

Abstract
Essentials.
Classification of VWD includes quantitative (types 1 and 3) and qualitative (type 2) variants.
New assays of VWF‐platelet binding may improve accuracy in diagnosis.
Collagen binding represents an additional function of VWF not measured by current tests.
Treatment options for VWD include desmopressin, plasma‐derived, and recombinant VWF.
Clinically, von Willebrand disease (VWD) presents as mucosal bleeding caused by a decreased quantity or quality of von Willebrand factor (VWF). Diagnosis of VWD requires careful consideration of patient specific factors, bleeding symptoms, and laboratory results. Patients with borderline low VWF levels remain challenging, given that low VWF is not necessarily a guarantee of bleeding, but is present in many patients with symptoms, and treatment of low VWF may improve bleeding. Laboratory diagnosis of VWD is complex and no single test can determine the presence or absence of functional VWF. Historically, VWF binding to platelet GPIbα was measured by the ristocetin cofactor assay (VWF:RCo); a new assay using platelet GPIbα in the absence of ristocetin (VWF:GPIbM) is gradually replacing the VWF:RCo due to improved accuracy in diagnosis. VWF binding to collagen is a separate function, and requires specific testing to determine if a collagen binding defect is present. Regardless of these laboratory complexities, clinicians can empirically treat VWD to alleviate bleeding symptoms by raising VWF levels through desmopressin or VWF concentrate. Recombinant VWF is now available, but clinicians may need to add an initial dose of FVIII when treating emergency bleeds.
Keywords: bleeding, hemostasis, platelets, von Willebrand disease, von Willebrand factor
1. BIOLOGY OF VON WILLEBRAND DISEASE
von Willebrand factor (VWF) is a large hemostatic protein produced in endothelial cells and megakaryocytes. VWF functions to bind platelets to exposed collagen in areas of endothelial injury to promote normal hemostasis. VWF also contributes to the fibrin clot by serving as a shuttle for the coagulation protein factor VIII (FVIII), preventing its degradation by plasma proteases. The three functional aspects of VWF can be described as platelet binding, collagen binding, and FVIII binding (Figure 1).
Figure 1.
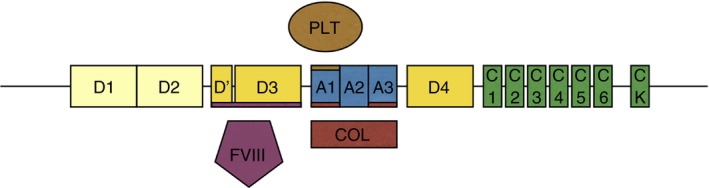
The structure of VWF. The D1 and D2 domains make up the VWF propeptide and are cleaved in the production of mature VWF. The D′ and D3 domains bind factor VIII (FVIII), the A1 domain binds platelets (PLT) and collagen (COL), and the A3 domain also binds collagen. VWF, von Willebrand factor
von Willebrand disease (VWD) is thought to be the most common bleeding disorder, affecting up to 0.1% of the population.1 Patients with VWD typically present with mild to severe mucosal bleeding including epistaxis, menorrhagia, gingival bleeding, bruising, and gastrointestinal bleeding. Personal and family history of bleeding, along with appropriate laboratory findings, is sufficient to make a diagnosis of VWD.2 Laboratory testing for VWD is complex and physicians do not consistently perform the same battery of tests on all patients. The purpose of this article is to provide an overview and discussion of some issues around the diagnosis of VWD.
2. CLASSIFICATION OF VON WILLEBRAND DISEASE
VWD is classified into three major types: type 1 VWD, type 2 VWD, and type 3 VWD. Within these three types there are two kinds of pathophysiology, those due to decreased quantity of VWF and those due to decreased quality of VWF. Table 1 summarizes typical laboratory findings for each type. Types 1 and 3 VWD are classified as a decreased quantity of VWF, meaning the concentration of plasma VWF is not high enough to prevent bleeding symptoms. Type 1 VWD patients have a decreased plasma concentration of VWF, while type 3 VWD patients generally have an undetectable plasma VWF concentration.
Table 1.
Screening assays for VWD and expected results for each type
| Assay | Type 1 | Type 2A | Type 2B | Type 2M | Type 2N | Type 3 | Reference range |
|---|---|---|---|---|---|---|---|
| VWF:Ag | Low | Low | Low | Normal/low | Normal/low | Undetectable | 50‐240 IU/dL |
| VWF:GPIbMa or VWF:RCoa | Low | Very low | Very low | Low | Normal/Low | Undetectable | 50‐240 IU/dL |
| VWF:GPIbM/VWF:Ag or VWF:RCo/VWF:Ag | Normal | Low | Low | Low | Normal | – | >0.6 |
| VWF:CB | Low | Very low | Very low | Normal/low | Normal/low | Undetectable | 50‐240 IU/dL |
| VWF:CB/VWF:Ag | Normal | Low | Low | Normal/low | Normal | – | >0.6 |
| FVIII:C | Normal/low | Normal/low | Normal/low | Normal/low | Very low | Very low | 60‐190 IU/dL |
| FVIII:C/VWF:Ag | Normal | Normal | Normal | Normal | Low | – | >0.7 |
FVIII:C, factor VIII; VWF, von Willebrand factor; VWF:Ag, VWF antigen; VWF:CB, VWF collagen binding; VWF:GPIbM, VWF binding to mutant (gain of function) GPIb; VWF:RCo, VWF ristocetin cofactor activity
VWF:GPIbM has replaced the VWF:RCo in some centers, but VWF:RCo or any VWF platelet‐dependent activity assay could be used here as well.
Type 1 VWD is usually autosomal dominant with variable penetrance among families, and accounts for 70‐80% of VWD cases.3 A specific type 1 VWD subgroup includes those with increased clearance of VWF, also called type 1C VWD.4 Type 1 VWD patients have a decreased VWF antigen (VWF:Ag), decreased ability of VWF to bind platelets (measured by either decreased binding to platelet GPIb through the VWF:GPIbM assay or decreased ristocetin‐induced binding to platelets by the ristocetin cofactor activity assay), and decreased ability to bind collagen (VWF:CB). The VWF propeptide will be elevated in type 1C.5 However, activity to antigen ratios will be normal since the VWF protein produced still maintains normal function.
With regard to type 3 VWD, some have used this category to include those with minimal residual VWF levels (ie, <5 IU/dL).6 Others consider any detectable VWF to represent type 1 VWD, albeit a more severe phenotype, and reserve the label of type 3 VWD for those with completely undetectable VWF levels.2, 7 The distinction is relevant to the inheritance, as type 1 is considered to be autosomal dominant and type 3 autosomal recessive, but not to treatment, as any patient with VWF levels <10 IU/dL will likely require treatment with a VWF concentrate. This discussion will be echoed below under “treatment of VWD” but there is a divergence between biologists whose interest is the pathophysiology of the disease and clinicians whose main concern remains the required treatment.
Type 2 VWD is classified as a decreased function of VWF, where VWF does not perform one or more of its native functions properly. Type 2 VWD is further divided into sub‐types: 2A, 2B, 2M, and 2N.2 The dysfunctions in type 2 VWD can be a loss of function, as seen in types 2A, 2M, and 2N, or a gain of function as seen in type 2B, where VWF binds platelets with increased affinity. Type 2 VWD represents differences in the degree and type of protein dysfunction, but all type 2 variants are linked by the fact that any VWF protein made is lacking in a critical function and in general will require treatment with a VWF concentrate.
In a normal physiologic state VWF is found in the plasma in various sizes, but notably including high molecular weight multimers (HMWM). The multimeric form of VWF is disrupted in types 2A and 2B of VWD (Figures 2A and B), decreasing the function of VWF. Normally, upon endothelial injury VWF's A1 and A3 domains bind to collagen, and the resultant shear stress changes the conformation of VWF to allow VWF binding to platelet GPIbα.8 In this way VWF acts as a bridge to bind platelets to exposed collagen on injured endothelium, acting as a first responder in coagulation. However, since platelets and collagen both bind to the A1 domain of VWF, a variant in this domain can lead to multiple dysfunctions. For example, in type 2M VWD a patient may experience decreased platelet binding and collagen binding (Figure 2C).
Figure 2.
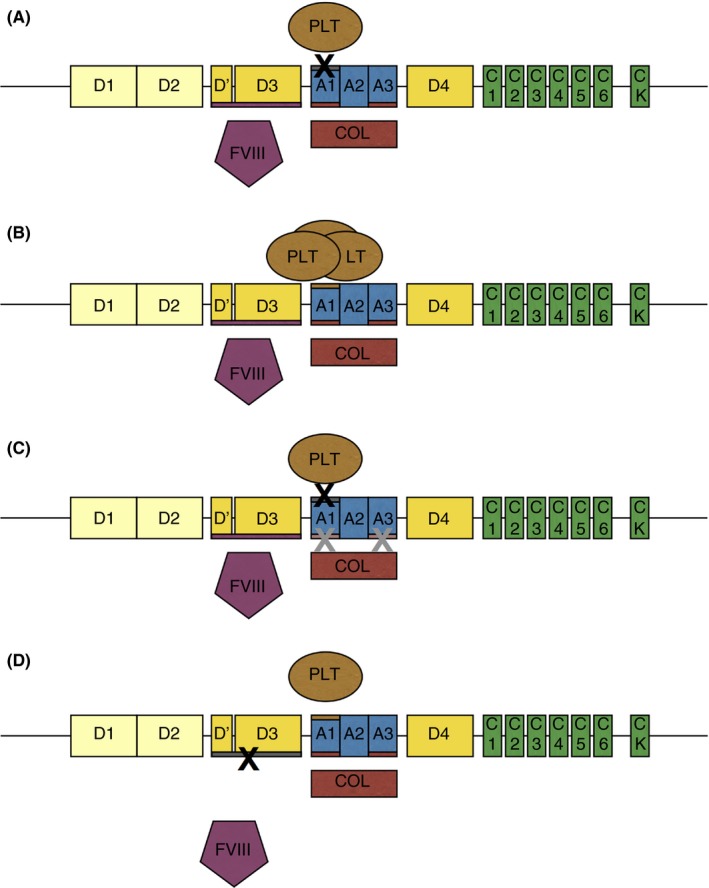
Type 2 VWD Variants. Panel A: Type 2A VWD has variants in the A2 domain, causing decreased platelet binding ability and loss of HMWM, and can also have variants in the N and C terminal multimerization domains. Panel B: Type 2B VWD has variants in the A1 domain which increase platelet (PLT) binding to VWF and result in clearance of the VWF:platelet complex. Panel C: Type 2M VWD has variants in the A1 and A3 domains, which cause decreased platelet (PLT) binding and may or may not cause decreased collagen (COL) binding. Panel D: Type 2N VWD has variants in the D′ and D3 domains which cause decreased factor VIII (FVIII) binding to VWF and subsequent FVIII clearance from circulation. HMWM, high molecular weight multimers; VWD, von Willebrand disease
Type 2A VWD is inherited as an autosomal dominant trait and accounts for 10‐20% of VWD cases.7 Mutations in the A2 domain, N terminal D3 domain, and C terminus (see Figure 2A) affect the ADAMTS13 cleavage site, prevent the formation of multimers, and/or cause potential intracellular retention of VWF.2, 9 The ADAMTS13 cleavage site is on exon 28 between p.Y1605 and p.M1606, and variants in this region can result in type 2A VWD.9 These patients will have a decreased VWF:Ag, a low VWF:GPIbM/VWF:Ag ratio, a low VWF:CB/VWF:Ag ratio, and an absence of HMWM. Platelet binding (VWF:PB) and low‐dose ristocetin‐induced platelet aggregation (LD‐RIPA) on these patients would be absent.7 Genetic testing may reveal a known type 2A variant and help solidify the diagnosis. Unfortunately, interpretation of novel variants is complicated by the known normal variability in the VWF gene.10
Type 2B VWD is inherited as an autosomal dominant trait and accounts for 5% of VWD cases.7 Most type 2B mutations occur on exon 28 in the A1 domain of VWF, which contains the binding site for platelet GPIbα as shown in figure 2A.11 The pathogenic change in the A1 domain in type 2B VWD causes VWF to more readily bind platelets, acting as a gain of function mutation, and resulting in increased uptake and destruction of the VWF‐platelet complex.12 In some cases the destruction of platelets in type 2B VWD can cause thrombocytopenia.13 The sequestration of the VWF‐platelet complexes also results in a loss of HMWM. These patients will have a decreased VWF:Ag, a low VWF:GPIbM/VWF:Ag ratio, a low VWF:CB/VWF:Ag ratio, an absence of HMWM, and an increased VWF:PB or an increased LD‐RIPA.2
Type 2M VWD is inherited as an autosomal dominant trait and makes up less than 5% of VWD cases.7 Many type 2M variants have been shown to cause a loss of function that interferes with VWF's ability to bind platelet GPIbα as shown in Figure 2C. Some of these variants have been shown to cause VWF misfolding as the reason for dysfunction.14 These patients will have a decreased VWF:Ag, a low VWF:GPIbM/VWF:Ag ratio, and normal distribution of HMWM. The present classification system also includes collagen binding defects in the type 2M category. Several variants in the VWF A3 domain, which binds collagen types I and III, have been reported, although most are also associated with low VWF:Ag.15, 16, 17 VWF A1 domain variants that affect binding to collagen types IV and VI have also been reported.18
Type 2N VWD is inherited as an autosomal recessive trait, making it the second least common type of VWD. Type 2N VWD can be caused by inheritance of two type 2N variants, or by co‐inheritance of one type 2N variant and one type 1 variant. Type 2N mutations can primarily be found in the VWF D′ and D3 domains as shown in Figure 2D, between p.S764 and p.R1035, which contain the binding site for FVIII.9 Without the proper protection afforded by VWF, FVIII is degraded in plasma. These patients may have a normal VWF:Ag with a normal distribution of multimers, but will have a low FVIII activity to VWF antigen ratio (FVIII:C/VWF:Ag), and low FVIII binding (VWF:FVIIIB). Because VWF levels may be normal, type 2N VWD is sometimes misdiagnosed as hemophilia A.19 A primary concern for clinicians is to ensure patients are not incorrectly diagnosed with mild hemophilia, and treated with only FVIII, which will not suffice to improve bleeding symptoms.
3. DIAGNOSIS OF VWD
Suspicion of VWD starts with a bleeding phenotype. Bleeding assessment tools (BATs) are questionnaires used to help clinicians evaluate a patient's history of bleeding symptoms and assign them a score based on the severity of those bleeding symptoms. BATs objectively score patients based on the severity, duration, and need for treatment for bleeding episodes. The International Society of Thrombosis and Hemostasis (ISTH) has created a comprehensive questionnaire (ISTH‐BAT) that can be used by researchers and clinicians.20 The normal score on the ISTH‐BAT is 0‐3 for adult males, 0‐5 for adult females, and 0‐2 for children. A positive or abnormal score would be ≥4 for males, ≥6 for females, and ≥3 for children, all of which should be considered further for bleeding disorders, including VWD.21
Bleeding assessment, either clinically or using a bleeding assessment tool, is critical when considering a diagnosis of VWD as some patients may have low VWF levels but lack bleeding symptoms and likely should not receive a diagnosis of VWD. The cutoff for type 1 VWD reference ranges is a topic of debate.22 Low VWF level alone does not indicate disease, but rather indicates a risk factor for bleeding. The normal range of VWF levels is set by most laboratories to include all levels within 2 standard deviations of the population mean. This arbitrarily excludes 5% of the population, half of whom will have levels below the lower limit of “normal”. Since 2.5% of the population does not have VWD, low levels of VWF alone should not constitute a diagnosis.
Patient‐specific factors should be considered when interpreting VWF levels, with a pre‐requisite presence of bleeding symptoms if VWD is being considered. For example, VWF levels are significantly affected by ABO blood groups. The average VWF:Ag for type O blood has been shown to be 30–50 IU/dL lower than A, B, or AB blood types, with some healthy controls in the 30‐40 IU/dL range.23 Many people who are blood group O will have low VWF, but this does not necessarily correspond to, or cause, bleeding symptoms.
The former NHLBI consensus guideline statement used a cut‐off of 30 IU/dL to declare the presence of VWD.7 UK guidelines have also adopted this cutoff.24 There may be a genetic reason for this, as the incidence of sequence variants in VWF increase with decreasing VWF:Ag, and there does appear to be a significant increase in frequency of such variants with VWF levels below 30 IU/dL.25, 26, 27 However, one confounder of many of the current studies is the predilection for inclusion of subjects with a pre‐existing diagnosis of VWD. In general, such subjects are diagnosed because of bleeding, and typically once a diagnosis of VWD is reached, clinicians stop their evaluation. Thus, some cases may have concomitant platelet defects or other coagulation defects that also contribute to bleeding.
The case for additional genetic modifiers of VWF is also growing stronger. While ABO blood group is the only modifier with substantial evidence in patients,23 several publications have implicated other genes in VWF pathophysiology including CLEC4M and STXBP5.28, 29 This raises the question of whether VWD is exclusively the province of variants in VWF, or the sum of all factors affecting VWF and coagulation in a given individual.
Several circumstances can complicate the phenotype of a VWD patient including race, age, and comorbidities. VWF levels have been shown to increase with age,30 which could mask a VWD phenotype in a patient that may still be at risk of bleeding. Similarly, inherited prothrombotic risk factors could mask a VWD phenotype. Factor V Leiden, a highly prevalent variant in the Caucasian population, reduces the ability of protein C to inactivate factor V, leading to a prothrombotic state. The factor V Leiden mutation has been shown to partially compensate for the low FVIII levels in patients with hemophilia, attenuating the bleeding phenotype.31 Similarly, patients with low VWF may have other conditions such as factor V Leiden masking a bleeding phenotype.
It has been argued that low VWF constitutes a risk factor for bleeding, but is not necessarily a single and sufficient cause.32 Therefore, patients could be classified and treated based on their presentation and concomitant risk factors for bleeding, rather than receiving a final and unchanging label of von Willebrand disease. It may therefore be prudent to consider other co‐inherited or acquired conditions when planning treatment for a patient with VWD, particularly type 1 VWD.
4. RISE OF THE VWF:GPIBM ASSAY (AND DEATH OF THE VWF:RCO?)
Previously VWF function was measured using the ristocetin cofactor activity assay (VWF:RCo).33 However, VWF:RCo has poor reproducibility between laboratories, giving a wide range of results and interpretations.34, 35 Additional platelet‐dependent VWF activity assays include using a monoclonal antibody to detect the GPIb site of VWF (VWF:Ab), and using a recombinant GPIb to induce ristocetin binding to VWF (VWF:GPIbR). The VWF:Ab assay does not necessarily test the function of the GPIb domain, rather the presence of it, but is an option in many laboratories.36 The VWF:GPIbR still requires optimal ristocetin concentration, and is therefore subject to some of the same limitations as the VWF:RCo, but may have improved coefficient of variation.37 Since testing the platelet binding ability of VWF is crucial to understanding VWF function and differentiating between quantitative or qualitative defects in VWF, high levels of variability have been a burden on clinicians in the process of diagnosing VWD. The inconsistencies and inadequacies in these assays, and the potential for misdiagnosis, have led to the novel VWF:GPIbM assay.
The VWF:GPIbM assay exploits of gain of function variants in the platelet GPIbα domain that accomplishes the same purpose as the ristocetin cofactor assay: measuring VWF's ability to bind platelets, but without the requirement for ristocetin.38 The name VWF:GPIbM refers to the use of GPIb rather than platelets and to the fact that the GPIb contains mutations (M) as per ISTH recommendations for VWF activity assay nomenclature.39 To properly evaluate VWF:GPIbM, results should be considered in context of VWF:Ag. If there is a discrepancy between VWF:Ag and VWF:GPIbM (ie, VWF:Ag is low and VWF:GPIbM is much lower), concern is raised for a qualitative dysfunction in VWF.
5. CLINICAL EVALUATION OF VWF COLLAGEN BINDING
Collagen binding (VWF:CB) can be tested via ELISA and measures one of the major functions of VWF. VWF:CB should be considered in proportion to VWF:Ag, and the VWF:CB/VWF:Ag ratio can help to distinguish between type 1 and 2 VWD. If VWF:Ag and VWF:CB are low in proportion to each other (VWF:CB/VWF:Ag ratio of approximately 1) the patient likely does not have a collagen binding defect. A decreased VWF:CB out of proportion to VWF:Ag (VWF:CB/VWF:Ag ratio <0.6) is indicative of a collagen binding defect (type 2M) and/or a multimer defect (types 2A and 2B).
Human type I and type III collagens are most commonly used in testing VWF:CB because of their sensitivity and specificity.40 Collagens I and III bind to the A3 domain of VWF.41 The VWF:CB assay can be used to determine if there are any binding defects between VWF and collagen I or III. Several mutations in the A3 domain have been shown to disrupt the VWF collagen interaction and lead to bleeding symptoms including p.Ser1731Thr,15 p.Trp1745Cys,16 p.Met1761Lys,42 p.Ser1783Ala,16 and p.His1786Asp.17
Type IV and type VI collagen also bind VWF, but via the A1 domain.43 The sequence variant p.Arg1399His within the A1 domain prevents VWF from binding type IV and VI collagen44 and has been reported to occur in up to 2% of the Caucasian population.45 In heterozygous form this would be expected to be asymptomatic, but the potential exists for individuals homozygous for this variant with completely absent collagen binding. While the clinical significance of this needs further evaluation, testing for binding defects in type IV or VI collagen is currently not common practice. Other A1 domain variants including p.1387Ile, p.1392Ala, p.1395Ala, and p.1402Pro have also been shown to abrogate VWF binding to collagen IV.44, 46 If a defect in VWF function is suspected due to increased bleeding, evaluation of VWF binding to platelets and collagen types I and III may not suffice.
6. TREATMENT OF VWD
The classification and diagnosis of VWD is complicated. As a biologist, the classification and laboratory differentiation of VWD is something that may be of significant interest and should be explored more thoroughly. However, as a clinician the important conclusion that needs to be reached is how to treat the patient. Figure 3 depicts a flowchart outlining possible treatment paths for patients depending on VWF laboratory assay results, with the understanding that such testing is being performed on a symptomatic patient.
Figure 3.
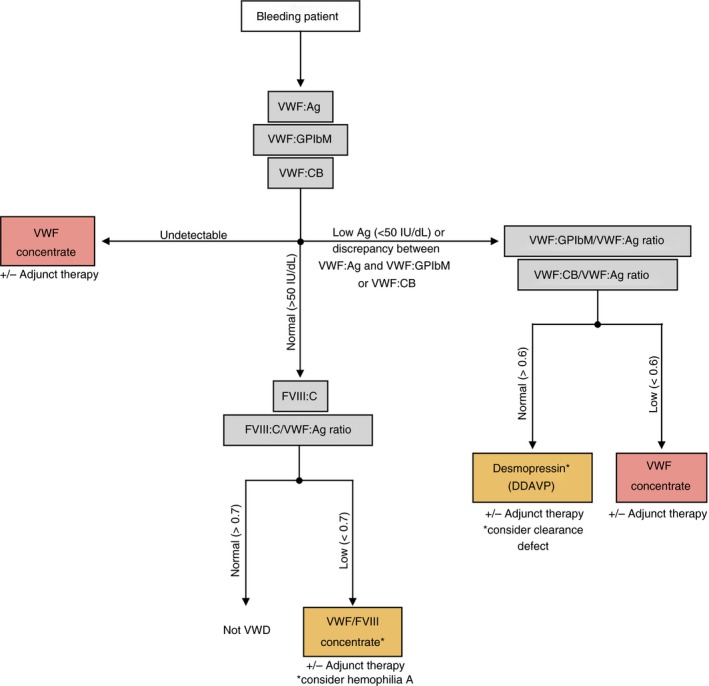
Treatment scheme for VWD. The gray boxes indicate laboratory assays and the red and orange boxes indicate treatment options. Treatments in the orange boxes should only be used after considering the other possible disorders indicated by an asterisk. Patients who do not fit the diagnostic criteria for VWD but are experiencing bleeding symptoms should be considered for collagen binding defects, platelet function defects, or other mild bleeding disorders. Clinicians should also consider adjunct therapy as needed to alleviate patient symptoms. VWD, von Willebrand disease
If a patient is experiencing bleeding symptoms and has a decreased VWF level, empiric treatment can be provided to raise VWF levels. Options include either desmopressin or replacement of VWF. In addition, adjunct therapies such as antifibrinolytics or hormone therapy for heavy menstrual bleeding may be effective.
Desmopressin (DDAVP) is an analog of vasopressin that causes the rapid release of VWF and FVIII. DDAVP is used to treat type 1 VWD, with a 2‐4–fold increase in VWF and a 3‐6–fold increase in FVIII within 30‐60 minutes.47, 48 Since patients with type 2 VWD do not have functional VWF, treatment with DDAVP is not usually adequate to alleviate bleeding symptoms as it only increases the concentration of dysfunctional VWF in the blood. Type 3 VWD patients do not produce VWF, therefore DDAVP would not produce a rise in VWF levels.
Human plasma‐derived VWF can be used as replacement therapy for patients lacking sufficient VWF levels or VWF function. In the United States, VWD is typically treated with either Humate‐P49 or Wilate.50 Both products also contain plasma derived FVIII, although at different ratios (VWF:FVIII 2.4:1 for Humate‐P and 1:1 for Wilate). In Europe, a plasma‐derived VWF with virtually no FVIII is also available, marketed as Wilfactin.51 The latter product does pose slight challenges for emergency treatment in VWD patients whose FVIII levels are also low, in that emergency treatment likely needs to include both VWF and FVIII. For surgical treatment, the VWF concentrate needs to be administered about 12 hours prior to surgery, to allow the newly provided VWF to stabilize endogenous FVIII levels.51 The other option is to provide both VWF and FVIII for the first dose, with the knowledge that subsequent FVIII dosing will not be needed since in the presence of normal VWF the endogenous FVIII production should be sufficient.
Recombinant VWF (rVWF, Vonvendi), a synthetically produced VWF, is another replacement therapy option in the treatment of type 1 (severe), type 2, and 3 VWD.52 As with Wilfactin, rVWF does not contain FVIII, so in patients with low FVIII levels emergency treatment will require additional administration of FVIII and surgical treatment will either require both VWF and FVIII or a scheduled preoperative dose of VWF the day before surgery.
Most type 1 patients will respond to desmopressin. A trial should still be performed as there remain select patients with no or less than ideal desmopressin responses. If the VWF:Ag is less than 20‐30 IU/dL, consideration should also be given to a clearance defect and either a late timepoint assessed following desmopressin or analysis of the VWF propeptide. Practically speaking, however, most type 1 patients with levels <10‐20 IU/dL will require treatment with a VWF concentrate regardless as even a 3‐fold increase in their VWF level will still be insufficient for surgery.
Most type 2 patients will require treatment with a VWF concentrate for most types of bleeds, as desmopressin may raise the level of VWF, but does not correct the underlying dysfunction. There may still be a role for desmopressin in type 2 VWD for minor bleeds such as epistaxis if the patient does experience clinical improvement, although type 2B VWD is still considered a relative contraindication for desmopressin treatment.
7. CONCLUSIONS
All laboratory testing for VWD should be considered in the context of the patient's bleeding symptoms and family history. Laboratory assays are not sufficient and bleeding is a prerequisite to diagnose VWD. Screening tests for VWD can help clinicians to detect the presence of a quantitative deficiency in VWF or determine the need for further testing. Laboratory testing for VWD is complex and results should be considered in the context of each individual patient. From a biology perspective, the complexities of VWD types merit investigation. From a clinical perspective, empiric treatment improves bleeding symptoms in VWD and use of desmopressin as compared to a VWF concentrate may not require complete phenotyping.
AUTHOR CONTRIBUTIONS
D.A. Keesler wrote the manuscript. V.H. Flood edited the manuscript.
RELATIONSHIP DISCLOSURE
V. Flood reports grants from National Institute of Health during the conduct of the study; as well as personal fees from CSL Behring and from Shire outside the submitted work. D. Keesler has nothing to disclose.
ACKNOWLEDGEMENTS
The authors would also like to acknowledge the numerous VWD researchers, both those cited here and those unable to be cited due to space constraints.
Keesler DA, Flood VH. Current issues in diagnosis and treatment of von Willebrand disease. Res Pract Thromb Haemost. 2018;2:34–41. 10.1002/rth2.12064
Funding Information
This work was funded by the National Institutes of Health (HL126810) and the MACC Fund Center for Cancer and Blood Disorders
REFERENCES
- 1. Bowman M, Hopman WM, Rapson D, Lillicrap D, James P. The prevalence of symptomatic von willebrand disease in primary care practice. J Thromb Haemost. 2010;8:213–6. [DOI] [PubMed] [Google Scholar]
- 2. Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von willebrand disease: a report of the subcommittee on von willebrand factor. J Thromb Haemost. 2006;4:2103–14. [DOI] [PubMed] [Google Scholar]
- 3. Robertson J, Lillicrap D, James PD. Von Willebrand disease. Pediatr Clin North Am. 2008;55:377–92, viii‐ix. [DOI] [PubMed] [Google Scholar]
- 4. Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006;108:3344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM‐1VWD). Blood. 2008;111:4979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bowman M, Tuttle A, Notley C, et al. The genetics of canadian type 3 von Willebrand disease: further evidence for co‐dominant inheritance of mutant alleles. J Thromb Haemost. 2013;11:512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence‐based diagnosis and management guidelines, the national heart, lung, and blood institute (NHLBI) expert panel report (USA). Haemophilia. 2008;14:171–232. [DOI] [PubMed] [Google Scholar]
- 8. Goto S, Salomon DR, Ikeda Y, Ruggeri ZM. Characterization of the unique mechanism mediating the shear‐dependent binding of soluble von Willebrand factor to platelets. J Biol Chem. 1995;270:23352–61. [DOI] [PubMed] [Google Scholar]
- 9. Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev. 2010;24:123–34. [DOI] [PubMed] [Google Scholar]
- 10. Bellissimo DB, Christopherson PA, Flood VH, et al. VWF mutations and new sequence variations identified in healthy controls are more frequent in the african‐american population. Blood. 2012;119:2135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fujimura Y, Titani K, Holland LZ, et al. Von Willebrand factor. A reduced and alkylated 52/48‐kDa fragment beginning at amino acid residue 449 contains the domain interacting with platelet glycoprotein ib. J Biol Chem. 1986;261:381–5. [PubMed] [Google Scholar]
- 12. Ruggeri ZM, Pareti FI, Mannucci PM, Ciavarella N, Zimmerman TS. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand's disease. N Engl J Med. 1980;302:1047–51. [DOI] [PubMed] [Google Scholar]
- 13. Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood. 2009;113:526–34. [DOI] [PubMed] [Google Scholar]
- 14. Tischer A, Madde P, Moon‐Tasson L, Auton M. Misfolding of vWF to pathologically disordered conformations impacts the severity of von Willebrand disease. Biophys J. 2014;107:1185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ribba AS, Loisel I, Lavergne JM, et al. Ser968Thr mutation within the A3 domain of von Willebrand factor (VWF) in two related patients leads to a defective binding of VWF to collagen. Thromb Haemost. 2001;86:848–54. [PubMed] [Google Scholar]
- 16. Riddell AF, Gomez K, Millar CM, et al. Characterization of W1745C and S1783A: 2 novel mutations causing defective collagen binding in the A3 domain of von willebrand factor. Blood. 2009;114:3489–96. [DOI] [PubMed] [Google Scholar]
- 17. Flood VH, Lederman CA, Wren JS, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost. 2010;8:1431–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Flood VH, Schlauderaff AC, Haberichter SL, et al. Crucial role for the VWF A1 domain in binding to type IV collagen. Blood. 2015;125:2297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gupta M, Lillicrap D, Stain AM, Friedman KD, Carcao MD. Therapeutic consequences for misdiagnosis of type 2N von Willebrand disease. Pediatr Blood Cancer. 2011;57:1081–3. [DOI] [PubMed] [Google Scholar]
- 20. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8:2063–5. [DOI] [PubMed] [Google Scholar]
- 21. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH‐BAT: adult and pediatric data from the merging project. Haemophilia. 2014;20:831–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sadler JE. Slippery criteria for von Willebrand disease type 1. J Thromb Haemost. 2004;2:1720–3. [DOI] [PubMed] [Google Scholar]
- 23. Gill JC, Endres‐Brooks J, Bauer PJ, Marks WJ Jr, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood. 1987;69:1691–5. [PubMed] [Google Scholar]
- 24. Laffan MA, Lester W, O'Donnell JS, et al. The diagnosis and management of von Willebrand disease: a united kingdom haemophilia centre doctors organization guideline approved by the british committee for standards in haematology. Br J Haematol. 2014;167:453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. James PD, Notley C, Hegadorn C, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109:145–54. [DOI] [PubMed] [Google Scholar]
- 26. Goodeve A, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand disease (MCMDM‐1VWD). Blood. 2007;109:112–21. [DOI] [PubMed] [Google Scholar]
- 27. Flood VH, Christopherson PA, Gill JC, et al. Clinical and laboratory variability in a cohort of patients diagnosed with type 1 VWD in the United States. Blood. 2016;127:2481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rydz N, Swystun LL, Notley C, et al. The C‐type lectin receptor CLEC4M binds, internalizes, and clears von Willebrand factor and contributes to the variation in plasma von Willebrand factor levels. Blood. 2013;121:5228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sanders YV, van der Bom JG, Isaacs A, et al. CLEC4M and STXBP5 gene variations contribute to von Willebrand factor level variation in von Willebrand disease. J Thromb Haemost. 2015;13:956–66. [DOI] [PubMed] [Google Scholar]
- 30. Konkle BA. Von Willebrand factor and aging. Semin Thromb Hemost. 2014;40:640–4. [DOI] [PubMed] [Google Scholar]
- 31. Franchini M, Lippi G. Factor V leiden and hemophilia. Thromb Res. 2010;125:119–23. [DOI] [PubMed] [Google Scholar]
- 32. Sadler JE. Von Willebrand disease type 1: a diagnosis in search of a disease. Blood. 2003;101:2089–93. [DOI] [PubMed] [Google Scholar]
- 33. Howard MA, Sawers RJ, Firkin BG. Ristocetin: a means of differentiating von Willebrand's disease into two groups. Blood. 1973;41:687–90. [PubMed] [Google Scholar]
- 34. Kitchen S, Jennings I, Woods TA, Kitchen DP, Walker ID, Preston FE. Laboratory tests for measurement of von Willebrand factor show poor agreement among different centers: results from the United Kingdom national external quality assessment scheme for blood coagulation. Semin Thromb Hemost. 2006;32:492–8. [DOI] [PubMed] [Google Scholar]
- 35. Meijer P, Haverkate F. An external quality assessment program for von Willebrand factor laboratory analysis: an overview from the european concerted action on thrombosis and disabilities foundation. Semin Thromb Hemost. 2006;32:485–91. [DOI] [PubMed] [Google Scholar]
- 36. Salem RO, Van Cott EM. A new automated screening assay for the diagnosis of von Willebrand disease. Am J Clin Pathol. 2007;127:730–5. [DOI] [PubMed] [Google Scholar]
- 37. Vanhoorelbeke K, Cauwenberghs N, Vauterin S, Schlammadinger A, Mazurier C, Deckmyn H. A reliable and reproducible ELISA method to measure ristocetin cofactor activity of von Willebrand factor. Thromb Haemost. 2000;83:107–13. [PubMed] [Google Scholar]
- 38. Patzke J, Budde U, Huber A, et al. Performance evaluation and multicentre study of a von willebrand factor activity assay based on GPIb binding in the absence of ristocetin. Blood Coagul Fibrinolysis. 2014;25:860–70. [DOI] [PubMed] [Google Scholar]
- 39. Bodo I, Eikenboom J, Montgomery R, et al. Platelet‐dependent von willebrand factor activity. nomenclature and methodology: communication from the SSC of the ISTH. J Thromb Haemost. 2015;13:1345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Favaloro EJ. An update on the von Willebrand factor collagen binding assay: 21 years of age and beyond adolescence but not yet a mature adult. Semin Thromb Hemost. 2007;33:727–44. [DOI] [PubMed] [Google Scholar]
- 41. Pareti FI, Niiya K, McPherson JM, Ruggeri ZM. Isolation and characterization of two domains of human von Willebrand factor that interact with fibrillar collagen types I and III. J Biol Chem. 1987;262:13835–41. [PubMed] [Google Scholar]
- 42. Keeling D, Beavis J, Marr R, Sukhu K, Bignell P. A family with type 2M VWD with normal VWF:RCo but reduced VWF:CB and a M1761K mutation in the A3 domain. Haemophilia 2012;18:e33. [DOI] [PubMed] [Google Scholar]
- 43. Rand JH, Patel ND, Schwartz E, Zhou SL, Potter BJ. 150‐kD von Willebrand factor binding protein extracted from human vascular subendothelium is type VI collagen. J Clin Invest. 1991;88:253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Flood VH, Gill JC, Christopherson PA, et al. Critical von Willebrand factor A1 domain residues influence type VI collagen binding. J Thromb Haemost. 2012;10:1417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sadler JE, Ginsburg D. A database of polymorphisms in the von Willebrand factor gene and pseudogene. for the consortium on von Willebrand factor mutations and polymorphisms and the subcommittee on von Willebrand factor of the scientific and standardization committee of the international society on thrombosis and haemostasis. Thromb Haemost. 1993;69:185–91. [PubMed] [Google Scholar]
- 46. Doruelo AL, Haberichter SL, Christopherson PA, et al. Clinical and laboratory phenotype variability in type 2M von Willebrand disease. J Thromb Haemost. 2017;15:1559–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Federici AB, Mazurier C, Berntorp E, et al. Biologic response to desmopressin in patients with severe type 1 and type 2 von Willebrand disease: results of a multicenter European study. Blood. 2004;103:2032–8. [DOI] [PubMed] [Google Scholar]
- 48. Castaman G, Lethagen S, Federici AB, et al. Response to desmopressin is influenced by the genotype and phenotype in type 1 von Willebrand disease (VWD): results from the European study MCMDM‐1VWD. Blood. 2008;111:3531–9. [DOI] [PubMed] [Google Scholar]
- 49. Miesbach W, Krekeler S, Wolf Z, Seifried E. Clinical use of haemate(R) P in von Willebrand disease: a 25‐year retrospective observational study. Thromb Res. 2015;135:479–84. [DOI] [PubMed] [Google Scholar]
- 50. Windyga J, von Depka‐Prondzinski M, European Wilate(R) Study Group . Efficacy and safety of a new generation von Willebrand factor/factor VIII concentrate (wilate(R)) in the management of perioperative haemostasis in von Willebrand disease patients undergoing surgery. Thromb Haemost. 2011;105:1072–9. [DOI] [PubMed] [Google Scholar]
- 51. Borel‐Derlon A, Federici AB, Roussel‐Robert V, et al. Treatment of severe von Willebrand disease with a high‐purity von Willebrand factor concentrate (wilfactin): a prospective study of 50 patients. J Thromb Haemost. 2007;5:1115–24. [DOI] [PubMed] [Google Scholar]
- 52. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von willebrand factor in severe von Willebrand disease. Blood. 2015;126:2038–46. [DOI] [PMC free article] [PubMed] [Google Scholar]