

Abstract
3,3′,4,4′,5-pentachlorobiphenyl (PCB126), a dioxin-like PCB, elicits toxicity through a wide array of noncarcinogenic effects, including metabolic syndrome, wasting, and nonalcoholic fatty-liver disease. Previously, we reported decreases in the transcription of several enzymes involved in gluconeogenesis, before the early onset of lipid accumulation. Hence, this study was aimed at understanding the impact of resultant decreases gluconeogenic enzymes on growth, weight, and metabolism in the liver, upon extended exposure. Male Sprague Dawley rats (75–100 g), fed a defined AIN-93G diet, were injected (ip) with single dose of soy oil (5 ml/kg body weight; n = 14) or PCB126 (5 µmol/kg; n = 15), 28 days, prior euthanasia. A subset of rats from each group were fasted for 12 h (vehicle [n = 6] and PCB126 [n = 4]). Rats only showed significant weight loss between days 14 and 28 (p < .05) and some mortality (p = .0413). As in our previous studies, the expression levels of enzymes involved in gluconeogenesis (Pepck-c, G6Pase, Sds, Pc, and Ldh-A) and glycogenolysis (Pygl) were strongly downregulated. The decreased expression of these enzymes in PCB126-treated rats after a 12 h fast decreased hepatic glucose production from glycogen and gluconeogenic substrates, exacerbating the hypoglycemia. Additionally, PCB126 caused hepatic steatosis and decreased the expression of the transcription factor Pparα and its targets, necessary for fatty-acid oxidation. The observed metabolic disruption across multiple branches of fasting metabolism resulted from inhibition in the activation of enzyme AMPK and transcription factor CREB signaling, necessary for “sensing” energy-deprivation and the induction of enzymes that respond to the PCB126 triggered fuel crisis in liver.
Keywords: polychlorinated biphenyls, carbohydrate metabolism, lipid metabolism, intermediary metabolism, liver, metabolic disruption
Polychlorinated biphenyls (PCBs) are broadly classified as dioxin-like and nondioxin-like congeners based on their mechanistic similarities in toxicity to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin, by activating the aryl hydrocarbon receptor (AhR) (Ahlborg and Hanberg, 1994). PCB126 (3,3′,4, 4′,5-pentachlorobiphenyl), a potent AhR agonist and a dioxin-like toxicant with a toxic equivalency factor (TEF) of 0.1 was recently upgraded to Group 1 Human Carcinogen by the International Agency for Research on Cancer (Lauby-Secretan et al., 2013, 2016). Upon ligand binding and activation by PCB126, the cytosolic nuclear transcription factor AhR, translocates into the nucleus and induces changes in expression of genes and proteins that eventually cause toxicity (Bandiera et al., 1982; Okey, 2007). PCB126 exposure mainly occurs through food, however other minor routes of exposure through air and water have also been described (Ampleman et al., 2015). PCB126 is resistant to metabolic elimination and bio-accumulates in tissues such as liver and adipose.
Exposure and bioaccumulation of persistent organic pollutants (POPs), including dioxin-like PCBs is proving to be significant risk factor for chronic metabolic diseases that pose a great threat to public health (Ruzzin, 2012; Ruzzin et al., 2010; Thayer et al., 2012). Considering new evidence that environmental exposure to POPs is associated with a broader range of metabolic diseases other than obesity, the definition of “obesogen” is now expanded to “metabolism disrupting chemical” (Grun and Blumberg, 2009; Heindel et al., 2015). This facilitates the classification of chemicals that exclusively target liver metabolism and cause liver lipid abnormalities, such as fatty liver or various forms of steatohepatitis (eg, nonalcoholic vs alcoholic) (Heindel et al., 2017; Kaiser et al., 2012; Wahlang et al., 2013). The body burden of dioxin-like PCBs such as PCB126 is significantly associated with nonalcoholic fatty-liver disease (NAFLD) along with a combined risk for obesity, diabetes, and hypertension (Cave et al., 2010; Donat-Vargas et al., 2014; Everett et al., 2007, 2011; Gauthier et al., 2014; Perkins et al., 2016; Silverstone et al., 2012).
Besides the role of liver in detoxification of xenobiotics, the liver is the principal tissue/site for glucose and lipid homeostasis and responds to various hormones to maintain energy homeostasis during both post- and preprandial metabolism. During the fed state, the liver responds to insulin by synthesizing glycogen (glycogenesis) as a reserve for glucose and increasing lipogenesis (Granner, 2015; O’Brien and Granner, 1996). In contrast, fasting induces glycogenolysis, gluconeogenesis and fatty-acid oxidation, in response to glucagon. Although glycogen acts as early substrate for glucose production during early fasting (glycogenolysis), lactate, pyruvate, glycerol, and certain amino acids are substrates for endogenous hepatic glucose production (HGP) during longer hours of fasting (gluconeogenesis) (Owen et al., 2002; Pilkis and Granner, 1992). Furthermore, liver also meets energy requirements through β-oxidation of fatty acids during fasting. The fasting physiology of liver is orchestrated by the activation of multiple transcription factors and their coactivators that control the specific and timely transcription of functional enzymes necessary for fasting metabolism (Rui, 2014). Toxicant induced perturbations of these tightly regulated transcriptional programs in the liver likely results in metabolic dyshomeostasis that leads to NAFLD and consequential metabolic syndrome (Cave et al., 2016; Heindel et al., 2017). The most studied genes induced by dioxin-like chemicals are the cytochrome P-450 monooxygenases, which respond rapidly to the presence of pollutants (Dalton et al., 2002; Puga et al., 2009; Swanson, 2002). However, the significance of changes in the expression of several other genes and their role in the observed toxicity is less understood (Boverhof et al., 2006; Forgacs et al., 2013; Lo et al., 2011; Ovando et al., 2010; Safe et al., 1998). Despite clear pathological outcomes with compromised liver function, a significant knowledge gap exists in the understanding of the key events in mechanisms of steatosis (Kaiser et al., 2012).
Several animal studies using rodent models reported hypoglycemia, wasting disorders, altered lipid profiles and ectopic lipid deposition in the liver (fatty liver) upon repeated exposure to multiple doses of dioxin-like chemicals (NTP, 2006; Seefeld and Peterson, 1984; Viluksela et al., 1998; Weber et al., 1995). However, in our previous studies to understand the roles of oxidative stress and micronutrient alteration in causing fatty-liver induction, rats injected with a single dose of PCB126 (5 μmol/kg) did not show any changes in growth, feed consumption or general overall health compared with the control animals after 14 days (Gadupudi et al., 2016a,b; Klaren et al., 2015; Lai et al., 2011, 2013). In a separate study specifically aimed at understanding the time-course of PCB126-induced liver steatosis using the same dose and time, we reported early transcriptional downregulation of several enzymes necessary for gluconeogenesis and fatty-acid oxidation with no changes in bodyweight or feed consumption (Gadupudi et al., 2016a). Since gluconeogenesis and fatty-acid oxidation are fasting-relevant biological processes, in the current study we asked the question(s) of whether this dose of PCB126 and decrease in the transcription of gluconeogenic and fatty-acid oxidation enzymes would (1) impact the normal growth of the animal and also impact (2) intermediate metabolism during fasting. To this end, we have injected male rats, with single dose of PCB126 (5 μmol/kg) and monitored their growth over 4 weeks of exposure. To test the effects of fasting, a subset of these PCB126 exposed and control animals were restricted from feeding for 12 h, prior to euthanasia.
MATERIALS AND METHODS
Animal studies
All experiments were conducted with the approval from Institutional Animal Care and Use Committee of the University of Iowa. Male Sprague Dawley (SD) rats weighing 75–100 g at an age of 4–5 weeks were fed a defined AIN-93G diet purchased from Envigo (Indianapolis, Indiana) and acclimatized to the powdered diet and animal housing for 1 week. Each animal was individually housed in wire hanging cages to monitor feed consumption and food wastage randomly assigned, with free access to feed and water and in a controlled environment of 22°C in a 12-h light-dark cycle. The PCB126 used in the injections was prepared by an improved Suzuki-coupling method, obtained from the Synthesis Core, University of Iowa Superfund Research Program (Lehmler and Robertson, 2001). Detailed synthesis and characterization information about this compound can be found in the supplementary materials (Supplementary Figs. 9 and 10). Soy oil, used to prepare the PCB126 and vehicle injections, was purchased from Envigo and was the same oil used to formulate the diet. Animals received a single ip injection of soy oil (5 ml/kg body weight; n = 14) or PCB126 at a dose 5 µmol/kg (1.63 mg/kg body weight; n = 15) body weight after 1 week of acclimatization (Supplementary Figure 1). This dose of PCB126 was used previously in several studies with no observable effects on growth or overt toxicity (Gadupudi et al., 2016a; Lai et al., 2011, 2013; Robertson et al., 2007). The weights of the animals and feed consumed were monitored every alternate day until the end of the study. Four weeks after injection and 12 h prior to euthanasia, feed was withdrawn from a subgroup of the PCB126-injected (n = 4) and the vehicle-only-injected rats (n = 6) for an overnight fast (Supplementary Figure 1). All the animals in both “fed” and “fasted” groups were euthanized in the following morning, using carbon-dioxide asphyxiation followed by thoracotomy. After euthanasia, whole blood was obtained from the heart and processed as described below. The livers were harvested and flash frozen in liquid nitrogen and stored at –80°C until further analysis. For histopathological analysis, slices of liver tissue were formalin-fixed in cassettes for preparing paraffin blocks or frozen over liquid nitrogen after embedding in optimal cutting temperature compound for frozen sections.
Blood collection and serum analysis
Whole blood samples from the hearts were collected into S-Monovette tubes designed for harvesting serum, after euthanasia. The blood was allowed to clot in the tubes and the serum fractions were separated by centrifugation at 1500 g for 10 min at 4°C. The serum samples were aliquoted and frozen at −80°C until further analysis. The measurements of serum glucose and other parameters were measured by Comparative Clinical Pathology Services LLC (CPath, Missouri).
Histology and special stains
Sections of liver were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned and routinely processed. Sections were stained with hematoxylin and eosin (H&E). Additional sections were also stained with periodic acid-Schiff (PAS) for measuring changes in glycogen (Sheehan and Hrapchak, 1987). All the sections were examined for necrosis, inflammation and additional histopathology; diagnosed and scored by a board certified veterinary pathologist.
Lipid staining and quantification
Sections prepared from frozen Optimal cutting temperature blocks of liver snippets were used for Oil-red-O (ORO) staining (Luna, 1992). Briefly, liver sections of 5-µm thickness were mounted on slides and allowed to air dry for 1–2 h. The sections were then fixed in 10% formalin and washed with tap water. The slides were dried and stained with (0.5% w/v) ORO in isopropanol for 15–30 min. Slides were washed and subsequently processed for hematoxylin counter staining. All the stained sections were examined by a board certified veterinary pathologist.
Glycogen and glucose measurements
Flash frozen rat liver tissue (approximately 50 mg) was homogenized (15 s intervals, 3 times) in ice-cold phosphate buffered saline (PBS) and sonicated with a probe sonicator on ice for further lysis (5 s intervals, 3 times). The samples were then centrifuged at 10 000 × g for 20 min at 4°C. In total 50 µl of supernatant was aliquoted and stored at −80°C to determine the protein concentrations. The remaining supernatant was boiled for 10 min at 70°C in a water bath to inactivate enzymes and subsequently stored at −80°C until further use.
For quantification, the frozen samples were thawed and diluted with 1× PBS (1:25). In total 30 µl of each sample was added to a clear bottom microtiter plate in duplicate along with either 70 µl of diluted amyloglucosidase (Sigma, St. Louis, Missouri; A1602) solution in sodium citrate buffer (0.15% v/v) or sodium citrate buffer alone to determine glycogen and glucose concentrations, respectively. In total 30 µl of both glucose (Sigma, St. Louis, Missouri; GAGO-20) and glycogen standards (Cayman Chemicals, Ann Arbor, Michigan; 700481) in the concentration range of 0–0.16 mg/ml prepared in 1× PBS were also incubated in separate wells. The samples and standards were incubated for 30 min at 47°C to facilitate the cleavage of glycogen polymers into glucose units by amyloglucosidase. Consequently, 100 µl of glucose oxidase (GO) reagent (Sigma, St. Louis, Missouri; GAGO-20), purchased from Sigma-Aldrich, was added to all samples and standards and incubated at 37°C for 1 h; 100 µl of 12 N sulfuric acid was then added to each well to terminate the reaction and the absorbance was measured at 540 nm using a plate reader (Tecan Inc, North Carolina). Free hepatic glucose levels in liver samples were calculated using a glucose standard curve. Total hepatic glucose, representing free glucose in addition to glucose generated by the hydrolysis of glycogen, was determined using the amyloglucosidase-treated samples. Hepatic glycogen levels were calculated by subtracting the absorbance of the free glucose from the absorbance of the total glucose following glycogen hydrolysis. Both glycogen and glucose concentrations, determined from their respective standard curves, were normalized to total protein quantified by Bradford assay (Biorad, California).
Gene expression analysis
Total RNA from each of the rat liver samples were extracted using the RNeasy extraction kit from Qiagen Inc (Valencia, California). Briefly, 20–30 mg of liver tissue was homogenized and RNA was extracted as described in the manufacturer’s protocol. Absorbance of the isolated RNA was determined spectrophotometrically at 260 and 280 nm. RNA samples with purity ratios (A260/A280) between 1.8 and 2.0 were used for generating complementary DNA (cDNA) employing a high-capacity cDNA reverse transcription kit from Applied Biosystems Inc. (Foster City, California), as described. The real-time quantitative PCR analysis was performed at an optimized cDNA template concentration of 50 ng, using an SYBR Green Master Mix kit supplied by Applied Biosystems Inc. The primers used to measure the transcript levels of various genes are listed in Supplementary Table 1 and were synthesized by Integrated DNA Technologies Inc. (Coralville, Iowa). Each sample was analyzed in duplicate. The amplification reaction was carried out with an Eppendorf RealPlex2 Mastercycler (Hamburg, Germany) using a program that started at 95°C for 10 min followed by 40 cycles of 2 step PCR cycle at 95°C for 15 s and 60°C for 1 min. Subsequently, a melting curve analysis was also performed, to check for any primer dimers. The transcript levels of all the quantified genes were normalized to the transcript levels of the reference gene hypoxanthine-guanine phosphoribosyl transferase (Hprt1) and relative expression was calculated and represented on the Y-axis. The expression level of each gene in a given sample was normalized to the mean of the biological control group of each study (oil vehicle, 5 ml/kg). The final transcript levels were quantified relative to the normalized transcript levels of the control group, using the Pfaffl method (Pfaffl, 2001).
Protein isolation and semiquantitative analysis
Liver snippets (approximatley 50 mg) were homogenized in lysis buffer (500 µl) containing phosphatase and protease inhibitors. The homogenates were sonicated for 2 min (in rounds of 10 s sonication/20 sec rest cycle) and incubated on ice for 30 min. Supernatants were collected after centrifugation at 10 000 rpm for 20 min at 4°C and stored at −20°C. Total protein was quantified by the Bradford assay as described by manufacturer (Biorad, California). Proteins were resolved by denaturing 10% SDS-polyacrylamide gel electrophoresis and transferred by wet blotting onto PVDF membranes (Millipore, Massachusetts). The membranes were blocked with 5% nonfat milk or 5% BSA in 25 mm Tris-HCl (pH 7.4), 150 mm NaCl and 0.1% Tween-20 (TBST) at room temperature for 1 h, and incubated with primary antibody overnight (1:1000) at 4°C and then with secondary antibody diluted in nonfat milk or BSA (1:3000) at room temperature for 1 h, as recommended by the supplier. The secondary antibodies conjugated with horseradish peroxidase were used to visualize the blots with a chemiluminescence Supersignal West Pico and West Femto kit (ThermoFisher Scientific, Massaschusssets) and observed on a LI-COR Odyssey chemiluminescent imager. For stripping of the antibodies bound to the blot, the blots were incubated with Restore western blot stripping buffer (Thermo Scientific, Massachusetts) for 30 min at 55°C with agitation and then washed liberally for 5 times with TBST. The membranes were probed with the primary antibodies to the following: Rabbit monoclonal antibodies against 5′ AMP-activated protein kinase (AMPK) phosphorylated at T172 (2535; Cell Signaling Technology, Massachussets), Total AMPK (2532; Cell Signaling Technology, Massachussets, and generously provided by Dr Dale Abel), cAMP response element-binding protein (CREB) phosphorylated at S133 (9198; Cell Signaling Technology, Massachusetts), CREB (9197; Cell Signaling Technology, Massachussets), goat polyclonal antibody against PEPCK-C (gift of Dr. Daryl J. Granner), rabbit polyclonal antibody against the β-actin (ab8227; Abcam, Massachusetts). The specificity of CREB phosphorylation (pCREB) and CREB antibodies was validated by probing for the protein levels of CREB and pCREB in unstimulated (−ve control) and stimulated (+ve control) neuronal cell extracts from SK-N-MC cells (Cell Signaling Technology, Massachussets), untreated or treated with Forskolin (30 µM), IBMX (0.5 mM) for 30 min to enhance CREB phosphorylation. Similarly, the specificities of antibodies against total AMPK and the T172 specific phosphorylated form of AMPK were validated by probing for protein levels of pAMPK and AMPK in serum starved (+ve control) and unstarved (−ve control) muscle cell extracts from C2C12 cells (Cell Signaling Technology, Massachussets). Pictures of the immunoblots of the various proteins were quantified for band intensities using ImageJ analysis and normalized to the loading control β-actin.
Statistics
The survival curves were generated using Kaplan and Meier method and compared with both log-rank and Gehan-Wilcoxon tests. The differences across the control and various treatment groups were analyzed using a 2-way ANOVA followed by Tukey-Kramer post hoc test. The outliers in the sample distribution were determined using Grubs test and eliminated, if applicable. Only results with significant differences (p < .05) were reported. All the error bars represent SEM. All the plots of graphs and statistical analyses were performed using GraphPad Prism software (GraphPad Software, Inc, California).
RESULTS
Effects of PCB126 on Survival, Growth, and Feed Consumption
The PCB126 (5 µmol/kg) and vehicle-injected rats were monitored for general growth and health by measuring their feed consumption and body weight gain. Consistent with our previous studies, the rats did not show any immediate changes or acute symptoms in the initial 2 weeks after PCB126 injection (Gadupudi et al., 2016a). However, the animals began to show significant (p < .05) and persistent weight loss between 14 and 28 days, at the end of the study (Figure 1A). Coincident with changes in weight loss at 14 day-postinjection, total feed consumption decreased and became statistically significant (p < .05) at 18 days of exposure (Figure 1B), and remained so until the end of the study. The substantial changes in weight and feed consumption after PCB126 exposure, led to decreased feed efficiency [Total weight gain (g)/Total feed consumed (g)] from 16 days to the end (Figure 1C). Increasing time of PCB126 exposure, prolonged weight loss, decreased feed consumption, and efficiency, led ultimately to the mortality of some rats (Figure 1D). There were 4 deaths before the end of the study, with the first incidence of death at 23 days. The survival curve of PCB126 exposed animals was significantly (p = .0413) different to the control group (Figure 1D).
Figure 1.

Effect of PCB126 on body weight gain, feed consumption, feed efficiency and survival. Body weight gain percentage (A), total feed consumed (B), total feed efficiency (total body weight gain/total feed consumed) (C), and survival (D) in PCB126 (5 µmol/kg) exposed rats (red/gray line, n = 12) were compared with vehicle (black line, n = 11). The survival curve was significantly different (Kaplan and Meier method; * represents p < .05).
Effects of PCB126 on Animal and Liver Weight After Fasting
To understand the effects of PCB126 on fasting metabolism, the designated subgroups of both the PCB126- and vehicle-treated rats exposed for 28 days were fasted for 12 h overnight before euthanasia. The final animal weights before the fasting of animals were measured (Figure 2A). PCB126 treatment induced a significant weight loss in all the animals compared with the control; however, there were no significant weight differences between the respective groups of control and PCB126-treated rats before fasting (Figure 2A). After the 12 h fast, there was significant weight loss (−3.5% of the total weight) in control animals (Figure 2B; black bars, α represents p < .05 with respect to the fed-control group). Interestingly, PCB126-treated animals that had reduced feed consumption also showed similar decreases in their % body weight (−3.3% of the total weight) upon overnight fasting (Figure 2B; gray bars, β represents p < .05 with respect to the fed PCB126 group). Hence, there were no significant changes in body weight compared with control animals in the fasted group (ns represents nonsignificant). In the fed group of control animals there was normal increase in mean body weight (1.1% of body weight/day). The fed group of PCB126-treated animals, showed a slight decrease in the mean body weight (−0.25% of body weight/day). PCB126 animals are skewed towards weight loss, while the vehicle-treated animals showed weight gain in their fed states.
Figure 2.

Effect of PCB126 on body and liver weights after fasting. The final weights (A) and % change in animal weights/day were (B) in both fed and fasted groups are shown (* = p < .05; α = p < .05 compared with fed-vehicle-treated animals; β = p < .05 compared with fed-PCB126-treated animals). PCB126 treatment caused Significant changes in relative liver weight (C) after PCB126 exposure were compared with the control rats (* = p < .05). PCB126 induced the transcript levels of Cyp1a1 (2-way ANOVA-Tukey; * represents p < .05).
Weights of livers from PCB126-treated rats with respect to body weight were significantly increased (Figure 2C; gray bars, * represents p < .05). There were significant relative liver weight changes in PCB126-treated rats that were fasted. Although not significant, the mean relative liver weight (Figure 2C; black bars) and also wet liver weight (Supplementary Figure 6) of fasted control animals showed a decrease (approximately 10% of fed-control animals), possibly because of the depletion of glycogen upon fasting. PCB126 is a potent AhR agonist and classically induces the transcript levels of cytochrome P-450, family 1, subfamily A, polypeptide 1 (Cyp1a1) along with other transcriptional targets. PCB126 significantly induced the transcript levels of Cyp1a1 (approximately 400-fold) in the PCB126-treated liver (p < .05). The increased Cyp1a1 levels are a measure of PCB126-induced AhR activity. There were no significant changes in Cyp1a1 transcript levels after fasting (Figure 2D). Along with liver weights, the thymus weights were also measured as a marker of PCB126 induced toxicity. Consistent with our previous findings of reduced thymus weights in PCB126 exposure after 14 days (Lai et al., 2010), the thymuses in this study were so involuted they were difficult to excise and were significantly decreased in weight (Supplementary Figure 2).
PCB126 Induces Vacuolation and Other Pathological Changes in Liver
Histological examinations of H&E stained liver sections showed mixed micro-vesicular and macro-vesicular hepatocellular vacuolation diffusely (Figs. 3B and 3D), consistent with our previous findings after 14 days of PCB126 exposure (Klaren et al., 2015; Wang et al., 2016). These vacuolar changes upon PCB126 exposure were consistent with previously documented hydropic degeneration and lipid accumulation. In this study, extended exposure to PCB126 for 28 days resulted in the finding of significant other gross pathologies as well. The PCB126 rat livers appeared diffusely distributed with multiple, small zones of hepatocellular loss/necrosis which contained acellular lightly eosinophilic material and rare necrotic cellular debris. Portal zones had mild fibrosis intermixed with inflammatory cells, primarily lymphocytes, and plasma cells and biliary hyperplasia in liver sections from PCB126-treated rats. A similar inflammatory infiltrate was present scattered randomly throughout some of the liver sections. Also scattered randomly were individual necrotic cells which had hypereosinophilic cytoplasm and pyknotic nuclei. In terms of gross liver pathology confirmed by H&E staining, there were no obvious differences across the fed and fasted liver sections of PCB126-treated rats. The H&E liver sections of control rats of fed (Figure 3A) and fasted (Figure 3C) groups are also shown. The tissues were examined by a pathologist and representative images of each treatment group are presented (Figure 3). Further, the necrosis and inflammation for scored for severity by a pathologist. PCB126 exposed livers showed significant increases in the mean scores of necrosis (Supplementary Figure 7A) and inflammation. Livers from PCB126-exposed rats showed significant increases in the mean scores of necrosis (Supplementary Figure 7B) across the replicates (1-way ANOVA-Tukey; p < .05). Additional fasting for 12 h did not impact the necrosis or inflammation scores in the livers of PCB126-treated rats (Supplementary Figure 7).
Figure 3.
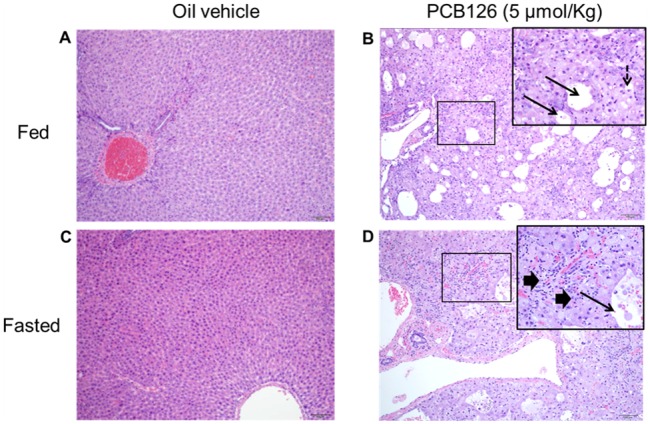
Liver pathology (H&E staining). PCB126 treatment caused vacuolation in rat livers (B and D) compared with the vehicle-treated rats (A and C). The pathology after PCB126 exposure was similar in both fed and fasted animals. Arrows pointing to areas of hepatic necrosis caused by tissue clearing dotted arrows point to hepatocellular vacuolation, arrowheads pointing out inflammation.
Exposure to PCB126 Decreases Serum and Hepatic Glucose Levels
Serum glucose levels of the rats treated with PCB126 were significantly decreased in both fed and fasted animals (Figure 4A, gray bars). There were no significant differences in fed and fasted control animals (Figure 4a, black bars). This lack of difference between fed and fasted groups of control animals after 12 h of fasting is expected, because of the induction of gluconeogenesis in the liver. Activation of gluconeogenic genes in the liver during fasting increases HGP and hence maintains homeostatic glucose levels in the blood. To understand liver glucose production, hepatic glucose levels were colorimetrically determined as described. PCB126 exposure results in decreased hepatic glucose levels in both fed and fasted states (Figure 4B, gray bars; p < .05). The hepatic glucose levels in the fasted control rats were decreased compared with fed controls (Figure 4B, black bars; p < .05). The decrease in hepatic glucose levels in fasted group can be attributed to lack of glucose import during feed withdrawal. Two-way ANOVA analysis of the treatment groups shows a significant effect of both the PCB126 treatment (row factor), fasting (column factor), and significant interaction of PCB126 (p = .0139) in further lowering the fasting state hepatic glucose levels.
Figure 4.

Effect of PCB126 on serum and hepatic glucose levels. Glucose levels in serum (A) and liver extracts (B) of PCB126-treated (5 µmol/kg) rats (gray bars, n = 4–7 animals/group) were compared with the vehicle control (black bars, n = 6–8 animals/group). Two-way ANOVA analysis of PCB126 treatment × fasting showed a significant interaction (p = .0139) (2-way ANOVA-Tukey; * represents p < .05 and α represents p < .05 compared with fed- and vehicle-treated animals).
PCB126 Decreases the Transcript Levels of Genes Necessary for Gluconeogenesis
Liver plays a critical role in maintaining homeostatic glucose levels by regulating glucose storage and glucose production during fed and starved states, respectively. Because of the decreased hepatic free glucose levels and hypoglycemia observed in this study and our previous shorter term studies, the transcripts of gluconeogenic genes were quantified (Gadupudi et al., 2016a). Consistent with our previous observations, we observed a significant decrease in the transcript levels of Pepck-c (* represents p < .05) also referred to as Pck1 in livers of PCB126 treated-fed rats (Figure 5A). Upon fasting, the PCB126-treated rats, contrary to our expectations, displayed a transient rise the transcript levels (Figure 5A), despite decreased hepatic glucose levels (Figure 4B). G6Pase is another important enzyme known for its role in gluconeogenesis. Similar to Pepck-c, the transcript levels of G6pase were found to be decreased by PCB126 in the fed state compared with their levels in fasting rats (Figure 5B). Although Pepck-c and G6pase are important enzymes of cataplerotic reactions during HGP, several anaplerotic enzymes such as Pc, Sds and Ldh-A are involved in replenishment of TCA cycle intermediates for gluconeogenesis (Kornberg, 1966; Owen et al., 2002; Su et al., 1990). Pc, Sds, and LdhA use pyruvate, gluconeogenic amino acid serine, and lactate, respectively, as metabolic precursors for generating TCA cycle intermediates. Similar to our findings with transcript levels of Pepck-c and G6pase, the transcript levels of Sds, Pc, and LdhA were found to be decreased more so in the fed state than fasted state after PCB126 exposure (*represents p < .05). The transcript levels of Glut2/Slc2a2 was quantified as predictor of glucose export during gluconeogenesis and were found to be decreased (Figure 5F) upon PCB126 exposure (Thorens, 1996; Thorens and Mueckler, 2010). Overall, the transcript levels of several genes necessary for gluconeogenesis are decreased in the liver, after exposure to PCB126. Although there is an increase in the transcripts of some of the gluconeogenic enzymes after fasting the PCB126-treated animals for 12 h, no significant replenishment in the serum glucose (Figure 4A) or hepatic glucose levels (Figure 4B) was observed.
Figure 5.

PCB126 and the transcript levels of genes involved in gluconeogenesis. The transcript levels of (A) Phosphoenol pyruvate carboxykinase-1 (soluble Pepck-c), (B) glucose-6-phosphotase (G6Pase), (C) serine dehydratase (Sds), (D) pyruvate carboxylase (Pc), (E) lactate dehydrogenase (Ldh-A) involved in gluconeogenesis and the (F) glucose transporter (Glut2) necessary for glucose export in the liver were quantified (2-way ANOVA-Tukey; * represents p < .05).
PCB126 Diminishes the Phosphorylation of Hepatic CREB and Its Targets
Energy requirement during fasting prompts the transcription of several gluconeogenic genes, which were found to be decreased as shown earlier. Several gluconeogenic enzymes, including the rate-limiting enzyme PEPCK-C, are regulated and transcribed by activation of a transcription factor CREB. Protein levels of PEPCK-C were hence measured semi-quantitatively using an immunoblot (n = 4, Supplementary Figs. 3 and 8C). PCB126 treatment reduced the total protein levels of PEPCK-C in the livers of fed rats (Figure 6A;Supplementary Figure 8C). It is interesting to see an increase in the PEPCK-C levels in the control-fasted rats, compared with the fed controls (Figure 6A; Supplementary Figure 8C). This marks the sensitivity in the expression of PEPCK-C in response to fasting (Supplementary Figure 8C; p < .1). The levels of PEPCK-C in the PCB126-treated-fasted rats were slightly induced upon fasting, compared with PCB126-treated fed rats. This is consistent with the mRNA expression data of various gluconeogenic genes (Figure 5).
Figure 6.

Effect of PCB126 on phosphorylation of CREB1 and the transcriptional coactivators involved in gluconeogenesis. The protein levels of pCREB on serine (S133) (B; top panel), the total protein levels of CREB1 (B; middle panel) and its transcript levels (C) measured. β-actin was used as the loading control (B; bottom panel). Forskolin stimulated and unstimulated SK-N-MC neuronal cell extracts were used as positive and negative control for validating the pCREB antibody. The protein levels of PEPCK-C, the transcriptional target regulated by CREB were also measured (A). The transcriptional coactivators of CREB1, Pgc1α (D), Crem (E), Icer (F), auto-regulated transcriptional targets of CREB1 phosphorylation were also quantified (* represents p < .05).
The activation of CREB1, which transcribes Pepck-c and other genes, is marked by the phosphorylation of a serine-133 residue (pCREB1 [S133]). Hence, the protein levels of pCREB were semiquantitatively measured (Figure 6B and Supplementary Figure 8B). Similar to its target PEPCK-C, the levels of pCREB were diminished by PCB126 in the fed rat livers compared with the fed controls. Upon fasting, the PCB126-treated rats demonstrated a transient increase compared with fed-PCB126-treated rats. pCREB levels in fasted controls were normal (Figure 6B, top panel). The protein (Figure 6B, middle panel) or transcript levels (Figure 6C) of total CREB1 were not altered. All the immunoblots were performed in replicates (n = 4, Supplementary Figure 4) and normalized to β-actin levels (Figs. 6A and 6B, bottom panel). The ratio of the band intensities (n = 4) of pCREB/total CREB were quantified (Supplementary Figure 8B). Exposure to PCB126 suppressed the activated levels of pCREB, compared with their vehicle-treated-fasted livers (Supplementary Figure 8B).
CREB1-mediated regulation of fasting and fed metabolism is coregulated by transcriptional coactivators, such as PGC1α, CREM and ICER. These coactivators interact with CREB1 to form stable transcription complexes that assist or repress the transcription of gluconeogenic genes. While PGC1α and CREM are coactivators, ICER is corepressor. All these coactivators/corepressors are synthesized under the transcriptional regulatory loops of CREB1 in response to fed/fasted or hormonal stimuli. The down regulation in the transcripts of the functional coactivators Pgc1α, Crem, and Icer in both and fed/fasted states of PCB126-treated rats (Figs. 6D–F) further marks the reduced activation of CREB1.
PCB126 Interrupts Glycogenolysis
Glycogen is an important source of energy stored at high concentrations in hepatocytes. During fasting, glucose is derived from glycogen by glycogenolysis. To understand the effects of PCB126 on fasting, glycogen content in the liver was analyzed by qualitatively staining for glycogen with PAS stain. In the fed control animals adequate levels of glycogen were apparent, as shown by the purple-magenta color of the stain (Figure 7A, top-left). In the fasted control animals, no glycogen levels were detected as indicated by the absence of any staining with PAS (Figure 7a, bottom-left). In the fed PCB126-treated animals, patches of PAS staining were observed (Figure 7A, top-right), demonstrating the presence of polymerized glycogen. The fasted PCB126-treated animals did not show any glycogen stain as well (Figure 7a, bottom-right). For a quantitative analysis, the glycogen levels were measured by indirect colorimetry (Figure 7B) as described in the methods section. The quantitative measurements were consistent with the findings of PAS staining. Despite low levels of glucose in serum after PCB126 exposure, it is interesting to find glycogen in the respective rat livers (Figure 7B), even more so than the fasted-control rat livers. To corroborate the observed decrease in glycogenolysis, the transcript levels of glycogen phosphorylase (Pygl), the rate-limiting enzyme involved in glycogenolysis were measured. PCB126 exposure significantly decreased the transcript levels of Pygl in both fed and fasted states (Figure 7C), thereby suggesting its role in the impaired glycogenolysis.
Figure 7.

Effect of PCB126 exposure on glycogenolysis. The presence of glycogen in the liver sections was evaluated by PAS staining. The magenta color indicates the presence of glycogen (A). The glycogen levels in liver extracts were measured colorimetrically (B). ^represents glycogen concentrations calculated by subtracting free glucose concentrations from the total glucose levels after glycogen hydrolysis in liver extracts (See methods). Transcript levels of Pygl, a rate-limiting enzyme involved in glycogenolysis were quantified by qPCR (C). (2-way ANOVA-Tukey; * represents p < .05; α represents p < 0.05 compared with fed-vehicle-treated rats).
PCB126 Decreases the Transcripts of Genes Involved in Fatty-Acid Oxidation and Inhibits the Activation AMPK
PCB126 is well-known to induce hepatic steatosis through accumulation of lipid-filled vacuoles in hepatocytes. In this 28 days exposure study, ORO staining was performed to depict lipid accumulation. In the fed-control animals, little or no staining of ORO was observed. In the fasted state control animals, tiny, punctate lipid droplets were found in the periportal hepatocytes, but not in hepatocytes in other zones (Figure 8, bottom-left panel). This is indicative of the peripheral lipid mobilization into the liver for fatty-acid oxidation during fasting. This is known to be transient and changes with fed/fasted states. In PCB126-treated animals, the lipid droplets are variable sized and distributed throughout the liver, but are most abundant and large in the portal zones of the hepatic lobule (Figure 8A, top-right and bottom-right panels). With predominant steatosis, the changes in lipid accumulation across fed and fasted states were indistinguishable. We and others previously observed the effects of PCB126 on the protein levels of PPARα, regarded as a master regulator of β-oxidation (Gadupudi et al., 2016a; Robertson et al., 2007). In this prolonged exposure study, we observed the levels of Pparα transcripts to be lowered by PCB126 in the fed state, but were induced upon fasting. Some of the transcriptional targets of Pparα, such as Acox1 and Hmgcs2, also showed similar results (Figs. 8C–E). However, some of the well-known transcriptional targets of Pparα, such as Cpt1a, Cyp4a2 were found to be unaltered or increased by PCB126 treatment (data not shown). These changes suggest that effects on Pparα may be consequential and subjective to fed or fasted states. Taking into consideration the diminished CREB activation during gluconeogenesis, as well as lowered glycogenolysis and fatty-acid oxidation; the levels of AMPK as a unified target for the consequent pathologies were measured semi-quantitatively using immunoblot (Supplementary Figure 8A). Although the fed controls had less phosphorylation of AMPK, the fasted control livers showed marked activation of AMPK through phosphorylation at the threonine position (pAMPK [T172]). The PCB126 treatment significantly decreased the pAMPK levels in both fed and fasted livers. Not only were pAMPK levels reduced (Figure 8B, top panel), but total AMPK levels (Figure 8B, middle panel) were also decreased, compared with the levels of loading control β-actin (Figure 8B, bottom panel). The changes in the pAMPK or AMPK levels were performed in replicate (n = 4, Supplementary Figure 5) and the relative ratio of pAMPK/AMPK were quantified (Supplementary Figure 8A). PCB126 decreased the pAMPK levels compared with their levels in normal fasted livers (Supplementary Figure 8A)
Figure 8.

Effect of PCB126 exposure on lipid accumulation and the transcript levels of genes involved in β-oxidation, the expression of pAMPK. Liver sections were stained for lipid droplets (red) with ORO. The accumulation of lipid droplets in periportal regions is stained red (A, right-top and bottom panels). Vehicle-treated-fed rat livers did not stain for lipid (A, left-top panel), while the fasted animals (A, left-bottom) showed mild accumulation in periportal regions represented by punctate red staining (dotted circle). The protein levels of the energy sensing enzyme AMPK (B, middle panel) and its phosphorylated form pAMPK (T172) (B, top panel) were measured by immunoblot. β-actin was used as the loading control (B, bottom panel). PCB126 exposure diminished the transcript levels and Pparα (C), and its target genes Acox1 (D) and Hmgcs2 (E).
Decreased Activation of AMPK by PCB126 Dysregulates the Ameliorating Functions of Adiponectin and Fgf21
Adiponectin is a hormone secreted by adipose tissue that plays an important role in regulating liver glucose production and fatty-acid oxidation. Adiponectin is known to increase β-oxidation and glucose production by the activation of AMPK through adiponectin receptors in the liver and hence to protect from metabolic derangements. PCB126 treatment results in a significant increase in the serum concentration of adiponectin, in order to protect from PCB126-induced steatosis and hypoglycemia (Figure 9A). Although not significant, there is a slight decrease in the transcript levels of liver adiponectin receptor (adipor2) in the PCB126-treated rat liver extracts (Figure 9C). In order to stimulate adiponectin secretion, the liver secretes Fgf21 as a “stress signal” that communicates with adipose tissue during nutrient deprivation and fasting. Concomitant with PCB126-induced metabolic stress in the liver, there is significant induction in the transcript levels of Fgf21 (Figure 9B). Despite the increased expression of adiponectin and fgf21, the decrease in the activation of AMPK by PCB126 dysregulates their ameliorating action on metabolic homeostasis in liver.
Figure 9.

PCB126 and the levels adiponectin and Fgf21. Exposure to PCB126 increases the protein levels of adiponectin in the serum (A) and the transcript levels of Fgf21 (B) in the liver extracts. The transcript levels of adiponectin receptor (Adipor2) in liver were measured (C) (2-way ANOVA-Tukey; * represents p < .05).
DISCUSSION
Previous studies showed acute exposures to a single dose of PCB126 (14 days) caused significant decreases in serum glucose levels without any alterations in feed consumption or body weight (Gadupudi et al., 2016a; Klaren et al., 2015; Lai et al. 2010). Several enzymes involved in gluconeogenesis were found to be down regulated despite PCB126-induced hypoglycemia (Gadupudi et al., 2016a,b; Petriello et al., 2016). We and others also reported that PCB126-mediated decrease in the phosphorylation of CREB, may lead to reduced transcription of fasting enzymes such as PEPCK-C in the liver (Gadupudi et al., 2016b; Zhang et al., 2012). Based on inhibitory effects of PCB126 on the fasting metabolism, this study was performed to evaluate effects of PCB126 on HGP and steatosis, following a sub-acute exposure for 28 days.
Exposure to PCB126 for the extended time period of 28 days resulted in decreased body weight gain that developed into weight loss (Figure 1A). The cause of weight loss seems to stem from overall loss in organ weights, including bones, rather than 1 specific target organ (eg, adipose or skeletal muscle). Along with effects of PCB126 on growth, the chemical-induced decrease in feed consumption (Figure 1B) and efficiency (Figure 1C) could also contribute to the overall shrinkage in animal size. Despite the overall “wasting” observed in PCB126-exposed animals (Figure 1A), the body weight loss caused by their overnight fasting was not significantly different from the control-fasted animals (Figure 2B). However, PCB126 treated-fed animals continued to persistently lose weight without any fasting (median % change in animal weight/day = approximately 0.2%). PCB126 exposure resulted in significant increase of relative liver weights, because of lipid accumulation and hydropic vacuolation. Noteworthy is the decrease in the liver weight (approximately 10% wet weight) of the control animals upon fasting (Supplementary Figure 5), because of glycogen depletion (Figure 7). The longer exposure to PCB126 in this study resulted in the onset of inflammation and necrosis in the liver (Figure 3 and Supplementary Figure 7). Consistent with our previous studies on PCB126, there was predominant lipid vacuolation in the periportal regions (Figure 8A).
Fasting metabolism in the liver maintains energy homeostasis through the activation of its 3 arms, namely glycogenolysis, gluconeogenesis, and β-oxidation (Figure 10) (Rui, 2014). Although the former 2 arms use carbohydrates, certain gluconeogenic amino acids or glycerol as fuel sources, the later uses lipids (Kalhan et al., 2001; Rui, 2014). Although there appears to be no strict order in the activation of these pathways, it is a precept that glycogenolysis occurs before gluconeogenesis and fatty-acid oxidation, during long hours of fasting. The key lies in the timely and tightly regulated activation of nuclear receptors that induce the transcription of enzymes involved in these pathways (Heindel et al., 2017; Hong et al., 2014; Rui, 2014). We and others previously identified alterations in the expression of enzymes that regulate fasting metabolism upon exposure to PCB126 and other dioxin-like compounds (Gadupudi et al., 2016a; Hsia and Kreamer, 1985; Seefeld et al., 1984; Viluksela et al., 1995, 1997, 1998; Weber et al., 1995). Thus, in the current study we analyzed the pathological manifestations of PCB126 on these 3 arms of fasting.
Figure 10.

PCB126 inhibits the activation of AMPK-CREB signal transduction and disrupts liver metabolism. Exposure to PCB126 decreases the protein levels and the phosphorylation of AMPK to further inhibit the gluconeogenesis, glycogenolysis and fatty-acid oxidation necessary for physiological homeostasis, during energy depletion.
Our previous time-course study with PCB126 showed a significant decrease in blood glucose levels within 9 h of exposure (Gadupudi et al., 2016a). With no changes in feed consumption (contributor of blood glucose), the observed hypoglycemia was attributed to decreased HGP caused by a decrease in the levels of the rate-limiting enzyme, PEPCK-C (Gadupudi et al., 2016b). Measurement of hepatic glucose levels in the current study showed decreased hepatic free glucose levels in fed state that further worsened upon fasting in PCB126-treated rats (Figure 4A) (2-way ANOVA; interaction [p < .01]). Decreased HGP, thus corroborates the decreased serum glucose levels and hypoglycemia (Figure 4A). Consistent with the decreased HGP, the transcript levels of the enzymes Pepck-c, G6Pase, Sds, Pc, and Ldh-A, involved in gluconeogenesis were also found to be decreased by PCB126. Functionally, the enzymes “translated” from these genes are either anaplerotic or cataplerotic in generating the reaction products that are precursor metabolites during glucose production (Owen et al., 2002). The enzymes Sds, Pc, and LdhA synthesize oxaloacetate by conversion of their substrates serine, pyruvate, and lactate, respectively, and contribute to anaplerotic flux of the TCA cycle (Kornberg, 1966; Owen et al., 2002; Su et al., 1990). On the other hand, Pepck-c and G6pase direct the metabolite flux of the TCA cycle towards glucose production through cataplerosis (Burgess et al., 2004).
The decreased transcript levels of several enzymes in gluconeogenesis that are not under the direct transcriptional control of AhR suggest the disruption of a common regulatory switch. Hepatic nuclear transcription factor CREB is activated through phosphorylation during fasting (pCREB1) (Herzig et al., 2001). The activated pCREB facilitates the transcription of several enzymes especially those involved in gluconeogenesis (Gadupudi et al., 2016a; Mayr and Montminy, 2001; Robertson et al., 2007). The activated pCREB also transcribes several coactivator proteins such as PGC1α, that interact with CREB1 itself and peroxisome proliferator activated receptors to aid the transcription of enzymes involved in gluconeogenesis and fatty-acid oxidation (Herzig et al., 2001, 2003). The activated pCREB, along with functional genes such as Pepck-c, also transcribes several of its isoforms such as CREM and ICER that play regulatory roles in either feedback activation/amplification or inhibition by binding to the promoters of target genes during fed/fasted cycles of the liver (Everett et al., 2013; Servillo et al., 2002). Although CREM isoform increases transcription of more genes involved in fasting and cellular stress responses, ICER is a truncated isoform that competes with original CREB and inhibits its transcriptional response by competitively occupying the promoters (Servillo et al., 2002). PCB126 exposure resulted in decreased phosphorylation of CREB in fed states, with a transient rise in pCREB levels upon fasting (Figure 6B). However, the transient increase in pCREB levels in fasted group of PCB126-treated rats did not aid in the replenishment of hepatic glucose levels. Coincident with pCREB levels, the transcript levels of the coactivator Pgc1α and the coregulatory factors of Crem and Icer were also decreased (Figure 6). These inhibitory effects of PCB126 on CREB and its battery of genes explain the decreased gluconeogenesis.
In addition to its effects on gluconeogenesis, PCB126 exposure also leads to lipid accumulation and disrupted glycogen metabolism. We and others previously identified decreased protein and transcript levels of PPARα along with decreased peroxisomal fatty-acid oxidation (Gadupudi et al., 2016a; Robertson et al., 2007). PPARα is known to be the master regulator of β-oxidation and controls the expression of enzymes for fatty-acid oxidation in peroxisomes, mitochondria and endoplasmic reticulum (microsomes) (Kersten, 2014; Kersten et al., 1999; Mandard et al., 2004). Consequent to decreased PPARα levels, some of its transcriptional targets such as Acox1 and Hmgcs2 are also decreased. However, its transcriptional targets Cpt1a and Cyp4a2 involved in mitochondrial and microsomal fatty-acid oxidation are not decreased (not shown). Therefore, the exclusive effects of PCB126 on PPARα mediated fatty-acid oxidation seem equivocal. Besides, the complexity across multiple stimuli and subcellular locations (organelles) that drive the activation of fatty-acid oxidation may also lead to compensatory mechanisms (Reddy and Rao, 2006).
Given the observed effects of PCB126 across multiple pathways of fasting metabolism, a common target that generally regulates fasting metabolism seems more plausible. Thus, the levels of pAMPK were measured (Figure 8B). AMPK is a well-known energy sensing protein-kinase that is activated by increased Adenosine monophosphate (AMP)/Adenosine triphosphate (ATP) or Adenosine diphosphate (ADP)/ATP ratios caused by low energy states such as fasting (Figure 10) (Hardie et al., 2012). Although not structurally alike AMP, several xenobiotic compounds including therapeutic drugs (eg, metformin), antioxidant polyphenols (eg, resveratrol) and toxicants such as vinyl chloride are implicated in altering AMPK activation (Guardiola et al., 2016; Hardie et al., 2012; Viollet et al., 2012). Activated pAMPK is well-known to switch-on the transcriptional regulation of enzymes necessary for catabolic pathways, such as β-oxidation of fatty acids, gluconeogenesis and glycogenolysis, while shutting-down the anabolic processes such as lipogenesis and glycogenesis. AMPK activation in the liver during fasting is known to increase fatty-acid oxidation and HGP through direct or indirect transcriptional regulation of target enzymes (Canto and Auwerx, 2010). In addition, hormones such as FGF21 and adiponectin, secreted by liver and adipose tissues, respectively, during energy crisis, bind to their target receptors and improve metabolic homeostasis by downstream activation of AMPK (Combs and Marliss, 2014; Hasenour et al., 2013; Potthoff, 2017; Potthoff et al., 2009; Salminen et al., 2017). Phosphorylated AMPK supports HGP by stimulating glycogenolysis and CREB phosphorylation necessary for gluconeogenesis through the aid of complex cell signaling coactivators, whose roles are under investigation (Koo et al., 2005; Thomson et al., 2008). Although the mechanisms are unclear, it has been proposed that AMPK activation may play subsidiary roles in PPARα-induced β-oxidation either by a direct interaction or through indirect effects on its coactivators such as PGC1α (Bronner et al., 2004; Canto and Auwerx, 2010; Vega et al., 2000). Nevertheless, the overlapping roles of AMPK and PPARα are both essential for fatty-acid oxidation. To this end, the inhibition of pCREB and its target enzymes necessary for HGP observed in the livers of PCB126-treated rats correspond to decreased levels of total AMPK and activated pAMPK. The disruption in AMPK and CREB activation by PCB126; thus leads to robust inhibition of gluconeogenesis and also partly affects fatty-acid oxidation. Despite the protective induction of adiponectin and FGF21 during PCB126-induced hepatic stress, the inhibition of the signaling-intermediate AMPK seems to resist their ameliorative effects on liver function (Figure 9).
Pathological investigations in this study showed increased lipid accumulation as well as mild fibrotic lesions in the periportal zones of hepatic lobules. Periportal hepatocytes or zone 1 hepatocytes are known to express more gluconeogenic and fatty-acid oxidation enzymes. Previous studies showed increased expression and activity of AMPK and CREB mediated expression of PEPCK-C in the periportal hepatocytes during fasting (Jungermann and Kietzmann, 1996; Park et al., 1993; Witters et al., 1994). The proclivity of PCB126-induced peri-portal lesions in this study further confirms the disruption of AMPK and CREB in the inhibition of fasting metabolism in the liver. While the pathological effects were mostly observed in the periportal regions, the expression of AhR, and its transcriptional target CYP1A1 are previously shown to be more active in peri-venous regions (Braeuning et al., 2006; Lindros et al., 1998). The contrasting localizations of the target receptor of PCB126, AhR, in peri-venous regions and the observed peri-portal lesions of fasting metabolism, warrants further understanding of the roles of AhR-mediated toxicity across the functionally polarized hepatocytes in the liver.
In conclusion, these studies show that dioxin-like congener PCB126 is a metabolic disruptor chemical, which disrupts energy homeostasis by its inhibitory effects on intermediate cellular metabolism. Exposure to PCB126 results in a fuel crisis caused by decreased HGP and the resultant hypoglycemia. PCB126 inhibits the activation of AMPK and CREB proteins, necessary for the transcriptional regulation of several functional enzymes necessary for metabolism of gluconeogenic substrates, glycogen and fatty-acids during fasting (Figure 10). Interestingly, we show here that the observed fuel crisis stems from persistent inhibition of AMPK and CREB under normal/base-line conditions, which leads to failure of energy sensing and robust induction of enzymes during fasting or energy-deprivation.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
This work was supported by National Institute of Environmental Health Sciences (P42 ES013661) awarded to L.W.R. G.S.G. recognizes the Iowa Superfund Training Core for training and financial support. The opinions expressed are solely those of the authors.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr Xueshu Li and Dr Hans-Joachim Lehmler of the University of Iowa Superfund Synthesis Core for synthesizing, characterizing and supplying PCB126, Dr Daryl K. Granner for providing the antibody against Pepck-C; Total AMPK antibody was generously provided by Dr Dale Abel. We also thank Dr Matt C. Cave of the University of Louisville for helpful discussions of research directions, and Ms Susanne Flor and members of our laboratory and the Comparative Pathology Laboratory, University of Iowa, for their expert help with the animal studies.
REFERENCES
- Ahlborg U. G., Hanberg A. (1994). Toxic equivalency factors for dioxin-like PCBs. Environ. Sci. Pollut. Res. Int. 1, 67–68. [DOI] [PubMed] [Google Scholar]
- Ampleman M. D., Martinez A., DeWall J., Rawn D. F., Hornbuckle K. C., Thorne P. S. (2015). Inhalation and dietary exposure to PCBs in urban and rural cohorts via congener-specific measurements. Environ. Sci. Technol. 49, 1156–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandiera S., Safe S., Okey A. B. (1982). Binding of polychlorinated biphenyls classified as either phenobarbitone-, 3-methylcholanthrene- or mixed-type inducers to cytosolic Ah receptor. Chem. Biol. Interact. 39, 259–277. [DOI] [PubMed] [Google Scholar]
- Boverhof D. R., Burgoon L. D., Tashiro C., Sharratt B., Chittim B., Harkema J. R., Mendrick D. L., Zacharewski T. R. (2006). Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in Sprague Dawley rats and C57BL/6 mice. Toxicol. Sci. 94, 398–416. [DOI] [PubMed] [Google Scholar]
- Braeuning A., Ittrich C., Kohle C., Hailfinger S., Bonin M., Buchmann A., Schwarz M. (2006). Differential gene expression in periportal and perivenous mouse hepatocytes. Febs J. 273, 5051–5061. [DOI] [PubMed] [Google Scholar]
- Bronner M., Hertz R., Bar-Tana J. (2004). Kinase-independent transcriptional co-activation of peroxisome proliferator-activated receptor alpha by AMP-activated protein kinase. Biochem. J. 384, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S. C., Hausler N., Merritt M., Jeffrey F. M., Storey C., Milde A., Koshy S., Lindner J., Magnuson M. A., Malloy C. R. et al. , (2004). Impaired tricarboxylic acid cycle activity in mouse livers lacking cytosolic phosphoenolpyruvate carboxykinase. J. Biol. Chem. 279, 48941–48949. [DOI] [PubMed] [Google Scholar]
- Canto C., Auwerx J. (2010). AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol. Life Sci. 67, 3407–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M., Appana S., Patel M., Falkner K. C., McClain C. J., Brock G. (2010). Polychlorinated biphenyls, lead, and mercury are associated with liver disease in American adults: nHANES 2003-2004. Environ. Health Perspect. 118, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M. C., Clair H. B., Hardesty J. E., Falkner K. C., Feng W., Clark B. J., Sidey J., Shi H., Aqel B. A., McClain C. J. et al. , (2016). Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta 1859, 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs T. P., Marliss E. B. (2014). Adiponectin signaling in the liver. Rev. Endocr. Metab. Disord. 15, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton T. P., Puga A., Shertzer H. G. (2002). Induction of cellular oxidative stress by aryl hydrocarbon receptor activation. Chem. Biol. Interact. 141, 77–95. [DOI] [PubMed] [Google Scholar]
- Donat-Vargas C., Gea A., Sayon-Orea C., Carlos S., Martinez-Gonzalez M. A., Bes-Rastrollo M. (2014). Association between dietary intakes of PCBs and the risk of obesity: The SUN project. J. Epidemiol. Commun. Health 68, 834–841. [DOI] [PubMed] [Google Scholar]
- Everett C. J., Frithsen I., Player M. (2011). Relationship of polychlorinated biphenyls with type 2 diabetes and hypertension. J. Environ. Monit. 13, 241–251. [DOI] [PubMed] [Google Scholar]
- Everett C. J., Frithsen I. L., Diaz V. A., Koopman R. J., Simpson W. M. Jr., Mainous A. G. 3rd, and, (2007). Association of a polychlorinated dibenzo-p-dioxin, a polychlorinated biphenyl, and DDT with diabetes in the 1999-2002 National Health and Nutrition Examination Survey. Environ. Res. 103, 413–418. [DOI] [PubMed] [Google Scholar]
- Everett L. J., Lay J., Lukovac S., Bernstein D., Steger D. J., Lazar M. A., Kaestner K. H. (2013). Integrative genomic analysis of CREB defines a critical role for transcription factor networks in mediating the fed/fasted switch in liver. BMC Genomics 14, 337.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgacs A. L., Dere E., Angrish M. M., Zacharewski T. R. (2013). Comparative analysis of temporal and dose-dependent TCDD-elicited gene expression in human, mouse, and rat primary hepatocytes. Toxicol. Sci. 133, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadupudi G. S., Klaren W. D., Olivier A. K., Klingelhutz A. J., Robertson L. W. (2016a). PCB126-induced disruption in gluconeogenesis and fatty acid oxidation precedes fatty liver in male rats. Toxicol. Sci. 149, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadupudi G. S., Klingelhutz A. J., Robertson L. W. (2016b). Diminished phosphorylation of CREB is a key event in the dysregulation of gluconeogenesis and glycogenolysis in PCB126 hepatotoxicity. Chem. Res. Toxicol. 29, 1504–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier M. S., Rabasa-Lhoret R., Prud'homme D., Karelis A. D., Geng D., van Bavel B., Ruzzin J. (2014). The metabolically healthy but obese phenotype is associated with lower plasma levels of persistent organic pollutants as compared to the metabolically abnormal obese phenotype. J. Clin. Endocrinol. Metab. 99, E1061–E1066. [DOI] [PubMed] [Google Scholar]
- Granner D. K. (2015). In pursuit of genes of glucose metabolism. J. Biol. Chem. 290, 22312–22324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grun F., Blumberg B. (2009). Endocrine disrupters as obesogens. Mol. Cell Endocrinol. 304, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardiola J. J., Beier J. I., Falkner K. C., Wheeler B., McClain C. J., Cave M. (2016). Occupational exposures at a polyvinyl chloride production facility are associated with significant changes to the plasma metabolome. Toxicol. Appl. Pharmacol. 313, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie D. G., Ross F. A., Hawley S. A. (2012). AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenour C. M., Berglund E. D., Wasserman D. H. (2013). Emerging role of AMP-activated protein kinase in endocrine control of metabolism in the liver. Mol. Cell Endocrinol. 366, 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel J. J., Blumberg B., Cave M., Machtinger R., Mantovani A., Mendez M. A., Nadal A., Palanza P., Panzica G., Sargis R. et al. , (2017). Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 68, 3–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel J. J., vom Saal F. S., Blumberg B., Bovolin P., Calamandrei G., Ceresini G., Cohn B. A., Fabbri E., Gioiosa L., Kassotis C. et al. , (2015). Parma consensus statement on metabolic disruptors. Environ. Health 14, 54.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S., Hedrick S., Morantte I., Koo S. H., Galimi F., Montminy M. (2003). CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature 426, 190–193. [DOI] [PubMed] [Google Scholar]
- Herzig S., Long F., Jhala U. S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P. et al. , (2001). CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183. [DOI] [PubMed] [Google Scholar]
- Hong S. H., Ahmadian M., Yu R. T., Atkins A. R., Downes M., Evans R. M. (2014). Nuclear receptors and metabolism: From feast to famine. Diabetologia 57, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia M. T., Kreamer B. L. (1985). Delayed wasting syndrome and alterations of liver gluconeogenic enzymes in rats exposed to the TCDD congener 3, 3', 4, 4'-tetrachloroazoxybenzene. Toxicol. Lett. 25, 247–258. [DOI] [PubMed] [Google Scholar]
- Jungermann K., Kietzmann T. (1996). Zonation of parenchymal and nonparenchymal metabolism in liver. Annu. Rev. Nutr. 16, 179–203. [DOI] [PubMed] [Google Scholar]
- Kaiser J. P., Lipscomb J. C., Wesselkamper S. C. (2012). Putative mechanisms of environmental chemical-induced steatosis. Int. J. Toxicol. 31, 551–563. [DOI] [PubMed] [Google Scholar]
- Kalhan S. C., Mahajan S., Burkett E., Reshef L., Hanson R. W. (2001). Glyceroneogenesis and the source of glycerol for hepatic triacylglycerol synthesis in humans. J. Biol. Chem. 276, 12928–12931. [DOI] [PubMed] [Google Scholar]
- Kersten S. (2014). Integrated physiology and systems biology of PPARalpha. Mol Metab 3, 354–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S., Seydoux J., Peters J. M., Gonzalez F. J., Desvergne B., Wahli W. (1999). Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 103, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaren W. D., Gadupudi G. S., Wels B., Simmons D. L., Olivier A. K., Robertson L. W. (2015). Progression of micronutrient alteration and hepatotoxicity following acute PCB126 exposure. Toxicology 338, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo S. H., Flechner L., Qi L., Zhang X., Screaton R. A., Jeffries S., Hedrick S., Xu W., Boussouar F., Brindle P. et al. , (2005). The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1111. [DOI] [PubMed] [Google Scholar]
- Kornberg H. (1966). Anaplerotic sequences and their role in metabolism. Essays Biochem. 2, 1–31. [Google Scholar]
- Lai I., Chai Y., Simmons D., Luthe G., Coleman M. C., Spitz D., Haschek W. M., Ludewig G., Robertson L. W. (2010). Acute toxicity of 3, 3', 4, 4', 5-pentachlorobiphenyl (PCB 126) in male Sprague-Dawley rats: Effects on hepatic oxidative stress, glutathione and metals status. Environ. Int. 36, 918–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai I. K., Chai Y., Simmons D., Watson W. H., Tan R., Haschek W. M., Wang K., Wang B., Ludewig G., Robertson L. W. (2011). Dietary selenium as a modulator of PCB 126-induced hepatotoxicity in male Sprague-Dawley rats. Toxicol. Sci. 124, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai I. K., Klaren W. D., Li M., Wels B., Simmons D. L., Olivier A. K., Haschek W. M., Wang K., Ludewig G., Robertson L. W. (2013). Does dietary copper supplementation enhance or diminish PCB126 toxicity in the rodent liver? Chem. Res. Toxicol. 26, 634–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauby-Secretan B., Loomis D., Baan R., El Ghissassi F., Bouvard V., Benbrahim-Tallaa L., Guha N., Grosse Y., Straif K. (2016). Use of mechanistic data in the IARC evaluations of the carcinogenicity of polychlorinated biphenyls and related compounds. Environ. Sci. Pollut. Res. Int. 23, 2220–2229. [DOI] [PubMed] [Google Scholar]
- Lauby-Secretan B., Loomis D., Grosse Y., El Ghissassi F., Bouvard V., Benbrahim-Tallaa L., Guha N., Baan R., Mattock H., Straif K. and Cancer, W. H. O. I. A. f. R. o. (2013). Carcinogenicity of polychlorinated biphenyls and polybrominated biphenyls. Lancet Oncol. 14, 287–288. [DOI] [PubMed] [Google Scholar]
- Lehmler H. J., Robertson L. W. (2001). Synthesis of polychlorinated biphenyls (PCBs) using the Suzuki-coupling. Chemosphere 45, 137–143. [DOI] [PubMed] [Google Scholar]
- Lindros K. O., Oinonen T., Kettunen E., Sippel H., Muro-Lupori C., Koivusalo M. (1998). Aryl hydrocarbon receptor-associated genes in rat liver: Regional coinduction of aldehyde dehydrogenase 3 and glutathione transferase Ya. Biochem. Pharmacol. 55, 413–421. [DOI] [PubMed] [Google Scholar]
- Lo R., Celius T., Forgacs A. L., Dere E., MacPherson L., Harper P., Zacharewski T., Matthews J. (2011). Identification of aryl hydrocarbon receptor binding targets in mouse hepatic tissue treated with 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 257, 38–47. [DOI] [PubMed] [Google Scholar]
- Luna L. G. (1992). Histopathologic Methods and Color Atlas of Special Stains and Tissue Artifacts. Johnson Printers, Downers Grove, Illinois. [Google Scholar]
- Mandard S., Muller M., Kersten S. (2004). Peroxisome proliferator-activated receptor alpha target genes. Cell Mol. Life Sci. 61, 393–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B., Montminy M. (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609. [DOI] [PubMed] [Google Scholar]
- NTP. (2006). NTP toxicology and carcinogenesis studies of 3, 3', 4, 4', 5-pentachlorobiphenyl (PCB 126) (CAS No. 57465-28-8) in female Harlan Sprague-Dawley rats (Gavage Studies). Natl. Toxicol. Prog. Tech. Rep. Ser. (520), 4–246. [PubMed] [Google Scholar]
- O’Brien R. M., Granner D. K. (1996). Regulation of gene expression by insulin. Physiol. Rev. 76, 1109–1161. [DOI] [PubMed] [Google Scholar]
- Okey A. B. (2007). An aryl hydrocarbon receptor odyssey to the shores of toxicology: The Deichmann Lecture, International Congress of Toxicology-XI. Toxicol. Sci. 98, 5–38. [DOI] [PubMed] [Google Scholar]
- Ovando B. J., Ellison C. A., Vezina C. M., Olson J. R. (2010). Toxicogenomic analysis of exposure to TCDD, PCB126 and PCB153: Identification of genomic biomarkers of exposure to AhR ligands. BMC Genomics 11, 583.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen O. E., Kalhan S. C., Hanson R. W. (2002). The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412. [DOI] [PubMed] [Google Scholar]
- Park E. A., Gurney A. L., Nizielski S. E., Hakimi P., Cao Z., Moorman A., Hanson R. W. (1993). Relative roles of CCAAT/enhancer-binding protein beta and cAMP regulatory element-binding protein in controlling transcription of the gene for phosphoenolpyruvate carboxykinase (GTP). J. Biol. Chem. 268, 613–619. [PubMed] [Google Scholar]
- Perkins J. T., Petriello M. C., Newsome B. J., Hennig B. (2016). Polychlorinated biphenyls and links to cardiovascular disease. Environ. Sci. Pollut. Res. Int. 23, 2160–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petriello M. C., Hoffman J. B., Sunkara M., Wahlang B., Perkins J. T., Morris A. J., Hennig B. (2016). Dioxin-like pollutants increase hepatic flavin containing monooxygenase (FMO3) expression to promote synthesis of the pro-atherogenic nutrient biomarker trimethylamine N-oxide from dietary precursors. J. Nutr. Biochem. 33, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilkis S. J., Granner D. K. (1992). Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu. Rev. Physiol. 54, 885–909. [DOI] [PubMed] [Google Scholar]
- Potthoff M. J. (2017). FGF21 and metabolic disease in 2016: A new frontier in FGF21 biology. Nat. Rev. Endocrinol. 13, 74–76. [DOI] [PubMed] [Google Scholar]
- Potthoff M. J., Inagaki T., Satapati S., Ding X., He T., Goetz R., Mohammadi M., Finck B. N., Mangelsdorf D. J., Kliewer S. A. et al. , (2009). FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. U.S.A. 106, 10853–10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puga A., Ma C., Marlowe J. L. (2009). The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 77, 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy J. K., Rao M. S. (2006). Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G852–G858. [DOI] [PubMed] [Google Scholar]
- Robertson L. W., Berberian I., Borges T., Chen L. C., Chow C. K., Glauert H. P., Filser J. G., Thomas H. (2007). Suppression of peroxisomal enzyme activities and cytochrome P450 4A isozyme expression by congeneric polybrominated and polychlorinated biphenyls. PPAR Res 2007, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L. (2014). Energy metabolism in the liver. Compr. Physiol. 4, 177–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzin J. (2012). Public health concern behind the exposure to persistent organic pollutants and the risk of metabolic diseases. BMC public health 12, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzin J., Petersen R., Meugnier E., Madsen L., Lock E. J., Lillefosse H., Ma T., Pesenti S., Sonne S. B., Marstrand T. T. et al. (2010). Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ Health Perspect 1184, 465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S., Wang F., Porter W., Duan R., McDougal A. (1998). Ah receptor agonists as endocrine disruptors: Antiestrogenic activity and mechanisms. Toxicol. Lett. 102-103, 343–347. [DOI] [PubMed] [Google Scholar]
- Salminen A., Kauppinen A., Kaarniranta K. (2017). FGF21 activates AMPK signaling: Impact on metabolic regulation and the aging process. J. Mol. Med. (Berl.) 95, 123–131. [DOI] [PubMed] [Google Scholar]
- Seefeld M. D., Corbett S. W., Keesey R. E., Peterson R. E. (1984). Characterization of the wasting syndrome in rats treated with 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 73, 311–322. [DOI] [PubMed] [Google Scholar]
- Seefeld M. D., Peterson R. E. (1984). Digestible energy and efficiency of feed utilization in rats treated with 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 74, 214–222. [DOI] [PubMed] [Google Scholar]
- Servillo G., Della Fazia M. A., Sassone-Corsi P. (2002). Coupling cAMP signaling to transcription in the liver: Pivotal role of CREB and CREM. Exp. Cell Res. 275, 143–154. [DOI] [PubMed] [Google Scholar]
- Sheehan D. C., Hrapchak B. B. (1987). Theory and Practice of Histotechnology, 2 ednBattelle Press, Columbus, Ohio. [Google Scholar]
- Silverstone A. E., Rosenbaum P. F., Weinstock R. S., Bartell S. M., Foushee H. R., Shelton C., Pavuk M. (2012). Polychlorinated biphenyl (PCB) exposure and diabetes: Results from the Anniston Community Health Survey. Environ. Health Perspect. 120, 727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y., Kanamoto R., Miller D. A., Ogawa H., Pitot H. C. (1990). Regulation of the expression of the serine dehydratase gene in the kidney and liver of the rat. Biochem. Biophys. Res. Commun. 170, 892–899. [DOI] [PubMed] [Google Scholar]
- Swanson H. I. (2002). DNA binding and protein interactions of the AHR/ARNT heterodimer that facilitate gene activation. Chem. Biol. Interact. 141, 63–76. [DOI] [PubMed] [Google Scholar]
- Thayer K. A., Heindel J. J., Bucher J. R., Gallo M. A. (2012). Role of environmental chemicals in diabetes and obesity: a National Toxicology Program workshop review. Environ Health Perspect. 1206, 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson D. M., Herway S. T., Fillmore N., Kim H., Brown J. D., Barrow J. R., Winder W. W. (2008). AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J. Appl. Physiol. (1985) 104, 429–438. [DOI] [PubMed] [Google Scholar]
- Thorens B. (1996). Glucose transporters in the regulation of intestinal, renal, and liver glucose fluxes. Am. J. Physiol. 270, G541–G553. [DOI] [PubMed] [Google Scholar]
- Thorens B., Mueckler M. (2010). Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 298, E141–E145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega R. B., Huss J. M., Kelly D. P. (2000). The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 20, 1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viluksela M., Stahl B. U., Birnbaum L. S., Rozman K. K. (1997). Subchronic/chronic toxicity of 1, 2, 3, 4, 6, 7, 8-heptachlorodibenzo-p-dioxin (HpCDD) in rats. Part II. Biochem. Effect Toxicol. Appl. Pharmacol. 146, 217–226. [DOI] [PubMed] [Google Scholar]
- Viluksela M., Stahl B. U., Birnbaum L. S., Rozman K. K. (1998). Subchronic/chronic toxicity of a mixture of four chlorinated dibenzo-p-dioxins in rats. II. Biochem. Effect Toxicol. Appl. Pharmacol. 151, 70–78. [DOI] [PubMed] [Google Scholar]
- Viluksela M., Stahl B. U., Rozman K. K. (1995). Tissue-specific effects of 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin (TCDD) on the activity of phosphoenolpyruvate carboxykinase (PEPCK) in rats. Toxicol. Appl. Pharmacol. 135, 308–315. [DOI] [PubMed] [Google Scholar]
- Viollet B., Guigas B., Sanz Garcia N., Leclerc J., Foretz M., Andreelli F. (2012). Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. (Lond) 122, 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B., Beier J. I., Clair H. B., Bellis-Jones H. J., Falkner K. C., McClain C. J., Cave M. C. (2013). Toxicant-associated steatohepatitis. Toxicol. Pathol. 41, 343–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Klaren W. D., Wels B. R., Simmons D. L., Olivier A. K., Wang K., Robertson L. W., Ludewig G. (2016). Dietary Manganese Modulates PCB126 Toxicity, Metal Status, and MnSOD in the Rat. Toxicol. Sci. 150, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber L. W., Lebofsky M., Stahl B. U., Smith S., Rozman K. K. (1995). Correlation between toxicity and effects on intermediary metabolism in 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin-treated male C57BL/6J and DBA/2J mice. Toxicol. Appl. Pharmacol. 131, 155–162. [DOI] [PubMed] [Google Scholar]
- Witters L. A., Gao G., Kemp B. E., Quistorff B. (1994). Hepatic 5'-AMP-activated protein kinase: Zonal distribution and relationship to acetyl-CoA carboxylase activity in varying nutritional states. Arch. Biochem. Biophys. 308, 413–419. [DOI] [PubMed] [Google Scholar]
- Zhang W., Sargis R. M., Volden P. A., Carmean C. M., Sun X. J., Brady M. J. (2012). PCB 126 and other dioxin-like PCBs specifically suppress hepatic PEPCK expression via the aryl hydrocarbon receptor. PLoS One 7, e37103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.