

Abstract
Morphologically unique silica nanoparticles can be used as effective templates to prepare silica–metal hybrid nanomaterials, which are highly applicable in a variety of areas. Mesoporous silica nanoparticles, which have high surface areas and an abundance of pores, can be used to synthesize mesoporous silica core–metal shell nanostructures with catalytically active sites. In this work, dendritic fibrous nanosilica (DFNS) with a high surface area is successfully employed as a template to synthesize DFNS/Au hybrid nanomaterials. Au nanodots are initially synthesized through the selective reduction of Au ions on the surface of the DFNS after surface modification to form DFNS/Au dots. A seed‐mediated growth method is used to controllably grow Au nanoparticles on the DFNS/Au dots to generate DFNS core–Au nanoparticles shell nanohybrids (DFNS/Au NPs) and DFNS core–Au layer shell nanohybrids (DFNS/Au layers). The catalytic activities of DFNS/Au NPs and DFNS/Au layers in the 4‐nitrophenol reduction reaction are compared.
Keywords: 4-nitrophenol reduction, Au layer, core–shell nanostructures, dendritic fibrous nanosilica, silica–metal hybrid nanoparticle
1. Introduction
In the fabrication of silica–metal hybrid nanoparticles, the use of pre‐synthesized silica as a template provides a variety of advantages, such as non‐toxicity, biocompatibility, chemical stability, porosity, and facile control of the size of the target materials.1 Silica is also frequently used as a support material for metallic nanocatalysts to enhance their catalytic activity and recovery.2 Morphologically unique silica nanoparticles can be available for effective templates to prepare unique silica–metal nanoparticle hybrids.3 The internal changes induced by applying selective etching processes to the silica cores can increase the size of internal spaces or pores without disturbing the basic spherical morphologies of the hybrids.1h, 1j, 4 In addition, mesoporous silica nanoparticles have also been employed as templates to prepare mesoporous silica core–metal shell nanostructures.5 Their high surface area and abundance of pores can improve the active sites in these hybrids, which ultimately enhance the catalytic activities of the hybrids compared to their metallic counterparts.
Recently, synthetic methods for the preparation of silica nanoparticles with many branches and wrinkles [dendritic fibrous nanosilica (DFNS)] have been reported.6 These silica nanoparticles show fibrous external morphologies with high surface area and great stability under high temperature and pressure.6, 7 Simple surface modification of these particles by replacing the hydroxy (−OH) groups with other functional groups can be carried out to generate efficient nanocatalysts for a variety of organic reactions.8 More importantly, the synthesis of Au, Pd, and Pt nanoparticles on the surface of DFNS can lead to the generation of unique silica–metal hybrid nanosystems.7a, 9 Obtaining hybrid materials by using DFNS as the core and growing metallic nanoparticles on the shell has clear advantages.7, 8, 9, 10 Firstly, the high surface area of DFNS can provide abundant space for the metallic nanoparticles. This enables a relatively large number of metallic nanoparticles to be efficiently deposited on a limited volume of silica. Secondly, the void spaces around the metallic nanoparticles within the final hybrids, which are able to effectively generate yolk–shell morphologies, can be used for a variety of applications, such as carriers for the delivery of other useful materials. Third, metallic nanoparticles located between the wrinkles can be protected from the formation of aggregates under some specific conditions.
Among the various DFNS/metallic hybrid nanostructures that have been reported, we are particularly interested in forming Au shells. In general, the synthetic process of forming a Au shell on a silica nanoparticle core includes a surface modification step in which the hydroxy (−OH) group of the silica particles is replaced by an amine group (−NH2) or another functional group that can interact efficiently with Au.9a, 11 The Au layer is then generated by in situ reduction of Au precursors9a, 9d, 12 or the attachment of pre‐synthesized Au nanoparticles on the silica surface.9h, 13 To control the number or size of the Au nanoparticles on the surface of the dendritic silica, it is common to change the amount of Au ion or reducing agents used.14 However, the formation of continuous Au layer on the surface of DFNS and the generation of significant space between the Au and silica particles has not been reported.
In this work, we report the synthesis of Au nanoparticles and Au layers on the surface of DFNS/Au hybrids: DFNS core–Au nanoparticles shell nanohybrids (DFNS/Au NPs) and DFNS core–Au layer shell nanohybrids (DFNS/Au layers). The structure of the Au nanoparticles and Au layers in the DFNS/Au hybrids can be effectively controlled. Different catalytic behavior is observed in the 4‐nitrophenol reduction reaction, depending on the structure of the DFNS/Au hybrids.
2. Results and Discussion
DFNS is synthesized by following procedures previously reported in the literature with some modifications.6 In particular, the current synthesis is carried out at room temperature without microwave assistance, resulting in a simpler procedure. Cetyltrimethylammonium bromide (CTAB) is used as a shape‐directing agent as well as a surfactant, which leads to the formation of multiple branches (wrinkles) on the surface of the DFNS. The synthesized DFNS are calcined to remove organic molecules and CTAB. Figure S1 shows the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of the synthesized DFNS, which clearly exhibit a spherical morphology with many branches and wrinkles. The average diameter of the synthesized DFNS is 250.59±24.78 nm (Figure S2, based on the evaluation of more than 100 particles). The Brunauer–Emmett–Teller (BET) surface area, total pore volume, and average pore size of the DFNS are 378.48 m2 g−1, 1.49 cm3 g−1, and 15.46 nm, respectively (Figure S3).
The synthetic protocol for the DFNS/Au NPs and DFNS/Au layers begins with the preparation of Au nanodots, which are used as seed particles to grow larger Au nanoparticles or Au layers. The Au nanodots preferentially form on the DFNS surface to produce DFNS/Au dots. In this process, the synthesized DFNS is washed and re‐dispersed in water, and then APTES is added to the reaction mixture. The surface of the DFNS can be modified with amine groups (−NH2), which facilitate the access of the Au ion and allow the Au nanodots to selectively grow on the silica surface through a reduction process; this selective growth via surface modification has also been confirmed in other reports (Figure S4).8, 9 Figures 1 a and 1 b are the bright‐field and dark‐field TEM images of the DFNS/Au dots, respectively, which clearly demonstrate the presence and wide distribution of the Au nanodots over the DFNS surface. The average size of the Au nanodots in the DFNS/Au dots is 2.34±0.64 nm (more than 100 nanodots are evaluated from the enlarged images, Figure S8 i). The energy‐dispersive X‐ray spectroscopy (EDS) results are shown in Figures 1 c (oxygen) and 1 d (gold), and demonstrate that the Au nanodots are completely dispersed over the whole DFNS structure.
Figure 1.

TEM images of DFNS/Au dots: a) bright‐field TEM image; b) dark‐field TEM image; c, d) EDS mapping elemental analysis showing the oxygen (c) and gold (d) content of the DFNS/Au dots.
To grow Au nanoparticles and Au layers on the surface of the DFNS, we employ a modified seed‐mediated synthetic method. Pre‐synthesized DFNS/Au dots (Figure 1) are used as seed nanoparticles, and two different growth solutions are prepared for the stepwise mixing procedure to avoid rapid growth reactions (Figure S5). The nanoparticles that are generated through the stepwise seed‐mediated growth methods are analyzed by using SEM and TEM (Figures 2, S6, and S7). By using different amounts of HAuCl4, the size of the Au nanoparticles can be effectively controlled (Figure 2). The average sizes of the Au nanoparticles in Figure 2 are a) 5.93±0.76 nm, b) 7.49±0.82 nm, c) 9.25±1.12, and d) 11.17±1.14 nm, showing a gradual increase with the increase of the amount of HAuCl4 used, that is, the ratio of HAuCl4 in the growth solution/HAuCl4 in the DFNS/Au dots (based on the evaluation of more than 100 nanoparticles, Figure S8). Importantly, as the amount of HAuCl4 is increased, the Au nanoparticles partially merge together (Figure 2 e) and finally form a continuous layer structure as a shell (Figure 2 f). Owing to the fusion process, the size of the Au nanoparticles in Figures 2 e and 2 f cannot be measured exactly, although parts of the layer still retain their spherical morphology. The powder X‐ray diffraction (PXRD) patterns (Figure S9) confirm that DFNS/Au hybrid nanomaterials comprise an amorphous SiO2 and crystalline Au phase, as indicated by diffraction peaks indexed to the fcc structure of metallic Au (JCPDS card no. 01–089–3697).
Figure 2.
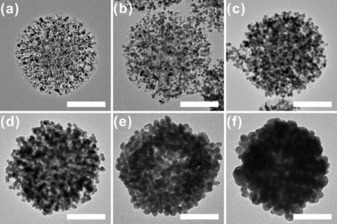
TEM images of DFNS/Au nanohybrids synthesized by using various HAuCl4(aq) concentrations (scale bar: 100 nm): a) 1.25 mm, b) 2.5 mm, c) 5 mm, d) 10 mm, e) 20 mm, and f) 30 mm HAuCl4(aq) (the same volume of the second growth solution, 0.8 mL, is used in all cases). Based on the synthesis ratio (molar ratio=amount of HAuCl4 in the growth solution/ amount of HAuCl4 in the DFNS/Au dots), the reaction ratios can be assigned as: a) 7, b) 15, c) 30, d) 61, e) 123, and f) 246.
The growth of Au nanoparticles on the DFNS surface can be followed easily by using UV/Vis spectroscopy (Figure 3). An aqueous solution of the DFNS/Au dots exhibits a resonance at around 510 nm, which is largely embedded in the broad absorption of silica over the whole region (Figure 3 i). The Au nanoparticles grown from the DFNS/Au dots clearly show extinction peaks, with maximum at 547 nm (Figure 3 ii). This is reasonable, as spherical Au nanoparticles with diameters around 10 nm generally exhibit a surface plasmon resonance (SPR) band at approximately 510–535 nm.15 A gradual increase in the intensity of the extinction peaks is observed as the amount of Au ion used in the growth reaction increases (Figures 3 iii–3 v). Interestingly, the peak maxima are largely red‐shifted in Figures 3 vi and 3 vii, which is attributed to the generation of the Au layers around the DFNS. This is mainly because of the formation of anisotropic Au materials with non‐spherical shapes (Au layers) through the fusion of the spherical Au nanoparticles on the surface of DFNS. The redshifts in the SPR of the Au nanomaterials and Au layers, which are strongly dependent on the shape of nanomaterials, have been well reported.16, 17
Figure 3.
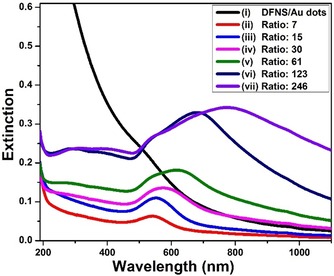
UV/Vis spectra of aqueous solutions of DFNS/Au dots and DFNS/Au nanohybrids synthesized using various HAuCl4(aq) concentrations: ii) 1.25 mm, iii) 2.5 mm, iv) 5 mm, v) 10 mm, vi) 20 mm, and vii) 30 mm HAuCl4(aq) (the same volume of the second growth solution, 0.8 mL, is used in all cases). According to the synthesis ratio (molar ratio=amount of HAuCl4 in the growth solution/ amount of HAuCl4 in the DFNS/Au dots), the reaction ratio can be assigned as: ii) 7, iii) 15, iv) 30, v) 61, vi) 123, and vii) 246.
Additional experiments are conducted to investigate the effect of the surfactants and AgNO3 in the synthesis of the DFNS/Au layers. Figure 4 shows the SEM images of the hybrids synthesized by varying the experimental conditions from the method used for the DFNS/Au layers (ratio: 246). Figure 4 a shows the DFNS/Au layers (synthesized as a control experiment), in which Au nanoparticles completely surround the DFNS surface. When AgNO3 is not used in the synthesis, the thickness of the Au layer becomes very irregular (Figure 4 b), mainly because the formation of the Au nanoparticles within the Au layer is not uniform. The presence of Ag+ critically affects the development of Au nanoparticles on the DFNS surface; such Ag+‐mediated behavior is often observed in the synthesis of other Au nanomaterials.18 When the Brij 35 surfactant is omitted from the synthesis, a smaller number of Au nanoparticles is formed on the DFNS surface compared to the conventional method (Figure 4 c). When the synthesis of DFNS/Au layers is attempted without CTAB, the growth of other Au materials separated from the DFNS surface is observed. The changes in the optical properties caused by the various synthesis conditions can be confirmed by UV/Vis spectroscopy measurements (Figure S10). Importantly, when either AgNO3 or Brij 35 are not used, only the SPR signals at around 520–530 nm, originating from the growth of spherical Au nanoparticles, are observed. Under these experimental conditions, the fusion process of Au nanoparticles to form an Au layer on the DFNS surface may not be favored. Based on the microscopic images and spectral results, it is believed that the combined use of CTAB and Brij 35, along with the use of AgNO3, is critical for the growth of the DFNS/Au layers.
Figure 4.

SEM images of DFNS/Au nanohybrids synthesized by using various reaction conditions (scale bar: 250 nm): DFNS/Au hybrids synthesized a) using the original synthesis method (ratio: 246), b) without AgNO3 (aq), c) without Brij 35, and d) without CTAB. The HAuCl4 concentration is 30 mm in each case, and other experimental conditions are kept the same.
During the growth of DFNS/Au hybrids, DFNS acts as a template for the generation of the Au layer. To investigate whether the morphological features of DFNS play a pivotal role in the formation of DFNS/Au hybrids, spherical silica nanoparticles with non‐branched (non‐wrinkled) surfaces are synthesized separately through the Stöber method (spherical nanosilica, SNS).19 The SNS are synthesized to have an average particle size (258.12±15.43 nm, based on the evaluation of more than 100 nanoparticles, Figure S11) similar to the mean particle size of the DFNS [250.59±24.78 nm (Figure S1)] to eliminate the effect of particle size (Figure 5 a). The much lower BET surface area of the SNS (11.55 m2 g−1) compared to that of the DFNS (378.48 m2 g−1) indicates that there are almost no pores or wrinkles in the particles (Figure S12). In the growth of Au nanoparticles on the surface of the SNS, all other experimental conditions are kept the same as in the conventional DFNS/Au hybrids synthesis. Figure 5 b shows the SNS after the growth of Au nanodots on their surfaces (SNS/Au dots); these nanodots then act as seeds for the further growth of Au nanoparticles. Figures 5 c–5 h show the SEM images of Au nanoparticles grown by using SNS (SNS/Au NPs). The images clearly demonstrate that the Au nanoparticles grown on the SNS are not spherical, but have spiky morphologies with sharp tips and edges. This is mainly attributed to the preferred radial growth of one‐dimensional (spiky) Au materials from the seed Au nanodots on the spherical silica surface, in the presence of mixed surfactants (CTAB and Brij 35).20 The SPR peak of spherical Au nanoparticles is also absent in the UV/Vis spectroscopy measurements (Figure S13). More importantly, the Au nanoparticles are not evenly distributed over the surface of the SNS, and instead grow independently from the silica; therefore, no formation of a Au layer is observed. In the case of DFNS, Au nanodots can be broadly located between the wrinkles and protected from the external conditions. Growth of Au from DFNS/Au dots occurs more evenly on the silica surface than that of SNS/Au dots. These results show that the structural properties of the DFNS, which serve as a template, are a key factor for the growth of DFNS/Au layers.
Figure 5.

SEM images of SNS/Au nanohybrids synthesized using various HAuCl4(aq) concentrations (scale bar: 500 nm): a) SNS, b) SNS/Au dots, and c–h) SNS/Au NPs synthesized using c) 1.25 mm, d) 2.5 mm, e) 5 mm, f) 10 mm, g) 20 mm, and h) 30 mm HAuCl4(aq). The same volume of the second growth solution, 0.8 mL, was used in all cases.
The secondary growth of metallic materials on silica surfaces using similar porous silica nanoparticles as templates has resulted in excellent catalytic activity in a variety of reactions, owing to the high surface area and easy access of the reactants to the metals via the pores.21 The catalytic activity of the DFNS/Au hybrid nanomaterials in the 4‐nitrophenol (4‐NP) reduction reaction is also tested.14c, 22 In general, as the reduction reaction proceeds, the absorbance of 4‐NP peak at 400 nm gradually decreases, and the absorption peak at 295 nm corresponding to the formation of 4‐aminophenol increases. In this process, the synthesized Au nanoparticles can be used as nanocatalysts in the presence of NaBH4. After the addition of dispersions of DFNS/Au hybrids in aqueous solution, the changes in the absorption peaks are observed at 5 min intervals by using UV/Vis spectroscopy to measure the catalytic performance of each nanocatalyst (Figure S14). The conversion of 4‐NP based on the peaks at 400 nm is recorded by the change of nanocatalysts (Figure 6 and Table S1). It is clear that the nanomaterials with more Au on the DFNS surface show higher catalytic activities. However, after the formation of the Au layer by fusion of the Au NPs, the catalytic activities of the DFNS/Au layers are not greatly affected by further increases in the amount of Au ion used. The rate constants, which are calculated based on the slope of the linear fit of −ln(C t C 0 −1) versus time, are shown in Figure S15 and Table S2 (where C t is the absorption of 4‐NP at a given reaction time, t, and C 0 is the initial absorption of 4‐NP at t=0). The calculated rate constant for 4‐NP reduction by the DFNS/Au layers is around 160×10−3 min−1, which is higher than those determined for the DFNS/Au NPs (23.8, 40.3, and 125.3×10−3 min−1 for the nanoparticles shown in Figures 2 a–2 c, respectively).
Figure 6.
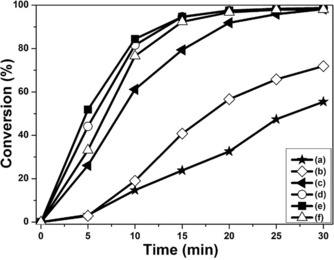
4‐NP conversion by a) DFNS/Au dots, (b to f) DFNS/Au nanohybrids synthesized: b) ratio: 7, c) ratio: 15, d) ratio: 30, d) ratio: 61, e) ratio: 123, and f) ratio: 246. 4‐NP conversion is calculated as follows: Conversion (%)= (1−C t C 0 −1)×100, where C t is the absorption of 4‐NP at a given reaction time, t, and C 0 is the initial absorption by 4‐NP at t=0.
3. Conclusions
In summary, dendritic fibrous nanosilica (DFNS) with a high surface area is synthesized and successfully employed as a template to fabricate DFNS/Au hybrid nanomaterials. DFNS/Au dots are initially synthesized by the selective reduction of Au ions on the surface after surface modification of the DFNS with amine group. A seed‐mediated growth method is used to grow Au nanoparticles on the DFNS/Au dots to generate DFNS core–Au nanoparticles shell nanohybrids (DFNS/Au NPs). The size of the Au nanoparticles of the DFNS/Au NPs is readily controlled by increasing the amount of Au ion added to the synthesis. Above a certain concentration of Au ion, DFNS core–Au layer shell nanohybrids (DFNS/Au layers) are synthesized by the fusion of the Au NPs on the surface. The catalytic activity of the DFNS/Au hybrids in 4‐nitrophenol reduction reaction are compared, and the activity is found to increase as the amount of Au ion increases, with DFNS/Au layers showing the best catalytic activity.
Experimental Section
Reagents
Cetyltrimethylammonium bromide [CTAB, CH3(CH2)15 N(CH3)3Br, 99+%, Acros organic], polyoxyethylene glycol dodecyl ether [(C2H4O)23C12H25OH, Brij 35, Acros Organics], silver nitrate (AgNO3, 99+%, Sigma–Aldrich), sodium borohydride (NaBH4, 99.99 %, Sigma–Aldrich), tetraethylorthosilicate (TEOS, 99 %, Sigma–Aldrich), polyvinylpyrolidone [(C6H9NO)n, PVP10, average molecular weight 10 000, Sigma–Aldrich], 3‐(aminopropyl)triethoxysilane (APTES, 98 %, Sigma–Aldrich), hydrogen tetrachloroaurate trihydrate (HAuCl4⋅3 H2O, 99.9 %, Sigma–Aldrich), cyclohexane (C6H12, 99 %, Sigma–Aldrich), l‐ascorbic acid (C6H8O6, 99+%, Sigma–Aldrich), urea [CO(NH2)2, 98 %, Sigma–Aldrich], ammonium hydroxide (NH4OH, 28–30 wt % ammonia, Sigma–Aldrich), 1‐penthanol [CH3(CH2)3CH2OH, 98 %, Alfa Aesar], cyclohexane (C6H12, 99 %, Sigma–Aldrich), HCl, HNO3, acetone, and ethyl alcohol are used as received. All stock solutions are freshly prepared before each reaction. Prior to use, all glassware was washed with Aqua Regia (3:1 ratio by volume of HCl and HNO3; Caution: Aqua Regia is highly toxic and corrosive and must be handled in a fume hood with proper personal protection equipment) and rinsed thoroughly with nanopure water.
Synthesis of DFNS
DFNS is prepared following a reported protocol with a slight modification.6 Typically, 2.5 g of TEOS (0.012 mol) is dissolved in a mixture of cyclohexane (30 mL) and 1‐penthanol (1.5 mL). The TEOS precursor solution is added dropwise into an aqueous solution (30 mL) of CTAB (1.0 g, 0.0027 mol) and urea (0.6 g, 0.01 mol). The reaction mixture is vigorously stirred for 2 h at room temperature before being transferred into a Teflon‐lined stainless‐steel autoclave and kept in a temperature‐controlled oven at 120 °C for 6 h. After naturally cooling to room temperature, the resultant white solids are collected by centrifugation at 3500 rpm for 5 min, washed several times with solvents (acetone and nanopure water), and dried at 60 °C for 24 h. Finally, the collected powder is calcined at 550 °C for 6 h to obtain DFNS.
Synthesis of DFNS Core–Au Nanodots (DFNS/Au dots)
The as‐synthesized DFNS is initially functionalized with amine groups using APTES as follows: 30 mg of DFNS is dispersed in 1 mL of nanopure water, to which 0.03 mL APTES and 0.1 mL NH4OH (aq) (28 %) are sequentially added. The reaction mixture is stirred for 12 h at room temperature. Afterwards, the solid powder (regarded as amine‐functionalized DFNS) is collected by centrifugation (13 500 rpm, 5 min) and thoroughly rinsed with nanopure water to remove unreacted APTES. The amine‐functionalized DFNS is re‐dispersed in nanopure water for further use.
For the synthesis of DFNS/Au dots, an aqueous suspension of amine‐functionalized DFNS (1 mL, 30 mg mL−1) is added into a solution of 0.3 mL of HAuCl4 (aq) (7.5 μmol, 0.025 m), and the reaction mixture is stirred for 30 min at room temperature. Afterward, the product is centrifuged (13500 rpm, 5 min) and re‐dispersed in 1 mL of nanopure water. Next, 0.5 mL NaBH4 (aq) (in ice bath) (0.05 mmol, 0.1 m) is added to the mixture. The reaction mixture is further stirred for 12 h at room temperature. Upon the completion of the reaction, the resultant DFNS/Au dots nanoparticles are centrifuged and purified by three repeated cycles of centrifugation (13 500 rpm, 5 min) and re‐dispersed in nanopure water for the further use.
Synthesis of DFNS core–Au (Nanoparticles or Layer) Shell Nnaohybrids (DFNS/Au NPs or DFNS/Au Layers)
DFNS/Au NPs are synthesized by the growth of Au onto the as‐prepared DFNS/Au dots seed through a seed‐mediated method. To grow the DFNS/Au NPs, two growth solutions are prepared [solution (A): a mixture of Brij 35 (aq) (1 mL, 200 mm), CTAB (aq) (1 mL, 100 mm), nanopure water (0.2 mL), HAuCl4 (aq) (80 μL, 10 mm), AgNO3 (aq) (10 μL, 10 mm), and l‐ascorbic acid (aq) (30 μL, 100 mm); solution (B): a mixture of Brij 35 (aq) (10 mL, 200 mm), CTAB (aq) (10 mL, 100 mm), distilled water (2 mL), HAuCl4 (aq) (800 μL, 10 mm), AgNO3 (aq) (300 μL, 10 mm) and ascorbic acid (aq) (300 μL, 100 mm)]. The formation of DFNS/Au NPs are initiated by adding the DFNS/Au dots seed solution (200 μL) to solution (A). Next, 200 μL of this mixture is quickly added to solution (B). The resultant reaction mixture is then shaken vigorously for more than 1 min, followed by aging for 14 h. Upon completion of the reaction, the resultant DFNS/Au NPs are collected and purified by three repeated cycles of centrifugation (13 500 rpm, 5 min) and re‐dispersed in nanopure water. The formation of DFNS/Au hybrids are controlled by varying the concentration of HAuCl4 (aq) used (1.25 mm, ratio: 7; 2.5 mm, ratio: 15; 5 mm, ratio: 30; 10 mm, ratio: 61; 20 mm, ratio: 123; and 30 mm, ratio: 246) [the ratios are calculated by the comparison of mole number of HAuCl4 used in the growth of Au NPs to Au dots (DFNS/Au dots)]. With higher amount of HAuCl4 (aq), the thickness of gold layers can be manipulated.
Synthesis of SNS and SNS Core–Au Shell Nanoparticles (SNS/Au NPs)
SNS, which is not morphologically porous or branched (wrinkled), is synthesized by using the Stöber method.19 Typically, 2.5 g TEOS (0.012 mol) and 4.59 mL NH4OH (aq) (28 %) are added to the mixed solvent (61 mL ethanol and 4.34 mL nanopure water). After being sonicated for 30 min, the reaction mixture is stirred for 12 h at room temperature. Upon completion of the reaction, the obtained product is collected by centrifugation (4 000 rpm, 10 min), washed several times with ethanol, nanopure water, and then air‐dried for 24 h at 60 °C. The resulting sample is calcined at 550 °C for 6 h, and then collected for the further use.
In the growth of Au nanoparticles on the surface of the SNS (SNS/Au NPs), all other experimental conditions are kept the same as in the conventional DFNS/Au hybrids synthesis. The SNS core–Au shell nanodots (SNS/Au dots) are prepared first, and used as seed nanoparticles for the further growth of Au nanoparticles through a seed‐mediated method to obtain SNS/Au NPs.
Catalysis of 4‐Nitrophenol Reduction
A suspension of DFNS/Au hybrids in deionized water (1 mL) is added to a mixture of deionized water (2 mL), 4‐nitrophenol (aq) (4‐NP, 1.7 mL, 0.2 mm), and NaBH4 (aq) (1 mL, 15 mm). The resultant mixture is swirled well, and transferred to a quartz cuvette. The color of the solution changes gradually from yellowish to clear as the reaction proceeds. UV/Vis spectra are recorded at 5 min intervals to monitor reaction progress. For comparison, DFNS/Au dots are used with all other experimental parameters unchanged, unless stated otherwise.
Characterization
The nanoparticles are imaged by using a Hitachi S‐4800 scanning electron microscope and a JEOL JEM‐2010 Luminography (Fuji FDL‐5000) Ultramicrotome (CRX) transmission electron microscope. High‐resolution transmission electron microscopy (HRTEM) and energy‐dispersive X‐ray (EDX) analysis were measured by using a JEOL JEM‐2100F microscope. Samples were prepared for TEM by concentrating the nanoparticle mixture through centrifugation (two times, 10 min, 6,000 rpm), followed by resuspension in nanopure water (100 μL) and immobilization of 10 μL portions of the solution on TEM grids (Ted Pella, Inc. Formvar/Carbon 400 mesh, copper coated). UV/Vis spectra were recorded using a UV‐1800 (Shimadzu, UV/Vis spectrophotometer). Gas adsorption isotherms are obtained by using BELSORP‐mini II (BEL Japan, Inc.). The gases used throughout adsorption experiments are highly pure (99.999 %). All samples are activated by thoroughly rinsing, followed by drying under vacuum for 24 h prior to the gas sorption measurements.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF‐2015R1A4A1041631 and NRF‐2016R1A2B4009281).
W. Byoun, S. Jung, N. M. Tran, H. Yoo, ChemistryOpen 2018, 7, 349.
References
- 1.
- 1a. Moore C. J., Montón H., O'Kennedy R., Williams D. E., Nogués C., Lynam C. C., Gubala V., J. Mater. Chem. B 2015, 3, 2043–2055; [DOI] [PubMed] [Google Scholar]
- 1b. Hoshikawa Y., Yabe H., Nomura A., Yamaki T., Shimojima A., Okubo T., Chem. Mater. 2010, 22, 12–14; [Google Scholar]
- 1c. Kim S. M., Jeon M., Kim K. W., Park J., Lee I. S., J. Am. Chem. Soc. 2013, 135, 15714–15717; [DOI] [PubMed] [Google Scholar]
- 1d. Lu J., Fan Y., Howard M. D., Vaughan J. C., Zhang B., J. Am. Chem. Soc. 2017, 139, 2964–2971; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Fedorenko S. V., Bochkova O. D., Mustafina A. R., Burilov V. A., Kadirov M. L. K., Holin C. V., Nizameev I. R., Skripacheva V. V., Menshikova A. Y., Antipin I. S., Konovalov A. I., J. Phys. Chem. C 2010, 114, 6350–6355; [Google Scholar]
- 1f. Rimer J. D., Lobo R. F., Vlachos D. G., Langmuir 2005, 21, 8960–8971; [DOI] [PubMed] [Google Scholar]
- 1g. Asefa T., Tao Z., Chem. Res. Toxicol. 2012, 25, 2265–2284; [DOI] [PubMed] [Google Scholar]
- 1h. Liu X., Jiao Z., Song T., Wu M., Zhang H., J. Colloid Interface Sci. 2017, 490, 497–504; [DOI] [PubMed] [Google Scholar]
- 1i. Lee I., Albiter M. A., Zhang Q., Ge J., Yin Y., Zaera F., Phys. Chem. Chem. Phys. 2011, 13, 2449–2456; [DOI] [PubMed] [Google Scholar]
- 1j. Fang X., Zhao X., Fang W., Chen C., Zheng N., Nanoscale 2013, 5, 2205–2218; [DOI] [PubMed] [Google Scholar]
- 1k. Li S., Pasc A., Fierroa V., Celzard A., J. Mater. Chem. A 2016, 4, 12686–12713; [Google Scholar]
- 1l. Zhou M., Liu Y., Chenbc J., Yang X., J. Mater. Chem. A 2015, 3, 1068–1076; [Google Scholar]
- 1m. Pak J., Yoo H., J. Mater. Chem. A 2013, 1, 5408–5413. [Google Scholar]
- 2.
- 2a. Ma Z., Han H., Xue J., J. Nanosci. Nanotechnol. 2009, 9, 3188–3192; [DOI] [PubMed] [Google Scholar]
- 2b. Jeong U., Joo J. B., Kim Y., RSC Adv. 2015, 5, 55608–55618; [Google Scholar]
- 2c. Wu Y., Su D., Qin D., ChemNanoMat 2017, 3, 245–251; [Google Scholar]
- 2d. Wu T., Jiang T., Hu B., Han B., He J., Zhou X., Green Chem. 2009, 11, 798–803; [Google Scholar]
- 2e. Liu Y., Qiao B., Li X., Le X., Zhang W., Ma J., J. Mol. Catal. A: Chem. 2015, 406, 65–71; [Google Scholar]
- 2f. Sharma R. K., Sharma S., Dalton Trans. 2014, 43, 1292–1304; [DOI] [PubMed] [Google Scholar]
- 2g. Banerjee B., Singuru R., Kundu S. K., Dhanalaxmi K., Bai L., Zhao Y., Reddy B. M., Bhaumik A., Mondal J., Catal. Sci. Technol. 2016, 6, 5102–5115; [Google Scholar]
- 2h. Cornejo A., Fuks G., Martínez-Merino V., Sarobe Í., Gil M. J., Philippot K., Chaudret B., Delpech F., Nayral C., New J. Chem. 2014, 38, 6103–6113; [Google Scholar]
- 2i. Zhang C., Zhou Y., Zhang Y., Zhao S., Fang J., Sheng X., New J. Chem. 2017, 41, 11089–11096; [Google Scholar]
- 2j. Petrov A., Lehmann H., Finsel M., Klinke C., Weller H., Vossmeyer T., Langmuir 2016, 32, 848–857. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Che S., Garcia-Bennett A. E., Yokoi T., Sakamoto K., Kunieda H., Terasaki O., Tatsumi T., Nat. Mater. 2003, 2, 801–805; [DOI] [PubMed] [Google Scholar]
- 3b. Wan Y., Zhao D., Chem. Rev. 2007, 107, 2821–2860; [DOI] [PubMed] [Google Scholar]
- 3c. Lu F., Wu S.-H., Hung Y., Mou C.-Y., Small 2009, 5, 1408–1413; [DOI] [PubMed] [Google Scholar]
- 3d. Wu S.-H., Moua C.-Y., Lin H.-P., Chem. Soc. Rev. 2013, 42, 3862–3875; [DOI] [PubMed] [Google Scholar]
- 3e. Du X., He J., Nanoscale 2011, 3, 3984–4002; [DOI] [PubMed] [Google Scholar]
- 3f. Huang M., Liu L., Wang S., Zhu H., Wu D., Yu Z., Zhou S., Langmuir 2017, 33, 519–526. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Du P., Zhao X., Zeng J., Guo J., Liu P., Appl. Surf. Sci. 2015, 345, 90–98; [Google Scholar]
- 4b. Tang Y., Hu H., Zhang M. G., Song J., Nie L., Wang S., Niu G., Huang P., Lu G., Chen X., Nanoscale 2015, 7, 6304–6310; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Liu J., Zhang B., Luo Z., Ding X., Li J., Dai L., Zhou J., Zhao X., Ye J., Cai K., Nanoscale 2015, 7, 3614–3626; [DOI] [PubMed] [Google Scholar]
- 4d. Wang Y., Huang H.-Y., Yang L., Zhang Z., Ji H., Sci. Rep. 2016, 6, 25468; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Wang S., Zhang M., Zhang W., ACS Catal. 2011, 1, 207–211; [Google Scholar]
- 4f. Yue Q., Li J., Zhang Y., Cheng X., Chen X., Pan P., Su J., Elzatahry A. A., Alghamdi A., Deng Y., Zhao D., J. Am. Chem. Soc. 2017, 139, 15486–15493; [DOI] [PubMed] [Google Scholar]
- 4g. Wang X., Feng J., Bai Y., Zhang Q., Yin Y., Chem. Rev. 2016, 116, 10983–11060. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Jankiewicz B. J., Jamiola D., Choma J., Jaroniec M., Adv. Colloid Interface Sci. 2012, 170, 28–47; [DOI] [PubMed] [Google Scholar]
- 5b. Chaudhuri R. G., Paria S., Chem. Rev. 2012, 112, 2373–2433. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Polshettiwar V., Cha D., Zhang X., Basset J. M., Angew. Chem. Int. Ed. 2010, 49, 9652–9656; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9846–9850; [Google Scholar]
- 6b. Maity A., Polshettiwar V., ChemSusChem 2017, 10, 3866–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Dhiman M., Chalke B., Polshettiwar V., ACS Sustainable Chem. Eng. 2015, 3, 3224–3230; [Google Scholar]
- 7b. Bayal N., Singh B., Singh R., Polshettiwar V., Sci. Rep. 2016, 6, 24888; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Singh R., Bapat R., Qin L., Feng H., Polshettiwar V., ACS Catal. 2016, 6, 2770–2784; [Google Scholar]
- 7d. Dhiman M., Polshettiwar V., J. Mater. Chem. A 2016, 4, 12416–12424; [Google Scholar]
- 7e. Bayal N., Singh R., Polshettiwar V., ChemSusChem 2017, 10, 2182–2191; [DOI] [PubMed] [Google Scholar]
- 7f. Patil U., Fihri A., Emwas A.-H., Polshettiwar V., Chem. Sci. 2012, 3, 2224–2229. [Google Scholar]
- 8.
- 8a. Fihri A., Bouhrara M., Patil U., Cha D., Saih Y., Polshettiwar V., ACS Catal. 2012, 2, 1425–1431; [Google Scholar]
- 8b. Bouhrara M., Ranga C., Fihri A., Shaikh R. R., Sarawade P., Emwas A.-H., Hedhili M. N., Polshettiwar V., ACS Sustainable Chem. Eng. 2013, 1, 1192–1199. [Google Scholar]
- 9.
- 9a. Yang H., Li S., Zhang X., Wang X., Ma J., J. Mater. Chem. A 2014, 2, 12060–12067; [Google Scholar]
- 9b. Sadeghzadeh S. M., Catal. Sci. Technol. 2016, 6, 1435–1441; [Google Scholar]
- 9c. Sadeghzadeh S. M., Green Chem. 2015, 17, 3059–3066; [Google Scholar]
- 9d. Dhiman M., Chalke B., Polshettiwar V., J. Mater. Chem. A 2017, 5, 1935–1940; [Google Scholar]
- 9e. Kundu P. K., Dhiman M., Modak A., Chowdhury A., Polshettiwar V., Maiti D., ChemPlusChem 2016, 81, 1142–1146; [DOI] [PubMed] [Google Scholar]
- 9f. Sadeghzadeh S. M., Zhiani R., Emrani S., RSC Adv. 2017, 7, 24885–24894; [Google Scholar]
- 9g. Gautam P., Dhiman M., Polshettiwar V., Bhanage B. M., Green Chem. 2016, 18, 5890–5899; [Google Scholar]
- 9h. Qureshi Z. S., Sarawade P. B., Hussain I., Zhu H., Al-Johani H., Anjum D. H., Hedhili M. N., Maity N., D′Elia V., Basset J.-M., ChemCatChem 2016, 8, 1671–1678; [Google Scholar]
- 9i. Singh R., Belgamwar R., Dhiman M., Polshettiwar V., J. Mater. Chem. B 2018, 6, 1600–1604. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Park J. C., Song H., Nano Res. 2011, 4, 33–49; [Google Scholar]
- 10b. Liu J., Qiao S. Z., Chen J. S., Lou X. W. D., Xing X., Lu G. Q. M., Chem. Commun. 2011, 47, 12578–12591; [DOI] [PubMed] [Google Scholar]
- 10c. Purbia R., Paria S., Nanoscale 2015, 7, 19789–19873; [DOI] [PubMed] [Google Scholar]
- 10d. Fang X., Liu S., Zang J., Xu C., Zheng M.-S., Dong Q.-F., Sun D., Zheng N., Nanoscale 2013, 5, 6908–6916. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Kim D., Zuidema J. M., Kang J., Pan Y., Wu L., Warther D., Arkles B., Sailor M. J., J. Am. Chem. Soc. 2016, 138, 15106–15109; [DOI] [PubMed] [Google Scholar]
- 11b. Caruso F., Adv. Mater. 2001, 13, 11–22. [Google Scholar]
- 12. Pham T., Jackson J. B., Halas N. J., Lee T. R., Langmuir 2002, 18, 4915–4920. [Google Scholar]
- 13. Liu A., Wang G., Wang F., Zhang Y., Coord. Chem. Rev. 2017, 336, 28–42. [Google Scholar]
- 14.
- 14a. Westcott S. L., Oldenburg S. J., Lee T. R., Halas N. J., Langmuir 1998, 14, 5396–5401; [Google Scholar]
- 14b. Pak J., Yoo H., Microporous Mesoporous Mater. 2014, 185, 107–112; [Google Scholar]
- 14c. Mai H. D., Seo K., Choi S., Yoo H., RSC Adv. 2015, 5, 18977–18982. [Google Scholar]
- 15.
- 15a. Link S., El-Sayed M. A., J. Phys. Chem. B 1999, 103, 8410–8426; [Google Scholar]
- 15b. Ghosh S. K., Pal T., Chem. Rev. 2007, 107, 4797–4862. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Link S., Mohamed M. B., El-Sayed M. A., J. Phys. Chem. B 1999, 103, 3073–3077; [Google Scholar]
- 16b. Willets K. A., Duyne R. P. V., Annu. Rev. Phys. Chem. 2007, 58, 267–297; [DOI] [PubMed] [Google Scholar]
- 16c. Burda C., Chen X., Narayanan R., El-Sayed M. A., Chem. Rev. 2005, 105, 1025–1102. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Oldenburg S. J., Averitt R. D., Westcott S. L., Halas N. J., Chem. Phys. Lett. 1998, 288, 243–247; [Google Scholar]
- 17b. Averitt R. D., Sarkar D., Halas N. J., Phys. Rev. Lett. 1997, 78, 4217–4220. [Google Scholar]
- 18.
- 18a. van der Zande B. M. I., Böhmer M. R., Fokkink L. G. J., Schönenberger C., J. Phys. Chem. B 1997, 101, 852–854; [Google Scholar]
- 18b. Kim F., Song J. H., Yang P., J. Am. Chem. Soc. 2002, 124, 14316–14317; [DOI] [PubMed] [Google Scholar]
- 18c. Niidome Y., Nishioka K., Kawasaki H., Yamada S., Chem. Commun. 2003, 2376–2377; [DOI] [PubMed] [Google Scholar]
- 18d. Sau T. K., Murphy C. J., Langmuir 2004, 20, 6414–6420; [DOI] [PubMed] [Google Scholar]
- 18e. Tsung C.-K., Kou X., Shi Q., Zhang J., Yeung M. H., Wang J., Stucky G. D., J. Am. Chem. Soc. 2006, 128, 5352–5353; [DOI] [PubMed] [Google Scholar]
- 18f. Orendorff C. J., Murphy C. J., J. Phys. Chem. B 2006, 110, 3990–3994; [DOI] [PubMed] [Google Scholar]
- 18g. Li N., Zhao P., Astruc D., Angew. Chem. Int. Ed. 2014, 53, 1756–1789; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1784–1818. [Google Scholar]
- 19. Stöber W., Fink A., Bohn E., J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar]
- 20. Choi S., Moon Y., Yoo H., J. Colloid Interface Sci. 2016, 469, 269–276. [DOI] [PubMed] [Google Scholar]
- 21. Zhan G., Zeng H. C., Coord. Chem. Rev. 2016, 320–321, 181–192. [Google Scholar]
- 22.
- 22a. Lee J., Park J. C., Song H., Adv. Mater. 2008, 20, 1523–1528; [Google Scholar]
- 22b. Moon Y., Mai H. D., Yoo H., ChemNanoMat 2017, 3, 196–203; [Google Scholar]
- 22c. Corma A., Concepción P., Serna P., Angew. Chem. 2007, 119, 7404–7407. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary