially diluted virus supernatant from HEK-293 cells or SPCS1 KO HEK-293 cells infected with JEV at an MOI of 0.01 for 12 h, 24 h, 48 h, and 72 h. After 6 h, 0.6 ml of 3% methyl cellulose (catalog number M6385; Sigma) was added to each well. Three days after infection, the supernatants were discarded, and cells were stained with 1 ml of a crystal violet-formaldehyde stain for 15 to 20 min. The resulting plaques were counted, and PFU per milliliter were calculated.
Establishment of stable cell lines and production of single-round infectious JEV RRPs.
The BJEV-CME cell line stably expressing the JEV C-prM-E protein was generated by the transfection of BHK-21 cells with plasmid pCAG-J-CME, as described previously (16). Briefly, a monolayer of BHK-21 cells was transfected with the pCAG-J-CME plasmid by using X-tremeGENE HP DNA transfection reagent (Roche Diagnostic GmbH, Mannheim, Germany). Two days later, the transfected cells were dispersed with trypsin and cloned by limiting dilution in 96-well plates with medium containing G418. The cloned cells were selected by an IFA with JEV E protein-specific mouse MAb 5E7. One clone, designated BJEV-CME, exhibited more-efficient expression of the E antigen and was therefore selected and maintained in G418-supplemented medium for further characterization and production of RRPs. For the production of JEV RRPs, BJEV-CME cells were transfected with plasmid pWNVrepdCME-GFP by using X-tremeGENE HP DNA transfection reagent. Four days after transfection, supernatants containing JEV RRPs were harvested and stored at −80°C. JEV RRPs were titrated on BJEV-CME cells by a plaque assay or a GFP focus assay.
Real-time reverse transcription-quantitative PCR (qRT-PCR).
HEK-293 cells and SPCS1 knockout HEK-293 cells were seeded into six-well plates and infected with JEV at an MOI of 0.5 or 1 when cells had formed a monolayer. Following incubation at 37°C with 5% CO2 for 2 h, infected cells were washed with PBS three times to remove noninfected virus from the culture fluid. Cells were collected 4 h, 6 h, 8 h, 10 h, and 12 h after infection with JEV, and RNA was extracted by using an RNeasy Plus minikit (catalog number 74104; Qiagen) according to the manufacturer's instructions. JEV RNA was reverse transcribed into cDNA by using Moloney murine leukemia virus (M-MLV) reverse transcriptase (catalog number 2641A; TaKaRa). Real-time PCR mixtures (20 μl) contained 10 μl Premix Ex Taq, 0.5 μl probe (12.5 pM), 1 μl primers (10 pM), and 1 μl cDNA template. Thermal cycling was performed by denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 10 s and annealing at 60°C for 40 s. Results were analyzed by using LightCycler480 software. The JEV E protein encoding cDNA sequence from GenBank (accession number U47032) was cloned into the pMD18-T plasmid and used as a standard for the quantification of JEV copy numbers. Primers 5′-AGGGGTTTTCAACTCCATAG-3′ and 5′-CATTAGCCCTTGTGTGATCC-3′ and TaqMan probe 5′–6-carboxyfluorescein (FAM)–TTCTGAAGGCACCACCAAACAC–Eclipse–3′ were used for viral genome quantification.
BiFC system.
The BiFC assay was performed as previously described (13). Briefly, the target protein pair was cloned into VN-tagged plasmid pCAG-VN and VC-tagged plasmid pCAG-VC, respectively, and recombinant plasmids were cotransfected into HEK-293T cells. The amount of DNA is about 100 ng in 10 μl DNA-lipid complex per well of a 96-well plate. The pair JEV NS1-VN and JEV NS2B-VC was set as a negative control (13). Twelve hours after transfection, cells were visualized, and images were captured by using a high-content screening microplate imaging reader (Operetta; PerkinElmer). The infection ratio of cells was calculated by using Acapella high-content imaging and analysis software.
Coimmunoprecipitation.
Transfected cells were washed three times with ice-cold PBS, and cells were lysed with ice-cold lysis buffer (catalog number L3412; Sigma). Cells were then incubated for 1 h on a shaker at 4°C and centrifuged for 15 min at 12,000 × g, and clarified lysates were incubated overnight on a shaker at 4°C with an EZview red anti-c-Myc affinity gel (catalog number E6654; Sigma). Incubated beads were washed twice with lysis buffer and three times with wash buffer (catalog number W0390; Sigma), 20 μl of 2× sample buffer was added to each sample, and samples were boiled for 10 min and analyzed by SDS-PAGE and Western blotting.
SDS-PAGE and Western blotting.
Protein samples were separated by 12.5% or 15% SDS-PAGE, transferred onto a polyvinylidene difluoride (PVDF) membrane, and incubated with 4% skim milk (catalog number P1622; Beijing Applygen Technologies Inc.) in PBS at room temperature for 2 h or overnight at 4°C. To visualize proteins by immunoblotting, the following primary antibodies were used: anti-myc tag mouse monoclonal antibody (catalog number MA1-980; ThermoFisher), anti-FLAG mouse monoclonal antibody (catalog number MA1-91878; ThermoFisher), anti-GAPDH mouse monoclonal antibody (catalog number 10494-1-AP; Proteintech), anti-GFP rabbit polyclonal antibody (catalog number A01388-40; GenScript), anti-GFP mouse monoclonal antibody (catalog number A00185-40; GenScript), and anti-E, anti-NS1, anti-PrM, and anti-NS2B mouse monoclonal antibodies. Primary antibodies were detected using Alexa Fluor 680 donkey anti-mouse IgG secondary antibodies (catalog number A10038; Thermo Fisher), by incubating membranes at a 1:15,000 dilution, and using IRDye 800CW donkey anti-rabbit IgG (catalog number 926-32213; Li-Cor), by incubating membranes at a 1:20,000 dilution, both for 1 h at room temperature. Following four washes with PBS, signal detection was performed by using a near-infrared fluorescence scanning imaging system (Odyssey; Li-Cor).
ACKNOWLEDGMENTS
We thank Shengbo Cao for providing the JEV NS2B monoclonal antibody. We thank the native-English-speaking scientists of Elixigen Company (Huntington Beach, CA) for language editing of our manuscript.
This work was supported by the National Program on Key Research Project of China (grant number 2016YFD0500403).
R.-H.H. and Z.-G.B. conceived and designed the experiments. L.M., R.-H.H., J.-W.Z., F.L., W.L., D.-M.Z., H.W., and Z.-G.B. performed the experiments. R.-H.H. and L.M. analyzed the data. R.-H.H. and L.M. wrote the paper.
REFERENCES
- 1.Chambers TJ, Hahn CS, Galler R, Rice CM. 1990. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- 2.Mukhopadhyay S, Kuhn RJ, Rossmann MG. 2005. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol 3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 3.Leung JY, Pijlman GP, Kondratieva N, Hyde J, Mackenzie JM, Khromykh AA. 2008. Role of nonstructural protein NS2A in flavivirus assembly. J Virol 82:4731–4741. doi: 10.1128/JVI.00002-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R. 2007. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J Biol Chem 282:8873–8882. doi: 10.1074/jbc.M609919200. [DOI] [PubMed] [Google Scholar]
- 5.Umareddy I, Chao A, Sampath A, Gu F, Vasudevan SG. 2006. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J Gen Virol 87:2605–2614. doi: 10.1099/vir.0.81844-0. [DOI] [PubMed] [Google Scholar]
- 6.Falgout B, Pethel M, Zhang YM, Lai CJ. 1991. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol 65:2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arias CF, Preugschat F, Strauss JH. 1993. Dengue 2 virus NS2B and NS3 form a stable complex that can cleave NS3 within the helicase domain. Virology 193:888–899. doi: 10.1006/viro.1993.1198. [DOI] [PubMed] [Google Scholar]
- 8.Chambers TJ, Nestorowicz A, Amberg SM, Rice CM. 1993. Mutagenesis of the yellow fever virus NS2B protein: effects on proteolytic processing, NS2B-NS3 complex formation, and viral replication. J Virol 67:6797–6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chambers TJ, Nestorowicz A, Rice CM. 1995. Mutagenesis of the yellow fever virus NS2B/3 cleavage site: determinants of cleavage site specificity and effects on polyprotein processing and viral replication. J Virol 69:1600–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Droll DA, Krishna Murthy HM, Chambers TJ. 2000. Yellow fever virus NS2B-NS3 protease: charged-to-alanine mutagenesis and deletion analysis define regions important for protease complex formation and function. Virology 275:335–347. doi: 10.1006/viro.2000.0488. [DOI] [PubMed] [Google Scholar]
- 11.Zuo Z, Liew OW, Chen G, Chong PC, Lee SH, Chen K, Jiang H, Puah CM, Zhu W. 2009. Mechanism of NS2B-mediated activation of NS3pro in dengue virus: molecular dynamics simulations and bioassays. J Virol 83:1060–1070. doi: 10.1128/JVI.01325-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu L, Takeda K, Markoff L. 2013. Protein-protein interactions among West Nile non-structural proteins and transmembrane complex formation in mammalian cells. Virology 446:365–377. doi: 10.1016/j.virol.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Li XD, Deng CL, Ye HQ, Zhang HL, Zhang QY, Chen DD, Zhang PT, Shi PY, Yuan ZM, Zhang B. 2016. Transmembrane domains of NS2B contribute to both viral RNA replication and particle formation in Japanese encephalitis virus. J Virol 90:5735–5749. doi: 10.1128/JVI.00340-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, Dowd KA, Pierson TC, Cherry S, Diamond MS. 2016. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 535:164–168. doi: 10.1038/nature18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki R, Matsuda M, Watashi K, Aizaki H, Matsuura Y, Wakita T, Suzuki T. 2013. Signal peptidase complex subunit 1 participates in the assembly of hepatitis C virus through an interaction with E2 and NS2. PLoS Pathog 9:e1003589. doi: 10.1371/journal.ppat.1003589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Ma L, Guo LP, Wang XL, Zhang JW, Bu ZG, Hua RH. 2017. West Nile virus infectious replicon particles generated using a packaging-restricted cell line is a safe reporter system. Sci Rep 7:3286. doi: 10.1038/s41598-017-03670-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofmann K, Stoffel W. 1993. Tmbase—a database of membrane spanning proteins segments. Biol Chem Hoppe Seyler 374:166. [Google Scholar]
- 18.Evans EA, Gilmore R, Blobel G. 1986. Purification of microsomal signal peptidase as a complex. Proc Natl Acad Sci U S A 83:581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalies KU, Hartmann E. 1996. Membrane topology of the 12- and the 25-kDa subunits of the mammalian signal peptidase complex. J Biol Chem 271:3925–3929. doi: 10.1074/jbc.271.7.3925. [DOI] [PubMed] [Google Scholar]
- 20.Xie X, Gayen S, Kang C, Yuan Z, Shi PY. 2013. Membrane topology and function of dengue virus NS2A protein. J Virol 87:4609–4622. doi: 10.1128/JVI.02424-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackenzie JM, Khromykh AA, Jones MK, Westaway EG. 1998. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 245:203–215. doi: 10.1006/viro.1998.9156. [DOI] [PubMed] [Google Scholar]
- 22.Kummerer BM, Rice CM. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J Virol 76:4773–4784. doi: 10.1128/JVI.76.10.4773-4784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie X, Zou J, Puttikhunt C, Yuan Z, Shi PY. 2015. Two distinct sets of NS2A molecules are responsible for dengue virus RNA synthesis and virion assembly. J Virol 89:1298–1313. doi: 10.1128/JVI.02882-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu RH, Tsai MH, Tsai KN, Tian JN, Wu JS, Wu SY, Chern JH, Chen CH, Yueh A. 2017. Mutagenesis of dengue virus protein NS2A revealed a novel domain responsible for virus-induced cytopathic effect and interactions between NS2A and NS2B transmembrane segments. J Virol 91:e01836-. doi: 10.1128/JVI.01836-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Apte-Sengupta S, Sirohi D, Kuhn RJ. 2014. Coupling of replication and assembly in flaviviruses. Curr Opin Virol 9:134–142. doi: 10.1016/j.coviro.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roosendaal J, Westaway EG, Khromykh A, Mackenzie JM. 2006. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J Virol 80:4623–4632. doi: 10.1128/JVI.80.9.4623-4632.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaufusi PH, Kelley JF, Yanagihara R, Nerurkar VR. 2014. Induction of endoplasmic reticulum-derived replication-competent membrane structures by West Nile virus non-structural protein 4B. PLoS One 9:e84040. doi: 10.1371/journal.pone.0084040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khromykh AA, Varnavski AN, Sedlak PL, Westaway EG. 2001. Coupling between replication and packaging of flavivirus RNA: evidence derived from the use of DNA-based full-length cDNA clones of Kunjin virus. J Virol 75:4633–4640. doi: 10.1128/JVI.75.10.4633-4640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Junjhon J, Pennington JG, Edwards TJ, Perera R, Lanman J, Kuhn RJ. 2014. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J Virol 88:4687–4697. doi: 10.1128/JVI.00118-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hua RH, Bu ZG. 2011. A monoclonal antibody against PrM/M protein of Japanese encephalitis virus. Hybridoma (Larchmt) 30:451–456. doi: 10.1089/hyb.2011.0027. [DOI] [PubMed] [Google Scholar]
- 32.Hua RH, Chen NS, Qin CF, Deng YQ, Ge JY, Wang XJ, Qiao ZJ, Chen WY, Wen ZY, Liu WX, Hu S, Bu ZG. 2010. Identification and characterization of a virus-specific continuous B-cell epitope on the PrM/M protein of Japanese encephalitis virus: potential application in the detection of antibodies to distinguish Japanese encephalitis virus infection from West Nile virus and dengue virus infections. Virol J 7:249. doi: 10.1186/1743-422X-7-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hua RH, Li YN, Chen ZS, Liu LK, Huo H, Wang XL, Guo LP, Shen N, Wang JF, Bu ZG. 2014. Generation and characterization of a new mammalian cell line continuously expressing virus-like particles of Japanese encephalitis virus for a subunit vaccine candidate. BMC Biotechnol 14:62. doi: 10.1186/1472-6750-14-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hua RH, Liu LK, Chen ZS, Li YN, Bu ZG. 2013. Comprehensive mapping antigenic epitopes of NS1 protein of Japanese encephalitis virus with monoclonal antibodies. PLoS One 8:e67553. doi: 10.1371/journal.pone.0067553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hua RH, Liu LK, Huo H, Li YN, Guo LP, Wang XL, Qin CF, Bu ZG. 2014. Comprehensive mapping of a novel NS1 epitope conserved in flaviviruses within the Japanese encephalitis virus serocomplex. Virus Res 185:103–109. doi: 10.1016/j.virusres.2014.03.001. [DOI] [PubMed] [Google Scholar]