

Abstract
Essentials.
Mutations in the RASGRP2 gene represent a new inherited platelet function disorder.
Report a five generation family with a novel frameshift mutation in RASGRP2 (p.F497Sfs*22).
Partial platelet activation defect and serious bleeding complications in homozygous patients.
Patients respond to recombinant Factor VIIa infusion but not platelet transfusions.
Background
Genetic variants in the RASGRP2 gene encoding calcium and diacylglycerol‐regulated guanine nucleotide exchange factor I (CalDAG‐GEFI) represent a new inherited bleeding disorder linked to major defects of platelet aggregation and activation of αIIbβ3 integrin. They are of major interest as CalDAG‐GEFI is receiving attention as a potential target for antiplatelet therapy for prevention and treatment of cardiovascular disorders including arterial thrombosis and atherosclerosis.
Objectives
To better understand the phenotypical and clinical profiles of patients with CalDAG‐GEFI deficiency.
Patients
We report a five‐generation family with a novel truncating CalDAG‐GEFI mutation detailing clinical management and phenotypic variability.
Results
Patients IV.6 & IV.4 manifested with episodes of serious mucocutanous bleeding or bleeding after surgery not responding to platelet transfusion but responding well to recombinant Factor VIIa infusions. Their blood counts and coagulation parameters were normal but platelet aggregation to ADP and collagen was defective. Further work‐up confirmed normal levels of αIIb and β3 in their platelets but decreased αIIbβ3 function. DNA analysis by whole exome sequencing within the BRIDGE‐BPD consortium (Cambridge, UK), allowed us to highlight a homozygous c.1490delT predicted to give rise to a p.F497Sfs*22 truncating mutation near to the C‐terminal domain of CalDAG‐GEFI. Sanger sequencing confirmed that both patients were homozygous for the c.1490delT and 3 out of 4 close family members were heterozygous.
Conclusions
A long‐term prospective study is warranted for full clinical exploration of CalDAG‐GEFI to understand the bleeding phenotyes and their management.
Keywords: bleeding syndrome, CalDAG‐GEFI, clinical management, inherited platelet disorder, RASGRP2 gene
1. INTRODUCTION
Platelet adhesion and aggregation to form a primary hemostatic plug is an essential step in stopping bleeding. Inherited disorders of platelet production and/or function lead to increased spontaneous or surgical mucocutaneous bleeding and greater mortality in patients harboring these defects.1, 2 Integrin αIIbβ3, formerly known as GPIIb/IIIa, is the adhesive protein receptor that in an activated state mediates platelet aggregation.3 αIIbβ3 undergoes conformational changes from a bent to an extended and open form in stimulated platelets; changes brought about by “inside‐out” signaling through GTPase Ras‐proximate‐1 (Rap1) promoted talin and kindlin interactions with β3.4 In a major pathway, activated phospholipase C causes increased intracellular Ca2+ that in turn activates calcium and diacylglycerol‐regulated guanine nucleotide exchange factor I (CalDAG‐GEFI) which then activates Rap1.5, 6 Significantly, the role of CalDAG‐GEFI extends to other integrins including those in white blood cells. The patho‐physiological importance of CalDAG‐GEFI is highlighted by studies on knockout mice, which exhibit marked defects in hemostasis, arterial thrombosis and atherosclerotic plaque formation; studies that have suggested CalDAG‐GEFI as a therapeutic target for thrombosis and atherosclerosis.7, 8, 9, 10 Abrogation of CalDAG‐GEFI function in man leads to a Glanzmann thrombasthenia (GT)‐like bleeding disorder and a major loss of platelet aggregation.11 Next generation sequencing technology has facilitated the identification of disease‐causing mutations in the RASGRP2 gene that encodes CalDAG‐GEFI; genetic variants that have segregated with a severe bleeding diathesis in families from France, Spain and Japan.12, 13, 14 However, the number of cases studied remains small and the description of clinical profiles within families having genetically altered CalDAG‐GEFI function is incomplete. Here we present the clinical management of a large family with Jamaican ancestry with unexpectedly variable phenotypes associated with a novel truncating CalDAG‐GEFI mutation.
2. PATIENTS AND FAMILY MEMBERS
Figure 1A gives the pedigree of a five‐generation consanguineous family of Jamaican origin, genotyped members are highlighted (+/+, +/− or −/−) with + representing presence of the mutation. Essential laboratory parameters are given in Table 1.
Figure 1.
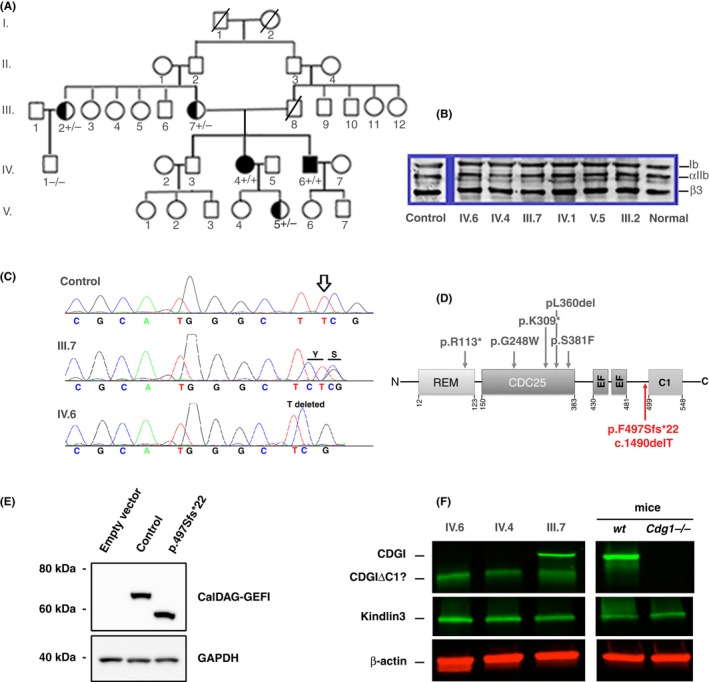
(A) Family pedigree of five generations. The genotyping of family members is shown when performed (+/+, black symbols; +/−, half‐black symbols; −/−, open symbols). Bar indicates deceased member. (B) Western blot of sodium‐dodecyl sulfate‐solubilized platelets from genotyped family members incubated after transfer with murine MoAbs to GPIb, αIIb and β3 with bound mouse immunoglobulin detected by chemiluminescence according to standard procedures.17 (C) RASGRP2 sequencing results showing the homozygous c.1490delT variation (p.F497Sfs*22) in propositus (bottom), heterozygous mother (middle) and the normal control sequence (top). (D) Schema showing the basic structure of the CalDAG‐GEFI protein and the positions of known disease‐causing genetic variants as reported in the literature.12, 13, 14 Our novel variant is in red. The different structural domains are the ras exchange motif (REM), the catalytic domain (CDC25), the calcium‐binding EF hands (EF), and the C1‐like regulatory domain with unknown function. (E) Representative western blot for CalDAG‐GEFI in GripTite 293 MSR cells transfected with vectors coding for the wild‐type and the p.F497Sfs*22 variant of RASGRP2. GAPDH (glyceraldehyde 3‐phosphate dehydrogenase) expression was used as a loading and electrophoretic transfer control.12 Note the presence of the faster migrating truncated band for the mutant protein. (F) Western blotting comparing the fluorescence detection of CalDAG‐GEFI in platelets from the index case (IV.6), his homozygous sister (IV.4); their heterozygous mother (III.7), wild‐type mice and Caldaggef1 knock‐out (Cdg1−/−) mice; technical details were as described.13 The intact CalDGAG‐GEFI protein is missing for the index case and his sister but present in mother and WT mice
Table 1.
Essential laboratory parameters
| Characteristic | IV.6 –index case | IV.4 sister |
|---|---|---|
| Age (years) | 51 | 55 |
| Gender | Male | Female |
| Blood counts | ||
| WBC (4.0‐10.5×103/μL) | 7.8 | 9.5 |
| Absolute neutrophil count (2‐6×103/μL) | 4.92 | 6.29 |
| Absolute lymphocyte count (2‐6×103/μL) | 1.96 | 2.63 |
| Monocyte count (0.06‐0.12×103/μL) | 0.55 | 0.47 |
| Hg (11.1‐14.6 g/dL) | 14.7 | 13.2 |
| Platelets (140‐400×103/μL) | 165 | 217 |
| PT (12.2‐14.9 s) | 13.8 | 13.4 |
| PTT (22.2‐35.5 s) | 28.7 | 31.2 |
| Fibrinogen level (139‐482 mg/dL) | 261 | 307 |
| D‐Dimer (<0.49 mg/L FEU) | 0.3 | 0.3 |
| Platelet aggregation assay | ||
| ADP | ||
| 10 μmol/L | 6% | 2% |
| 2 μmol/L | 0% | 1% |
| Collagen | ||
| 4 μg/mL | 1% | 11% |
| 1 μg/ml | 0% | 8% |
| TRAP | ||
| 14‐40 μmol/L | 68% | 75% |
| 8 μmol/L | 72% | 80% |
| PMA | ||
| 10 μmol/L | 75% | 78% |
| 2 μmol/L | 78% | 78% |
| Epinephrine 10 μmol/L | 2% | NA |
| Arachidonic acid 0.5 mmol/L | 0% | NA |
ADP, adenosine diphosphate; Hg, hemoglobin; PMA, phorbol 12‐myristate 13‐acetate; PT, prothrombin time; PTT, partial thromboplastin time; TRAP, thrombin receptor activating peptide.
2.1. Patient IV.6
During childhood, a Jamaican man, the index case IV.6 (+/+), now middle‐aged, experienced excessive gum bleeding and easy bruising leading to a suspected diagnosis of GT (see George et al.15, for the GT phenotype). His first major hemorrhage at age 11 after a dental extraction needed hospitalization, where he received platelet transfusions with no apparent benefit. He was then given fresh frozen plasma (FFP) and whole blood products. Excessive bleeding during elective hernia repair was controlled by surgical glue although he again received FFP and whole blood products. He presented at the University of Miami Medical Center at age 45 complaining of headaches and was found to have a 6×3.7×3.7 cm parafalcine meningioma with significant mass effect and edema requiring surgery. Platelet function testing confirmed severely impaired platelet aggregation particularly with ADP and collagen. Surgery began under platelet transfusion but continued excessive bleeding led to the use of recombinant factor VIIa (rFVIIa, NovoSeven) (Figure S1) a regimen that was continued 4.5 days post‐surgery. There was an episode of transient hypoxemia when the rFVIIa drip was started intraoperatively but it resolved spontaneously within minutes. Surgery went well with low blood loss (400 cc) without major perioperative complications except for transient paraplegia lasting for approximately 7 hours on post‐op day 1 but imaging studies did not show any evidence of intracranial bleeding and he was continued on the rFVIIa drip and aminocaproic acid. Recently he received rFVIIa plus platelets during hernia repair surgery with good results. He carries out an active professional life without any limitation as a banker and while currently experiencing occasional gum bleeding; other bleeding manifestations are absent. He has no history of infections or cardiac problems. However, another medical problem is poor wound healing with excessive scarring and abnormal keloid.
2.2. Patient IV.4
The affected sister of the index case (+/+), now aged 55, manifested extensive petechial rash at birth and early work‐up also led to a misdiagnosis of GT. She had severe epistaxis during childhood requiring frequent hospitalizations, nasal packing, and whole blood and platelet transfusions. Epistaxis stopped during adolescence but she experienced menorrhagia at menarche. Surgical bleeding during cholecystectomy and hysterectomy led to platelet transfusions but like her brother there was little benefit. During a recent appendectomy, she initially received platelets without benefit but abnormal bleeding stopped after the administration of rFVIIa. She currently has occasional gum/nose bleeding but is otherwise healthy with no history of abnormal wound healing, infections or cardiac problems.
2.3. Other genotyped family members
The 61‐year‐old elder brother (IV.3, not genotyped) of the index case experienced mild bleeding during a tooth extraction but this resolved with extra packing locally. He has not experienced major bleeding. The 82‐year‐old mother (patient III.7) (+/−) of the affected cases has no history of excessive bleeding, heavy periods or bleeding during deliveries. Neither of these family members have a history of wound healing, infections or cardiac problems. His father (patient III.8) (not genotyped) died at age 60; he was said not to have bleeding problems when young. Nevertheless, he died of bleeding complications from a gunshot injury to the abdomen. Also genotyped were a maternal aunt (III.2) (+/−), her son (IV.1) (−/−) and a niece (V.5) (+/−) of the index case (IV.6).
Interestingly, the maternal aunt (III.2) (+/−) and her son (IV.1) (−/−) have experienced thrombocytopenia that may have influenced bleeding. The aunt developed idiopathic thrombocytopenic purpura (ITP) after her third pregnancy at age 27. She now has platelet counts in the normal range (196×103/μL) but they fell to a nadir of 20‐30×103/μL during her ITP. For her son (IV.1) (−/−) severe thrombocytopenia was linked to an immuno‐allergic reaction after aspirin intake when he was young. None of the fifth‐generation children have bleeding problems but the one niece (V.5) (+/−) who was available for genotyping also had a low platelet count when she was 7 years old.
3. STUDY OVERSIGHT
DNA sequencing of the index case (IV.6) and other family members was performed in France according to the approved protocol from INSERM (RBM‐04‐14). The index case (IV.6) was included in the Cambridge BRIDGE‐BPD study for whole exome sequencing.2, 16 All participants (patients and unaffected family members) provided written informed consent.
4. GENETIC AND BIOLOGICAL STUDIES (RESULTS)
The index case (IV.6) and his sister (IV.4) were originally thought to have a variant form of GT but their platelets exhibited a full complement for αIIbβ3 and other major surface receptors by flow cytometry, a finding confirmed for αIIb and β3 by western blotting (WB) for all genotyped family members (Figure 1B). To further exclude a variant form of GT the index case was included in an international sequencing study for GT which failed to identify mutations in ITGA2B or ITGB3 encoding αIIbβ3 (case 29, Nurden et al., 2015).17 Nevertheless, his platelets and those of his sister showed a reduction in binding to 5‐30% of the normal levels of PAC‐1, a monoclonal antibody (MoAb) recognizing the active conformation of αIIbβ3 after platelet stimulation with ADP (10 μmol/L). A much decreased αIIbβ3 activation suggested a signaling defect. Platelet aggregation from IV.4 and IV.6 was retested in citrated platelet‐rich plasma with a range of agonists (Table 1). Noteworthy were significant aggregation with high doses of thrombin receptor activating peptide (TRAP‐14, acting through PAR‐1) (8 and 40 μmol/L) and a full response with phorbol 12‐myristate 13‐acetate (PMA) (2 and 10 μmol/L), a membrane‐permeable agonist reacting directly with protein kinase C and a diagnostic characteristic of CalDAG‐GEFI deficiency (Cifuni et al., 2008). Our examination of the results of the whole exome sequencing for IV.6 highlighted a homozygous c.1490delT within exon 13 of RASGRP2 predicted to give rise to a disease causing p.F497Sfs*22 truncating mutation (ENSEMBL Database; http://www.ensembl.org) near the C‐terminal regulatory domain of CalDAG‐GEFI (see Figure S2 for structure of RasGRP family members). Sanger sequencing confirmed that IV.4 and IV.6 were homozygous for the c.1490delT variant and that their mother (III.7), an aunt (III.2), and a niece (V.5) of the index case out of 4 available close family members were heterozygous for the mutation (sequencing illustrated for IV.6 and III.2 in Figure 1C). This segregation fits with the mutation being the cause of the platelet defect. The localisation of this novel mutation is compared to that of other reported variants12, 13, 14 for CalDAG‐GEFI in Figure 1D. This genetic variant of RGSP2 (rs752492512) was made public on the Cambridge‐based UK10K database (http://www.uk10k.org). It is unique in leading to the loss of the C‐terminal regulatory C1‐like domain of the protein. To examine further the truncating effect of the mutation, human embryonic kidney (HEK) cells (GripTite 293 MSR cell line) were transfected with the mutated CalDAG‐GEFI according to previously published protocols.12 Results confirmed the expression of the truncated protein (Figure 1E). Re‐examination of platelets from the index case (IV.6) and his homozygous sister (IV.4) for CalDAG‐GEFI expression by WB showed only a band with increased electrophoretic mobility, consistant with the predicted truncated form of CalDAG‐GEFI. Their mother (III.7), heterozygous for the mutation, showed both intact CalDAG‐GEFI and the higher mobility band (Figure 1F). Comparable blots for normal human platelets and another truncating mutation (R113X) abrogating CalDAG‐GEFI expression are shown in figure 4 of Lozano et al. (2016).
5. DISCUSSION
CalDAG‐GEFI‐mediated Rap1 activation is rapid and reversible and is necessary for full platelet activation and stable thrombus formation. Similar to other patients with mutations in RASGRP212, 13, 14 or animals with altered CalDAG‐GEFI‐Rap1 signaling,7, 18 platelet aggregation in our patients was partially impaired, with the strongest defect seen in response to activation with ADP or collagen. Since the first report from Canault et al.,12 two other groups have described mutations in RASGRP2 associated with an absence of CalDAG‐GEFI expression or function; 3 cases in 2 Spanish families and a young Japanese woman.13, 14 Severe bleeding is a consistent finding recapitulating the findings in knock‐out mouse models for CalDAG‐GEFI.7, 8 For the family described here, the index case and his sister, both with homozygous expression, have experienced spontaneous bleeding and excessive bleeding after surgery. We were surprised by the severity of the hemostatic defects in our patients, as mice expressing CalDAG‐GEFIΔC1 in blood cells exhibited a less severe bleeding phenotype than CalDAG‐GEFI knockout mice.8 However, the CalDAG‐GEFIΔC1 mice were generated by transplantation of retrovirally transduced bone marrow cells into lethally irradiated mice, i.e., an approach that may have led to chimeric mice with residual amounts of circulating platelets expressing intact CalDAG‐GEFI. It is also important to remember that defective function of CalDAG‐GEFI in endothelial cells and/or extravascular cells may contribute to the more severe hemostasis defect in our patients when compared to the chimeric mice. Lastly, our Western blotting studies suggest that the frameshift mutation in our patients affects both the size and the expression level of CalDAG‐GEFI in platelets. Less protein with impaired function would explain markedly impaired platelet function and moderate to severe bleeding in the patients. Intriguingly, both homozygous patients gained no observable benefit from platelet transfusions; but when rFVIIa was used hemostasis was restored. This was probably life‐saving for the index case (IV.6) who developed a meningioma that required major surgery (6 hours). While the development of a mengioma may be coincidental, it is of interest that CalDAG‐GEFI is highly expressed in the brain.19 The index case and his father experienced problems with wound repair namely abnormal scarring with excessive keloid. Pasvolsky et al.20 reported a case of Leukocyte Adhesion Deficiency III (LAD III) with defective expression of CalDAG‐GEFI manifesting poor wound healing; however, this was later corrected to a kindlin‐3 deficiency.21 Kindlin‐3 is normally expressed in the platelets of IV.6 and IV.4 (Figure 1F). Although mildly altered neutrophil function associated with decreased activity of β1 or β2 integrins has been reported for the Spanish cases of inherited CalDAG‐GEFI deficiency it was not so for the Japanese and French patients or for the present family.12, 13, 14 We did not perform in vitro studies to assess leukocyte function as none of our patients show increased suspectibity to infections or infectious complications. Clearly, the phenotype of CalDAG‐GEFI deficiency differs from Leukocyte Adhesion Deficiency type III where the loss of kindlin‐3, also essential for integrin activation, combines bleeding with infections.21, 22 Transient ITP occurred in one heterozygous family member (III.2), but thrombocytopenia in two other family members was less well defined and did not always segregate with the RASGRP2 genotype. In summary, we have reported the largest kindred so far associated with RASGRP2 mutations and the variety of associated phenotypes together with the observed lack of efficacity of platelet transfusions recommend the assembly of as much data as possible to define the phenotypes and treatment of bleeding in families with inherited CalDAG‐GEFI expression and/or function. This is particularly so as CalDAG‐GEFI is receiving increasing attention as a new target for antiplatelet and antithrombotic therapy for prevention and treatment of cardiovascular disorders.
Supporting information
ACKNOWLEDGMENTS
We thank Professor Willem Ouwehand (University of Cambridge, UK) for making freely available the results of the whole exome sequencing for the Index Case. Dr. Ahn receives funding from W H Coulter foundation.
RELATIONSHIP DISCLOSURES
None of the authors have any disclosures relevant to this paper.
Desai A, Bergmeier W, Canault M, et al. Phenotype analysis and clinical management in a large family with a novel truncating mutation in RASGRP2, the CalDAG‐GEFI encoding gene. Res Pract Thromb Haemost. 2017;1:128–133. 10.1002/rth2.12019
Funding Information
WB is supported by National Institutes of Health National Heart, Lung, and Blood Institute grant R01HL130404.
REFERENCES
- 1. George JN, Nurden AT, Phillips DR. Molecular defects in interactions of platelets with the vessel wall. N Engl J Med. 1984;311:1084–98. [DOI] [PubMed] [Google Scholar]
- 2. Lentaigne C, Freson K, Laffan MA, Turro E, Ouwehand WH; BRIDGE‐BPD Consortium and the ThromboGenomics Consortium . Inherited platelet disorders: towards DNA‐based diagnosis. Blood. 2016;127:2814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coller BS, Shattil SA. The GPIIb/IIIa (integrin αIIbβ3) odyssey: a technology‐driven saga of a receptor with twists, turns, and even a bend. Blood. 2008;112:3011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cifuni SM, Wagner DD, Bergmeier W. CalDAG‐GEFI and protein kinase C represent alternative pathways leading to activation of integrin alphaIIbbeta3 in platelets. Blood. 2008;112:1696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stefanini L, Roden RC, Bergmeier W. CalDAG‐GEFI is at the nexus of calcium‐dependent platelet activation. Blood. 2009;114:2506–14. [DOI] [PubMed] [Google Scholar]
- 7. Crittenden JR, Bergmeier W, Zhang Y, et al. CalDAG‐GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10:982–6. [DOI] [PubMed] [Google Scholar]
- 8. Stolla M, Stefanini L, Roden RC, et al. The kinetics of αIIbβ3 activation determines the size and stability of thrombi in mice: implications for antiplatelet therapy. Blood. 2011;117:1005–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boulaftali Y, Owens AP III, Beale A, et al. CalDAG‐GEFI deficiency reduces atherosclerotic lesion development in mice. Arterioscler Thromb Vasc Biol. 2016;36:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piatt R, Paul DS, Lee RH, et al. Mice expressing low levels of CalDAG‐GEFI exhibit markedly impaired platelet activation with minor impact on hemostasis. Arterioscler Thromb Vasc Biol. 2016;36:1838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118:5996–6005. [DOI] [PubMed] [Google Scholar]
- 12. Canault M, Ghalloussi D, Grosdidier C, et al. Human CalDAG‐GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J Exp Med. 2014;211:1349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lozano ML, Cook A, Bastida JM, et al. Novel mutations in RASGRP2, which encodes CalDAG‐GEFI, abrogate Rap1 activation, causing platelet dysfunction. Blood. 2016;128:1282–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kato H, Nakazawa Y, Kashiwagi H, et al. Human CalDAG‐GEFI deficiency confers severe bleeding tendency and delayed αIIbβ3 activation velocity. Blood. 2016;128:2729–33. [DOI] [PubMed] [Google Scholar]
- 15. George JN, Caen JP, Nurden AT. Glanzmann's thrombasthenia: the spectrum of clinical disease. Blood. 1990;75:1383–95. [PubMed] [Google Scholar]
- 16. Greene D, BioResource NIHR, Richardson S, Turro E. Phenotype similarity regression for identifying the genetic determinants of rare diseases. Am J Hum Genet. 2016;8:490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nurden AT, Pillois X, Fiore M, et al. Expanding the mutation spectrum of the αIIbβ3 integrin in Glanzmann thrombasthenia: screening of the ITGA2B and ITGB3 genes in a large international cohort. Hum Mutat. 2015;36:548–61. [DOI] [PubMed] [Google Scholar]
- 18. Boudreaux MK, Schmutz S, French PS. Calcium diacylglycerol guanine nucleotide exchange factor I (CalDAG‐GEFI) gene mutations in a thrombopathic Simmental calf. Vet Pathol. 2007;44:932–5. [DOI] [PubMed] [Google Scholar]
- 19. Crittenden JR, Dunn DE, Meralli FI, et al. CalDAG‐GEFI down‐regulation in the striatum as a neuroprotective change in Huntington's disease. Hum Mol Genet. 2010;19:1756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pasvolsky R, Feigelson SW, Kilic SS, et al. LAD‐III syndrome is associated with defective expression of the Rap‐1 activator CalDAG‐GEF1 in lymphocytes, neutrophils and platelets. J Exp Med. 2007;204:1571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Svenssen L, Howarth K, McDowall A, et al. Leukocyte adhesion deficiency‐III is caused by mutations in kindlin3 affecting integrin activation. Nat Med. 2009;15:306–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin‐3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials