

Abstract
AIM
To assess the effects of hepatitis E virus (HEV) on the production of type I interferons (IFNs) and determine the underlying mechanisms.
METHODS
We measured the production of interferon (IFN)-alpha and -beta (-α/β) in genotype 3 HEV-infected C3A cells at different time points (0, 8, 12, 24, 48, 72 and 120 h) by enzyme-linked immunosorbent assay (ELISA). The expression levels of IFN-stimulated gene (ISG)15 in HEV-infected C3A cells at different time points were tested by western blotting. The plasmid-expressing open reading frame 3 (ORF3) or control plasmids (green fluorescent protein-expressing) were transfected into C3A cells, and the levels of IFN-α/β and ISG15 were evaluated, respectively. Furthermore, the plasmid-expressing ISG15 or small interfering RNA-inhibiting ISG15 was transfected into infected C3A cells. Then, the production of IFN-α/β was also measured by ELISA.
RESULTS
We showed that genotype 3 HEV could enhance the production of IFN-α/β and induce elevation of ISG15 in C3A cells. HEV ORF3 protein could enhance the production of IFN-α/β and the expression of ISG15. Additionally, ISG15 silencing enhanced the production of IFN-α/β. Overexpression of ISG15 resulted in the reduction of IFN-α/β.
CONCLUSION
HEV may promote production of IFN-α/β and expression of ISG15 via ORF3 in the early stages, and increased ISG15 subsequently inhibited the production of IFN-α/β.
Keywords: Open reading frame 3, Interferon-stimulated gene 15, Interferon-alpha, Hepatitis E virus, Interferon-beta
Core tip: To date, few studies have investigated the role of ISG15 in hepatitis E virus (HEV) infection and the impact of ISG15 on interferons (IFN) production. This study showed that HEV could inhibit the level of type I IFN through regulating the expression of IFN-stimulated gene (ISG)15, which may attenuate the IFN response, allowing for successful infection of their hosts. The present study enhances the understanding of the interaction between ISG15 and HEV in the host innate immune response, which may provide useful insight for the development of new antiviral drugs and antiviral strategies.
INTRODUCTION
Hepatitis E virus (HEV) infection is one of the most common causes of acute hepatitis or sporadic acute hepatitis in many parts of the world[1]. At least 20 million HEV infections occur annually, which are mostly self-limiting and have different clinical manifestations, such as asymptomatic, acute liver failure and rare chronicity[2,3]. In some cases, HEV infections can cause up to 30% mortality in pregnant women[4] and may result in chronic hepatitis in immunocompromised individuals, such as those receiving organ transplants or chemotherapy, and individuals infected with human immunodeficiency virus[5-7]. Currently, at least four genotypes exist among HEV strains, of which HEV genotypes 1 and 2 are obligate human pathogens and HEV genotypes 3 and 4 are mostly zoonotic[8]. Recently, a study found that a genotype 3 HEV isolated from a chronically infected patient could be adapted to grow in human C3A hepatoma cells[9].
HEV has a 7.2-kb single-stranded, positive-sense RNA genome containing three open reading frames (ORFs)[10]. The ORF1 protein comprises the functional domains of virions. ORF2 is the major structural protein of virions and encodes the viral capsid protein. And, ORF3 protein, a small molecule phosphoprotein, is involved in viral particle secretion[11]. A recent study found that chronic HEV infections were almost exclusively caused by the zoonotic genotype 3 HEV strains[12].
Type I interferons (IFNs) are key components of innate immunity and known to be the first-line defense against virus infection. IFN exerts many biological effects, such as antiviral and antitumor activity, and immune regulation by activating hundreds of downstream IFN-stimulated genes (ISGs)[13,14]. One of the most abundantly IFN-induced proteins is ISG15, which is encoded and expressed as a ubiquitin-like protein. ISG15 is covalently attached to target proteins through a C-terminal LRLRGG motif, in a process called ISGylation. ISGylation could alter protein properties directly by addition of ISG15, as well as by reducing the degradation of the target protein by competing with ubiquitin conjugation[15]. Previous studies have shown that ISG15 is critical for control of certain viral and bacterial infections and is linked to the process of budding or egress for many RNA virus families[16-18].
As few studies have focused on the role of ISG15 in HEV infection, we investigated whether HEV infection could regulate the expression of ISG15 and the impact of ISG15 on IFN production during HEV infection in the present study, which is of great significance to expand our understanding of the interaction between ISG15 and HEV pathogenesis.
MATERIALS AND METHODS
Cells and virus
C3A is a derivative of HepG2 cells (hepatoma cells). C3A cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA, United States) and cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (Gibco, Thermo Scientific, Waltham, MA, United States), penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37 °C with 5% CO2. The genotype 3 Kernow-C1 HEV infectious cDNA clone was a kind gift from the National Institute of Diagnostics and Vaccine Development in Infectious Diseases (NIDVD, Xiamen, China). The ATG start codon of ORF3 in Kernow-C1 HEV was altered to GTG to construct an HEV ORF3 mutant, which led to the loss of ORF3 protein expression.
Reagents and plasmids
Anti-ISG15 antibody was purchased from Signalway Antibody (42509, 1:1000 dilution; Pearland, TX, United States). AntiHEV ORF3-specific antibody was purchased from Abbiotec (250688, 1:1000; San Diego, CA, United States). Primary antibody against β-actin (#BM0627, 1:400 dilution) was purchased from Boster (Wuhan, China). We constructed a plasmid-expressing ORF3 (ORF3) as well as a plasmid-expressing ISG15 (ISG15). ORF3 and ISG15 fragments were cloned into the pEGFP-N1 vector (green fluorescent protein (GFP)-expressing, GenBank accession #55762) by digesting with Xho I and Hind III to construct the pORF3-EGFP plasmid (ORF3) and pISG15-EGFP plasmid (ISG15). All the clones were confirmed by restriction digestion and DNA sequencing.
HEV infection
Th cDNA clones were linearized with BglII, and the genomic RNA was transcribed using the MEGAscript Kit (Ambion, Thermo Scientific). C3A cells cultured in a six-well plate were transfected with capped RNA transcripts from Kernow-C1 HEV infectious cDNA clone using DMRIE-C reagent (Invitrogen, Grand Island, NY, United States) following the manufacturer’s protocol. The ratio of RNA to DMRIE-C was 2 μg:8 μL. After 6 h, the medium in the six-well plate was removed and replaced with serum-free medium. The cells and cell culture supernatants were harvested at different time points (0, 8, 12, 24, 48, 72 and 120 h) and used in experiments.
Cell transfection
C3A cells were plated in six-well microplates and transfected with a plasmid-expressing ORF3-EGFP (ORF3) or the HEV mutant, leading to the loss of ORF3 protein expression (ΔORF3) using Lipofectamine TM 3000 Transfection Reagent (Invitrogen, San Diego, CA, United States) for 48 h. Then, the cells and cell culture supernatants were harvested at different time points (0, 8, 12, 24, 48, 72 and 120 h) and used in experiments.
Small interfering RNA-mediated knockdown
The small interfering RNAs (siRNAs) targeted against ISG15 were purchased from Santa Cruz Biotechnologies (Dallas, TX, United States). C3A cells were transiently transfected with 20 nmol/L of siRNA using the DMRIE-C reagent (Invitrogen) according to the manufacturer’s protocol. Primers are shown in Table 1.
Table 1.
Primers used in this study
| Name | Primer | Sequence |
| β-actin | Forward | 5’- AGCGAGCATCCCCCAAAGTT -3’ |
| Reverse | 5’- GGGCACGAAGGCTCATCATT -3’ | |
| ISG15 | Forward | 5’- GCGCAGATCACCCAGAAGAT -3’ |
| Reverse | 5’- GTTCGTCGCATTTGTCCACC -3’ | |
| siRNA1 | Forward | 5’- UGUCGGUGUCAGAGCUGAATT -3’ |
| Reverse | 5’- UUCAGCUCUGACACCGACATT -3’ | |
| siRNA2 | Forward | 5’- GAUGCUGGCGGGCAACGAATT -3’ |
| Reverse | 5’- UUCGUUGCCCGCCAGCAUCTT -3’ | |
| siRNA3 | Forward | 5’- UGAGCACCGUGUUCAUGAATT -3’ |
| Reverse | 5’- UUCAUGAACACGGUGCUCATT -3’ | |
| Negative control | Forward | 5’- UUCUCCGAACGUGUCACGUTT -3’ |
| Reverse | 5’- ACGUGACACGUUCGGAGAATT -3’ |
Enzyme-linked immunosorbent assay
The concentrations of IFN-alpha and -beta (-α/β) were evaluated in cell culture supernatants using a human IFN-α/β ELISA kit (Joyee Biotechnics, Shanghai, China). All samples were tested in triplicate.
Western blot analysis
Cells were lysed in RIPA buffer (Beyotime Biotechnology, Shanghai, China), and the protein concentration was determined using an enhanced BCA protein assay kit (Beyotime Biotechnology). Ten micrograms of each protein sample were separated on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and subsequently transferred to polyvinylidene difluoride membranes. Samples were then incubated with specific primary antibodies overnight, following incubation with an antimouse secondary antibody solution for 2 h at room temperature. The blotted membranes were exposed to film, and the protein bands were visualized by a scanner system.
Real-time reverse transcriptase-polymerase chain reaction
Total RNA from C3A cells was isolated using Trizol (Thermo Scientific) and reverse-transcribed into cDNA using the TaqMan Reverse Transcription Reagents (Thermo Scientific). The mRNA level of ISG15 was quantified by real-time PCR using SYBR master mix (Thermo Scientific). The PCR amplification conditions included 1 cycle of 95 °C for 2 min, and 40 cycles of 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 30 s; the Roche Light Cycler 480 II Real-Time PCR System was used. Threshold cycle (CT) values were determined by RT-PCR and normalized by the housekeeping gene β-actin. The mRNA abundance was calculated using the 2−ΔΔCt method and expressed as fold-change. Primers are shown in Table 1.
Statistical analysis
All the data are presented as mean ± SD. Statistical differences between the samples and the controls were assessed by the Student’s t-test. P < 0.05 was considered statistically significant.
RESULTS
HEV infection enhances the production of IFN-α/β and induces elevation of ISG15 protein in C3A cells
In this study, C3A cells were transfected with HEV RNA. To determine whether HEV infection could affect the production of type I IFN, the concentrations of IFN-α/β were evaluated in cell culture supernatants at different time points (0, 8, 12, 24, 48, 72 and 120 h) by ELISA. The levels of IFN-α/β in the HEV infection group were significantly increased at each time point (P < 0.05), and the levels of IFN-α/β reached the maximum at 24 h, while no significant changes were observed in the control group (Figure 1A).
Figure 1.
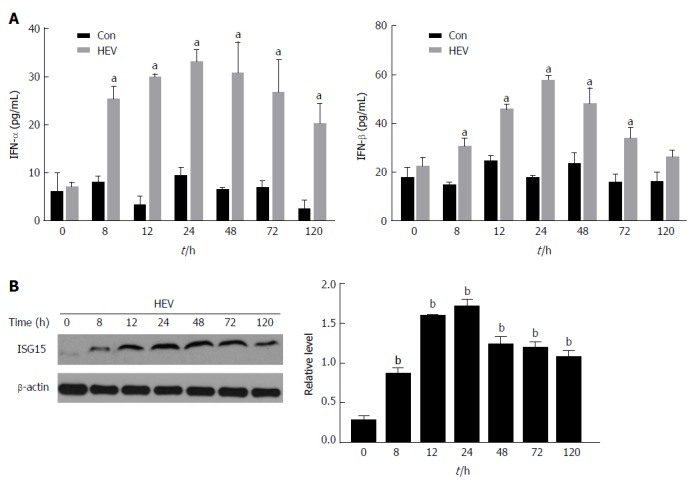
Hepatitis E virus infection enhances the production of Interferon-α and -β and induces elevation of Interferon-stimulated gene 15 protein in C3A cells. A: The concentrations of IFN-α and β were evaluated in cell culture supernatants at different time points (0, 8, 12, 24, 48, 72 and 120 h) by enzyme-linked immunosorbent assay; B: The expression of ISG15 protein replication was detected at different time points (0, 8, 12, 24, 48, 72 and 120 h) by western blotting. The data are expressed as mean ± standard error of the mean. aP < 0.05, bP < 0.01. HEV: Hepatitis E virus; IFN: Interferon; ISG: IFN-stimulated gene.
Next, we identified whether HEV could induce ISG15. ISG15 protein expression was detected at different time points by western blotting. It showed HEV enhanced the expression of ISG15 (P < 0.01) and ISG15 protein expression reach the maximum at 24 h as compared to the control group (Figure1B).
HEV ORF3 protein enhances the production of IFN-α/β and the expression of ISG15 protein in C3A cells
To investigate how HEV enhances the production of IFN-α/β and induces elevation of ISG15, we constructed a plasmid-expressing ORF3-EGFP (ORF3) and the HEV mutant leading to the loss of ORF3 protein expression (ΔORF3). C3A cells were transfected with ORF3 or ΔORF3 respectively, and the production of IFN-α/β and the expression of ISG15 protein were detected. The production of IFN-α/β was significantly increased at each time point in the group transfected with ORF3 plasmid (ORF3 group) compared to the ORF3-lacking group (ΔORF3 group) (P < 0.05). In addition, the concentrations of IFN-α/β increased the maximum at 24 h (Figure 2A). The ORF3 group could induce higher ISG15 protein expression than ΔORF3 group (P < 0.01). ISG15 protein was poorly expressed in the early stage, from 0 h to 12 h, and significantly increased at 24 h, reaching the maximum at 72 h (Figure 2B).
Figure 2.
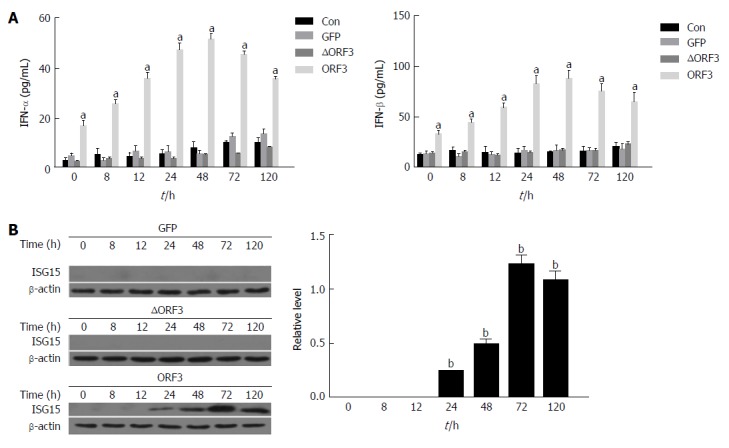
Hepatitis E virus open reading frame 3 protein enhances the production of Interferon-α and β and the expression of Interferon-stimulated gene 15 protein in C3A cells. C3A cells were transfected with 2 μg of either pORF3-EGFP (ORF3) or pEGFP-N1 empty vector (GFP), and the HEV mutant, leading to the loss of ORF3 protein expression (ΔORF3) for 48 h, untreated cells were used as the control (Con). Then, the cells and cell culture supernatants were harvested at different time points (0, 8, 12, 24, 48, 72 and 120 h). A: The concentrations of IFN-α and β were evaluated in cell culture supernatants at different time points (0, 8, 12, 24, 48, 72, 120 h) by enzyme-linked immunosorbent assay; B: The expression of ISG15 protein was detected at different time points (0, 8, 12, 24, 48, 72 and 120 h) by western blotting. The data are expressed as mean ± standard error of the mean. aP < 0.05, bP < 0.01. GFP: Green fluorescent protein; HEV: Hepatitis E virus; IFN: Interferon; ISG: IFN-stimulated gene; ORF: Open reading frame.
ISG15 silencing enhances the production of IFN-α/β in C3A cells
To investigate the levels of IFN-α/β when ISG15 was inhibited, we first constructed three siRNAs that could inhibit the expression of ISG15 and then transfected them into C3A cells, respectively. Then, the mRNA and protein levels of ISG15 were detected to determine which siRNA had the best inhibitory effect (Figure 3A and B). The siRNA2 was found to have the best inhibitory effect on ISG15. Next, the siRNA2 was transfected into C3A cells that had been pretreated with HEV RNA. The expression of IFN-α was significantly increased in the HEV-infected C3A cells transfected with siISG15 compared to the HEV-infected cells (P < 0.05). Consistently, the expression of IFN-β showed the same trend (P < 0.01) (Figure 3C).
Figure 3.
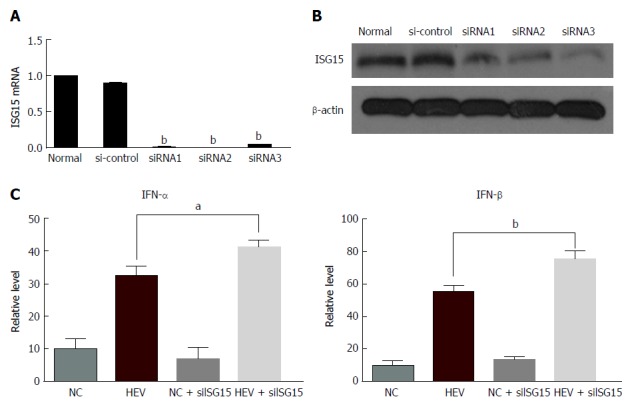
Interferon-stimulated gene 15 silencing enhances the production of Interferon-α/β in C3A cells. Three siRNA inhibiting ISG15 (siISG15) and control-siRNA (si-control) were constructed, and C3A cells were cotransfected with 20 nmol/L ISG15-targeted siRNA (siISG15) or control-siRNA (si-control) along with HEV RNA. A: The mRNA level of ISG15 was estimated by reverse transcription-PCR; B: The protein level of ISG15 was estimated by western blotting. Next, the siRNA2 was transfected into C3A cells that had been pretreated with HEV RNA, and the impact of siISG15 on IFN was investigated; C: The level of IFN-α/β was tested by enzyme-linked immunosorbent assay. The data are expressed as mean ± standard error of the mean. aP < 0.05, bP < 0.01. HEV: Hepatitis E virus; IFN: Interferon; ISG: IFN-stimulated gene; si: Small interfering.
Overexpression of ISG15 results in the reduction of IFN-α/β in C3A cells
To investigate the effect of ISG15 overexpression on IFN-α/β, the plasmid -expressing ISG15 was constructed and transfected into C3A cells. The level of ISG15 was detected by western blotting. The cells transfected with ISG15 plasmid had a considerably higher ISG15 level than the control cells (Figure 4A). Then, the production of IFN-α/β was tested when cells were transfected with ISG15 plasmid and HEV RNA. The level of IFN-α/β was decreased when the cells were treated with HEV and ISG15 plasmid (P < 0.05) (Figure 4B), which indicated over-expression of ISG15 may result in reduction of IFN-α/β.
Figure 4.
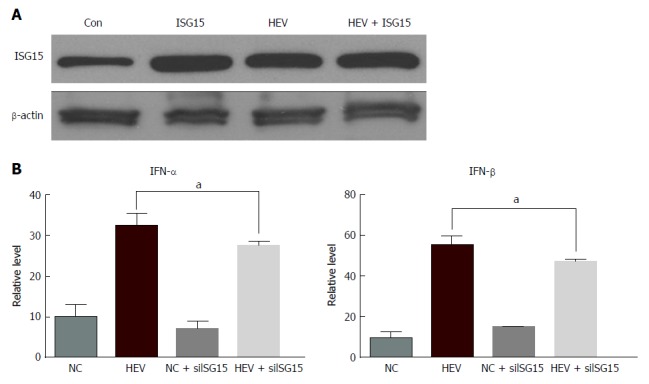
Over-expression of Interferon-stimulated gene 15 results in the reduction of Interferon-α/β in C3A cells. C3A cells were transfected with 2 mg ISG15-EGFP plasmid (ISG15) along with HEV RNA. A: ISG15 protein expression was detected by western blotting; B: The levels of IFN-α/β were detected by enzyme-linked immunosorbent assay. aP < 0.05. HEV: Hepatitis E virus; IFN: Interferon; ISG: IFN-stimulated gene.
DISCUSSION
The innate immune system is the first line of defense against invading pathogens. Type I IFNs, such as IFN-α/β, make up part of the innate immunity system and are critical for innate immunity against viral infection. ISG15, a member of the ISGs induced by IFN, has an important role in the regulation of antiviral immunity. In this study, we demonstrated that HEV infection could enhance the production of IFN-α/β and induce elevation of ISG15 protein. ORF3 protein may be responsible for the enhancement of IFN-α/β and ISG15 by HEV. Furthermore, ISG15 silencing enhanced the production of IFN-α/β in C3A cells. Over-expression of ISG15 resulted in the reduction of IFN-α/β in C3A cells. Collectively, these results indicated that HEV could induce the production of IFN and ISG15, and the increased ISG15 in turn reduced the level of IFN, which revealed a possible correlation between HEV infection and IFN production by regulating the expression of ISG15.
In our study, we first found that HEV infection could enhance the production of IFN-α/β and induce the elevation of ISG15 protein. A previous study demonstrated that most viruses have the ability to regulate the IFN response during infection[19]. When cells are infected by viruses, such as influenza viruses, the activated host innate immune response will lead to secretion of type I IFN. Then, the induced IFN exerts its antiviral activity[20]. Over the last decade, numerous studies have reported that ISG15 plays a crucial role against viral infection[21,22]. However, few studies have investigated the effects of HEV on ISG15. In the present study, we found that the expression of ISG15 and the IFN level were simultaneously increased during HEV infection, which indicated that HEV could induce ISG15 expression in C3A cells. Recent studies have revealed that ISG15, as an innate immune protein with broad spectrum antiviral activity, continues to accumulate rapidly when the host is infected by a virus[23], which is consistent with the findings in this research.
HEV consists of three ORFs. Genotype 1 HEV strain could inhibit type I IFN induction by ORF1 products[24]. However, the ORF3 products of genotype 1 HEV have been reported to enhance the production of IFN[25]. Because we found that genotype 3 HEV could induce the level of IFN, we next tested the impact of HEV ORF3 on IFN. We found that HEV ORF3 could increase the levels of IFN-α/β, which indicates that HEV ORF3 is responsible for the regulation of IFN. Similarly, we investigated whether HEV ORF3 could affect the expression of ISG15. As we expected, the level of ISG15 was increased by HEV ORF3. Previous studies have shown that ORF3 protein is associated with the egress of HEV[26]. As ISG15 is also associated with the process of egress for many RNA virus families[27], we supposed that HEV ORF3 may regulate the egress of HEV through ISG15, which requires further experiments to confirm.
Furthermore, the possible correlation between enhanced levels of IFN and ISG15 during HEV infection was investigated. ISG15 silencing enhanced the production of IFN-α/β, and over-expression of ISG15 resulted in the reduction of IFN-α/β in C3A cells, indicating that ISG15 is a negative regulator of IFN. It has been indicated that the absence of intracellular ISG15 in the ISG15-deficient patients prevents the accumulation of USP18, which is a potent negative regulator of IFN-α/β signaling, thus resulting in the enhancement and amplification of IFN-α/β responses and increase of viral resistance in humans[15,28].
In addition, it was found that ISG15 over-expression inhibited induction of IFN-β by HCV infection[29]. Evidence revealing high hepatic ISG15 levels was associated with low antiviral IFN-response during the early phase of antiviral therapy[30], supporting the notion that ISG15 is a negative regulator of the IFN system. However, this raises the question of why the levels of IFN-α/β could be reduced by HEV infection via ISG15. As we know, the IFN production is always enhanced to promote an antiviral state in an infected host, but viruses have evolved many strategies to antagonize the induction of IFNs. As there is little knowledge about how HEV inhibits induction of IFN, the result that the level of IFN could be inhibited by HEV via ISG15 will enhance our fundamental knowledge of the mechanisms of induction and evasion of type I IFN responses by HEV.
In conclusion, the findings of the present study showed that HEV could inhibit the level of type I IFN through regulating the expression of ISG15, which may attenuate the IFN response, allowing for successful infection of the host. The present study enhances the understanding of the interaction between ISG15 and HEV in the host innate immune response, which may provide useful insight for the development of new antiviral drugs and antiviral strategies.
ARTICLE HIGHLIGHTS
Research background
Hepatitis E virus (HEV) infection is one of the most common causes of acute hepatitis or sporadic acute hepatitis in the world. At least 20 million HEV infections occur annually, which may result in chronic hepatitis in immunocompromised individuals. However, the mechanism of HEV pathogenesis remains obscure.
Research motivation
Over the last decade, numerous studies have reported that interferon (IFN)-stimulated gene (ISG)15 plays a crucial role against viral infection. However, few studies have investigated the effects of HEV on ISG15.
Research objectives
In the present study, we investigated whether HEV infection could regulate the expression of ISG15 and the impact of ISG15 on IFN production during HEV infection, which is of great significance to expand our understanding of the interaction between ISG15 and HEV pathogenesis.
Research methods
In this study, C3A cells were first transfected with genotype 3 HEV RNA. The production of IFN-alpha and -beta (IFN-α/β) at different time points (0, 8, 12, 24, 48, 72 and 120 h) were measured by enzyme-linked immunosorbent assay (ELISA). The expression levels of ISG15 in HEV-infected C3A cells at different time points were tested by western blotting. Then, C3A cells were transfected with plasmid-expressing open reading frame 3 (ORF3) or control plasmids, the levels of IFN-α/β and ISG15 was evaluated, respectively. Next, the plasmid-expressing ISG15 or small interfering RNA-inhibiting ISG15 was transfected into infected C3A cells. The production of IFN-α/β was also measured by ELISA.
Research results
In this study, we demonstrated that HEV infection could enhance the production of IFN-α/β and induce elevation of the ISG15 protein. ORF3 protein may be responsible for the enhancement of IFN-α/β and ISG15 by HEV. Furthermore, ISG15 silencing enhanced the production of IFN-α/β in C3A cells. Over-expression of ISG15 resulted in the reduction of IFN-α/β in C3A cells.
Research conclusions
The findings of the present study showed that HEV could inhibit the level of type I IFN through regulating the expression of ISG15, a finding which may enhance the understanding of the interaction between ISG15 and HEV in the host innate immune response, and provide useful insight for the development of new antiviral drugs and antiviral strategies.
Research perspectives
The future research will focus on whether HEV ORF3 could regulate the egress of HEV through ISG15.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C, C
Grade D (Fair): D
Grade E (Poor): 0
Supported by the National Natural Science Foundation of China, No. 81570540.
Institutional review board statement: The study was reviewed and approved by the Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology Institutional Review Board.
Conflict-of-interest statement: All authors declare that they have no conflict of interest.
Data sharing statement: No additional data are available.
ARRIVE guidelines statement: The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 30, 2018
First decision: April 11, 2018
Article in press: April 23, 2018
P- Reviewer: Domagalski K, Ierardi E, Inoue K, Tomizawa M S- Editor: Gou ZM L- Editor: Filipodia E- Editor: Yin SY
Contributor Information
Min Wang, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei Province, China.
Ying Huang, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou 510700, Guangdong Province, China.
Man He, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei Province, China.
Wen-Ju Peng, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei Province, China.
De-Ying Tian, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei Province, China. dytian@tjh.tjmu.edu.cn.
References
- 1.Kamar N, Bendall R, Legrand-Abravanel F, Xia NS, Ijaz S, Izopet J, Dalton HR. Hepatitis E. Lancet. 2012;379:2477–2488. doi: 10.1016/S0140-6736(11)61849-7. [DOI] [PubMed] [Google Scholar]
- 2.Dalton HR, Bendall R, Ijaz S, Banks M. Hepatitis E: an emerging infection in developed countries. Lancet Infect Dis. 2008;8:698–709. doi: 10.1016/S1473-3099(08)70255-X. [DOI] [PubMed] [Google Scholar]
- 3.Hoofnagle JH, Nelson KE, Purcell RH. Hepatitis E. N Engl J Med. 2012;367:1237–1244. doi: 10.1056/NEJMra1204512. [DOI] [PubMed] [Google Scholar]
- 4.Shalimar, Acharya SK. Hepatitis e and acute liver failure in pregnancy. J Clin Exp Hepatol. 2013;3:213–224. doi: 10.1016/j.jceh.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalton HR, Bendall RP, Keane FE, Tedder RS, Ijaz S. Persistent carriage of hepatitis E virus in patients with HIV infection. N Engl J Med. 2009;361:1025–1027. doi: 10.1056/NEJMc0903778. [DOI] [PubMed] [Google Scholar]
- 6.Kamar N, Garrouste C, Haagsma EB, Garrigue V, Pischke S, Chauvet C, Dumortier J, Cannesson A, Cassuto-Viguier E, Thervet E, et al. Factors associated with chronic hepatitis in patients with hepatitis E virus infection who have received solid organ transplants. Gastroenterology. 2011;140:1481–1489. doi: 10.1053/j.gastro.2011.02.050. [DOI] [PubMed] [Google Scholar]
- 7.Geng Y, Zhang H, Huang W, J Harrison T, Geng K, Li Z, Wang Y. Persistent hepatitis e virus genotype 4 infection in a child with acute lymphoblastic leukemia. Hepat Mon. 2014;14:e15618. doi: 10.5812/hepatmon.15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song YJ, Park WJ, Park BJ, Lee JB, Park SY, Song CS, Lee NH, Seo KH, Kang YS, Choi IS. Hepatitis E virus infections in humans and animals. Clin Exp Vaccine Res. 2014;3:29–36. doi: 10.7774/cevr.2014.3.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shukla P, Nguyen HT, Torian U, Engle RE, Faulk K, Dalton HR, Bendall RP, Keane FE, Purcell RH, Emerson SU. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc Natl Acad Sci U S A. 2011;108:2438–2443. doi: 10.1073/pnas.1018878108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Y, Zhao C, Tian Y, Xu N, Wang Y. Characteristics and Functions of HEV Proteins. Adv Exp Med Biol. 2016;948:17–38. doi: 10.1007/978-94-024-0942-0_2. [DOI] [PubMed] [Google Scholar]
- 11.Ahmad I, Holla RP, Jameel S. Molecular virology of hepatitis E virus. Virus Res. 2011;161:47–58. doi: 10.1016/j.virusres.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamar N, Rostaing L, Izopet J. Hepatitis E virus infection in immunosuppressed patients: natural history and therapy. Semin Liver Dis. 2013;33:62–70. doi: 10.1055/s-0033-1338115. [DOI] [PubMed] [Google Scholar]
- 13.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- 15.Zhang D, Zhang DE. Interferon-stimulated gene 15 and the protein ISGylation system. J Interferon Cytokine Res. 2011;31:119–130. doi: 10.1089/jir.2010.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao C, Hsiang TY, Kuo RL, Krug RM. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc Natl Acad Sci U S A. 2010;107:2253–2258. doi: 10.1073/pnas.0909144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giannakopoulos NV, Arutyunova E, Lai C, Lenschow DJ, Haas AL, Virgin HW. ISG15 Arg151 and the ISG15-conjugating enzyme UbE1L are important for innate immune control of Sindbis virus. J Virol. 2009;83:1602–1610. doi: 10.1128/JVI.01590-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okumura A, Lu G, Pitha-Rowe I, Pitha PM. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc Natl Acad Sci USA. 2006;103:1440–1445. doi: 10.1073/pnas.0510518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katze MG, He Y, Gale M Jr. Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 20.García-Sastre A. Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 2011;162:12–18. doi: 10.1016/j.virusres.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skaug B, Chen ZJ. Emerging role of ISG15 in antiviral immunity. Cell. 2010;143:187–190. doi: 10.1016/j.cell.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeon YJ, Yoo HM, Chung CH. ISG15 and immune diseases. Biochim Biophys Acta. 2010;1802:485–496. doi: 10.1016/j.bbadis.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao C, Collins MN, Hsiang TY, Krug RM. Interferon-induced ISG15 pathway: an ongoing virus-host battle. Trends Microbiol. 2013;21:181–186. doi: 10.1016/j.tim.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nan Y, Yu Y, Ma Z, Khattar SK, Fredericksen B, Zhang YJ. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J Virol. 2014;88:11924–11932. doi: 10.1128/JVI.01935-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nan Y, Ma Z, Wang R, Yu Y, Kannan H, Fredericksen B, Zhang YJ. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J Virol. 2014;88:8696–8705. doi: 10.1128/JVI.01228-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagashima S, Takahashi M, Jirintai, Tanaka T, Yamada K, Nishizawa T, Okamoto H. A PSAP motif in the ORF3 protein of hepatitis E virus is necessary for virion release from infected cells. J Gen Virol. 2011;92:269–278. doi: 10.1099/vir.0.025791-0. [DOI] [PubMed] [Google Scholar]
- 27.Chen BJ, Lamb RA. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology. 2008;372:221–232. doi: 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature. 2015;517:89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnaud N, Dabo S, Akazawa D, Fukasawa M, Shinkai-Ouchi F, Hugon J, Wakita T, Meurs EF. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011;7:e1002289. doi: 10.1371/journal.ppat.1002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Broering R, Zhang X, Kottilil S, Trippler M, Jiang M, Lu M, Gerken G, Schlaak JF. The interferon stimulated gene 15 functions as a proviral factor for the hepatitis C virus and as a regulator of the IFN response. Gut. 2010;59:1111–1119. doi: 10.1136/gut.2009.195545. [DOI] [PubMed] [Google Scholar]