

Abstract
Condition‐dependent genetic interactions can reveal functional relationships between genes that are not evident under standard culture conditions. State‐of‐the‐art yeast genetic interaction mapping, which relies on robotic manipulation of arrays of double‐mutant strains, does not scale readily to multi‐condition studies. Here, we describe barcode fusion genetics to map genetic interactions (BFG‐GI), by which double‐mutant strains generated via en masse “party” mating can also be monitored en masse for growth to detect genetic interactions. By using site‐specific recombination to fuse two DNA barcodes, each representing a specific gene deletion, BFG‐GI enables multiplexed quantitative tracking of double mutants via next‐generation sequencing. We applied BFG‐GI to a matrix of DNA repair genes under nine different conditions, including methyl methanesulfonate (MMS), 4‐nitroquinoline 1‐oxide (4NQO), bleomycin, zeocin, and three other DNA‐damaging environments. BFG‐GI recapitulated known genetic interactions and yielded new condition‐dependent genetic interactions. We validated and further explored a subnetwork of condition‐dependent genetic interactions involving MAG1,SLX4, and genes encoding the Shu complex, and inferred that loss of the Shu complex leads to an increase in the activation of the checkpoint protein kinase Rad53.
Keywords: condition‐dependent, DNA barcode, en masse, genetic interaction, sequencing
Subject Categories: Genome-Scale & Integrative Biology, Methods & Resources, Network Biology
Introduction
The importance of condition‐dependent genetic interactions
Genetic interactions, defined by a surprising phenotype that is observed when mutations in two genes are combined (Mani et al, 2008), are powerful tools to infer gene and pathway functions (Baryshnikova et al, 2010; Ideker & Krogan, 2012). Of the genetic interactions currently known in any species, the vast majority were found using Synthetic Genetic Array (SGA) technology in Saccharomyces cerevisiae (Bandyopadhyay et al, 2010; Costanzo et al, 2010, 2016; van Leeuwen et al, 2016) and these studies have yielded a rich landscape of genetic interactions. The sign of genetic interaction (defined to be negative when mutants are synergistically deleterious, and positive when the combination is less severe than would be expected from independent effects) provides clues about whether the genes act in parallel or in a concerted or serial fashion. Measuring similarity between genetic interaction profiles, both at the level of single genes and of clusters of genes, has revealed a hierarchical map of eukaryotic gene function (Costanzo et al, 2010, 2016). However, the vast majority of genetic interaction mapping has been conducted under a single standard culture condition.
The importance and qualitative nature of gene function can change with environmental fluctuation, so that a complete understanding of genetic interactions will require mapping under multiple conditions. For example, pairs of DNA repair genes had 2–4 times more genetic interactions between DNA repair genes under MMS treatment compared with rich media alone (St Onge et al, 2007; Bandyopadhyay et al, 2010; Ideker & Krogan, 2012), so that a plethora of condition‐dependent genetic interactions remain to be uncovered via gene × gene × environment studies.
Current genetic interaction discovery technologies
Essentially every large‐scale genetic interaction mapping strategy in S. cerevisiae uses a genetic marker system developed for the SGA technique, which works by mating a single‐gene deletion query strain with an array of different single‐gene deletion strains from the Yeast Knockout Collection (YKO) (Giaever et al, 2002). The SGA system provides genetic markers by which mated diploids can be subjected to a series of selections to ultimately yield haploid double mutants. In “standard” SGA mapping, the fitness of the resulting double mutants is determined by statistical analysis of the images from each plate, yielding cell growth estimates for each separately arrayed strain (Tong & Boone, 2005). SGA has also been used to study genetic interactions within functionally enriched gene groups (Collins et al, 2006) and has been applied to detect environment‐dependent interactions (St Onge et al, 2007; Bandyopadhyay et al, 2010). For example, St Onge et al (2007) used the SGA markers to generate all pairwise double mutants between 26 DNA repair genes in yeast. The authors cultured each double mutant individually in microplates and monitored cell density over time to infer the fitness of double mutants and thereby identify genetic interactions in the presence and absence of MMS.
Others have measured genetic interactions via competition‐based fitness measurements in liquid cultures, adding fluorescent markers for tracking cell viability, and using robotic manipulation to inoculate and measure cell growth (DeLuna et al, 2008; Garay et al, 2014). A recent technique called iSeq incorporated barcodes into single‐mutant strains, such that pairs of barcodes identifying corresponding pairs of deleted genes could be fused by Cre‐mediated recombination (Jaffe et al, 2017). The authors demonstrated the method, showing that a pool corresponding to nine gene pairs could be sequenced to monitor competitive growth of double mutants en masse in different environments (Jaffe et al, 2017). Cre‐mediated approaches have been used similarly to map protein–protein interactions (Hastie & Pruitt, 2007; Yachie et al, 2016; Schlecht et al, 2017).
For each of the above genetic interaction methods, double mutants were generated by individual mating of two specific yeast strains, requiring at least one distinct location for each double‐mutant strain on an agar or microwell plate and necessitating robotic strain manipulation to achieve large scale. By contrast, other methods to map genetic interactions generated double mutants in a “one‐by‐many” fashion. For example, diploid‐based synthetic lethality analysis on microarrays (dSLAM) (Pan et al, 2004) disrupted a single “query” gene by homologous recombination via transformation of a marker into a pool of diploid heterozygous deletion strains bearing the SGA marker. After selecting for double‐mutant haploids from such a one‐by‐many haploid double‐mutant pool, barcodes were PCR‐amplified from extracted double‐mutant DNA and hybridized to microarrays to infer the relative abundance and fitness of each double mutant. Another method, genetic interaction mapping (GIM) (Decourty et al, 2008), generated a one‐by‐many pool of barcoded double mutants by en masse mating a single query strain to a pool of haploid gene deletion strains. Like dSLAM, GIM inferred strain abundance and fitness via barcode hybridization to microarrays. Despite the efficiency of generating one‐by‐many double‐mutant pools, a matrix involving thousands of query strains would require thousands of such pools to be generated.
Each of the above methods has advantages and disadvantages. For example, measuring a growth time‐course for each double‐mutant strain provides high‐resolution fitness measurements (St Onge et al, 2007; Garay et al, 2014), but scalability is low. Standard SGA is high‐throughput, but requires specialized equipment for robotic manipulation, and these manipulations must be repeated to test genetic interactions in new environments. The iSeq method shares the scaling challenge of SGA in strain construction, in that it requires many pairwise mating operations; however, once a double‐mutant pool has been generated, it represents a promising strategy for measurement of competitive pools in different environments. The dSLAM and GIM methods allow generation of one‐by‐many pools, which reduces the number of mating operations, but both methods require customized microarrays as well as pool generation and microarray hybridization steps for every query mutation in the matrix.
Barcode fusion genetics to map genetic interactions (BFG‐GI)
Here, we describe BFG‐GI, which borrows elements from several previous approaches. Like iSeq, BFG‐GI requires generation of barcoded single‐mutant strains, with only minimal use of robotics. To generate double‐mutant pools, BFG‐GI uses the SGA marker system and, like the GIM strategy, BFG‐GI employs en masse mating. Unlike GIM and all other previous genetic interaction mapping strategies, BFG‐GI employs many‐by‐many “party mating” to generate all double mutants for a matrix of genes in a single mating step. All successive steps—including barcode fusion, sporulation, selection of haploid double mutants, and measurement of relative strain abundance—are also conducted en masse. We show that double mutants can be generated and monitored in competitive pools using BFG‐GI. Like iSeq, BFG‐GI infers double‐mutant fitness in competitively grown strain pools using next‐generation sequencing of fused barcodes, and BFG‐GI double‐mutant pools can be aliquoted and stored. Aliquots can be thawed later and challenged under specific environments (e.g., drugs) to detect condition‐dependent genetic interactions without having to regenerate the double‐mutant strains.
We assessed BFG‐GI by mapping genetic interactions of DNA repair‐related genes under multiple DNA‐damaging conditions, revealing many condition‐dependent interactions and a discovery that perturbation of the Shu complex leads to increased activation of the Rad53 checkpoint protein kinase.
Results
BFG‐GI experimental design overview
The first step in the BFG‐GI process is generating uniquely barcoded donor and recipient strains with complementary mating types. Each donor and recipient strain contains a unique barcode locus. In the donor strain, this barcode is flanked by two distinct site‐specific recombination sites (loxP/2272 sites), while in the recipient strain, both recombination sites lie on the same side of the unique recipient barcode. After the mating step, these sites mediate barcode fusion via the Cre/Lox system, yielding chimeric barcode sites that uniquely identify specific deletion combinations. We created donors by crossing individual gene deletion strains from the YKO collection with proDonor strains that contained newly constructed pDonor plasmids (Figs 1A and EV1, and Materials and Methods). We generated recipient strains by crossing individual gene deletion strains from the SGA query collection with proRecipient strains (Figs 1B and EV2, and Materials and Methods). Haploid selection of double mutants followed mating of donor and recipient strains, sporulation, and in vivo fusion of barcodes using Cre/Lox recombination (Fig 1C).
Figure 1. BFG‐GI pipeline summary.
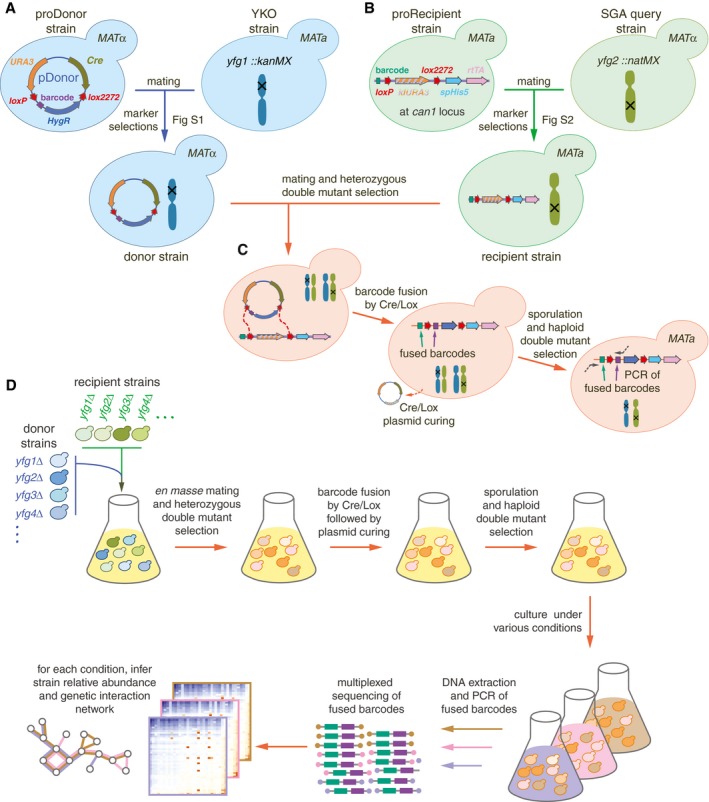
- Construction of donors with unique barcodes representing each gene deletion in parental strains from the YKO collection.
- Construction of recipients also with unique barcodes representing genes of interest in parental strains from the SGA query collection. Pairs of recombination sites (loxP and lox2272) were located at the barcode loci of donor and recipient strains to enable in vivo intracellular fusion of barcode pairs at the recipient barcode locus.
- Donors and recipients were mated with each other to generate heterozygous diploid double mutants, and barcodes were fused in vivo by the Cre/Lox system. The relic plasmid remaining in donors after Cre/Lox recombination was counter‐selected after barcode fusion. Sporulation was induced to select for the MAT a progeny and haploid double mutants.
- BFG‐GI was conducted en masse to generate “many‐by‐many” pools for a set of 26 DNA repair and 14 neutral genes. The resulting pool of haploid double mutants was stored as aliquots of glycerol stock. Thawed aliquots were used to inoculate media containing different chemical agents (“drugs”). Genomic DNA was extracted and fused barcodes were amplified and sequenced to monitor double‐mutant abundance and to infer genetic interactions. Details of donor and recipient strain construction are shown in Figs EV1 and EV2, respectively. Media details are shown in Fig EV3.
Figure EV1. Donor toolkit construction.
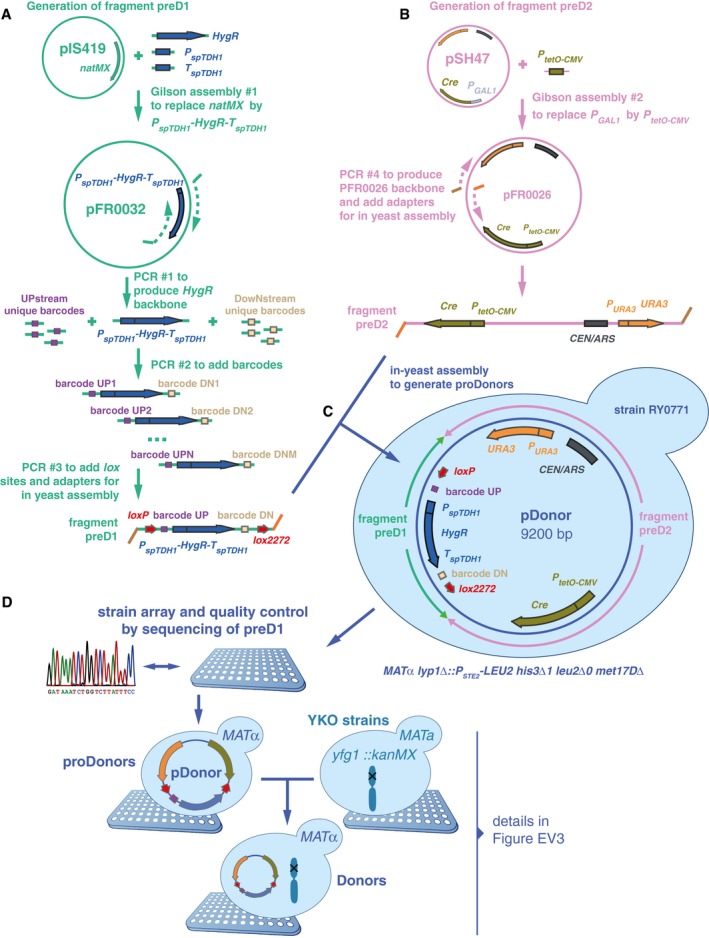
- Two fragments were built to generate proDonor plasmids. The first, preD1, contained loxP/lox2272 sites flanking two 20‐bp unique barcodes and a hygromycin resistance marker. In this study, only the upstream barcode was used for further steps, and for simplification, the downstream barcode was omitted from Fig 1.
- The second, preD2, contained the Cre recombinase driven by the doxycycline‐inducible tetO‐CMV, and a URA3 marker.
- The two fragments were assembled in vivo in yeast to generate pDonors.
- pDonors were arrayed and Sanger‐sequenced to confirm the integrity of the preD1 fragment. ProDonors with confirmed preD1 fragments were mated with YKO strains to generate strains carrying both a uniquely barcoded pDonor and a gene deletion of interest. Then, they were sporulated and the haploid MATalpha progeny were selected using the mating type maker indicated in panel (C). Details on selective media are shown in Fig EV3.
Figure EV2. Recipient toolkit construction.
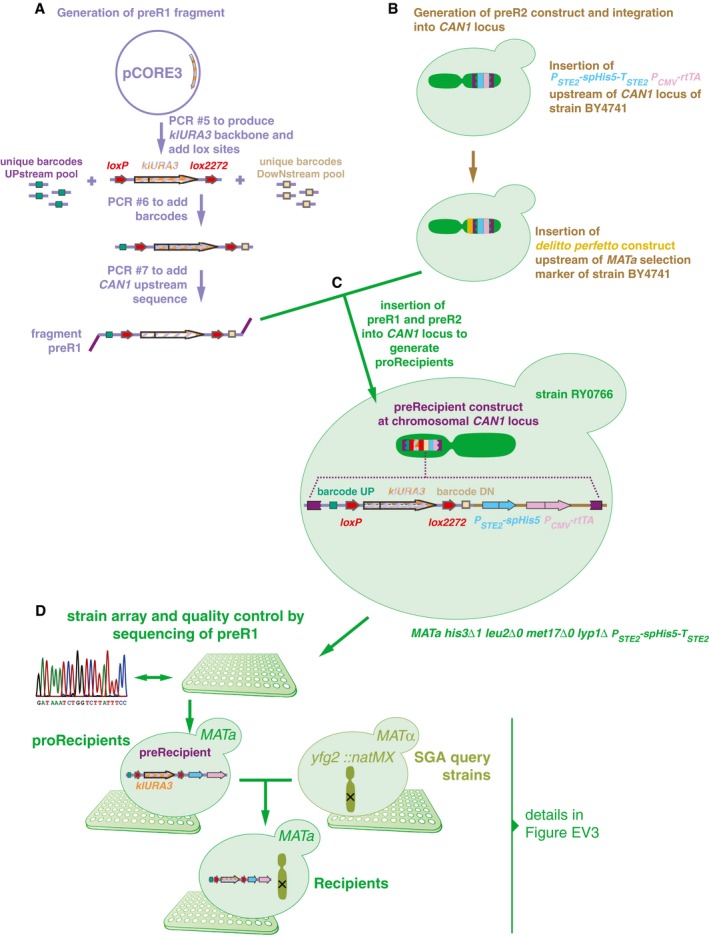
- Two constructs were built to generate recipients. The first fragment, preR1, contained loxP/lox2272 sites flanking a klURA3 marker and two 20‐bp unique barcodes flanking these loci. In this study, only the upstream barcode was used for further steps, and for simplification, the downstream barcode was omitted from Fig 1.
- The second construct, preR2, contained the can1Δ::P STE2 ‐spHis5‐T STE2 mating type marker.
- The two fragments were assembled in vivo using a derivative of the delitto perfetto construct.
- Resulting proRecipients were arrayed and Sanger‐sequenced to confirm integrity of preR1 loci. ProRecipients with confirmed preR1 loci were mated with SGA query strains to generate strains carrying both a uniquely barcoded recipient construct and a gene deletion of interest. Then, they were sporulated and the haploid MAT a progeny were selected using the mating type maker indicated in panel (C). Details on selective media are shown in Fig EV3.
We confirmed that barcode fusion was successful using two control strains carrying markers at likely‐neutral loci. Specifically, we crossed a MATalpha Donor hoΔ::kanMX to a MAT a Recipient ylr179cΔ::natMX and induced Cre/Lox recombination to fuse their barcodes. After sporulation and selection of the MATalpha haploid double‐mutant progeny (Materials and Methods), we extracted genomic DNA, amplified barcode fusions by PCR, and confirmed their integrity by Sanger sequencing (Fig 1C).
To scale up the BFG‐GI process, we optimized mating and sporulation steps to generate double mutants with unique barcodes that had been fused en masse (Materials and Methods). We selected hundreds of double mutants using a series of marker selection steps in a many‐by‐many fashion. Intermediate selection steps allowed us to fuse barcodes representing each donor and recipient parental pair within each double‐mutant cell (Fig 1D and Materials and Methods).
Once we generated the pool of fused‐barcode double mutants, aliquots were stored at −80°C for future experiments. Amplification and next‐generation sequencing of fused barcodes in the pool allowed us to infer the relative abundance of each double mutant in each condition of interest (Fig 1D and Materials and Methods). In addition to haploid double‐mutant pools, we sequenced fused barcodes from the heterozygous diploid double‐mutant pools and used those as reference (“time zero”) controls for fitness and genetic interaction calculations (Materials and Methods).
BFG‐GI measures strain abundances within a heterogeneous population
We first evaluated the ability of BFG‐GI to accurately detect the abundance of pooled double‐mutant strains. To generate reference data for this evaluation, we used the array‐based SGA strategy to generate 2,800 double mutants by individual mating of barcoded BFG‐GI strains, subsequently inducing barcode fusion via the Cre/Lox system. The purpose of this experiment was to assess the extent to which quantifying growth via fused‐barcode sequencing of pooled strains could recapitulate the measurements of growth in individual cell patches (as in conventional SGA). We recorded patch sizes, scraped plates to pool all double‐mutant cells, extracted genomic DNA, and sequenced the fused barcodes (Materials and Methods). The resulting numbers of sequencing reads for each strain were strongly correlated with the corresponding colony sizes (r = 0.92; Fig 2A). Importantly, colonies that were very small or absent often corresponded to double mutants with very few or no sequencing reads. These results show that BFG‐GI detects the abundance of specific double mutants in pools of cells, with results comparable to an array‐based method.
Figure 2. BFG‐GI quality control and benchmarking.
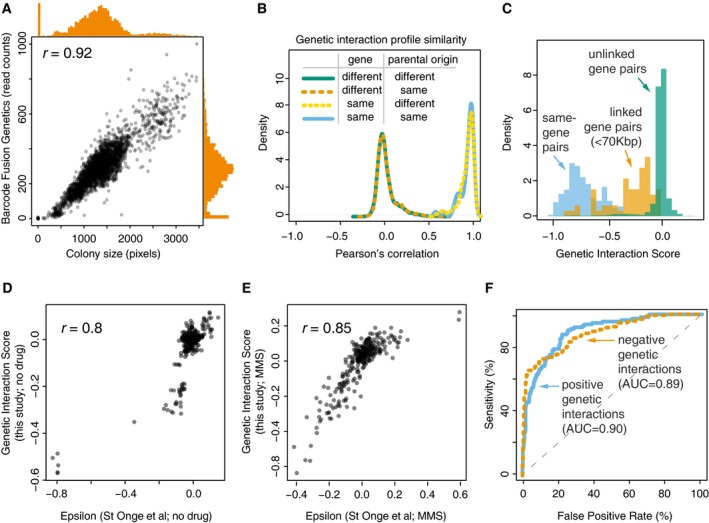
- Correlation between two measures of cell abundance (colony size and next‐generation‐sequencing‐based quantification of fused barcodes) for BFG‐GI double‐mutant strains. Histograms show distribution of abundance in the two measurements. Peaks in the histograms representing data points in the bottom‐left corner of the scatter plot indicate that absent and very small colonies produced few or no sequencing reads.
- Density plots for BFG‐GI genetic interaction score (GIS) correlation between replicates of the same gene, with same or different parental origin, or pairs of different genes. Only replicates with a GIS correlation > 0.5 were retained for further analyses.
- Histograms comparing the GIS distribution for “same‐gene pairs” (which are expected to behave like synthetic lethals given the SGA double‐mutant selection process) with that for linked‐ and unlinked‐gene pairs.
- Comparison of BFG‐GI‐inferred genetic interactions in haploid double‐mutant media without MMS with genetic interactions identified using similar media (St Onge et al, 2007).
- Comparison of BFG‐GI‐inferred genetic interactions in haploid double‐mutant media containing MMS with genetic interactions previously identified in similar media (St Onge et al, 2007).
- Benchmarking of BFG‐GI genetic interactions against the St. Onge et al (2007) dataset. Note that “false positives” may be real interactions that were not found in the benchmark study.
Generating a DNA repair‐focused double‐mutant strain pool
To test whether BFG‐GI can accurately map genetic interactions, we generated a double‐mutant pool focused on DNA repair genes and compared BFG‐GI results to those of other validated genetic interaction assays. We began by generating donor and recipient strains by crossing 35 YKO (yfg1Δ::kanMX, MAT a) single‐gene deletion strains to 65 BFG‐GI proDonor strains, and 38 SGA query (yfg2Δ::natMX, MATalpha) single‐gene deletion strains to 71 BFG‐GI proRecipient strains. The set of deleted genes to which these strains correspond include 26 DNA repair genes from a previous condition‐dependent genetic interaction study (St Onge et al, 2007), as well as 14 likely neutral loci (i.e., the already‐disrupted HO locus, pseudogenes, and other loci for which single‐ and double‐mutant phenotypes have not been previously observed). Inclusion of neutral loci allowed us to infer single‐mutant fitness from pools of double mutants (Materials and Methods).
To generate haploid double mutants, donor and recipient cells were scraped from plates and all subsequent steps in the BFG‐GI pipeline were conducted en masse. First, the pools were combined for party mating. Seven selection steps followed mating, including four that correspond to those in the standard SGA procedure: heterozygous diploid selection, sporulation, MAT a progeny selection, and haploid double‐mutant selection. Additionally, before sporulation, we completed three selection steps to fuse barcodes and subsequently remove Cre to limit additional recombination events (Figs 1C and EV3). This generated a pool of 4,288 haploid double mutants, which was aliquoted and stored as frozen glycerol stock. Thawed samples were used to inoculate solid media appropriate for selecting haploid double‐mutant cells. The media was used alone, supplemented with dimethyl sulfoxide (DMSO) as a solvent control, or supplemented with one of seven drugs targeting DNA repair pathways (Table EV1). We extracted genomic DNA and amplified and sequenced fused barcodes to infer the relative abundance of each double mutant in each condition.
Figure EV3. Media details to generate BFG‐GI strains and pools.
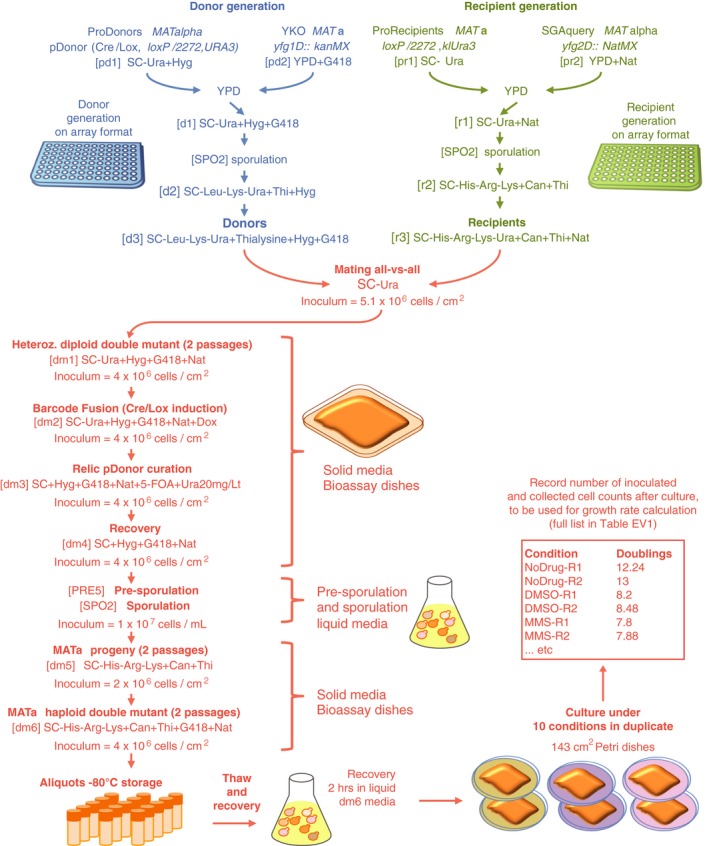
Donors, recipients, and double mutants used in BFG‐GI were generated as shown in Figs 1, EV1 and EV2. This figure shows media details, optimal inoculum cell densities, and incubation times for pool‐based cultures. All incubations were at 30°C for 24 h, except for mating (12 h at 23°C) and sporulation (12 days at 21°C). Sporulation was conducted in flasks with liquid media shaking at 200 rpm. We used the following reagent concentrations: G418 = 200 μg/ml; clonNat = 100 μg/ml; canavanine = 100 μg/ml; thialysine = 100 μg/ml; hygromycin = 200 μg/ml; and 5‐FOA = 1 mg/ml. Amino acid concentrations were as described in Tong and Boone (2005).
To evaluate assay reproducibility, we ran all BFG‐GI procedures in duplicate, starting from the mating step (technical replicates) and also barcoded multiple strains representing the same gene (biological replicates). Biological replicate strains had either the same or different parental strain origin (the parental strain for a given gene deletion might be from either the YKO or SGA query strain collection). Relative strain abundance was highly correlated between technical replicates (r > 0.95). Next, we used a multiplicative model to infer a genetic interaction score (GIS) from relative strain abundances, analogous to other methods based on strain growth (Materials and Methods). The relative strain abundance, GIS correlation between technical replicates was also high (r = 0.96).
Correlation of GIS profiles between biological replicates representing the same gene was generally high, with 85% of replicates showing GIS r > 0.5. We computationally excluded from analysis 21 biological replicates (six donors and 15 recipients) showing GIS r < 0.5. For the remaining strains, biological replicate profiles clearly showed higher correlation than did the profiles of replicates carrying deletions in different genes (Fig 2B). To understand factors contributing to poorly correlated replicate pairs, we sequenced the genomes of 20 strain pairs. Ten of those pairs corresponded to strains with GIS r < 0.5 and other 10 with GIS r > 0.5. We found that all 10 strain pairs with GIS r < 0.5 had chromosome V duplicated in one of the two strains, in agreement with the report of iSeq strains showing low strain profile reproducibility, owing to this same chromosome V duplication (Jaffe et al, 2017). Chromosome V contains the CAN1 locus, the locus at which both BFG‐GI recipients and iSeq strain constructs are inserted. By contrast, only three out of 10 strain pairs with r > 0.5 showed aneuploidies in just one strain in the pair (for these strains, the aneuploidies were also in chromosome V). All BFG‐GI strains showing aneuploidies were recipients. This suggests that future versions of BFG‐GI recipients for which selection markers are carried by plasmids may increase reproducibility, as we found for our Donor strains. Furthermore, we removed strains with poor representation in the heterozygous diploid pool, because GIS profiles from these strains yielded neutral scores even for controls (“same‐gene” pairs described below) that should behave like strong negative interactions, presumably due to poor statistical power to detect fitness effects (Fig EV4B). This included all replicates representing swc5Δ, which showed very low relative abundance in the sequencing results. Our final dataset consisted of 3,232 double mutants, with 59 Donors and 56 Recipients, representing 39 genes (25 DNA repair genes and 14 neutral genes; Fig EV4A and Table EV2). Finally, GIS measurements for technical and biological replicates (Table EV3) were combined into a single score for each gene pair (Table EV4; Materials and Methods).
Figure EV4. Calling genetic interactions.
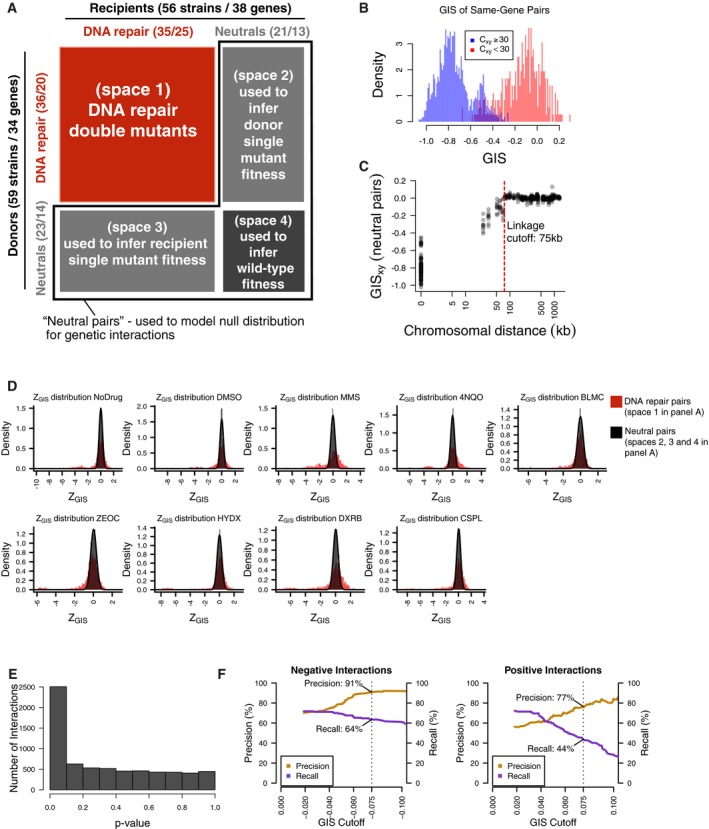
- Two collections of 59 donor strains (containing 34 unique knockouts) and 56 recipient strains (containing 38 unique gene knockouts) were crossed against each other in an all‐by‐all pooled format. Each strain contains a knockout at either a DNA repair gene or neutral locus. Double‐knockout strains were divided into four spaces based on the types of genes knocked out. Numbers in parentheses represent the number of strains and unique gene knockouts, respectively.
- Distribution of GIS amongst strain pairs containing the same gene, split by those which were well‐measured from the heterozygous diploid stage (C xy ≥ 30) and not well‐measured from the same stage (C xy < 30). Non‐well‐measured strains (72 out of 3,305) were excluded from analysis, and GIS was re‐calculated after their exclusion.
- Distribution of GIS in strains representing linked neutral pairs. Using the GIS profiles, an empirical cutoff of 75 kbp (red dashed line) was chosen to classify strains with knockout pairs on the same chromosome as either linked or unlinked. GIS was then re‐calculated based on this linkage criterion.
- Distribution of Z GIS calculated for DNA repair pairs (space 1 in panel A, red) and pairs involving well‐measured and unlinked neutral genes (spaces 2, 3, and 4 in panel A, black). Z GIS for pairs involving neutral genes were used to calculate a P‐value.
- Distribution of P‐values calculated by the null distribution in (D). P‐values were combined for multiple barcode replicates of each gene–gene pair and converted to FDR scores (see Materials and Methods). Barcode‐level P‐values are available in Table EV3, and gene‐level FDR scores are available in Table EV4.
- Benchmarks of BFG‐GI with data from St Onge et al (2007) for strains containing a significant genetic interaction (FDR < 0.01). Each graph shows precision and recall using the benchmark of St Onge et al (2007) as a function of an additional GIS effect‐size cutoff (left = negative interaction performance; right = positive interaction performance). Overlay text indicates performance at |GIS| = 0.075 (dashed lines), which was chosen as the effect‐size threshold.
We next assessed the ability of BFG‐GI to infer the fitness for three classes of double‐mutant strains. First, we measured the abundance of strains carrying two differently barcoded mutations corresponding to the same gene. Compound heterozygous diploids bearing a mutation at both loci for a given gene (e.g., mms4Δ::kanMX/mms4Δ::natMX) can survive in media supplemented with selective antibiotics; however, haploid cells derived from this parental diploid should not survive because they should only carry one locus for each gene and therefore only one of the two antibiotic resistance markers required to survive the selection. Thus, haploid strains for “same‐gene pairs” are expected to exhibit reduced fitness, behaving like synthetic lethal combinations, and be depleted from the pools. The calculated GIS agreed with this expectation (Fig 2C). Second, we assessed the abundance of double mutants representing pairs of linked genes (< 75 kbp apart; Fig EV4C). Independent segregation is reduced between linked genes, and as expected, our GIS indicated these double mutants were also depleted from the pools (Fig 2C). Third, we analyzed double mutants representing unlinked genes and we found that their GIS distribution is clearly distinguishable from same‐gene and linked‐gene pairs (Fig 2C).
Finally, we sought to compare BFG‐GI results against another dataset of genetic interactions (St Onge et al, 2007), both to obtain an overall evaluation of our method and as a way to calibrate our GIS thresholds for calling genetic interactions. We first compared BFG‐GI GISs with the epsilon scores reported by St Onge et al (2007) under both no‐drug and MMS conditions, for pairs of DNA repair genes that had been tested in both studies. We found that GIS and epsilon scores correlated well with each other in both no‐drug (r = 0.8) and MMS (r = 0.85) conditions (Fig 2D and E). Taking both conditions together, and using GIS thresholds with an estimated 5% false‐positive rate, BFG‐GI captured 56% of the positive genetic interactions reported by St. Onge et al and 66% of the negative genetic interactions (Fig 2F), while reporting an additional 23 positive and 20 negative interactions not reported by St Onge et al (2007).
Taken together, these results provide evidence that BFG‐GI offers a powerful means of generating double mutants by en masse mating and monitoring strain abundance in a multiplexed fashion to infer condition‐dependent genetic interactions.
BFG‐GI reveals condition‐dependent genetic interactions
Having determined that BFG‐GI can accurately detect genetic interactions, we analyzed the same double‐mutant pool under seven additional culture conditions to more broadly explore condition‐dependent genetic interactions (see Fig 3C legend for condition names and Table EV1 for details). To call positive and negative interactions, we first standardized GIS by the estimated error (Z GIS; Materials and Methods), and used the distribution of Z GIS amongst unlinked barcode pairs containing a neutral gene (“neutral pairs”; Fig EV4A) to estimate the false discovery rate (FDR) at each given Z GIS cutoff (Fig EV4D–E). To call interactions, we used both a Z GIS cutoff corresponding to FDR = 0.01 in each condition and an additional effect‐size cutoff (|GIS| > 0.075) to filter out interactions of high confidence but low magnitude. At these cutoffs, 91% of the called negative interactions and 77% of the called positive interactions were also observed in a previous study (St Onge et al, 2007), while 64% of the previously reported negative and 44% of the previously reported positive interactions were reproduced by BFG‐GI (Fig EV4F; Table EV4).
Figure 3. Condition‐dependent genetic interactions mapped by BFG‐GI.
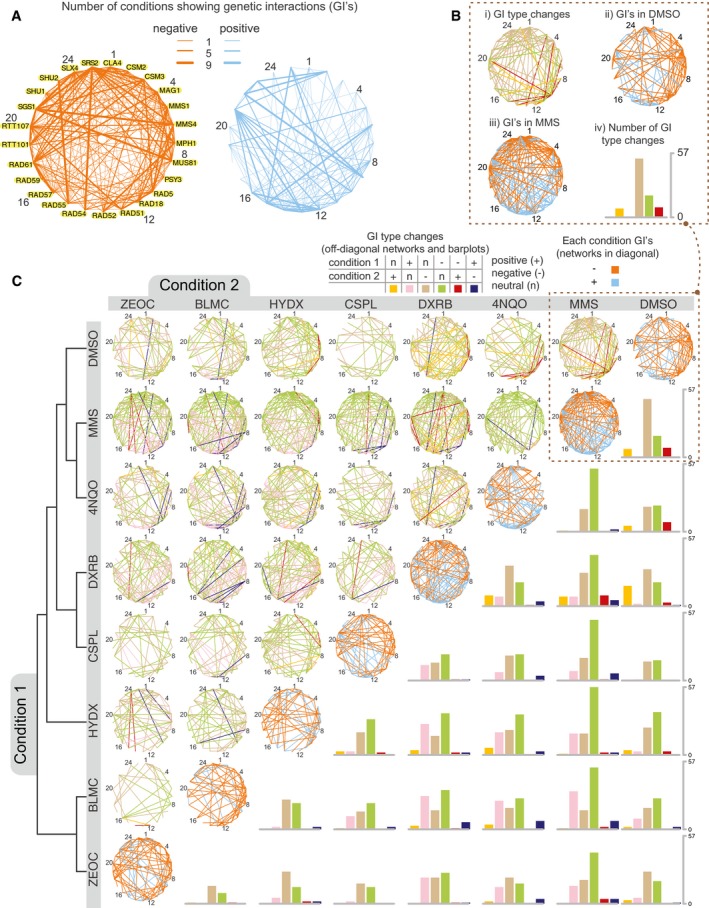
- Networks showing the number of conditions with a genetic interaction for each gene pair (using FDR < 0.01 and |GIS| > 0.075 as cutoffs). Numbers besides gene names are guides for the reader to locate nodes in networks of panels (B) and (C). Data for individual interactions are available in Tables EV3 and EV4.
- Networks in the diagonal (subpanels ii and iii) show genetic interactions for DMSO or MMS after applying the same criteria as in (A). The network in subpanel i shows significant genetic interaction changes (FDR < 0.01, |∆GIS| > 0.1) when comparing the DMSO and MMS treatments. Interaction types are positive (+), negative (−), or neutral (n). The barplot in subpanel iv summarizes the number of changes between interaction type in subpanel i.
- The networks are the same as described in (B) with additional drug conditions: cisplatin (CSPL), doxorubicin (DXRB), hydroxyurea (HYDX), zeocin (ZEOC), bleomycin (BLMC), and 4NQO. The no‐drug condition was omitted from this figure as it showed no significant condition‐dependent genetic interactions with DMSO. GIS profiles were hierarchically clustered using maximum distance and complete linkage, with the resulting dendrogram shown on the left. Data for individual differential interactions are available in Tables EV5 and EV6. This figure was generated with Cytoscape (Shannon et al, 2009) and R scripts (R Core Team, 2017).
Analyzing BFG‐GI results further, we found that all DNA repair genes showed at least one genetic interaction and that some genes showed markedly more interactions than others. For example, we found that the DNA helicase gene SGS1 yielded negative interactions with MMS4, MUS81, or SLX4 (all of which participate in template switching during break‐induced replication) in all nine conditions (Fig 3A, Table EV4). Another DNA helicase gene, SRS2, interacted negatively with both SGS1 and the DNA translocase gene RAD54 in all nine conditions. A third DNA helicase/ubiquitin ligase gene, RAD5, showed positive genetic interactions with SGS1 in six conditions. SGS1 and SRS2 are involved in error‐free DNA damage tolerance, while RAD5 is involved in recombinational repair of double‐strand breaks. These findings coincide with previous reports showing SGS1 and SRS2 centrality in DNA repair pathways in both unperturbed and MMS‐induced stress conditions (St Onge et al, 2007).
We next examined condition‐dependent changes in genetic interactions. First, genetic interaction differences between conditions were calculated (∆GIS). Then, using a similar approach to that which was used to call genetic interactions within each condition, ∆GIS was standardized by the estimated error (∆ZGIS), and the distribution of ∆ZGIS amongst neutral pairs was used to calculate an FDR for each differential interaction (Fig EV5A; Materials and Methods). At a ∆ZGIS cutoff corresponding to FDR = 0.01 and an effect‐size cutoff of |∆GIS| > 0.1, we identified 2,932 differential interactions amongst DNA damage genes and further considered only the subset of 2,335 differential interactions that changed between interaction type (i.e., between the three classes of positive, negative, and neutral) for further analysis. For any given pair of conditions, an average of 9% of all gene pairs exhibited differential interaction. For example, we found mus81Δ/rad5Δ displayed a negative genetic interaction in DMSO, a positive genetic interaction in MMS, and a significant difference between the two conditions. This change is shown as a red edge in Fig 3B, panel i, and agrees with a previous report (St Onge et al, 2007). By contrast, most changes in genetic interaction between DMSO and MMS were from neutrality in one condition to either a positive or negative genetic interaction in the other (Fig 3B, panels i and iv). Generalizing this observation to all pairwise condition comparisons, a large majority of significant differential genetic interactions were neutral in one condition and either positive or negative in the other (94%), and thus, only 6% of significant genetic interactions changed sign between conditions (Fig 3C and Table EV5).
Figure EV5. Calling differential genetic interactions.
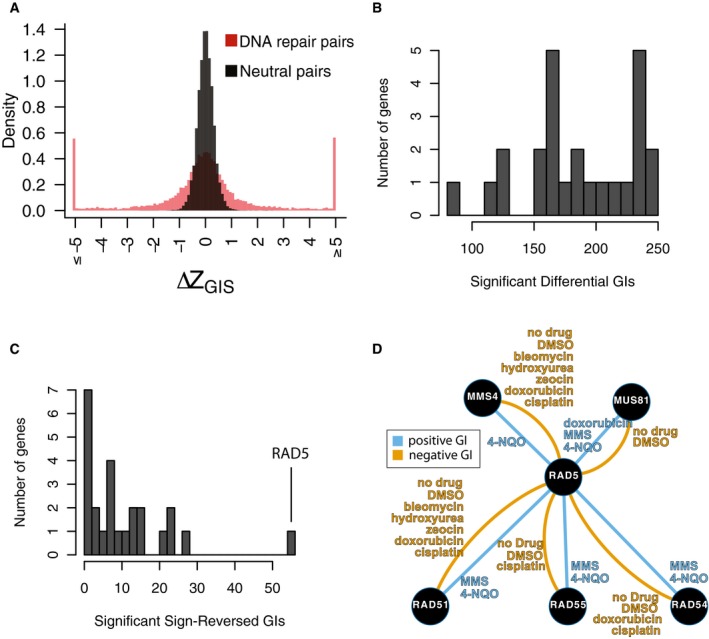
- Distribution of ∆Z GIS for neutral pairs compared to DNA repair pairs. The distribution amongst neutral pairs was used to calculate a P‐value for ∆Z GIS amongst DNA repair pairs, which was then converted to an FDR for each differential interaction (see Materials and Methods; Table EV6). An additional effect‐size cutoff of |∆GIS| > 0.1 was added to call differential genetic interactions in Fig 3 and Table EV5.
- Distribution of significant differential genetic interaction calls per gene.
- Distribution of significant differential genetic interaction calls involving a reversal of direction (i.e., from positive to negative or vice versa) by gene. RAD5 is involved in 47 differential genetic interactions with a reversal of direction.
- Summary of significant genetic interactions of RAD5 with MUS81, MMS4, RAD51, RAD54, or RAD55 in different conditions. Edges represent genetic interaction type and are labeled by conditions in which significant genetic interactions were found for the corresponding pair and direction.
Genes differed both in the total number of differential genetic interactions in which they participated (Fig EV5B) and in the number of their differential genetic interactions that involved a change in sign (Fig EV5C). Genetic interactions involving RAD5 were especially dynamic—RAD5 participated in 233 significant differential genetic interactions (out of 1,224 comparisons; Fig EV5B), and 55 of these involved sign reversals (Fig EV5C). Out of 55 sign‐reversed differential genetic interactions involving RAD5, 48 involved MMS4, MUS81, RAD51, RAD54, or RAD55 (Fig EV5D). MUS81 and MMS4 encode a heterodimer which cleaves nicked intermediates in recombinational DNA repair (Schwartz et al, 2012), while RAD51 binds ssDNA to facilitate homologous recombination and requires RAD54 and RAD55 for its activity (Sugawara et al, 2003). Genetic interactions with RAD5 were often positive for all five of these genes in 4NQO and MMS, and negative with all five in other tested conditions (Fig EV5D). These findings are consistent with previously reported negative interactions of RAD5 with these genes in MMS and positive interactions when no drug stress is added (St Onge et al, 2007; Table EV4). The dynamic interactions of RAD5 with these two gene groups may reflect the previously reported multifunctional nature of RAD5 and its ability to coordinate repair events and replication fork progression differently in response to different types of lesions (Choi et al, 2015).
We assessed similarity between growth conditions as measured by similarity between patterns of GIS profiles. As expected, the two conditions most similar to each other were no‐drug and DMSO, which also yielded no significant between‐condition differential interactions (Table EV5). A hierarchical clustering of conditions by their GIS profiles (Fig 3C) showed that pairs of drugs with similar mechanisms of action clustered together. For example, bleomycin and zeocin, which are members of the same family of glycopeptides that intercalate into DNA to induce double‐strand breaks (Claussen & Long, 1999), were grouped as nearest neighbors and also had the least number of differential interactions between any two drug pairs (26, compared to an average of 67 across all condition pairs).
Interestingly, MMS and 4NQO were also grouped as nearest neighbors. Although there were a large number of differential interactions between them (75), the vast majority (73) showed neutrality in one condition and negative genetic interaction in the other. MMS and 4NQO are members of different drug classes, but both are DNA alkylating agents (Xiao & Chow, 1998; Svensson et al, 2012). Both MMS and 4NQO cause checkpoint‐modulated fork stalling (Minca & Kowalski, 2011; Iyer & Rhind, 2017) that appears to facilitate replication of damaged templates allowing forks to quickly pass lesions (Iyer & Rhind, 2017). Furthermore, strains carrying deletion of genes involved in postreplication repair (PRR) processes, such as MMS2, RAD5, and UBC13, are significantly hypersensitive to both MMS and 4NQO (Lee et al, 2014), suggesting that PRR acts on both MMS and 4NQO lesions. DNA lesions caused by these drugs are typically corrected by either base‐excision repair (MMS) or nucleotide‐excision repair (4NQO), and these pathways are synergistic with each other in genetic backgrounds like mag1Δ (Xiao & Chow, 1998). We believe that these mechanistic similarities between MMS and 4NQO contributed to the similarity between their GIS profiles in comparison with those from other drugs we tested.
The most divergent condition pairs (those yielding the highest number of differential interactions) were MMS versus doxorubicin (104 changes) and MMS versus bleomycin (110 changes). These results are consistent with the fact that MMS, doxorubicin, and bleomycin have different mechanisms of action and cause DNA lesions that are repaired by different pathways.
A condition‐dependent subnetwork of MAG1, SLX4, and Shu complex genes
The Shu complex (a heterotetrameric protein complex consisting of Csm2, Psy3, Shu1, and Shu2) promotes Rad51 filament formation and homologous recombination during error‐free lesion bypass, double‐strand break repair, and meiosis (Mankouri et al, 2007; Ball et al, 2009; Bernstein et al, 2011; Godin et al, 2013; Sasanuma et al, 2013) (Fig 4A). Our BFG‐GI results indicated that genes encoding all four members of the Shu complex showed negative genetic interactions with both MAG1 and SLX4 during exposure to MMS. Additionally, the Shu complex genes interacted negatively with SLX4 during treatment with 4NQO, bleomycin, and zeocin (Fig 4B). Mag1 is a 3‐methyladenine DNA glycosylase that removes alkylated bases from DNA to initiate base‐excision repair (BER), thereby protecting cells against alkylating agents like MMS (Berdal et al, 1990; Chen et al, 1990). Slx4 promotes the activity of three structure‐specific endonucleases (Fricke & Brill, 2003; Flott et al, 2007; Toh et al, 2010; Gritenaite et al, 2014) and, upon exposure to MMS, plays a key role in down‐regulating phosphorylation of the checkpoint kinase Rad53 (Ohouo et al, 2013; Jablonowski et al, 2015). We generated double mutants for each Shu complex member in combination with either MAG1 or SLX4 and tested fitness on media containing DMSO or various genotoxins using spot dilution assays (Fig 4C). Our results validated the MAG1–Shu complex gene interactions in MMS that we detected with BFG‐GI, and are consistent with a previous study (Godin et al, 2016). The negative interactions between MAG1 and Shu complex members are explained (Godin et al, 2016) by the fact that these double mutants have simultaneously lost Mag1‐mediated BER (which directly removes alkylated bases) and have a diminished capacity for error‐free lesion bypass, a major pathway used during MMS‐induced blocks in DNA replication (Huang et al, 2013) (Fig 4A). Our spot dilution assays also confirmed that MAG1 interacts negatively with SLX4 during MMS treatment (Fig 4C). This result is also consistent with a previous study showing that BER is unlikely to be the major function of SLX4 (Flott et al, 2007). Of particular interest, we validated the BFG‐GI interactions between Shu complex members and SLX4 during treatment with MMS, 4NQO, bleomycin, and zeocin (Fig 4C).
Figure 4. Shu complex condition‐dependent genetic interactions with MAG1, SLX4, PPH3, and RAD53 .
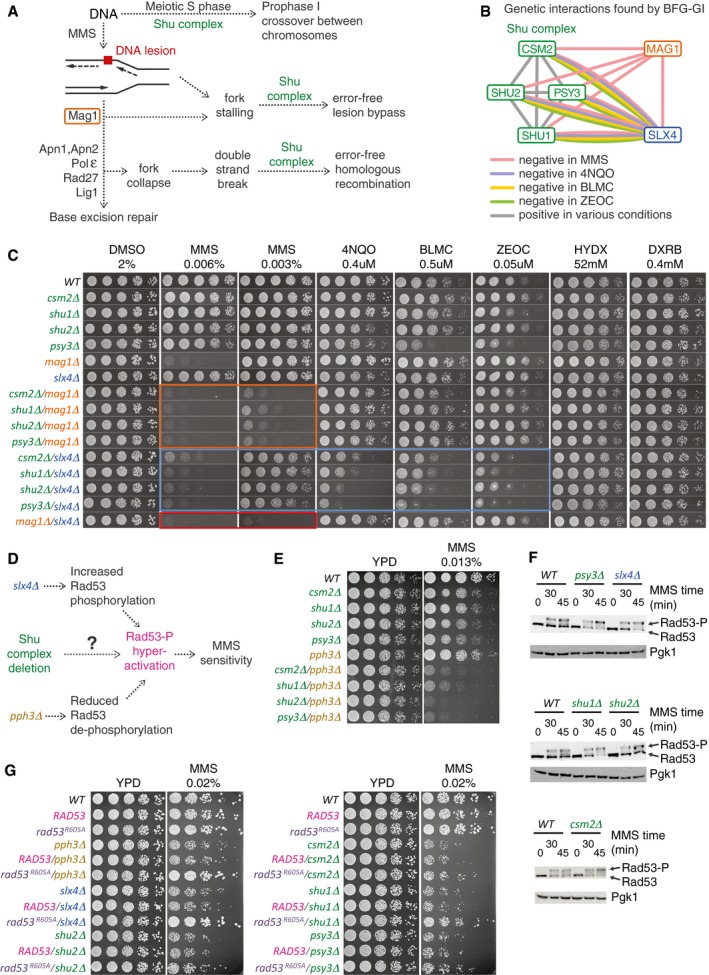
- Pleiotropic participation of the Shu complex in DNA replication and repair pathways.
- Network showing condition‐dependent genetic interactions inferred from BFG‐GI for the indicated conditions.
- Confirmation of interactions between the Shu complex, MAG1, and SLX4 using spot dilution assays including single and double mutants exposed to the indicated drugs for 48 h. Orange, blue, and red boxes indicate genetic interactions of Shu complex members with MAG1 and SLX4, and of MAG1 with SLX4, respectively.
- Schematic of potential functional connections between the Shu complex and SLX4. As with deletion of SLX4 or PPH3, deletion of Shu complex members may lead to hyperphosphorylation and hyperactivation of Rad53, resulting in increased sensitivity to MMS.
- Spot dilution assays showing genetic interactions of Shu complex genes/pph3Δ double mutants and corresponding single mutants exposed to MMS at the indicated concentration for 48 h.
- Western blot assays showing hyperphosphorylation of Rad53 in csm2Δ, psy3Δ, shu1Δ, and slx4Δ strains following treatment with 0.03% MMS. Note increased intensity of Rad53‐P bands compared with the Rad53 bands.
- Spot dilution assays of Shu complex mutants expressing a hypomorphic rad53‐R605A allele (rad53‐R605A‐6xHis‐3xFLAG‐kanMX6) compared with a wild‐type RAD53 allele (RAD53‐6xHis‐3xFLAG‐kanMX6). Cells were exposed to MMS at the indicated concentration for 60 h.
Source data are available online for this figure.
As the nature of the SLX4 interactions with genes encoding Shu complex proteins is unknown, we studied them in more detail. That there are negative genetic interactions between SLX4 and Shu complex members in MMS was unexpected, given that the Shu complex promotes error‐free lesion bypass (Mankouri et al, 2007; Ball et al, 2009; Xu et al, 2013; Godin et al, 2016) and SLX4 is epistatic to genes that regulate error‐free lesion bypass during MMS treatment (Flott et al, 2007). A major role for Slx4 under MMS conditions is down‐regulating phosphorylation and activation of Rad53, which occurs by Slx4 competing with Rad9 for binding to Dpb11 and consequently limiting the formation of Rad9–Dpb11 complexes that activate Rad53 (Pfander & Diffley, 2011; Ohouo et al, 2013; Cussiol et al, 2015; Jablonowski et al, 2015). Levels of phosphorylated Rad53 are also reduced by the presence of PPH3, which encodes the catalytic subunit of the protein phosphatase PP4 complex that binds and dephosphorylates Rad53 during MMS treatment (O'Neill et al, 2007). Deletions of either SLX4 or PPH3 or both genes result in hyperactivation of Rad53 and hypersensitivity to MMS (Jablonowski et al, 2015). This phenotype is suppressed by expression of a hypomorphic rad53‐R605A allele (Ohouo et al, 2013; Cussiol et al, 2015; Jablonowski et al, 2015). To determine whether the genetic interactions between SLX4 and Shu complex members (Fig 4C) reveal an unanticipated role for the Shu complex regulating activation of Rad53 (Fig 4D), we tested the sensitivity of pph3Δ/Shu complex double mutants to MMS using spot dilution assays. Combining pph3Δ with deletion of any of the Shu complex genes resulted in a dramatic increase in MMS sensitivity relative to the single mutants (Fig 4E), indicating negative genetic interactions similar to those seen between SLX4 and Shu complex members (Fig 4C), or between SLX4 and PPH3 (Jablonowski et al, 2015).
To assess MMS‐induced Rad53 activation in Shu complex mutants more directly, we monitored Rad53 phosphorylation (which is a proxy for Rad53 activation) using Western blot assays. Consistent with the role of SLX4 in dampening Rad53 activation (Ohouo et al, 2013; Balint et al, 2015; Jablonowski et al, 2015), slx4Δ cells challenged with MMS showed an increase in Rad53‐P levels relative to wild type (Fig 4F). Interestingly, three of the Shu complex mutants (csm2Δ, psy3Δ, and shu1Δ) also showed an increase in Rad53‐P levels upon treatment with MMS (Fig 4F), indicating that these Shu complex mutants, like slx4Δ and pph3Δ cells, display hyperactivated Rad53 under exposure to MMS. We asked whether the MMS sensitivity of Shu complex mutants could be suppressed by expression of the rad53‐R605A allele. Expression of rad53‐R605A, which is not effectively hyperactivated, suppresses the MMS sensitivity of slx4Δ and pph3Δ (Ohouo et al, 2013; Jablonowski et al, 2015). Similarly, the MMS sensitivity of csm2Δ, psy3Δ, shu1Δ, and shu2Δ mutants was partially suppressed by rad53‐R605A (Fig 4G). Together, our data indicate that deletions of genes encoding the Shu complex, as for Slx4 and Pph3, lead to an increase in Rad53 activation, in response to MMS treatment, as revealed by unique condition‐dependent genetic interactions detected by BFG‐GI.
Discussion
We developed a new technology, called BFG‐GI, in which pools of double‐mutant yeast strains corresponding to a matrix of target genes are generated en masse through many‐by‐many “party” mating. These pools are induced to form double‐mutant‐identifying chimeric barcodes by intracellular site‐specific recombination and assayed for growth via next‐generation sequencing. Aliquots of these pools can be stored and later cultured with different drugs to identify condition‐dependent genetic interactions. To our knowledge, BFG‐GI is the first method to generate haploid double‐mutant strains en masse for a many‐by‐many matrix of genes without the requirement for multiple mating steps, thus enabling large‐scale conditional genetic interaction mapping without extensive use of robotics.
BFG‐GI showed good agreement with a previous genetic interaction mapping method (St Onge et al, 2007). Quantitatively, our GISs show a correlation of r = 0.8–0.85 with the epsilon scores obtained in St Onge et al (2007). Considering only significant interactions, 91% of the negative and 77% of the positive interactions found by BFG‐GI were also observed by St Onge et al (2007), and 44–64% of St Onge et al (2007) interactions were reproduced by BFG‐GI. The contrast between the 0.01 FDR estimate and the validation rate by an orthogonal method suggests that the latter is a too‐conservative measure of precision and that many of the novel interactions are bona fide interactions despite not having been seen by St Onge et al (2007).
We detected and validated unanticipated interactions between SLX4 and Shu complex genes, which mirrored the genetic interactions observed between PPH3 and the Shu complex. We further found that presence of a functional Shu complex corresponded to reduced activation of Rad53 during MMS treatment.
By calculating similarity between the genetic interaction profiles of different drugs, we found that those with similar mechanisms of action, like zeocin and bleomycin, are considerably more alike than comparisons between compounds with different mechanisms of action, e.g., the comparison between MMS and either zeocin or bleomycin. This suggests the potential of BFG‐GI to shed light on drug mechanisms through measurement of gene–gene–environment interactions.
One advantage of BFG‐GI is its cost‐effectiveness. BFG‐GI uses fewer reagents and less robotic assistance than other technologies to map genetic interactions. Like other pool‐based technologies, BFG‐GI requires less media, plates, and drugs than array‐based technologies, resulting in a substantial cost advantage. For example, the amount of media used in 1,536 spot arrays on OmniTrays is reduced 50‐fold by studying the same number of gene pairs in pooled cultures with 4 × 106 cells/cm2 in 143‐cm2 Petri dishes, which is the optimal cell density we calculated for pooled double‐mutant selections (Materials and Methods). BFG‐GI is also more cost‐effective than other barcode‐sequencing technologies because in BFG‐GI, strains are pooled before the mating step, so that generating double mutants does not require robotic manipulation of strain arrays.
The reproducibility of BFG‐GI indicates that it is a robust technology. Technical replicates in BFG‐GI are highly reproducible, and 85% of the biological replicates correlated well with each other (GIS r > 0.5). The remaining 15% of biological replicates showing low correlations could be identified and removed computationally. We concur with the iSeq study (Jaffe et al, 2017) that aneuploidies in chromosome V are the main factor contributing to the replicates with low reproducibility. Chromosome V carries both CAN1 and URA3 loci, which were replaced by selection markers in the iSeq protocol (Jaffe et al, 2017), while CAN1 was replaced by the recipient constructs in BFG‐GI. Thus, de novo structural variation around these loci during strain construction could explain the low correlation between some pairs of biological replicates. This possibility is supported by our observation that almost all BFG‐GI strains showing GIS r < 0.5 were recipients, whereas donors—for which constructs are carried on plasmids—showed GIS r > 0.5. In the BFG‐GI protocol, once the donor and recipient barcodes are fused, the “relic” donor plasmid is counter‐selected with 5‐FOA to reduce the chance of undesired recombination events. We concur with Jaffe et al (2017) who suggest that future protocols using constructs located on plasmids, such as the one we used with the proDonor strains, or at other chromosomal loci could eliminate this issue. Despite this issue, the BFG‐GI method proved to be highly accurate when compared with previous benchmark studies.
Although this study focused on a relatively small matrix (34 × 38 genes), we elaborated on previous studies to optimize the two main bottlenecks of pooled cultures: mating (Soellick & Uhrig, 2001) and sporulation (Codon et al, 1995). We calculated that to cover a yeast genome‐scale matrix of 5,500 × 5,500 genes, with 1,000 representative cells for each cross, we would need ~3 × 1010 cells at each step along the BFG‐GI procedure. Furthermore, using the optimal conditions that we established for mating (22%) and sporulation (18%), an experiment covering all 5,500 × 5,500 crosses would need to culture pools in ~27 Bioassay 500‐cm2 dishes for mating and ~10 l of liquid media for sporulation. Thus, in principle, BFG‐GI could be extended to genome‐scale studies.
BFG‐GI is a flexible technique that can be used in the future to identify genetic interactions in many different settings. Generation of BFG‐GI proDonor and proRecipient strains is one of the most time‐consuming steps in our pipeline because it includes sequence verification of both loxP/lox2272 sites and barcodes. However, once generated, these proDonor and proRecipient “toolkits” can be used many times to create donor and recipient strains representing different genes with minimal robotic manipulation. We anticipate that BFG‐GI will be a valuable technology to map condition‐dependent genetic interactions in yeast and, as next‐generation sequencing costs continue to decrease, BFG‐GI can be expanded to interrogate pools of double mutants representing bigger sets of gene pairs, including full genome combinations, across multiple conditions.
Materials and Methods
Selected DNA repair and neutral gene strains
We retrieved strains representing 26 DNA repair genes whose null mutants were sensitive to MMS (St Onge et al, 2007) from the YKO and SGA query collections. Additionally, 14 other deemed‐neutral loci were selected, based on lack of evidence that their null mutations affected cell fitness (Table EV2). These 14 loci have few or no genetic interactions in genome‐scale screens (Costanzo et al, 2010), and we did not find growth defects upon deletion of any of them.
BFG‐GI toolkit strains
Donor toolkit construction
We constructed 60 donor strains by generating two DNA fragments with overlapping ends. These were co‐transformed into yeast where they recombined to generate pDonor constructs (Fig EV1). The first fragment, called preD1, contained the hygromycin resistance gene (HygR) driven by the Schizosaccharomyces pombe TDH1 promoter and terminator, a barcode locus bearing a 20‐bp unique barcode flanked by loxP/2272 sites, and flanking primer sites. First, we used Gibson assembly (Gibson, 2009) to produce plasmid pFR0032 with the P spTDH1 ‐HygR‐T spTDH1 backbone. Then, we used three consecutive PCRs to add barcodes, priming sites, loxP/2272 loci, and in‐yeast recombination adapters (Fig EV1A). The second fragment, preD2, contained the URA3 marker and Cre recombinase driven by P tetO‐CMV. We generated this fragment by Gibson assembly of pFR0026, followed by a PCR to add in‐yeast recombination adapters (Fig EV1B). Then, preD1 and preD2 fragments were co‐transformed into yeast strain RY0771 (derived from BY4742) and merged by in‐yeast assembly to generate pDonor plasmids (Fig EV1C). We arrayed transformant strains to extract DNA and sequenced the preD1 loci, and proceeded with those strains containing confirmed preD1 loci. We mated selected MATalpha proDonors with MAT a deletion strains of interest (i.e., DNA repair or neutral genes) from the YKO collection (Fig EV1D). A series of selective passages (Figs EV1D and EV3) resulted in Donor strains with the relevant genotype:
MATalpha lyp1Δ::P STE3 ‐LEU2 his3Δ1 leu2Δ0 met17Δ0 ura3Δ0 yfg1Δ::kanMX pDonor(P tetO‐CMV ‐Cre lox2272 P TDH1 ‐HygR‐T TDH1 barcode loxP P URA3 ‐URA3 CEN/ARS P AmpR ‐AmpR ori).
Recipient toolkit construction
We constructed 56 recipient strains using a method based on the previously described delitto perfetto construct (Storici & Resnick, 2006) to enhance homologous recombination of constructs as follows. First, we used consecutive PCRs to produce a fragment preR1, containing the Kluyveromyces lactis URA3 gene, flanked by loxP/2272 sites, 20‐bp unique barcodes, and a sequence complementary to the S. cerevisiae CAN1 locus (Fig EV2A). Second, we incorporated the P STE2 ‐spHis5‐T STE2 into the CAN1 locus of the strain BY4741. Then, the delitto perfetto construct was inserted upstream of the MAT a selection reporter of the same strain (Fig EV2B) to enhance homologous recombination of preR1 fragments. This generated a pool of RY0766 proRecipient strains (Fig EV2C). We isolated and arrayed monoclonal proRecipient strains and then sequenced and selected strains with intact preR1 loci. Selected MAT a proRecipients were mated with MATalpha strains of the SGA query collection representing DNA repair and neutral genes (Fig EV1D). A series of selective passages (Figs EV2D and EV3) resulted in recipient strains with the relevant genotype:
MAT a his3Δ1 leu2Δ0 met17Δ0 lyp1Δ ura3Δ0 can1Δ::barcode loxP klURA3 lox2272 P STE2 ‐spHis5‐T STE2 P CMV ‐rtTA I‐SceI P GAL1 ‐ISceI yfg2::natMX
Generation of BFG‐GI double mutants
We took several steps to reduce the chance of undesired strains in BFG‐GI from taking over pooled cultures. This included optimization of both mating and sporulation, and adapting protocols and molecular constructs that have been reported to improve the selection of the MAT a double‐mutant progeny in SGA. Mating and sporulation are the two primary population bottlenecks when generating haploid double mutants by meiotic segregations. As described below, we sought to optimize cultures at these stages to maintain a pool complexity which was large enough to interrogate all desired gene–gene combinations. Optimizing these two processes is also important to reduce potential jackpot effects in pool cultures (i.e., to avoid strains with genetic anomalies to take over the entire pool growth).
Mating optimization for en masse BFG‐GI
We focused on optimization of cell density for en masse party mating because previous evidence shows cell density influences mating efficiency (Soellick & Uhrig, 2001). We determined the optimal cell density for en masse party mating by inoculating mating Petri dishes with a mixture of two neutral strains (MATalpha Donor hoΔ:: kanMX, and MAT a Recipient ylr179cΔ::natMX) at cell densities varying from 3 × 108 to 3 × 109 per dish. After generating mating mixtures, we took samples at 0 and 12 h of incubation at 23°C, and inoculated plates with either non‐selective or heterozygous diploid double‐mutant selective media and counted colony‐forming units (CFUs). The ratio of CFUs in non‐selective versus selective media indicated that inoculating a 58‐cm2 Petri dish with 3 × 108 cells of mating mixture resulted in 22% mating efficiency. In contrast, 1 × 109 cells of mating mixture resulted in 13% mating efficiency, and 3 × 109 cells of mating mixture resulted in 3% mating efficiency. Hence, we used 5.1 × 106 cells of mating mixture per cm2 of plate for further en masse party matings.
To generate pools of double mutants, we arrayed BFG‐GI donors and recipients in their respective selective media and cultured at 30°C for 48 h (Fig EV3). We made one pool for each mating type by scraping cells from plates into liquid media and normalized cell densities with 1 M sorbitol to have equal number of cells per strain (5 × 108 cells per ml) for each pool. Then, we lightly sonicated cells to disrupt clumps (Branson microtip sonicator, 10% duty cycle, output 2, 25 bursts, pause of 3 s, and a second 25 burst). We mixed the two pools together by stirring them in a flask for 10 min. Finally, we inoculated two Bioassay dishes (500 cm2) with 2.59 × 109 cells each of the mating mixture, and mating cultures were incubated for 12 h at 23°C (Fig EV3).
Generation of heterozygous diploid double mutants, induction of barcode fusion, and pDonor elimination
Generation of heterozygous diploid double mutants required passaging the mating progeny every 24 h into fresh selective media. Passages included selection of heterozygous diploid double mutants, induction of the Cre/Lox system with doxycycline, counter‐selection of the relic pDonor with 5‐FOA, and recovery from 5‐FOA counter‐selection to increase sporulation efficiency (Fig EV3).
Sporulation optimization for en masse BFG‐GI
We used cultures recovered from 5‐FOA counter‐selection to inoculate liquid PRE5 pre‐sporulation media (Codon et al, 1995) for 2 h at 30°C to induce exponential growth, then spun down the cells, and transferred them to SPO2 sporulation media (Codon et al, 1995) supplemented with histidine, leucine, methionine, and uracil to mask BFG‐GI strain auxotrophies at concentrations used in the SGA sporulation protocol (Tong & Boone, 2005). We incubated sporulation cultures at 21°C for 12 days. This resulted in ~18% sporulation efficiency, as evaluated by counting CFUs in non‐selective and selective media and tetrad visualization. Shorter incubation periods reduced the sporulation efficiency (~4% at 5 days, ~13% at 7 days).
Selection of MATa haploid double mutants with fused barcodes
We selected MAT a haploid progeny from sporulation cultures, followed by haploid double‐mutant selection (Fig EV3). Aliquots were stored in glycerol at −80 degrees for future use. We used the STE2 and STE3 promoters currently used for SGA to select for haploid cells, as markers with these promoters have been reported to perform better than earlier alternatives (e.g., MFA1/MFA2 promoters) (Tong & Boone, 2007). We used these constructs to first select the MAT a progeny from sporulation cultures and then the haploid double mutants. Using STE2/STE3 promoters, optimizing mating and sporulation, and using an intermediate MAT a selection step between sporulation and haploid double‐mutant selection together likely reduced the number of mitotic crossover survivors and jackpot mutation effects in our pools.
Exposure of pooled cultures to drugs
Before challenging haploid double‐mutant pools to drugs, we identified the appropriate drug concentration for our experiment by exposing a neutral BFG‐GI haploid double mutant (hoΔ::kanMX/ylr179cΔ::natMX) in growth assay liquid cultures to various drug concentrations. We selected drug doses corresponding to 20% of the minimal inhibitory concentration for the neutral test strain (Table EV1). To expose mutant strains to drugs, we thawed frozen haploid double‐mutant pools, allowed the pools to recover for 2 h in haploid double‐mutant liquid media at 30°C, and then used 1 × 109 cells of this culture to inoculate 143‐cm2 Petri dishes containing solid media supplemented with each DNA repair drug. We cultured pools at 30°C for 24 h and then collected samples to sequence fused barcodes and thus infer the abundance of each double‐mutant.
Generation of BFG‐GI double mutants in an array format
Mating and selecting donor and recipient strains in an array format was similar to the pool‐based en masse party mating assay described above, but in this case, we used robotic assistance to pairwise mate each donor with an array of recipients. We completed all steps, including sporulation, on solid media, and imaged the final haploid double‐mutant selection plates. We scraped cells from the final selection plates to sequence the fused‐barcode population which allowed us to compare cell patch sizes with numbers of sequencing reads.
Next‐generation sequencing and mapping of fused barcode pairs
The BFG‐GI technology relies on the Cre/Lox system to recombine the complementary donor and recipient loxP/lox2272 sites that serve to introduce the donor barcode adjacent to the recipient barcode (Fig 1). We multiplex‐sequenced the fused barcodes from pools of cells using the following steps: (i) genomic DNA extraction using glass beads and phenol/chloroform; (ii) PCR amplification of the 325‐bp barcode fusion product including the two 20‐bp barcodes and the multiplexing sequencing adapters (one index for each condition, for each technical replicate); (iii) concentration and gel purification of amplicons using 2% E‐Gel EX agarose 2% (Invitrogen), DNA Clean & Concentrator Kit (Zymo Research), and MinElute Gel Extraction Kit 50 (Qiagen); (iv) normalization of DNA libraries using Qubit Fluorometric Quantitation (Invitrogen); (v) mix of libraries at equal concentrations; (vi) quantification of the pooled DNA library mix by qPCR; and (vii) sequencing by Illumina 75‐cycle NextSeq paired‐end technology, including 25 cycles for each barcode and 6 cycles for the multiplex index. We mapped sequencing *.fastq files against the library of expected barcode sequences using the program Segemehl (v0.1.7, ‐A 85) and custom scripts; 97% of all sequencing reads mapped to expected barcodes.
Whole‐genome sequencing and detection of chromosome duplications
Ten strain pairs with one strain with GIS r < 0.5 and another with GIS r > 0.5 with other replicates for the same gene were selected for genome sequencing. Genomic DNA from 20 strains was extracted via cell wall disruption with Zymolyase 100T 10 mg/ml (Amsbio) and purification using AMPure beads (Agilent). gDNA was quantified with Quant‐iT PicoGreen dsDNA assay kit (Invitrogen) and normalized to 2 ng/μl for DNA fragmentation and library normalization with a Nextera XT DNA Library Prep Kit, using a transposase (Tn5) for tagmentation. A limited‐cycle PCR was used to add Illumina sequencing adapters and indices i5 and i7. PCR amplicons with size between 400 and 800 bp were gel‐purified using a 2% E‐Gel EX agarose 2% (Invitrogen) and MinElute Gel Extraction kit (Qiagen). Whole‐genome sequencing was conducted on an Illumina NextSeq 500 using a HighOutput 150 cycles v2 kit with 40× coverage. Sequencing results were mapped against the reference genome UCSC sacCer3 (SGD vR64.1.1), corrected for GC content, and chromosomal duplications detected with the HMMcopy R package (Ha et al, 2012).
Retesting double‐mutant construction and spot dilution assays
We generated double‐mutant strains for retesting in spot dilution assays by mating single‐mutant MATalpha SGA queries with MAT a YKO collection strains, the exceptions being the MAT a RAD53 (MBS1437) and rad53‐R605A (MBS1440) strains with the RAD53 loci linked C‐terminally to a 6xHis‐3xFLAG‐kanMX6 tag and resistance marker (Ohouo et al, 2013). Next, we induced sporulation of heterozygous diploid double mutants as we did for BFG‐GI strains. To confirm segregation of kanMX and natMX markers, we manually dissected haploid double mutants from tetrads and verified segregation using both selective media and PCR. Sanger sequencing confirmed the proper identity of residue 605 in intact RAD53 and rad53‐R605A strains. We grew strains overnight to saturation in liquid media, diluted them 1:10, and then used 1:5 serial dilutions for the spot assays. All cultures used YPD media supplemented with indicated drug concentrations.
Defining a genetic interaction score (GIS)
In an exponential growth model, the frequency of a double‐mutant strain s xy in a given condition at a time t () represents its total growth from an initial number as a proportion of the total growth of all other strains in the pool:
Note: Before calculating frequency, we add a pseudocount of 0.5 to the count of every strain in our analysis to avoid a zero denominator in several calculations.
Here, g xy is inversely related to the doubling time of strain s xy and g xy t effectively represents the number of doublings of strain s xy. Units for t can be chosen arbitrarily. In this model, a frequency at t = 0 evaluates as:
To remove the unknown term, we define :
We note that the term is the ratio between the initial and final number of cells in the pool and can be calculated by the total number of generations of pool growth (gen pool):
Therefore, g xy t can be calculated as:
To calculate g wt t, we take the mean g xy t of all neutral‐neutral pairs:
We then obtain the relative growth rate w xy of each strain compared to the wild type by dividing their number of doublings. In a constant exponential growth model, this metric is independent of time. In practice, g represents the average growth rate over the measured time period.
To estimate the single‐mutant fitness w x and w y for a given pair, we use the mean estimate of x or y combined with neutral genes.
We then define the genetic interaction score (GIS) as the difference between w xy and the product of w x with w y:
Because there is uncertainty in w, it is possible to calculate w < 0 for w x, w y, or w xy. Such values are assigned as 0 when performing the GIS calculation.
Normalizing genetic interactions and calculating P‐values
To assign a threshold for positive and negative genetic interactions, several additional steps are performed. GIS xy is converted to a standard score by calculating how many standard deviations GIS xy is from 0 given an estimate of GIS xy uncertainty ().
To calculate , we identify various sources of uncertainty. Another way to state GIS xy is as such:
We then define an error model to calculate the standard error σ for each term used in this calculation:
: This is estimated globally for each condition as the median difference between w xy t between the R1 and R2 technical replicates for all strains. We note that this error model only captures the general expected error between two separate runs of the same biological sample.
, , : Each of these g values is calculated by taking the mean of multiple strains. We use the variation of growth estimates in these strains (i.e., the standard deviation) as the uncertainty.
The delta method for approximating the propagation of measurement uncertainty is used to combine , , and into . This formula is also used for obtaining the other error estimates reported (i.e., , , ).
To assign a P‐value for each interaction, we then analyze the distribution of Z GIS in all unlinked neutral‐neutral and neutral‐DNA damage pairs (hereafter called “neutral pairs”), as few or no genetic interactions are expected to take place in this space. We model as a normal distribution (Fig EV4D shows the empirical and fitted normal distribution for each condition to validate this decision) and use the pnorm function in R to calculate and for each pair. We then combine these single‐tailed tests into a two‐tailed value:
p neutral represents the probability that a score as extreme as or more would be found amongst neutral pairs.
Combining multiple biological replicates and calculating a FDR
We consolidated multiple measurements of w x , w y , w xy , GIS xy, as well as , , , and p neutral from multiple barcode pairs into a single value for each gene pair. GIS xy values were weighted by the inverse of estimated squared error () and averaged to obtain . Similarly, w x , w y , w xy were averaged by the same weight (w) to obtain their corresponding gene‐wise value. , , , were obtained using the propagation of uncertainty when calculating a weighted average:
was calculated using and :
Finally, a gene‐wise p neutral was calculated using Stouffer's method weighted by w. The gene‐wise p neutral values were then converted to FDR neutral using the qvalue function in the qvalue R package.
Calling differential genetic interactions
For each gene pair, we calculated ∆GIS and ∆Z for all pairwise comparisons (a–b) amongst the tested conditions. was calculated as , and was calculated as:
For each pair of conditions, ∆Z was calculated for all unlinked neutral‐neutral and neutral‐DNA damage pairs (“neutral pairs”) to create a null distribution for ∆Z neutral. was then calculated for each pair from the ∆Z neutral distribution in the same manner as calculating p neutral. values were then converted to using the qvalue function in the qvalue R package.
Data availability
Raw and normalized sequencing measurements and GIS for each gene pair are available in Tables EV2, EV3, EV4 EV5 and EV6, and Code EV1, written in R (R Core Team, 2017), allows to generate Tables EV3, EV4 EV5 and EV6 from Table EV2. Any modifications post‐publication will have been documented at https://github.com/a3cel2/BFG_GI_stats.
Author contributions
FPR, JJD‐M, JCM, and ACo (Atina Coté) conceived the project; ACo, AK, and SO constructed pilot strains. ACo, PB, CW, and JR constructed final BFG‐GI strains and performed pilot mating experiments. JJDM, FS, YZ, DAP, and GG optimized mating, marker selection, and sporulation protocols. JJD‐M, FS, ACo, MG, and MV performed array and pool cultures and sequencing of fused barcodes. JJD‐M and JCM performed computational mapping of barcodes. JJD‐M and ACe (Albi Celaj) performed scoring of genetic interactions. JJD‐M and MG performed aneuploidy experiments, and JW analyzed the results. JJD‐M, AB, and BH performed Shu complex‐related experiments. DD and GWB provided advice on DNA repair pathways. JJD‐M, FPR, GWB, AB, and ACe wrote the manuscript. FPR supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Code EV1
Review Process File
Source Data for Figure 4
Acknowledgements
We are grateful for helpful comments from Yong Lu, Michael Principato, Ramamurthy Mani, and Meng Xiao He at the outset of this project, to Brenda Andrews and Charles Boone and members of their laboratories for providing reagents and insightful comments, and to many Roth Lab members for support and feedback throughout this project. We gratefully acknowledge support by the Canadian Excellence Research Chairs (CERC) Program (to FPR), Canadian Institutes of Health Research (MOP‐79368 to GWB, and FDN143343 to DD), National Human Genome Research Institute of the National Institutes of Health (NIH/NHGRI) HG004756, and by an individual NRSA award (HG004825) to JCM. FPR was also supported by a NIH/NHGRI Center of Excellence in Genomic Science (HG004233), by NIH/NHGRI Grant HG001715, and by the One Brave Idea Foundation.
Mol Syst Biol. (2018) 14: e7985
References
- Balint A, Kim T, Gallo D, Cussiol JR, Bastos de Oliveira FM, Yimit A, Ou J, Nakato R, Gurevich A, Shirahige K, Smolka MB, Zhang Z, Brown GW (2015) Assembly of Slx4 signaling complexes behind DNA replication forks. EMBO J 34: 2182–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LG, Zhang K, Cobb JA, Boone C, Xiao W (2009) The yeast Shu complex couples error‐free post‐replication repair to homologous recombination. Mol Microbiol 73: 89–102 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Mehta M, Kuo D, Sung M‐K, Chuang R, Jaehnig EJ, Bodenmiller B, Licon K, Copeland W, Shales M, Fiedler D, Dutkowski J, Guénolé A, van Attikum H, Shokat KM, Kolodner RD, Huh W‐K, Aebersold R, Keogh M‐C, Krogan NJ et al (2010) Rewiring of genetic networks in response to DNA damage. Science 330: 1385–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Kim Y, Ding H, Koh J, Toufighi K, Youn J‐Y, Ou J, San Luis B‐J, Bandyopadhyay S, Hibbs M, Hess D, Gingras A‐C, Bader GD, Troyanskaya OG, Brown GW, Andrews B, Boone C, Myers CL (2010) Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods 7: 1017–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdal KG, Bjørås M, Bjelland S, Seeberg E (1990) Cloning and expression in Escherichia coli of a gene for an alkylbase DNA glycosylase from Saccharomyces cerevisiae; a homologue to the bacterial alkA gene. EMBO J 9: 4563–4568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein KA, Reid RJD, Sunjevaric I, Demuth K, Burgess RC, Rothstein R (2011) The Shu complex, which contains Rad51 paralogues, promotes DNA repair through inhibition of the Srs2 anti‐recombinase. Mol Biol Cell 22: 1599–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Derfler B, Samson L (1990) Saccharomyces cerevisiae 3‐methyladenine DNA glycosylase has homology to the AlkA glycosylase of E. coli and is induced in response to DNA alkylation damage. EMBO J 9: 4569–4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, Batke S, Szakal B, Lowther J, Hao F, Sarangi P, Branzei D, Ulrich HD, Zhao X (2015) Concerted and differential actions of two enzymatic domains underlie Rad5 contributions to DNA damage tolerance. Nucleic Acids Res 43: 2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claussen CA, Long EC (1999) Nucleic acid recognition by metal complexes of bleomycin. Chem Rev 99: 2797–2816 [DOI] [PubMed] [Google Scholar]
- Codon AC, Gasent‐Ramirez JM, Benitez T (1995) Factors which affect the frequency of sporulation and tetrad formation in Saccharomyces cerevisiae Baker's yeasts. Appl Environ Microbiol 61: 1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Schuldiner M, Krogan NJ, Weissman JS (2006) A strategy for extracting and analyzing large‐scale quantitative epistatic interaction data. Genome Biol 7: R63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JLY, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M et al (2010) The genetic landscape of a cell. Science 327: 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, Pelechano V, Styles EB, Billmann M, van Leeuwen J, van Dyk N, Lin Z‐Y, Kuzmin E, Nelson J, Piotrowski JS, Srikumar T et al (2016) A global genetic interaction network maps a wiring diagram of cellular function. Science 353: aaf1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussiol JR, Jablonowski CM, Yimit A, Brown GW, Smolka MB (2015) Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. EMBO J 34: 1704–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decourty L, Saveanu C, Zemam K, Hantraye F, Frachon E, Rousselle J‐C, Fromont‐Racine M, Jacquier A (2008) Linking functionally related genes by sensitive and quantitative characterization of genetic interaction profiles. Proc Natl Acad Sci USA 105: 5821–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuna A, Vetsigian K, Shoresh N, Hegreness M, Colón‐González M, Chao S, Kishony R (2008) Exposing the fitness contribution of duplicated genes. Nat Genet 40: 676–681 [DOI] [PubMed] [Google Scholar]
- Flott S, Alabert C, Toh GW, Toth R, Sugawara N, Campbell DG, Haber JE, Pasero P, Rouse J (2007) Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single‐strand annealing mode of DNA repair in budding yeast. Mol Cell Biol 27: 6433–6445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke WM, Brill SJ (2003) Slx1‐Slx4 is a second structure‐specific endonuclease functionally redundant with Sgs1‐Top3. Genes Dev 17: 1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay E, Campos SE, González de la Cruz J, Gaspar AP, Jinich A, Deluna A (2014) High‐resolution profiling of stationary‐phase survival reveals yeast longevity factors and their genetic interactions. PLoS Genet 10: e1004168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau‐Danila A, Anderson K, André B, Arkin AP, Astromoff A, El‐Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A et al (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391 [DOI] [PubMed] [Google Scholar]
- Gibson DG (2009) Synthesis of DNA fragments in yeast by one‐step assembly of overlapping oligonucleotides. Nucleic Acids Res 37: 6984–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin S, Wier A, Kabbinavar F, Bratton‐Palmer DS, Ghodke H, Van Houten B, VanDemark AP, Bernstein KA (2013) The Shu complex interacts with Rad51 through the Rad51 paralogues Rad55‐Rad57 to mediate error‐free recombination. Nucleic Acids Res 41: 4525–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin SK, Zhang Z, Herken BW, Westmoreland JW, Lee AG, Mihalevic MJ, Yu Z, Sobol RW, Resnick MA, Bernstein KA (2016) The Shu complex promotes error‐free tolerance of alkylation‐induced base excision repair products. Nucleic Acids Res 44: 8199–8215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritenaite D, Princz LN, Szakal B, Bantele SCS, Wendeler L, Schilbach S, Habermann BH, Matos J, Lisby M, Branzei D, Pfander B (2014) A cell cycle‐regulated Slx4‐Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev 28: 1604–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, Chin S‐F, Turashvili G, Hirst M, Caldas C, Marra MA, Aparicio S, Shah SP (2012) Integrative analysis of genome‐wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple‐negative breast cancer. Genome Res 22: 1995–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie AR, Pruitt SC (2007) Yeast two‐hybrid interaction partner screening through in vivo cre‐mediated binary interaction tag generation. Nucleic Acids Res 35: e141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Piening BD, Paulovich AG (2013) The preference for error‐free or error‐prone postreplication repair in Saccharomyces cerevisiae exposed to low‐dose methyl methanesulfonate is cell cycle dependent. Mol Cell Biol 33: 1515–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Krogan NJ (2012) Differential network biology. Mol Syst Biol 8: 565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer DR, Rhind N (2017) Replication fork slowing and stalling are distinct, checkpoint‐independent consequences of replicating damaged DNA. PLoS Genet 13: e1006958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonowski CM, Cussiol JR, Oberly S, Yimit A, Balint A, Kim T, Zhang Z, Brown GW, Smolka MB (2015) Termination of replication stress signaling via concerted action of the Slx4 scaffold and the PP4 phosphatase. Genetics 201: 937–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe M, Sherlock G, Levy SF (2017) iSeq: a new double‐barcode method for detecting dynamic genetic interactions in yeast. G3 7: 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, St Onge RP, Proctor MJ, Wallace IM, Nile AH, Spagnuolo PA, Jitkova Y, Gronda M, Wu Y, Kim MK, Cheung‐Ong K, Torres NP, Spear ED, Han MK, Schlecht U, Suresh S, Duby G, Heisler LE, Surendra A, Fung E et al (2014) Mapping the cellular response to small molecules using chemogenomic fitness signatures. Science 344: 208–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen J, Pons C, Mellor JC, Yamaguchi TN, Friesen H, Koschwanez J, Ušaj MM, Pechlaner M, Takar M, Ušaj M, VanderSluis B, Andrusiak K, Bansal P, Baryshnikova A, Boone CE, Cao J, Cote A, Gebbia M, Horecka G, Horecka I et al (2016) Exploring genetic suppression interactions on a global scale. Science 354: aag0839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani R, St Onge RP, Hartman JL, Giaever G, Roth FP (2008) Defining genetic interaction. Proc Natl Acad Sci USA 105: 3461–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri HW, Ngo H‐P, Hickson ID (2007) Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1‐Rmi1‐Top3. Mol Biol Cell 18: 4062–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minca EC, Kowalski D (2011) Replication fork stalling by bulky DNA damage: localization at active origins and checkpoint modulation. Nucleic Acids Res 39: 2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB (2013) DNA‐repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 493: 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill BM, Szyjka SJ, Lis ET, Bailey AO, Yates JR, Aparicio OM, Romesberg FE (2007) Pph3‐Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc Natl Acad Sci USA 104: 9290–9295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Yuan DS, Xiang D, Wang X, Sookhai‐Mahadeo S, Bader JS, Hieter P, Spencer F, Boeke JD (2004) A robust toolkit for functional profiling of the yeast genome. Mol Cell 16: 487–496 [DOI] [PubMed] [Google Scholar]
- Pfander B, Diffley JFX (2011) Dpb11 coordinates Mec1 kinase activation with cell cycle‐regulated Rad9 recruitment. EMBO J 30: 4897–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; https://www.R-project.org/ [Google Scholar]
- Sasanuma H, Tawaramoto MS, Lao JP, Hosaka H, Sanda E, Suzuki M, Yamashita E, Hunter N, Shinohara M, Nakagawa A, Shinohara A (2013) A new protein complex promoting the assembly of Rad51 filaments. Nat Commun 4: 1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlecht U, Liu Z, Blundell JR, St Onge RP, Levy SF (2017) A scalable double‐barcode sequencing platform for characterization of dynamic protein‐protein interactions. Nat Commun 8: 15586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz EK, Wright WD, Ehmsen KT, Evans JE, Stahlberg H, Heyer WD (2012) Mus81‐Mms4 functions as a single heterodimer to cleave nicked intermediates in recombinational DNA repair. Mol Cell Biol 32: 3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soellick TR, Uhrig JF (2001) Development of an optimized interaction‐mating protocol for large‐scale yeast two‐hybrid analyses. Genome Biol 2: RESEARCH0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Onge RP, Mani R, Oh J, Proctor M, Fung E, Davis RW, Nislow C, Roth FP, Giaever G (2007) Systematic pathway analysis using high‐resolution fitness profiling of combinatorial gene deletions. Nat Genet 39: 199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Resnick MA (2006) The delitto perfetto approach to in vivo site‐directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol 409: 329–345 [DOI] [PubMed] [Google Scholar]
- Sugawara N, Wang X, Haber JE (2003) In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51‐mediated recombination. Mol Cell 12: 209 [DOI] [PubMed] [Google Scholar]
- Svensson JP, Fry RC, Wang E, Somoza LA, Samson LD (2012) Identification of novel human damage response proteins targeted through yeast orthology. PLoS ONE 7: e37368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh GW‐L, Sugawara N, Dong J, Toth R, Lee SE, Haber JE, Rouse J (2010) Mec1/Tel1‐dependent phosphorylation of Slx4 stimulates Rad1‐Rad10‐dependent cleavage of non‐homologous DNA tails. DNA Repair 9: 718–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Boone C (2005) Synthetic Genetic Array (SGA) analysis in Saccharomyces cerevisiae In Yeast protocols, Xiao W. (ed.), pp 171–192. Totowa, NJ: The Humana Press Inc; [Google Scholar]
- Tong AH, Boone C (2007) High‐throughput strain construction and systematic synthetic lethal screening in Saccharomyces cerevisiae . Methods Microbiol 36: 369–707 [Google Scholar]
- Xiao W, Chow BL (1998) Synergism between yeast nucleotide and base excision repair pathways in the protection against DNA methylation damage. Curr Genet 33: 92 [DOI] [PubMed] [Google Scholar]
- Xu X, Ball L, Chen W, Tian X, Lambrecht A, Hanna M, Xiao W (2013) The yeast Shu complex utilizes homologous recombination machinery for error‐free lesion bypass via physical interaction with a Rad51 paralogue. PLoS ONE 8: e81371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachie N, Petsalaki E, Mellor JC, Weile J, Jacob Y, Verby M, Ozturk SB, Li S, Cote AG, Mosca R, Knapp JJ, Ko M, Yu A, Gebbia M, Sahni N, Yi S, Tyagi T, Sheykhkarimli D, Roth JF, Wong C et al (2016) Pooled‐matrix protein interaction screens using barcode fusion genetics. Mol Syst Biol 12: 863 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Code EV1
Review Process File
Source Data for Figure 4
Data Availability Statement
Raw and normalized sequencing measurements and GIS for each gene pair are available in Tables EV2, EV3, EV4 EV5 and EV6, and Code EV1, written in R (R Core Team, 2017), allows to generate Tables EV3, EV4 EV5 and EV6 from Table EV2. Any modifications post‐publication will have been documented at https://github.com/a3cel2/BFG_GI_stats.