

Abstract
Stem cells are considered the origin of neoplasms in general, and malignant tumours in particular, and the stage at which the stem cells stop their differentiation determines the degree of malignancy. However, there is increasing evidence supporting an alternative paradigm. Tumours may develop by dedifferentiation from mature cells able to proliferate. Studies of gastric carcinogenesis demonstrate that mature neuroendocrine (NE) cells upon long-term overstimulation may develop through stages of hyperplasia, dysplasia, and rather benign tumours, into highly malignant carcinomas. Dedifferentiation of cells may change the histological appearance and impede the identification of the cellular origin, as seen with gastric carcinomas, which in many cases are dedifferentiated neuroendocrine tumours. Finding the cell of origin is important to identify risk factors for cancer, prevent tumour development, and tailor treatment. In the present review, we focus not only on gastric tumours, but also evaluate the role of neuroendocrine cells in tumourigenesis in two other foregut-derived organs, the lungs and the pancreas, as well as in the midgut-derived small intestine.
Keywords: carcinogenesis, neuroendocrine, stem cells, tumour
Introduction
Stem cells versus dedifferentiation
The number of cells of a specific type is tightly regulated by functional demand by growth-controlling signal substances from other, regulatory cells and by direct or indirect negative feedback of specific substances released from the particular cell type. At present, the prevailing theory is that tumours develop from stem cells that stop differentiating at a certain level.1 Benign tumours are composed of well-differentiated cells, but the ‘set point’ is changed allowing an increased cell number. According to the stem cell theory of carcinogenesis, the malignant process stops tumour cell differentiation at an earlier stage. Stem cells have the ability to divide as well as to differentiate. In addition, the partly differentiated daughter cells have both these capabilities, but during the further differentiation process the ability to divide may be lost. Neoplasia may develop when mutations affect the normal growth regulation. The malignancy of the resulting tumour depends on the degree of differentiation of the mutated cell, and the importance of the mutated gene in growth regulation. The belief that stem cells are the sole origin of neoplasia seems partly based on the concept that only stem cells have the ability to proliferate. Knowledge of the receptors on the mutated cell and the ligands regulating their proliferation will accordingly be crucial in understanding the carcinogenesis and for the prevention and treatment of tumours. The stem cell origin of for example, colorectal cancer has recently been challenged.2 Moreover, there are multiple examples of transformation of a certain cell type via hyperplasia and a rather benign tumour into a highly malignant tumour.3,4
An alternative to the theory of stem cells giving rise to all tumours, is the concept that all cells with the ability to divide may develop into tumours by dedifferentiation. The dedifferentiation theory of carcinogenesis prevailed in a period before the stem cell was suggested as the cell of origin (Sell S, stem cells and cancer, Springer Science, LCC 2009). According to the dedifferentiation theory, tumours become more malignant as cells lose their ability to differentiate through accumulation of mutations. Most mutations result in an altered amount of dysfunctional proteins, which in turn alter the cellular phenotype but seldom result in gain of new properties. The occurrence of common neuroendocrine (NE) markers in normal NE cells, in well-differentiated neuroendocrine tumours (NETs), and also to a lesser degree in NE carcinomas (NECs), is compatible with tumour development from mature NE cells.5–7 The general mechanism of tumourigenesis is similar in stem cells and dividing differentiated cells; mutations occur during cell division. Rapidly dividing cells are accordingly more prone to develop into tumours. Therefore, stimulation of proliferation either due to destruction of the cell by inflammation or due to an increased concentration of hormones having a positive trophic effect on that particular cell type, will increase the tumour risk. The consequence of the mutations occurring by chance depends on the gene affected and whether an inherent allelic mutation in the particular gene is already present. Alternatively, direct genotoxic agents may induce tumours. Whatever the cause of mutations of the mature cells of origin, the process of carcinogenesis will change the cells towards a dedifferentiated phenotype. In this process, it is pivotal to understand growth regulation of mature cells, which is then important for both the prevention and treatment of tumours. The question of stem cells versus dedifferentiated mature cells as the origin of neoplasia in general, was recently discussed in depth8 focusing on possible reprogramming of differentiated cells and the role of dedifferentiated cells (which were also named, reserve stem cells) in metaplasia and carcinogenesis as well. With respect to gastric cancer, the chief cells were discussed in particular, and dedifferentiation of the chief cells was claimed to be a consequence of parietal cell atrophy. We consider this a weakness in the theory, as it is not clear why the parietal cells should undergo a specific atrophy, or how the parietal cells control the differentiation of the chief cells.8 Very recently Hayakawa and co-workers published a review on the origin of gastric cancer mainly based on mice studies, concluding that most probably, cancers including gastric cancers, develop from stem cells.9 They, however, dismissed the theory of the so-called SPEM cells derived from chief cells, as the cells developing into gastric cancer. There is, however, accumulated knowledge about the NETs, suggesting that such tumours develop from mature cells. A NET may be defined as a tumour originating from NE cells where the growth regulation is only moderately disturbed, and thus these tumours are growing slowly, but nevertheless have the ability to metastasize.
NE cells and replication
The NE cells share many properties with neurons and endocrine cells (Figure 1), such as small vesicles containing the marker protein synaptophysin,10 as well as secretory granules with their specific proteins, the chromogranins.11 NE cells also have protrusions12,13 resembling axons, mediating signals to neurons as well as other effector cells.13 Together with neurons and endocrine cells, the NE cells represent a system for regulation in a multicellular organism. Based upon similarities in phenotype as well as signalling function, it was proposed that all these cell types in fetal life could originate from the neural crest of the neuroectoderm.14 In chimeric studies, however, Le Douarin and Teillet found that enteric ganglion cells originated from the neural crest, in contrast with enterochromaffin cells of the digestive tract.15 On the other hand, the very similar C cells of the thyroid may have their origin in the neural crest,16 although a more recent study suggests an endodermal origin.17 From studies describing differentiation of all cells in a crypt from a common stem cell, an endodermal origin of gastric endocrine cells has been suggested.18 Whatever the embryological origin, the NE cells of the digestive organs can replicate, as demonstrated in pancreatic beta-cells19 and gastric enterochromaffin-like (ECL) cells in the stomach.20 The function and proliferation of the ECL cells in the oxyntic gastric mucosa of rodents20 as well as man21 are regulated by gastrin. Since the ECL cells of the stomach have been extensively studied in animals as well as man, this knowledge will be used to discuss the cellular origin of NETs of the lungs and pancreas as well as the small intestine.
Figure 1.

The regulatory systems (the neural system, the NE cell system, and the endocrine organs) show morphological similarities and expression of secretory granules and small vesicles.
NE, neuroendocrine.
The stomach
The ECL cell produces and releases histamine taking part in the regulation of gastric acid secretion.22,23 It is the only cell of the gastric mucosa definitely possessing the gastrin receptor,24–26 and gastrin is the main regulator of its function as well as proliferation.18,20,27 Although gastrin is the most important regulator, pituitary adenylate cyclase-activating polypeptides (PACAPs)28 and the vagal nerves29 also play a role in the regulation of ECL cell proliferation. Chronic hypergastrinaemia induces a sequence of ECL cell hyperplasia through increasing dysplasia to ECL cell neoplasia.30,31 The ECL cell hyperplasia in conditions with hypergastrinaemia is a direct consequence of the long-term stimulation of that particular cell. Each cell division is accompanied by a certain risk of mutation, and it is even likely that accelerated proliferation will increase the mutation risk due to the reduced time of repair. Although some mutations may result in improved function, and thus contribute to evolution, most mutations cause a reduction or even loss of function. When mutations only affect genes involved in the regulation of replication, tumours of apparently normal ECL cells develop, that is gastric NETs (gNETs). Over time, these gNETs gain more mutations changing their phenotype and further increasing the proliferation rate; and thus, become more malignant. When a mutation by chance affects a crucial regulator of replication, a malignant tumour may emerge at an early stage. Such a mutation would be expected to occur more often in cells with accelerated proliferation (ECL cell hyperplasia or ECL cell gNETs), but may also occur in normal ECL cells at normogastrinaemia and thus cause gNETs type III32 or gastric neuroendocrine carcinoma (gNEC). Independently of its cause, long-term hypergastrinaemia induces ECL cell NETs in all species studied.33–37 ECL cell NETs most often show a rather benign behavior, but are nevertheless malignant as they can metastasize and in some cases develop into highly malignant neoplasms.5 A parallel may be seen in EC cell tumours of the small intestine, which for years may remain indolent, but sooner or later become highly malignant tumours with increased proliferation rates.38 During the indolent phase, NETs may appear dormant. However, the apparent dormancy most probably just reflects the extremely slow proliferation rate of well-differentiated NET cells since, to our knowledge, a complete lack of dividing tumour cells at any stage of NETs has never been described. The slow replication rate of normal NE cells is also reflected by the late acceptance of their ability to divide.39
ECL cell-derived tumours are considered rare, the dominating malignant tumour type of the stomach being gastric carcinomas. These tumours are all considered to be adenocarcinomas, and are divided according to Laurén into those growing with a glandular pattern, called the intestinal type, and those without such a pattern, called the diffuse type.40 However, dedifferentiation of neoplastic cells may change their histological appearance, making it difficult to establish their cellular origin. Tumour cells in carcinomas of the diffuse type often have NE and, more specifically, ECL cell differentiation.41 In-situ hybridization showed no expression of mucin mRNAs contradicting exocrine cell origin,42 but chromogranin A mRNA expression indicating NE origin. In fact, gastric carcinomas occurring in patients with pernicious anemia are found in the oxyntic area,43 similar to ECL cell carcinoids, and virtually all show NE/ECL cell differentiation.44 NE expression is also observed in intestinal-type adenocarcinomas. There is a recent description of a Spanish family where all individuals were homozygous for a mutation in the gene encoding the α-subunit of the gastric proton pump, developed ECL cell gNETs (five persons) from the age of 23 years, and one person also developed a more malignant tumour classified as an adenocarcinoma.7 Further histological analysis revealed a combined ECL gNET and carcinoma.45 It is therefore reasonable to believe that there is a continuous process starting with mature NE cells exposed to long-term overstimulation, developing via hyperplasia to neoplasia of variable malignancy (Figure 2).
Figure 2.
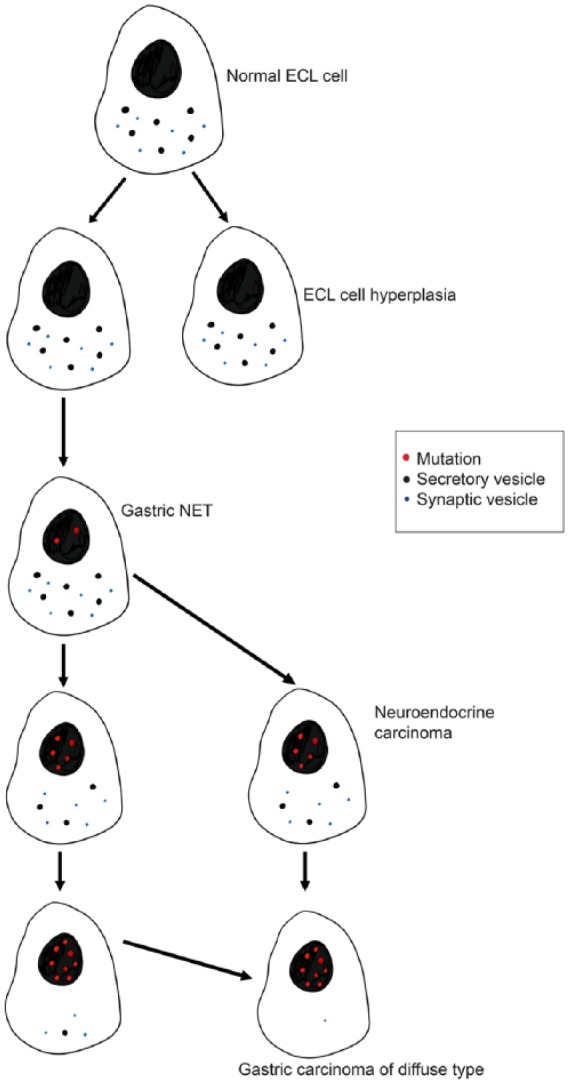
Gradual dedifferentiation of the ECL cell may result in the cancer cell of diffuse gastric carcinoma (Waldum and colleagues46).
ECL, enterochromaffin-like; NET, neuroendocrine tumour.
Mitoses represent a small, but definitive risk of mutation, which may be increased when proliferation is over-stimulated, reducing the time for repair. Patients with long-term hypergastrinaemia due to atrophic gastritis often develop multiple ECL cell NETs, which regress upon treatment with the gastrin receptor antagonist, netazepide.47 We recently showed that netazepide also reduced the density of ECL cells in the flat mucosa.48 Moreover, the reason why gastric carcinomas of the diffuse type according to Laurén40 were classified as adenocarcinomas, was Periodic acid–Schiff (PAS)-positivity believed to reflect mucin. However, PAS-positivity is not specific for mucin since PAS binds to glycoproteins/peptides in general.49 Furthermore, antibodies directed against glycoproteins/peptides are probably less specific than those raised against proteins/peptides, and for mucins in particular, there seems to be a great problem with antibody specificity,50 since it is difficult to purify the molecule for the use as antigen.51 Accordingly, in-situ hybridization seems to be a more specific method available for the detection of mucin-producing cells, and as noted above,42 mucin mRNA expression was not detected in gastric signet ring cell carcinoma. We have recently asked why the classification system relies on unspecific histochemistry, dismissing more specific methods.52 Rather recently, gastric adenocarcinomas were characterized molecularly by their mutations.53 However, mutations found in advanced carcinomas probably do not provide much information about the cell of origin, which is a crucial factor in the understanding of carcinogenesis. A molecular comparison between cancer cells and the normal mucosal cells with the ability to divide would be more adequate. Moreover, additional limitations of the classification of gastric cancers based upon mutation analysis have been addressed by others.54
A tumour apparently consisting of two different entities, an adenocarcinoma and another of a NE nature, has been described as mixed adeno-neuroendocrine carcinoma (MANEC). This term has recently been changed to mixed neuroendocrine-non-neuroendocrine neoplasms (MINEN). The two types of tumour cells may occur in a mixed pattern or they may be found partly separated, suggesting that two different tumours have merged.55 However, the two components in the same tumour have been reported to be monoclonal,56 and since the adenocarcinoma is more malignant, it is reasonable to suppose that this part has developed from the NE component and not vice versa. In a case report from Japan, a patient with long-term hypergastrinaemia secondary to treatment with a proton pump inhibitor, was reported to develop a MINEN with a NE component and a signet ring cell component.57 The NE component was, in contrast with the signet ring cells, reported to be positive for general NE markers like synaptophysin and chromogranin A.57 However, we have previously reported that the signet ring tumour cells of gastric carcinomas are positive for NE markers when applying methods with increased sensitivity,42,58 which may indicate a direct transition from a NE cell (probably of ECL cell origin in a hypergastrinaemic patient) to a carcinoma showing signet ring phenotype (Figure 3). Accordingly, not only the rather benign ECL cell NETs and the highly malignant gastric NECs,59,60 but also a considerable proportion of gastric carcinomas hitherto classified as adenocarcinomas, may origin from the ECL cells based upon numerous studies showing NE cell differentiation in tumour cells.6,41,42,44,58,61–65 A stem cell origin of these tumour cells would imply that the NE differentiation was due to re-differentiation, which seems unlikely given that mutations often lead to loss and not gain of function. In fact, the NE cancer cells of these tumours are the most differentiated tumour cells, and generally, tumours are classified according to the most differentiated tumour cells. The distinction between adenocarcinomas and NECs has for long been acknowledged to be difficult, as exemplified by reclassification from adenocarcinomas to NECs in human gastric tumours,66–69 as well as those occurring in rodents.69,70 Accordingly, the ECL cell seems to be crucial in gastric carcinogenesis, and therefore, the regulation of ECL cell proliferation is of great importance in gastric carcinogenesis and consequently in the prevention and treatment of such tumours at early stages. By its regulation of ECL cell proliferation, gastrin becomes important in gastric carcinogenesis and may even mediate the carcinogenic effect of gastric Helicobacter pylori (Hp) infection.71 It has been known for many decades that gastric carcinomas seldom develop in a stomach without gastritis.72 Hp was soon realized to be a major gastric carcinogen.73 However, in spite of intensive research for more than two decades, the mechanism by which Hp infection induces gastric cancer is unknown. Moreover, Hp infection itself does not seem to be a direct carcinogen, as Hp infection confined to the antral mucosa predisposes to duodenal ulcer disease74 but protects against gastric cancer.75 Even though it was recently demonstrated that Hp infection could reach the area of the stem cells (proliferation zone)76 there is hitherto no example of bacterial carcinogenesis due to internalization. On the other hand, there is convincing evidence that Hp infection has carcinogenic potential only after inducing atrophic gastritis in the oxyntic mucosa.77,78 Patients with Hp infection have reduced gastric acid secretion, reduced gastric acidity, and secondary hypergastrinaemia,77,78 which may be marked in contrast with the slight gastrin elevation in those with duodenal ulcer,79 which nevertheless is sufficient to induce hypersecretion of acid due to extreme sensitivity for gastrin.80 The increased gastric acidity, on the other hand, inhibits further gastrin release,81 thus preventing marked hypergastrinaemia.81,82 Furthermore, autoimmune gastritis may result in total atrophy of the oxyntic mucosa, resulting in anacidity and hypergastrinaemia, and predisposing to gNETs as well as gastric carcinomas.77 Thus, hypergastrinaemia is a common factor of the two major conditions predisposing to gastric cancer, Hp gastritis and autoimmune gastritis.82 In fact, hypergastrinaemia in patients with gastric carcinomas has been described already many years ago.83,84 Very recently we detected gastrin receptors by immunohistochemistry and by in-situ hybridization in normal, hyperplastic and neoplastic (NETs, NECs and adenocarcinoma) NE cells,85 suggesting that even malignant gastric carcinomas could respond to anti-gastrin therapy. In many ways, the role of gastrin in the oxyntic mucosa of the stomach may be compared to the role of estrogens in the mammary glands in terms of regulation of function (acid secretion and lactation, respectively), as well as growth affecting the development of neoplasia. This is partly based on the fact that gastric cancer seldom occurs in patients with duodenal ulcer, who very rarely have hypergastrinaemia, only an inappropriately increased gastrin levels, as well as the scarcity of breast cancer in males. We are well aware of the many reports on the role of cancer stem cells in carcinogenesis in general,86,87 including gastric carcinogenesis88 as well as the development of NE tumours.89 On the other hand, Quante and co-workers reported that a gastric progenitor cell from the oxyntic mucosa developed into mucus neck cells, parietal cells, and chief cells, but not NE cells.90 This finding indicates that there is a NE cell, progenitor or mature, being able to proliferate independently of the other cells in the oxyntic mucosa. The concept of a cancer stem cell being present already at an early stage is difficult to conceive, since it does not explain how the tumours become increasingly malignant over time.
Figure 3.

Mixed adenoneuroendocrine carcinoma (MANEC, now called MINEN) consisting of equal amounts of signet ring cells and neuroendocrine carcinoma cells, as illustrated by (A) Hematoxylin and eosin, × 40, (B) Chromogranin A, × 40, (C) Synaptophysin, × 40, and (D) Ki67, × 40.
Interestingly, NE cells of the upper gastrointestinal tract lack E-cadherin91 that may predispose these cells to become invasive and metastasize. Another feature of NE cells that may be important for their carcinogenic potential is their production of signal substances affecting the function and growth of neighboring cells and tissues. Thus, the ECL cell of the stomach produces histamine, and the EC cell of the small intestine produces serotonin, both substances having profound vascular effects such as increasing vascular permeability and stimulating angiogenesis,92 that conceivably could promote metastases. The discrepancy between a rather normal cellular appearance93 and the biological malignancy of NE tumours, which often tend to metastasize when the tumour is still small, may be explained by these properties of normal NE cells. Although there are many and strong indications of an important role of the ECL cell in subgroups of gastric carcinomas, their origin has still not been completely established. Nevertheless, given the very limited treatment options, this should not further delay clinical trials in patients with gastric carcinoma positive for the gastrin receptor.
NE cells and tumours outside the stomach
The small intestine
The EC cell gives rise to the classical NETs of the small intestine. The growth regulation of normal EC cells has been indirectly studied in the EC cell-derived tumour cell line KRJ-I,94 but no in vivo study has been performed. The regulation of EC cell function or growth is not fully understood. The EC cell produces serotonin, causing the classical carcinoid syndrome, as well as fibrosis of the heart valves as found in patients with EC cell NETs.95 The EC cell NETs are growing slowly, but over time they develop into more malignant tumours. This process has not been well studied, presumably because this would require repetitive tissue sampling, and since there may be heterogeneity between different metastases.96 However, a subset of small intestine NETs has somatic mutations in the CDKNB1 gene encoding p27.97 A subset of mature enteroendocrine cells has recently been shown to have stem cell properties.98 Karpathakis and colleagues recently performed integrated DNA-sequencing, DNA-methylation, and gene expression analysis on 97 small intestinal NETs from a cohort of 85 patients.99 The authors identified three subgroups of small intestine neuroendocrine tumours (SI-NETs) distinguished by molecular profiling, with different outcomes and progression-free survival (PFS) rates. The largest group (57%) was defined by chromosome 18 loss of heterozygosity.100 This group was associated with the presence of CDKN1B mutations and CpG island methylator phenotype (CIMP) negativity. These patients had the most favorable PFS, endpoint not reached at 10 years of follow up after resection, and an older age at diagnosis of 67 years. The second group (18%) was characterized by a high degree of CIMP positivity and the absence of arm-level copy-number changes.101 This group was associated with an intermediate PFS (56 months) and a younger age at diagnosis of 60 years. The third group was 26% and was characterized by the presence of multiple copy-number changes, a significantly poor PFS (21 months), and younger age at diagnosis of 54 years, suggesting a more aggressive clinical phenotype. These new data are in line with clinical observations that not all SI-NETs demonstrate slow benign growth with long PFS. Most recently, a study from the Uppsala group revealed that the mutY homologue (MUTYH)-DNA glycosylated gene was significantly enriched in SI-NET patients.102 MUTYH is involved in the protection of DNA exposed to oxidative stress, and has been shown to be involved in various cancers in humans and experimental animals. It has been suggested that this mutation, in the DNA-excision/repair pathway, might be involved in driving the tumourigenesis, thus causing both familial and sporadic SI-NETs. The occurrence of similar mutations in MUTYH has recently been published for pancreatic NETs. Interestingly, the multiple tumours occurring in patients with familial small intestinal NETs, were found to be polyclonal and originate from a subset of EC cells expressing intestinal stem cell genes.103 It is reasonable to presume that not only the enteroendocrine cells but also NE cells located in other parts of the gastrointestinal tract, including the stomach, have stem cell properties. Accordingly, it is conceivable that also tumours developing from NE cells in general may show variable phenotypes. The discrepancy between NETs in the small intestine and the appendix (both midgut and serotonin producing), in their ability to metastasize,104 may be related to the anatomical localization of the appendix.
Pancreas
The pancreas is composed of exocrine glands producing enzyme precursors, and duct cells secreting bicarbonate, as well as the endocrine islets of Langerhans with cells producing hormones, including insulin, glucagon, and somatostatin. The large majority of pancreatic neoplasms are thought to arise from acinar cells, whereas only 3–5% of the tumours are typically classified as NETs. The cellular origin of pancreatic NETs (pNETs) should be investigated based on knowledge on the replication and proliferation of normal NE cells. Furthermore, many adenocarcinomas contain NE cells, where NE cells have an uncertain role and prognostic importance. It has been questioned whether these cells could be normal NE cells trapped in a malignant tumour. Alternatively, they could be the most differentiated part of an otherwise dedifferentiated NET.
It has previously been debated how new pancreatic endocrine cells are formed.105 However, there is evidence from older 3H-thymidine incorporation studies,106 and from more recent genetic linage tracing studies, that adult pancreatic β-cells are formed by self-duplication rather than stem cell differentiation.19
The regulation of mature β-cell proliferation is thus of interest in the understanding of carcinogenesis. In patients with type 2 diabetes, the phenomenon of compensatory islet hyperplasia in response to insulin resistance107 is well known, but the underlying mechanisms of hyperplasia have until recently been obscure. Although the exact mechanisms in a liver–pancreas endocrine axis are not fully understood, there is evidence from mice models that liver-specific deficiency of the insulin receptor (IR) causes β-cell hyperplasia.108,109 More recently, the protein serpinB1,110 found to be abundantly expressed in the liver of mice with liver-specific IR deficiency, stimulates proliferation of pancreatic β-cells.
Similarly, mechanisms for the regulation of α-cell mass have been elucidated. Mice deficient of the glucagon receptor, develop hyperplasia of the α-cell,111 and in man, homozygous mutations of the glucagon receptor lead not only to hyperplasia, but also neoplasia of the glucagon-producing cell. Liver-specific deficiency of the glucagon receptor results in hypertrophy of the α-cell mass, suggesting that a liver-derived circulating growth factor may stimulate α-cell proliferation.112 Furthermore, mice lacking all proglucagon-derived peptides, including glucagon and glucagon-like peptide (GLP)-1, develop pancreatic NETs.113 It therefore seems plausible that one or several factors released from the liver cause α-cell proliferation and possibly also pNETs. An alternative hypothesis is that the α-cells themselves harbor a glucagon receptor with a negative trophic effect.
There is evidence that pNETs seem to be more prevalent due to increased use and better quality of abdominal imaging.114 A larger proportion of pNETs are nonfunctional,115,116 constituting the majority of tumours.117 The most common functional tumours are insulinomas, and while most are sporadic, some are associated with the multiple endocrine neoplasia type 1 (MEN1) syndrome.118 Patients with diabetes have a theoretically increased risk of insulinoma due to prolonged stimulation of β-cell proliferation, and this has been reported in some patients with diabetes.119–121 In a recent meta-analysis of epidemiological studies, diabetes is one of few risk factors found to be associated with pNETs,122 the others being heavy alcohol consumption, and a family history of cancer. Studies of mice with β-cell-specific MEN1 deletion have shown development of insulin expressing pancreatic tumours,123 which is a further demonstration of the islet cells as the origin of insulinomas.
In the fetal and neonatal pancreas there are gastrin-positive cells which disappear soon after birth and are not present in the adult normal pancreas.124 Gastrinomas are present in both the pancreas and the duodenum, and are the second most common functional pNET.117 Cell lineage studies of the transiently pancreatic gastrin-expressing cells have demonstrated that pancreatic gastrin-positive tumours derive from islet cells, and some of these co-express glucagon or insulin.125 Duodenal gastrinomas in patients with MEN1 are thought to develop from the mucosa of the small intestine, with diffuse and nodular hyperplasia of G-cells and microtumours of G-cells, and D-cells are frequently found.126 In the pancreas, microadenomatosis defined as multiple tumours up to 5 mm in diameter, is a feature of MEN1.127 These lesions frequently express glucagon and pancreatic polypeptide. Glucagonomas are exceedingly rare, and little is known about the risk factors for sporadic tumours in humans. However, α-cell hyperplasia is seen in patients with MEN1 and von Hippel–Lindau disease,128 and these patients also have an increased risk of developing glucagonomas. Studies of glucagon receptor deficient mice have demonstrated that these mice develop α-cell hyperplasia and eventually islet cell neoplasia.129 α-cell specific deletion of MEN1 also causes glucagonomas,130 but also some insulinomas and mixed type NETs.
The mutational landscape in human pancreatic NETs has recently been described.131,132 Among the somatic mutations most frequently found, were MEN1 mutations, but also mutations in genes related to mammalian target of rapamycin (mTOR) signalling (PTEN, DEPDC5, TSC1 and TSC2), DNA damage repair (MUTYH, CHEK2 and BRACA2), chromatin modification (SETD2 and MLL3), as well as altered telomere length (DAXX and ATRX) were frequently found. Finally, findings of hyperplasia preceding neoplasia in numerous murine models of pNETs have been reviewed,133 and it was suggested that primary alterations, such as MEN1 mutations or glucagon signalling inhibition, may be followed by the accumulation of mutations in hyperplastic endocrine cells, causing progression towards dysplasia and neoplasia. The evidence strongly supports a sequence from hyperplasia to benign and malignant NE neoplasia also in the pancreas, similar to what has been shown above for ECL cells in the stomach.
Evidence that neuroendocrine cells play a role in the development of pancreas adenocarcinoma is limited, as adenocarcinomas appear to originate from the exocrine pancreas and NETs from islet cells. The observation of tumours with mixed differentiation complicates this view. The mutational profile of mixed tumours has only been investigated in single cases, but was found to be common for both tumour compartments.134 It has been suggested that islet cells contribute to the development of ductal cancers. After studying the localization of the stem cell markers LGR5 and Nanog in the normal pancreas and adenocarcinomas, it was concluded that islet β-cells expressing LGR5 and Nanog markers are the initiating cells of pancreas cancer, and that these cells migrate from the islets to form the ductal cancerous tissue after mutation and de-differentiation.135 Experimental studies on tumour origin, supporting a role of islet cells in adenocarcinoma development, are few, but implantation of Kras and p16-mutated islet culture cells formed pancreatic duct adenocarcinomas in a hamster model. However, the majority of studies on the evolution of pancreatic adenocarcinomas suggest that adenocarcinomas derive from a non-neuroendocrine cell.136
The lungs
Pulmonary tumours are frequent, and tobacco smoking is the main etiological factor in lung carcinogenesis. Lung cancers are traditionally divided into small cell carcinomas (SCCs) and non-small cell carcinomas. The latter group is subdivided into adenocarcinomas, squamous cell carcinomas, and large cell carcinomas. SCCs are accepted to be NE tumours,137 and also large cell carcinomas show NE differentiation,138 while adenocarcinomas and squamous cell carcinomas are believed not to be related to NE cells. However, even squamous cell carcinomas and adenocarcinomas may express NE markers,139–141 and after treatment of adenocarcinomas with a tyrosine kinase inhibitor, small cell differentiation may occur.142 The cell of origin of the different types of pulmonary carcinomas has been disputed, and has still not been settled. There is consensus on the presence of an endodermal derived stem cell in the bronchial mucosa, which may be the origin of most cell types, and possibly also the NE cells. However, it is also clear that the NE cells of different organs have striking similarities, and that they in contrast to other cells may proliferate.19,20 This is also the case for NE cells of the lungs.143 Furthermore, NE cells of the bronchial tree often produce the same signal substances as NE cells of the stomach, for instance histamine.144 We have previously focused on the similarities between lung and gastric NE cells, and the contrast between the acceptance of NE cancers in the lungs and the reluctance to accept such tumours in the stomach.145 On the other hand, in contrast with the gastrointestinal tract, where the knowledge of stem cell location and regulation of proliferation has been greatly improved during the last decade,146 such information is more incomplete for the lungs. However, in multiple endocrine neoplasia I (MEN I), bronchial tree carcinoids (NETs) develop on the basis of NE cells.147 According to the World Health Organization, NETs of the lungs are termed carcinoids (typical and atypical). In order to prevent confusion, they are called carcinoids, with NETs in parenthesis, in this manuscript. There is also experimental evidence for a NE origin of SCCs.148 Based upon studies in mice, where key tumour suppressor genes were inactivated in different labelled pulmonary cell types, NE cells were speculated to be the most probable origin of SCCs. Recently, an immunohistochemical study indicated that SCCs originated from a NE progenitor cell, whereas more differentiated NE tumours could develop from more differentiated NE cells.149
Lung NE cells either occur as single cells in the mucosa or in clusters in so-called NE bodies. NE cells of the lungs have a receptor monitoring oxygen tension,150 and by release of regulatory substances they adjust ventilation and circulation to optimize oxygenation. Like NE cells of other organs, lung NE cells express secretory granules, which may be identified by chromogranins and also markers of synaptic vesicles like synaptophysin. Besides histamine, lung NE cells produce calcitonin gene related peptide (CGRP)151 and gastrin-releasing peptide.152 Furthermore, lung NE cells show neuron-like extensions137 similar to NE cells in the gastrointestinal tract.13,153
In the lungs, NE neoplasms make up an important proportion of the total pulmonary carcinomas with the phenotype of either small or large cell carcinoma. Smoking is an important cause of such cancers, but seems unrelated to nicotine since exposure by inhalation over a 2-year period did not affect NE growth in rats.154 Carcinoids (NETs) also occur in the airways, but a link between these carcinoids (NETs) and lung cancers has not been established,137,155 and pulmonary carcinoids do not seem to be induced by smoking.137,155 Nevertheless, mutational analysis has shown common molecular factors in pulmonary carcinoids (NETs) and NECs,156 and bronchial carcinoids (NETs) appear to develop from NE cell hyperplasia.157 We also studied the effect of long-term CO exposure in rats for 2 years, but did not detect any increase in NE cells or tumours in the lungs.158 The study hypothesis was that CO, by blocking O2 receptors on NE cells regulating the flow of air and blood locally, would stimulate the function and proliferation of that particular NE cell type. Presently, it must be acknowledged that the most important factors of tobacco smoke causing pulmonary cancer is still unknown, although many carcinogens have been identified.159
To summarize, there are indications on an important role of differentiated NE cells in the development of neoplasia of all organs, but particularly the stomach, as covered in this review (Table 1).
Table 1.
The role of NE cells in tumourigenesis in the organs discussed above.
| Organ and tumour type | Evidence for NE origin | References |
|---|---|---|
| Lung | ||
| NETs | +++ | |
| Carcinomas | ||
| Small cell | +++ | Bensch and colleagues137 |
| Large cell | +++ | Jiang and colleagues138 |
| Squamous cell | (+) | Linnoila and colleagues139 |
| Adenocarcinoma | (+) | Fresvig and colleagues,140 Sorhaug and colleagues141 |
| Stomach | ||
| NETs (ECL cell-derived) | +++ | |
| Gastric adenocarcinomas | ||
| Diffuse type | ++ | Waldum and colleagues,41 Sørdal and colleagues,42 Bakkelund and colleagues,58 Qvigstad and colleagues,61 Rogers and Murphy62 |
| Intestinal type | + | Waldum and colleagues, 6 Mjønes and colleagues85 |
| Pancreas | ||
| NETs (islet cell-derived) | +++ | |
| Adenocarcinoma | (+) | Pelosi and colleagues136 |
| Small intestine | ||
| NETs (EC cell-derived) | +++ | |
| Adenocarcinomas | - | |
++ clarified ++ firm evidence + some evidence (+) faint evidence - no indication.
NETs (previously called carcinoids).
ECL, enterochromaffin-like; NE, neuroendocrine; NET, neuroendocrine tumour.
Conclusion
To conclude, based upon studies of the stomach, but also other organs derived from the primitive gut, it has been established that NE cells are able to divide. Furthermore, continuous activation of their function also stimulates proliferation, which in the long term, through a sequence of hyperplasia, leads to neoplasia with variable degrees of malignancy. Knowledge on the regulation of proliferation of the specific NE cells may give information on how to prevent and treat tumours originating in that particular cell type. In early phases of tumourigenesis, antagonists of dominating trophic hormones may reverse tumour development. In the future, the stage of malignancy at which the tumour cells become independent of growth factors may be determined and tumour treatment tailored accordingly.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Helge L. Waldum, Department of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, N-7491, Norway Department of Gastroenterology and Hepatology, St. Olav’s University Hospital, Trondheim, Norway.
Kjell Öberg, Department of Endocrine Oncology Uppsala University and University Hospital, Uppsala, Sweden.
Øystein F. Sørdal, Department of Gastroenterology and Hepatology, St. Olav’s University Hospital, Trondheim, Norway
Arne K. Sandvik, Department of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, Norway Department of Gastroenterology and Hepatology, St. Olav’s University Hospital, Trondheim, Norway.
Bjørn I. Gustafsson, Department of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, Norway Department of Gastroenterology and Hepatology, St. Olav’s University Hospital, Trondheim, Norway.
Patricia Mjønes, epartment of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, Norway; Department of Pathology, St. Olav’s University Hospital, Trondheim, Norway.
Reidar Fossmark, Department of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, Norway; Department of Gastroenterology and Hepatology, St. Olav’s University Hospital, Trondheim, Norway.
References
- 1. Sell S. Stem cells and cancer. Springer Science, LCC, 2009, pp. 1–31. [Google Scholar]
- 2. Ray K. Colorectal cancer: on the origin of colonic tumours-outside the niche. Nat Rev Gastroenterol Hepatol 2015; 12: 62. [DOI] [PubMed] [Google Scholar]
- 3. Kloppel G, Anlauf M, Perren A, et al. Hyperplasia to neoplasia sequence of duodenal and pancreatic neuroendocrine diseases and pseudohyperplasia of the PP-cells in the pancreas. Endocr Pathol 2014; 25: 181–185. [DOI] [PubMed] [Google Scholar]
- 4. Janjua HG, Hogdall E, Linnemann D. Hyperplastic polyps of the colon and rectum - reclassification, BRAF and KRAS status in index polyps and subsequent colorectal carcinoma. APMIS 2015; 123: 298–304. [DOI] [PubMed] [Google Scholar]
- 5. Qvigstad G, Falkmer S, Westre B, et al. Clinical and histopathological tumour progression in ECL cell carcinoids (“ECLomas”). APMIS 1999; 107: 1085–1092. [DOI] [PubMed] [Google Scholar]
- 6. Waldum HL, Aase S, Kvetnoi I, et al. Neuroendocrine differentiation in human gastric carcinoma. Cancer 1998; 83: 435–444. [PubMed] [Google Scholar]
- 7. Calvete O, Reyes J, Zuniga S, et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumour. Hum Mol Genet 2015; 24: 2914–2922. [DOI] [PubMed] [Google Scholar]
- 8. Mills JC, Sansom OJ. Reserve stem cells: differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal 2015; 8: re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hayakawa Y, Sakitani K, Konishi M, et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell 2017; 31: 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wiedenmann B. Synaptophysin. A widespread constituent of small neuroendocrine vesicles and a new tool in tumor diagnosis. Acta Oncol 1991; 30: 435–440. [DOI] [PubMed] [Google Scholar]
- 11. Fischer-Colbrie R, Lassmann H, Hagn C, et al. Immunological studies on the distribution of chromogranin A and B in endocrine and nervous tissues. Neuroscience 1985; 16: 547–555. [DOI] [PubMed] [Google Scholar]
- 12. Larsson LI, Goltermann N, de Magistris L, et al. Somatostatin cell processes as pathways for paracrine secretion. Science 1979; 205: 1393–1395. [DOI] [PubMed] [Google Scholar]
- 13. Gustafsson BI, Bakke I, Tommeras K, et al. A new method for visualization of gut mucosal cells, describing the enterochromaffin cell in the rat gastrointestinal tract. Scand J Gastroenterol 2006; 41: 390–395. [DOI] [PubMed] [Google Scholar]
- 14. Pearse AG, Polak JM. Neural crest origin of the endocrine polypeptide (APUD) cells of the gastrointestinal tract and pancreas. Gut 1971; 12: 783–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Le Douarin NM, Teillet MA. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J Embryol Exp Morphol 1973; 30: 31–48. [PubMed] [Google Scholar]
- 16. Polak JM, Pearse AG, Le Lievre C, et al. Immunocytochemical confirmation of the neural crest origin of avian calcitonin-producing cells. Histochemistry 1974; 40: 209–214. [DOI] [PubMed] [Google Scholar]
- 17. Johansson E, Andersson L, Ornros J, et al. Revising the embryonic origin of thyroid C cells in mice and humans. Development 2015; 142: 3519–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thompson M, Fleming KA, Evans DJ, et al. Gastric endocrine cells share a clonal origin with other gut cell lineages. Development 1990; 110: 477–481. [DOI] [PubMed] [Google Scholar]
- 19. Dor Y, Brown J, Martinez OI, et al. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429: 41–46. [DOI] [PubMed] [Google Scholar]
- 20. Tielemans Y, Willems G, Sundler F, et al. Self-replication of enterochromaffin-like cells in the mouse stomach. Digestion 1990; 45: 138–146. [DOI] [PubMed] [Google Scholar]
- 21. Lamberts R, Creutzfeldt W, Stockmann F, et al. Long-term omeprazole treatment in man: effects on gastric endocrine cell populations. Digestion 1988; 39: 126–135. [DOI] [PubMed] [Google Scholar]
- 22. Hakanson R, Owman C. Concomitant histochemical demonstration of histamine and catecholamines in enterochromaffin-like cells of gastric mucosa. Life Sci 1967; 6: 759–766. [DOI] [PubMed] [Google Scholar]
- 23. Sandvik AK, Waldum HL, Kleveland PM, et al. Gastrin produces an immediate and dose-dependent histamine release preceding acid secretion in the totally isolated, vascularly perfused rat stomach. Scand J Gastroenterol 1987; 22: 803–808. [DOI] [PubMed] [Google Scholar]
- 24. Hakan SR, Owman C, Sjoberg NO. Cellular stores of gastric histamine in the developing rat. Life Sci 1967; 6: 2535–2543. [DOI] [PubMed] [Google Scholar]
- 25. Bakke I, Qvigstad G, Sandvik AK, et al. The CCK-2 receptor is located on the ECL cell, but not on the parietal cell. Scand J Gastroenterol 2001; 36: 1128–1133. [DOI] [PubMed] [Google Scholar]
- 26. Athmann C, Zeng N, Scott DR, et al. Regulation of parietal cell calcium signaling in gastric glands. Am J Physiol Gastrointest Liver Physiol 2000; 279: G1048–G1058. [DOI] [PubMed] [Google Scholar]
- 27. Brenna E, Waldum HL. Trophic effect of gastrin on the enterochromaffin like cells of the rat stomach: establishment of a dose response relationship. Gut 1992; 33: 1303–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oh DS, Lieu SN, Yamaguchi DJ, et al. PACAP regulation of secretion and proliferation of pure populations of gastric ECL cells. J Mol Neurosci 2005; 26: 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hakanson R, Vallgren S, Ekelund M, et al. The vagus exerts trophic control of the stomach in the rat. Gastroenterology 1984; 86: 28–32. [PubMed] [Google Scholar]
- 30. Hakanson R, Sundler F. Proposed mechanism of induction of gastric carcinoids: the gastrin hypothesis. Eur J Clin Invest 1990; 20(Suppl. 1): S65–S71. [DOI] [PubMed] [Google Scholar]
- 31. Solcia E, Fiocca R, Villani L, et al. Hyperplastic, dysplastic, and neoplastic enterochromaffin-like-cell proliferations of the gastric mucosa. Classification and histogenesis. Am J Surg Pathol 1995; 19(Suppl 1): S1–S7. [PubMed] [Google Scholar]
- 32. Rindi G, Luinetti O, Cornaggia M, et al. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma: a clinicopathologic study. Gastroenterology 1993; 104: 994–1006. [DOI] [PubMed] [Google Scholar]
- 33. Waldum HL, Fossmark R, Bakke I, et al. Hypergastrinaemia in animals and man: causes and consequences. Scand J Gastroenterol 2004; 39: 505–509. [DOI] [PubMed] [Google Scholar]
- 34. Havu N. Enterochromaffin-like cell carcinoids of gastric mucosa in rats after life-long inhibition of gastric secretion. Digestion 1986; 35(Suppl. 1): 42–55. [DOI] [PubMed] [Google Scholar]
- 35. Cui G, Qvigstad G, Falkmer S, et al. Spontaneous ECLomas in cotton rats (Sigmodon hispidus): tumours occurring in hypoacidic/hypergastrinaemic animals with normal parietal cells. Carcinogenesis 2000; 21: 23–27. [DOI] [PubMed] [Google Scholar]
- 36. Borch K, Renvall H, Liedberg G. Gastric endocrine cell hyperplasia and carcinoid tumors in pernicious anemia. Gastroenterology 1985; 88: 638–648. [DOI] [PubMed] [Google Scholar]
- 37. Jianu CS, Fossmark R, Viset T, et al. Gastric carcinoids after long-term use of a proton pump inhibitor. Aliment Pharmacol Ther 2012; 36: 644–649. [DOI] [PubMed] [Google Scholar]
- 38. Singh S, Hallet J, Rowsell C, et al. Variability of Ki67 labeling index in multiple neuroendocrine tumors specimens over the course of the disease. Eur J Surg Oncol 2014; 40: 1517–1522. [DOI] [PubMed] [Google Scholar]
- 39. Barrett P, Hobbs RC, Coates PJ, et al. Endocrine cells of the human gastrointestinal tract have no proliferative capacity. Histochem J 1995; 27: 482–486. [PubMed] [Google Scholar]
- 40. Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. an attempt at a histo-clinical classification. Acta Pathol Microbiol Scand 1965; 64: 31–49. [DOI] [PubMed] [Google Scholar]
- 41. Waldum HL, Haugen OA, Isaksen C, et al. Enterochromaffin-like tumour cells in the diffuse but not the intestinal type of gastric carcinomas. Scand J Gastroenterol Suppl 1991; 180: 165–169. [DOI] [PubMed] [Google Scholar]
- 42. Sordal O, Qvigstad G, Nordrum IS, et al. The PAS positive material in gastric cancer cells of signet ring type is not mucin. Exp Mol Pathol 2014; 96: 274–278. [DOI] [PubMed] [Google Scholar]
- 43. Zamcheck N, Grable E, Ley A, et al. Occurrence of gastric cancer among patients with pernicious anemia at the Boston City Hospital. N Engl J Med 1955; 252: 1103–1110. [DOI] [PubMed] [Google Scholar]
- 44. Qvigstad G, Qvigstad T, Westre B, et al. Neuroendocrine differentiation in gastric adenocarcinomas associated with severe hypergastrinaemia and/or pernicious anemia. APMIS 2002; 110: 132–139. [DOI] [PubMed] [Google Scholar]
- 45. Fossmark R, Calvete O, Mjones P, et al. ECL-cell carcinoids and carcinoma in patients homozygous for an inactivating mutation in the gastric H(+) K(+) ATPase alpha subunit. APMIS 2016; 124: 561–566. [DOI] [PubMed] [Google Scholar]
- 46. Waldum HL, Brenna E, Sandvik AK. Relationship of ECL cells and gastric neoplasia. Yale J Biol Med 1998; 71: 325–335. [PMC free article] [PubMed] [Google Scholar]
- 47. Fossmark R, Sordal O, Jianu CS, et al. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment Pharmacol Ther 2012; 36: 1067–1075. [DOI] [PubMed] [Google Scholar]
- 48. Sagatun L, Fossmark R, Jianu CS, et al. Follow-up of patients with ECL cell-derived tumours. Scand J Gastroenterol 2016; 51: 1398–1405. [DOI] [PubMed] [Google Scholar]
- 49. Tian H, Guan R, Salsi E, et al. Identification of the structural determinants for the stability of substrate and aminoacrylate external Schiff bases in O-acetylserine sulfhydrylase-A. Biochemistry 2010; 49: 6093–6103. [DOI] [PubMed] [Google Scholar]
- 50. Real FX, de Bolos C, Oosterwijk E. Polyclonal and monoclonal techniques. In: Methods in Molecular Biology, Vol 125: Glycoproteins Methods and Protocols. The mucins. Humana Press, 2010, pp. 353–386. [DOI] [PubMed] [Google Scholar]
- 51. Thornton DJ, Khan N, Sheehan JK. Separation and identification of mucins and their glycoforms. In: Methods in Molecular Biology, Vol 125: Glycoproteins Methods and Protocols. The mucins. Humana Press, 2010, pp. 77–85. [DOI] [PubMed] [Google Scholar]
- 52. Waldum HL, Sørdal OF. Classification of epithelial malignant tumors-the differentiation between adenocarcinomas and neuroendocrine carcinomas: why rely on nonspecific histochemistry and dismiss specific methods like immunohistochemistry and in situ hybridization? Appl Immunohistochem Mol Morphol 2016; 24: 309–312. [DOI] [PubMed] [Google Scholar]
- 53. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lim SM, Park HS, Kim S, et al. Next-generation sequencing reveals somatic mutations that confer exceptional response to everolimus. Oncotarget 2016; 7: 10547–10556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fujiyoshi Y, Kuhara H, Eimoto T. Composite glandular-endocrine cell carcinoma of the stomach. Report of two cases with goblet cell carcinoid component. Pathol Res Pract 2005; 200: 823–829. [DOI] [PubMed] [Google Scholar]
- 56. Scardoni M, Vittoria E, Volante M, et al. Mixed adenoneuroendocrine carcinomas of the gastrointestinal tract: targeted next-generation sequencing suggests a monoclonal origin of the two components. Neuroendocrinology 2014; 100: 310–316. [DOI] [PubMed] [Google Scholar]
- 57. Yamauchi H, Sakurai S, Nakazawa N, et al. A case of mixed adenoneuroendocrine carcinoma of the stomach with focal intestinal metaplasia and hypergastrinaemia. Int Surg 2015; 100: 562–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bakkelund K, Fossmark R, Nordrum I, et al. Signet ring cells in gastric carcinomas are derived from neuroendocrine cells. J Histochem Cytochem 2006; 54: 615–621. [DOI] [PubMed] [Google Scholar]
- 59. Matsui K, Jin XM, Kitagawa M, et al. Clinicopathologic features of neuroendocrine carcinomas of the stomach: appraisal of small cell and large cell variants. Arch Pathol Lab Med 1998; 122: 1010–1017. [PubMed] [Google Scholar]
- 60. Rindi G, Azzoni C, La Rosa S, et al. ECL cell tumor and poorly differentiated endocrine carcinoma of the stomach: prognostic evaluation by pathological analysis. Gastroenterology 1999; 116: 532–542. [DOI] [PubMed] [Google Scholar]
- 61. Qvigstad G, Sandvik AK, Brenna E, et al. Detection of chromogranin A in human gastric adenocarcinomas using a sensitive immunohistochemical technique. Histochem J 2000; 32: 551–556. [DOI] [PubMed] [Google Scholar]
- 62. Rogers LW, Murphy RC. Gastric carcinoid and gastric carcinoma. Morphologic correlates of survival. Am J Surg Pathol 1979; 3: 195–202. [DOI] [PubMed] [Google Scholar]
- 63. Nunobe S, Taniguchi H, Katai H, et al. A rare case of poorly differentiated endocrine cell carcinoma of the stomach with signet ring cell differentiation. Gastric Cancer 2010; 13: 131–134. [DOI] [PubMed] [Google Scholar]
- 64. Bartley AN, Rashid A, Fournier KF, et al. Neuroendocrine and mucinous differentiation in signet ring cell carcinoma of the stomach: evidence for a common cell of origin in composite tumors. Hum Pathol 2011; 42: 1420–1429. [DOI] [PubMed] [Google Scholar]
- 65. Martinsen TC, Skogaker NE, Fossmark R, et al. Neuroendocrine cells in diffuse gastric carcinomas: an ultrastructural study with immunogold labeling of chromogranin A. Appl Immunohistochem Mol Morphol 2010; 18: 62–68. [DOI] [PubMed] [Google Scholar]
- 66. Azzopardi JG, Pollock DJ. Argentaffin and argyrophil cells in gastric carcinoma. J Pathol Bacteriol 1963; 86: 443–451. [DOI] [PubMed] [Google Scholar]
- 67. Black WC, Haffner HE. Diffuse hyperplasia of gastric argyrophil cells and multiple carcinoid tumors. An historical and ultrastructural study. Cancer 1968; 21: 1080–1099. [DOI] [PubMed] [Google Scholar]
- 68. Partanen S, Syrjanen K. Argyrophilic cells in carcinoma of the female breast. Virchows Arch A Pathol Anat Histol 1981; 391: 45–51. [DOI] [PubMed] [Google Scholar]
- 69. Soga J, Tazawa MD, Ito H. Ultrastructural demonstration of specific secretory granules of Mastomys gastric carcinoids. Acta Med Biol (Niigata) 1969; 17: 119–124. [PubMed] [Google Scholar]
- 70. Waldum HL, Rorvik H, Falkmer S, et al. Neuroendocrine (ECL cell) differentiation of spontaneous gastric carcinomas of cotton rats (Sigmodon hispidus). Lab Anim Sci 1999; 49: 241–247. [PubMed] [Google Scholar]
- 71. Waldum HL, Hauso O, Sordal OF, et al. Gastrin may mediate the carcinogenic effect of helicobacter pylori infection of the stomach. Dig Dis Sci 2015; 60: 1522–1527. [DOI] [PubMed] [Google Scholar]
- 72. Morson BC. Intestinal metaplasia of the gastric mucosa. Br J Cancer 1955; 9: 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Parsonnet J, Friedman GD, Vandersteen DP, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991;325:1127–1131. [DOI] [PubMed] [Google Scholar]
- 74. Levi S, Beardshall K, Haddad G, et al. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet 1989; 1: 1167–1168. [DOI] [PubMed] [Google Scholar]
- 75. Hansson LE, Nyren O, Hsing AW, et al. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N Engl J Med 1996; 335: 242–249. [DOI] [PubMed] [Google Scholar]
- 76. Sigal M, Rothenberg ME, Logan CY, et al. Helicobacter pylori activates and expands Lgr5(+) stem cells through direct colonization of the gastric glands. Gastroenterology 2015; 148: 1392–1404. e21. [DOI] [PubMed] [Google Scholar]
- 77. Kokkola A, Sjoblom SM, Haapiainen R, et al. The risk of gastric carcinoma and carcinoid tumours in patients with pernicious anaemia. A prospective follow-up study. Scand J Gastroenterol 1998; 33: 88–92. [DOI] [PubMed] [Google Scholar]
- 78. Fossmark R, Sagatun L, Nordrum IS, et al. Hypergastrinaemia is associated with adenocarcinomas in the gastric corpus and shorter patient survival. APMIS 2015; 123: 509–514. [DOI] [PubMed] [Google Scholar]
- 79. Levi S, Dollery CT, Bloom SR, et al. Campylobacter pylori, duodenal ulcer disease, and gastrin. BMJ 1989; 299: 1093–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sandvik AK, Waldum HL. Rat gastric histamine release: a sensitive gastrin bioassay. Life Sci 1990; 46: 453–459. [DOI] [PubMed] [Google Scholar]
- 81. Walsh JH, Richardson CT, Fordtran JS. pH dependence of acid secretion and gastrin release in normal and ulcer subjects. J Clin Invest 1975; 55: 462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Waldum HL, Kleveland PM, Sordal OF. Helicobacter pylori and gastric acid: an intimate and reciprocal relationship. Therap Adv Gastroenterol 2016; 9: 836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. McGuigan JE, Trudeau WL. Serum and tissue gastrin concentrations in patients with carcinoma of the stomach. Gastroenterology 1973; 64: 22–25. [PubMed] [Google Scholar]
- 84. Rakic S, Milicevic MN. Serum gastrin levels in patients with intestinal and diffuse type of gastric cancer. Br J Cancer 1991; 64: 1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mjones P, Nordrum IS, Sordal O, et al. Expression of the cholecystokinin-B receptor in neoplastic gastric cells. Horm Cancer. Epub ahead of print 4 October 2017. DOI: 10.1007/s12672-017-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Woodward WA, Sulman EP. Cancer stem cells: markers or biomarkers? Cancer Metastasis Rev 2008; 27: 459–470. [DOI] [PubMed] [Google Scholar]
- 87. Klonisch T, Wiechec E, Hombach-Klonisch S, et al. Cancer stem cell markers in common cancers - therapeutic implications. Trends Mol Med. Epub ahead of print 3 September 2008. DOI: 10.1016/j.molmed.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 88. Saikawa Y, Fukuda K, Takahashi T, et al. Gastric carcinogenesis and the cancer stem cell hypothesis. Gastric Cancer 2010; 13: 11–24. [DOI] [PubMed] [Google Scholar]
- 89. Gaur P, Sceusi EL, Samuel S, et al. Identification of cancer stem cells in human gastrointestinal carcinoid and neuroendocrine tumors. Gastroenterology 2011; 141: 1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Quante M, Marrache F, Goldenring JR, et al. TFF2 mRNA transcript expression marks a gland progenitor cell of the gastric oxyntic mucosa. Gastroenterology 2010; 139: 2018–2027. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Waldum HL, Ringnes E, Nordbo H, et al. The normal neuroendocrine cells of the upper gastrointestinal tract lack E-cadherin. Scand J Gastroenterol 2014; 49: 974–978. [DOI] [PubMed] [Google Scholar]
- 92. Qin L, Zhao D, Xu J, et al. The vascular permeabilizing factors histamine and serotonin induce angiogenesis through TR3/Nur77 and subsequently truncate it through thrombospondin-1. Blood 2013; 121: 2154–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Al-Khafaji B, Noffsinger AE, Miller MA, et al. Immunohistologic analysis of gastrointestinal and pulmonary carcinoid tumors. Hum Pathol 1998; 29: 992–999. [DOI] [PubMed] [Google Scholar]
- 94. Siddique ZL, Drozdov I, Floch J, et al. KRJ-I and BON cell lines: defining an appropriate enterochromaffin cell neuroendocrine tumor model. Neuroendocrinology 2009; 89: 458–470. [DOI] [PubMed] [Google Scholar]
- 95. Gustafsson BI, Tommeras K, Nordrum I, et al. Long-term serotonin administration induces heart valve disease in rats. Circulation 2005; 111: 1517–1522. [DOI] [PubMed] [Google Scholar]
- 96. Yang Z, Tang LH, Klimstra DS. Effect of tumor heterogeneity on the assessment of Ki67 labeling index in well-differentiated neuroendocrine tumors metastatic to the liver: implications for prognostic stratification. Am J Surg Pathol 2011; 35: 853–860. [DOI] [PubMed] [Google Scholar]
- 97. Francis JM, Kiezun A, Ramos AH, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet 2013; 45: 1483–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gross S, Balderes D, Liu J, et al. Nkx2.2 is expressed in a subset of enteroendocrine cells with expanded lineage potential. Am J Physiol Gastrointest Liver Physiol 2015; 309: G975– G987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Karpathakis A, Dibra H, Pipinikas C, et al. Prognostic impact of novel molecular subtypes of small intestinal neuroendocrine tumor. Clin Cancer Res 2016; 22: 250–258. [DOI] [PubMed] [Google Scholar]
- 100. Cunningham JL, Diaz de Stahl T, Sjoblom T, et al. Common pathogenetic mechanism involving human chromosome 18 in familial and sporadic ileal carcinoid tumors. Genes Chromosomes Cancer 2011; 50: 82–94. [DOI] [PubMed] [Google Scholar]
- 101. Crona J, Gustavsson T, Norlen O, et al. Somatic mutations and genetic heterogeneity at the cdkn1b locus in small intestinal neuroendocrine tumors. Ann Surg Oncol 2015; 22(Suppl. 3): S1428–S1435. [DOI] [PubMed] [Google Scholar]
- 102. Dumanski JP, Rasi C, Bjorklund P, et al. A MUTYH germline mutation is associated with small intestinal neuroendocrine tumors. Endocr Relat Cancer. Epub ahead of print 20 June 2017. DOI: 10.1530/ERC-17-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sei Y, Feng J, Zhao X, et al. Polyclonal crypt genesis and development of familial small intestinal neuroendocrine tumors. Gastroenterology 2016; 151: 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Moertel CG, Weiland LH, Nagorney DM, et al. Carcinoid tumor of the appendix: treatment and prognosis. N Engl J Med 1987; 317: 1699–1701. [DOI] [PubMed] [Google Scholar]
- 105. Bonner-Weir S, Sharma A. Pancreatic stem cells. J Pathol 2002; 197: 519–526. [DOI] [PubMed] [Google Scholar]
- 106. Tsubouchi S, Kano E, Suzuki H. Demonstration of expanding cell populations in mouse pancreatic acini and islets. Anat Rec 1987; 218: 111–115. [DOI] [PubMed] [Google Scholar]
- 107. Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature 2001; 414: 788–791. [DOI] [PubMed] [Google Scholar]
- 108. Escribano O, Guillen C, Nevado C, et al. Beta-Cell hyperplasia induced by hepatic insulin resistance: role of a liver-pancreas endocrine axis through insulin receptor A isoform. Diabetes 2009; 58: 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Okada T, Liew CW, Hu J, et al. Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci U S A 2007; 104: 8977–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. El Ouaamari A, Dirice E, Gedeon N, et al. SerpinB1 promotes pancreatic beta cell proliferation. Cell Metab 2016; 23: 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gelling RW, Du XQ, Dichmann DS, et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A 2003; 100: 1438–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Longuet C, Robledo AM, Dean ED, et al. Liver-specific disruption of the murine glucagon receptor produces alpha-cell hyperplasia: evidence for a circulating alpha-cell growth factor. Diabetes 2013; 62: 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Takano Y, Kasai K, Takagishi Y, et al. Pancreatic neuroendocrine tumors in mice deficient in proglucagon-derived peptides. PLoS One 2015; 10: e0133812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hallet J, Law CH, Cukier M, et al. Exploring the rising incidence of neuroendocrine tumors: a population-based analysis of epidemiology, metastatic presentation, and outcomes. Cancer 2015; 121: 589–597. [DOI] [PubMed] [Google Scholar]
- 115. Vagefi PA, Razo O, Deshpande V, et al. Evolving patterns in the detection and outcomes of pancreatic neuroendocrine neoplasms: the Massachusetts General Hospital experience from 1977 to 2005. Arch Surg 2007; 142: 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cheema A, Weber J, Strosberg JR. Incidental detection of pancreatic neuroendocrine tumors: an analysis of incidence and outcomes. Ann Surg Oncol 2012; 19: 2932–2936. [DOI] [PubMed] [Google Scholar]
- 117. Zerbi A, Falconi M, Rindi G, et al. Clinicopathological features of pancreatic endocrine tumors: a prospective multicenter study in Italy of 297 sporadic cases. Am J Gastroenterol 2010; 105: 1421–1429. [DOI] [PubMed] [Google Scholar]
- 118. Placzkowski KA, Vella A, Thompson GB, et al. Secular trends in the presentation and management of functioning insulinoma at the Mayo Clinic, 1987-2007. J Clin Endocrinol Metab 2009; 94: 1069–1073. [DOI] [PubMed] [Google Scholar]
- 119. Cander S, Gul OO, Yildirim N, et al. A rare cause of hypoglycemia in a type 2 diabetic patient: insulinoma. J Diabetes Complications 2012; 26: 65–67. [DOI] [PubMed] [Google Scholar]
- 120. Maiza JC, Wantz C. Type 2 diabetes and insulinoma: an unusual association. Diabetes Metab 2015; 41: 432-433. [DOI] [PubMed] [Google Scholar]
- 121. Ravnik-Oblak M, Janez A, Kocijanicic A. Insulinoma induced hypoglycemia in a type 2 diabetic patient. Wien Klin Wochenschr 2001; 113: 339–341. [PubMed] [Google Scholar]
- 122. Leoncini E, Carioli G, La Vecchia C, et al. Risk factors for neuroendocrine neoplasms: a systematic review and meta-analysis. Ann Oncol 2016; 27: 68–81. [DOI] [PubMed] [Google Scholar]
- 123. Crabtree JS, Scacheri PC, Ward JM, et al. Of mice and MEN1: insulinomas in a conditional mouse knockout. Mol Cell Biol 2003; 23: 6075–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Larsson LI, Rehfeld JF, Sundler F, et al. Pancreatic gastrin in foetal and neonatal rats. Nature 1976; 262: 609–610. [DOI] [PubMed] [Google Scholar]
- 125. Bonnavion R, Teinturier R, Jaafar R, et al. Islet cells serve as cells of origin of pancreatic gastrin-positive endocrine tumors. Mol Cell Biol 2015; 35: 3274–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Anlauf M, Perren A, Henopp T, et al. Allelic deletion of the MEN1 gene in duodenal gastrin and somatostatin cell neoplasms and their precursor lesions. Gut 2007; 56: 637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Anlauf M, Perren A, Kloppel G. Endocrine precursor lesions and microadenomas of the duodenum and pancreas with and without MEN1: criteria, molecular concepts and clinical significance. Pathobiology 2007; 74: 279–284. [DOI] [PubMed] [Google Scholar]
- 128. Perigny M, Hammel P, Corcos O, et al. Pancreatic endocrine microadenomatosis in patients with von Hippel-Lindau disease: characterization by VHL/HIF pathway proteins expression. Am J Surg Pathol 2009; 33: 739–748. [DOI] [PubMed] [Google Scholar]
- 129. Yu R, Dhall D, Nissen NN, et al. Pancreatic neuroendocrine tumors in glucagon receptor-deficient mice. PLoS One 2011; 6: e23397 DOI: 10.1371/journal.pone.0023397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lu J, Herrera PL, Carreira C, et al. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 2010; 138: 1954–1965. [DOI] [PubMed] [Google Scholar]
- 131. Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011; 331: 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Scarpa A, Chang DK, Nones K, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017; 543: 65–71. [DOI] [PubMed] [Google Scholar]
- 133. Babu V, Paul N, Yu R. Animal models and cell lines of pancreatic neuroendocrine tumors. Pancreas 2013; 42: 912–923. [DOI] [PubMed] [Google Scholar]
- 134. Takano A, Hirotsu Y, Amemiya K, et al. Genetic basis of a common tumor origin in the development of pancreatic mixed acinar-neuroendocrine-ductal carcinoma: a case report. Oncol Lett 2017; 14: 4428–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Amsterdam A, Raanan C, Schreiber L, et al. LGR5 and Nanog identify stem cell signature of pancreas beta cells which initiate pancreatic cancer. Biochem Biophys Res Commun 2013; 433: 157–162. [DOI] [PubMed] [Google Scholar]
- 136. Pelosi E, Castelli G, Testa U. Pancreatic cancer: molecular characterization, clonal evolution and cancer stem cells. Biomedicines 2017; 5: pii: E65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bensch KG, Corrin B, Pariente R, et al. Oat-cell carcinoma of the lung. Its origin and relationship to bronchial carcinoid. Cancer 1968; 22: 1163–1172. [DOI] [PubMed] [Google Scholar]
- 138. Jiang SX, Kameya T, Shoji M, et al. Large cell neuroendocrine carcinoma of the lung: a histologic and immunohistochemical study of 22 cases. Am J Surg Pathol 1998; 22: 526–537. [DOI] [PubMed] [Google Scholar]
- 139. Linnoila RI, Mulshine JL, Steinberg SM, et al. Neuroendocrine differentiation in endocrine and nonendocrine lung carcinomas. Am J Clin Pathol 1988; 90: 641–652. [DOI] [PubMed] [Google Scholar]
- 140. Fresvig A, Qvigstad G, Halvorsen TB, et al. Neuroendocrine differentiation in bronchial carcinomas of classic squamous-cell type: an immunohistochemical study of 29 cases applying the tyramide signal amplification technique. Appl Immunohistochem Mol Morphol 2001; 9: 9–13. [PubMed] [Google Scholar]
- 141. Sorhaug S, Steinshamn S, Haaverstad R, et al. Expression of neuroendocrine markers in non-small cell lung cancer. APMIS 2007; 115: 152–163. [DOI] [PubMed] [Google Scholar]
- 142. Chen B, Hu B, Li W, et al. Transformation from NSCLC to SCLC: when did it happen? Lancet Oncol 2015; 16: e309. [DOI] [PubMed] [Google Scholar]
- 143. Stevens TP, McBride JT, Peake JL, et al. Cell proliferation contributes to PNEC hyperplasia after acute airway injury. Am J Physiol 1997; 272: L486–L493. [DOI] [PubMed] [Google Scholar]
- 144. Matsuki Y, Tanimoto A, Hamada T, et al. Histidine decarboxylase expression as a new sensitive and specific marker for small cell lung carcinoma. Mod Pathol 2003; 16: 72–78. [DOI] [PubMed] [Google Scholar]
- 145. Waldum HL, Rorvik H, Brenna E. The gastrointestinal tract and the lungs. Similarities with particular emphasis on the neuroendocrine cells. Sarcoidosis Vasc Diffuse Lung Dis 1996; 13: 63–65. [PubMed] [Google Scholar]
- 146. Tian H, Biehs B, Warming S, et al. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 2011; 478: 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bartsch DK, Albers MB, Lopez CL, et al. Bronchopulmonary neuroendocrine neoplasms and their precursor lesions in multiple endocrine neoplasia type 1. Neuroendocrinology 2016; 103: 240–247. [DOI] [PubMed] [Google Scholar]
- 148. Cheng CY, Nikitin AY. Neuroendocrine cells: potential cells of origin for small cell lung carcinoma. Cell Cycle 2011; 10: 3629–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Miskovic J, Brekalo Z, Vukojevic K, et al. Co-expression of TTF-1 and neuroendocrine markers in the human fetal lung and pulmonary neuroendocrine tumors. Acta Histochem 2015; 117: 451–459. [DOI] [PubMed] [Google Scholar]
- 150. Youngson C, Nurse C, Yeger H, et al. Oxygen sensing in airway chemoreceptors. Nature 1993; 365: 153–155. [DOI] [PubMed] [Google Scholar]
- 151. Dakhama A, Larsen GL, Gelfand EW. Calcitonin gene-related peptide: role in airway homeostasis. Curr Opin Pharmacol 2004; 4: 215–220. [DOI] [PubMed] [Google Scholar]
- 152. Sunday ME, Choi N, Spindel ER, et al. Gastrin-releasing peptide gene expression in small cell and large cell undifferentiated lung carcinomas. Hum Pathol 1991; 22: 1030–1039. [DOI] [PubMed] [Google Scholar]
- 153. Hauso O, Gustafsson BI, Waldum HL. Long slender cytoplasmic extensions: a common feature of neuroendocrine cells? J Neuroendocrinol 2007; 19: 739–742. [DOI] [PubMed] [Google Scholar]
- 154. Waldum HL, Nilsen OG, Nilsen T, et al. Long-term effects of inhaled nicotine. Life Sci 1996; 58: 1339–1346. [DOI] [PubMed] [Google Scholar]
- 155. Godwin JD, II, Brown CC. Comparative epidemiology of carcinoid and oat-cell tumors of the lung. Cancer 1977; 40: 1671–1673. [DOI] [PubMed] [Google Scholar]
- 156. Vollbrecht C, Werner R, Walter RF, et al. Mutational analysis of pulmonary tumours with neuroendocrine features using targeted massive parallel sequencing: a comparison of a neglected tumour group. Br J Cancer 2015; 113: 1704–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Miller RR, Muller NL. Neuroendocrine cell hyperplasia and obliterative bronchiolitis in patients with peripheral carcinoid tumors. Am J Surg Pathol 1995; 19: 653–658. [DOI] [PubMed] [Google Scholar]
- 158. Sorhaug S, Steinshamn S, Nilsen OG, et al. Chronic inhalation of carbon monoxide: effects on the respiratory and cardiovascular system at doses corresponding to tobacco smoking. Toxicology 2006; 228: 280–290. [DOI] [PubMed] [Google Scholar]
- 159. Morse MA, Hecht SS, Chung FL. Inhibition of tobacco-specific nitrosamine 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumors and DNA methylation in F344 rats and A/J mice by phenethyl isothiocyanate. Basic Life Sci 1990; 52: 345–350. [DOI] [PubMed] [Google Scholar]