

Abstract
Essentials.
Platelets can stop bleeding independently of integrin‐mediated aggregation in inflamed organs.
GPVI contributes to aggregation‐independent hemostasis in the inflamed skin.
GPVI supports sealing of neutrophil‐induced vascular breaches by nonaggregated platelets.
Evaluation of anti‐GPVI drugs should take into account the risk of inflammatory bleeding.
Glycoprotein VI (GPVI), the main platelet receptor for collagen, has been shown to play a central role in various models of thrombosis, and to be a minor actor of hemostasis at sites of trauma. These observations have made of GPVI a novel target for antithrombotic therapy, as its inhibition would ideally combine efficacy with safety. Nevertheless, recent studies have indicated that GPVI could play an important role in preventing bleeding caused by neutrophils in the inflamed skin and lungs. Remarkably, there is evidence that the GPVI‐dependent hemostatic function of platelets at the acute phase of inflammation in these organs does not involve aggregation. From a therapeutic perspective, the vasculoprotective action of GPVI in inflammation suggests that blocking of GPVI might bear some risks of bleeding at sites of neutrophil infiltration. In this review, we summarize recent findings on GPVI functions in inflammation and discuss their possible clinical implications and applications.
Keywords: bleeding, glycoprotein VI, inflammation, platelets, vascular integrity
1. GPVI IN PRIMARY HEMOSTASIS AND THROMBOSIS
Glycoprotein VI (GPVI) is a type I transmembrane protein of 58‐60 kDa that belongs to the immunoglobulin superfamily and whose expression is restricted to the megakaryocyte lineage.1 It was initially identified as a collagen receptor in the 1980s based on clinical evidence that platelets from patients presenting with mild bleeding disorders were specifically unresponsive to fibrillar collagen, a defect that was associated with a deficiency in GPVI.2, 3 The cloning of GPVI few years later.1, 4, 5 allowed the development of various experimental tools including genetically‐modified mice, recombinant forms of GPVI, and anti‐GPVI antibodies, which have all been crucial in dissecting GPVI structural organization and functions. Those tools helped define GPVI as the primary receptor for platelet/collagen interactions and subsequent platelet activation.6, 7, 8, 9 They have also made possible the demonstration that GPVI forms highly competent dimers at the platelet surface whose clustering upon adhesion to collagen further increases its avidity and downstream signaling.10, 11, 12, 13, 14 The signaling activity of GPVI depends on its physical association with the homodimeric γ chain common to Fc receptors (FcRγ), whose cytoplasmic domains each bears a signaling motif known as immunoreceptor tyrosine‐based activation motif (ITAM).15, 16 Upon ligand binding, the Src family kinases Lyn and Fyn mediate phosphorylation of the ITAM tyrosine residues that transmit activation signals.17
Although GPVI in its dimeric form binds collagen with a relatively high affinity,10, 18 it is considered as a signaling rather than an adhesion receptor, as its interaction with collagen is a prerequisite for the activation of platelet integrins, including that of the collagen receptor integrin α2β1.6, 7, 8, 19, 20 GPVI‐dependent signaling indeed ensures firm adhesion and aggregation through activation of platelets. In addition, it has been shown that GPVI interaction with immobilized collagen related peptide (CRP), a synthetic peptide that mimics the triple‐helical structure of collagen and specifically engages GPVI,21, 22 is transient and not sufficient for firm platelet adhesion under flow, thus arguing against a role for GPVI in triggering platelet adhesion to collagen.20, 23 Instead, it has been proposed that the interaction between GPIbα and collagen‐bound von Willebrand factor (VWF) would play a prominent role in initiating platelet adhesion to collagen, especially at higher shear, with GPVI‐dependent signaling taking over integrin activation subsequently.20, 23 Nonetheless, it should be noted that several studies have shown that GPVI permits platelet adhesion to fibrillar collagen irrespective of integrin activation in both static and flow conditions.6, 7, 23, 24, 25 Moreover, absence or inhibition of GPVI was shown to cause a drastic reduction in platelet adhesion to fibrillar collagen, despite normal levels of GPIbα.6, 7, 26, 27 Therefore, although GPIbα primes adhesion to collagen and integrin activation reinforces it and allows aggregation, there is evidence that GPVI can support platelet adhesion to fibrillar collagen. In recent years, laminins,28, 29 fibrin,30, 31 and fibronectin32, 33 have also been identified as adhesive and/or activating ligands for GPVI. The fact that GPVI has multiple ligands suggests that GPVI interactions with the injured or diseased vessel wall could be more complex than what can be deduced from its interactions with CRP or purified collagen alone.
Considering the central role of GPVI in mediating collagen‐induced platelet activation, as well as its ability to potentiate other platelet activation pathways,34, 35, 36 one could expect from GPVI to be a major player in primary hemostasis. However, although their platelets are highly refractory to collagen‐induced activation, patients with genetic or acquired GPVI deficiency only have a mild bleeding disorder.2, 3, 37, 38, 39, 40, 41 Those patients present a bleeding phenotype comparable to that of patients with moderate thrombocytopenia, which includes easy bruising, a prolonged bleeding time, and so‐called spontaneous bleeding events like petechiae and epistaxis. Autoantibodies to GPVI can be associated with idiopathic thrombocytopenic purpura,2, 37 which can be considered as a confounding factor in the setting of abnormal bleeding. Hence, it is worth noting that the bleeding phenotype of the first patient described with anti‐GPVI antibodies persisted despite correction of thrombocytopenia by steroid therapy.2 This indicated that the patient's bleeding tendency was a consequence of platelet dysfunction rather than of reduced platelet counts. Consistent with observations made in GPVI‐deficient patients, inhibition, immunodepletion, or genetic deletion of GPVI in animals only had minor consequences on primary hemostasis.42, 43 In contrast to its limited impact on primary hemostasis, GPVI deficiency conferred remarkable protection against thrombosis in a variety of in vitro and in vivo experimental models, including flow chamber‐based assays using human atherosclerotic plaque material.27, 33, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58 For these reasons, and despite some controversies, GPVI has been proposed as a promising target for antithrombotic therapy with reduced bleeding risk as compared to current antiplatelet therapies based on administration of aspirin and/or P2Y12 inhibitors.42, 43
The mild bleeding manifestations in patients with GPVI deficiency suggest that either GPVI plays a modest role in hemostasis or, more likely, that its absence is overcome by compensatory and/or redundant mechanisms. Among the many pathways of platelet activation, the protease‐activated receptors (PARs) for thrombin and the relatively newly discovered hem‐ITAM‐containing platelet activation receptor for podoplanin, C‐type lectin‐like receptor 2 (CLEC‐2), have both shown functional redundancy with GPVI in mouse models of hemostasis and thrombosis.59, 60 It has also been shown that the requirement for GPVI‐dependent signaling through Syk in platelet spreading on various matrix proteins can be bypassed by thrombin.35 Taken together, these data suggest that GPVI deficiency is compensated for in situations where vascular injury leads to significant thrombin generation and/or podoplanin exposure. Those situations likely encompass most traumatic vascular injuries, which might explain why GPVI patients do not present severe bleeding. Indeed, besides lymphatic endothelial cells, several types of podoplanin‐expressing cells (inflammatory macrophages,61, 62, 63, 64, 65, 66 fibroblasts65, 66) have been described in the vicinity of blood vessels and could therefore engage platelet CLEC‐2 through vascular breaches.
Around 10 years back, inflammation was identified as a cause of nontraumatic bleeding in thrombocytopenia.67 The fact that inflammation can cause bleeding in thrombocytopenia has hinted that spontaneous bleeding in patients with thrombocytopenia or platelet dysfunction may not always be “spontaneous” but rather evoked by unsuspected underlying inflammatory reactions. In addition, it has revealed a so far unnoticed and uncharacterized protective function of platelets in inflammation where they continuously prevent bleeding from inflamed vessels. Experiments in mouse models of inflammation have indicated that, surprisingly, the hemostatic function of platelets in inflammation can operate independently of platelet aggregation, the main and best known mechanism of action of platelets in hemostasis.68 As presented in further detail below, there is experimental evidence to suggest that there are situations where GPVI could play a predominant role in preventing inflammatory bleeding, notably in the skin. In the light of these findings, it is worth noting that inflammation‐induced bleeding in humans and mice can take the form of cutaneous petechiae,67, 69, 70 a manifestation shared by both thrombocytopenic and GPVI‐deficient patients.
2. PLATELETS AND GPVI IN INFLAMMATION
Platelets have long been considered primarily through the lens of hemostatic platelet plug formation and thrombosis, who both rely on integrin‐mediated platelet aggregation. They are, however, increasingly recognized as major actors of inflammatory reactions and immune responses.71, 72, 73, 74, 75, 76, 77 Platelets intervene at various stages of inflammatory reactions. Whereas platelets enhance the barrier function of the quiescent endothelium and stop bleeding in case of vessel injury,68 in the context of inflammation, they contribute to opening of endothelial junctions and thereby to edema formation.70, 78, 79, 80, 81, 82 In addition, platelets promote leukocyte recruitment and infiltration in many tissues and organs under a range of inflammatory conditions.26, 31, 70, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92 Platelets also regulate several effector functions of innate immune cells including degranulation,93, 94 oxidative activity,95, 96 phagocytosis,95 or the formation of neutrophil extracellular traps.83, 97 Besides these proinflammatory effects, platelets also act to limit inflammation‐associated collateral damage to the host by preventing bleeding in inflamed organs.68, 98 With respect to the role of GPVI in inflammation, GPVI has been involved in the recruitment of platelets and leukocytes to the inflamed vessel wall,26, 33, 80, 84, 94, 99 the regulation of vascular permeability80 and leukocyte activation,94, 100, 101 and in the prevention of inflammatory bleeding94, 102 (Table 1).
Table 1.
GPVI functions in inflammation. A non‐exhaustive list of the various effects of GPVI deficiency or blockade in models of inflammation. anti‐GBM: anti–glomerular basement membrane antibody; K/BxN serum: serum from K/BxN a mouse strain known to develop severe autoimmune inflammatory arthritis
| Inflammation model | Effect of GPVI deficiency or blockade | References |
|---|---|---|
| Acute dermatitis (IgG immune‐complex mediated) |
|
94, 102 |
| Peritonitis (IgG immune‐complex mediated) |
|
94 |
| Rheumatoid arthritis (K/BxN serum) |
|
80, 84 |
| Glomerulonephritis (anti‐GBM antibody) |
|
99 |
| Myocardial Ischemia‐Reperfusion (30 min long ligation of the left coronary artery) |
|
103, 104, 105 |
| Cerebral Ischemia‐Reperfusion (60 min long occlusion of middle cerebral artery) |
|
113, 116, 136 |
| Lung infection (Klebsiella pneumoniae) |
|
[148, 149] |
| Acute lung injury (LPS inhalation) |
|
102 |
| Atherosclerosis (ApoE‐/‐ mice) |
|
26, 33 |
The role of GPVI in platelet recruitment to the inflamed vasculature has been assessed and demonstrated in various models of inflammation including atherosclerosis in hypercholesterolemic animals,26, 33 rheumatoid arthritis,80, 84 myocardial ischemia‐reperfusion,103, 104, 105 and immune complex (IC)‐induced glomerulonephritis99 and dermatitis94 (Table 1).
In hypercholesterolemic mice and rabbits, GPVI was shown to mediate transient but not firm adhesion of platelets to atherosclerotic endothelium. Chronic inhibition of those GPVI‐dependent interactions with the diseased vessel wall by anti‐GPVI antibodies or GPVI competitive antagonists reduced lesion formation and corrected endothelial dysfunction in atherosclerotic arteries.26, 33 These results exemplify how, even in the absence of aggregation, GPVI engagement can contribute to inflammation and thereby to atheroprogression in experimental atherosclerosis.
Rheumatoid arthritis is another situation where GPVI promotes inflammation independently of aggregation. In a mouse model of autoimmune rheumatoid arthritis, platelets recruited to the inflamed joints promoted inflammation by increasing the permeability of the synovial microvasculature and the recruitment of neutrophils.80, 84 These proinflammatory effects of platelets involved the release of serotonin and microparticles in synovial fluid. Platelet serotonin and microparticles, respectively, induced endothelial gap formation in the synovial vasculature and the secretion of the neutrophil chemoattractant interleukin‐8 by synovial fibroblasts, thus amplifying arthritis. The contribution of various platelet activation pathways to the inflammatory functions of platelets in this model was investigated using genetically deficient mice and pharmacologic blockade. The G protein‐coupled receptors for thromboxane A2 (TP) and ADP (P2Y12), GPIbα, and integrin αIIbβ3 were all found to be dispensable for microparticles generation and development of joint inflammation, which instead relied on GPVI.84
The clinical observations of increased circulating platelet‐derived factors (e.g, serotonin, platelet factor 4, β‐thromboglobulin), their presence in urine, and the localization of platelets in glomerular capillaries in various forms of glomerulonephritis have suggested that platelets are also involved in the pathogenesis of human renal diseases.106, 107, 108 Like in rheumatoid arthritis, platelets are suspected of increasing vascular permeability and leukocyte infiltration in the inflamed glomerulus. They would also favor local cellular and matrix remodeling, thus further promoting glomerular injury and dysfunction. Although platelet aggregates have been described in biopsies from patients with glomerulonephritis, they are not always present. Several hypotheses have been proposed to explain the paucity of morphologically intact platelets in human glomerular lesions. These hypotheses include the possibility of a hit‐and‐run model where platelets interacting transiently with the vessel wall exit the glomerular circulation after releasing their granular content.107, 108, 109 An alternative explanation is that, having released their granules, platelets might become difficult to identify in transmission electron microscopy.107, 108, 109 Renal intravital microscopy in a mouse model of IC‐mediated glomerulonephritis has brought some insights on the interactions between platelets and the inflamed glomerulus vasculature.99 Platelet recruitment to the glomerulus started within minutes of inflammation initiation and increased progressively during the course of the reaction. Primary adhesion of platelets was unaffected by inhibition of GPIbα but was reduced by GPVI deficiency, thus indicating that early platelet recruitment was independent of GPIbα and mediated by GPVI. More precisely, GPVI initiated platelet adhesion by permitting platelet tethering but was not sufficient for subsequent stable adhesion, which required integrin αIIbβ3 activation. Platelets recruited to the inflamed glomerulus then stimulated neutrophil recruitment in an ADP and P‐selectin‐dependent way.99, 110 Interestingly, whereas platelets enhanced neutrophil recruitment in this model, the opposite was also true. In fact, neutrophil depletion prior to glomerulonephritis induction markedly reduced GPVI‐dependent platelet adhesion and accumulation within the glomerulus. This result was a first indication that there are situations where neutrophils unmask and/or provide binding sites for GPVI in the inflamed vasculature. Electron microscopy observations that alterations of the endothelial morphology occur very rapidly after glomerulonephritis initiation argue in favor of an increased exposure of subendothelial GPVI ligands to circulating platelets in the inflamed glomerulus.99
More recent data have suggested that besides regulation of endothelial permeability and leukocyte recruitment, prevention of inflammatory bleeding is another function that can depend on GPVI‐mediated platelet recruitment to the inflamed vasculature94, 102, 103 (Table 1). Studies based on the combination of mouse models of inflammation and severe thrombocytopenia (< less than 3% of normal mouse platelet count) have helped to demonstrate that platelets continuously secure the inflamed vasculature by preventing bleeding from the very onset of inflammation.67, 68 This protective action of platelets was reported in many different models of acute inflammation including various models of dermatitis,67, 70, 94, 102, 111, 112, 113 IC‐mediated glomerulonephritis,114 endotoxin‐ and bacteria‐induced acute lung injury,61, 67, 102, 111, 113, 115 and myocardial and cerebral ischemia‐reperfusion injury,67, 103, 113, 116 as well as in models of viral infection117, 118 and solid tumors.119, 120, 121 Strikingly, in several of these models, prevention of inflammatory bleeding was maintained in mice compromised for platelet aggregation at sites of traumatic vessel injury.67, 102, 113 In particular, in contrast to thrombocytopenic mice, mice lacking GPIbα or integrin αIIbβ3 showed no bleeding in IC‐mediated dermatitis or solid tumors.67, 113, 119 Moreover, platelets lacking the thrombin receptor PAR4 and treated with inhibitors of the P2Y12 and TXA2 activation pathways were fully capable of ensuring hemostasis during IC‐mediated dermatitis and LPS‐induced lung inflammation when transfused to thrombocytopenic mice.102 Conversely, transfusion of platelets lacking GPVI, CLEC‐2, or their common downstream transducer SLP‐76, failed to restore hemostasis in the inflamed skin and lungs of thrombocytopenic mice.102 Taken together, these results have indicated that GPVI and CLEC‐2 could play a predominant role in aggregation‐independent inflammation‐associated hemostasis.122 Nevertheless, because the role of these receptors was suggested on the basis of transfusion experiments in which platelet counts were only partially restored, it is conceivable that relative thrombocytopenia might have sensitized recipients to inflammatory bleeding, thus magnifying the impact of GPVI and CLEC‐2 deficiency. Using GPVI−/− mice, which have normal platelet count, the role of GPVI in limiting bleeding in IC‐induced dermatitis has since been confirmed and investigated further.94
Intravital microscopy analysis of the skin vasculature during IC‐induced dermatitis showed that, like in models of IC‐induced glomerulonephritis,99, 114 platelet recruitment to the inflammation site started within minutes of reaction induction. Early platelet recruitment occurred without signs of thrombosis and in the form of transient or firm adhesion of individual platelets to the vessel wall and adherent neutrophils in post‐capillary venules. Remarkably, platelet recruitment was prevented by neutrophil depletion and reduced, but not abolished, in GPVI−/− mice compared to their wild‐type littermates.94 These results indicated that GPVI contributed to prevention of inflammatory bleeding by mediating, in part, the recruitment of platelets at sites of neutrophil infiltration. They also implied that infiltrating neutrophils unmasked binding sites for GPVI on the vessel wall, which was confirmed by the presence of endothelial gaps and the adhesion of microspheres coated with a chimeric form of GPVI in post‐capillary venules where neutrophils accumulated and extravasated. The fact that GPVI deficiency did not completely abrogate platelet recruitment to inflamed skin vessels supported the idea that, as previously hinted by transfusion experiments,102 redundant and/or compensatory mechanisms made up in part for the loss of GPVI. Consistent with these results, cutaneous inflammatory bleeding was less severe in GPVI−/− mice than in mice immunodepleted for platelets.
How platelets recruited through GPVI at sites of neutrophil infiltration prevent bleeding independently of aggregation? Dampening of neutrophil histotoxic activities represents a mechanism of action by which platelets could limit injury and bleeding. This possibility has however been dismissed on the grounds that markers of neutrophil activation during IC‐mediated inflammation were reduced in thrombocytopenic and GPVI−/− mice as compared to wild‐type mice.94 These data argued that platelets stimulated rather than prevent neutrophil activation during IC‐mediated inflammation, and that GPVI contributed to this effect. They also added to growing indications that GPVI can regulate innate immune cell activation and function,100, 123 an effect that might involve the interaction of GPVI with one of its newly identified ligand, EMMPRIN, a surface receptor widely expressed in human tumors but also platelets, monocytes, and activated lymphocytes.101, 124
Considering that GPVI binds various components of the subendothelial matrix, it is plausible that single platelets or sheets of platelets stop bleeding by covering small areas where the basement membrane gets exposed and disrupted by neutrophils. The role of GPVI in this inflammation‐associated hemostatic process might not be restricted to platelet adhesion as it was shown that defective ITAM signaling, either due to a genetic deficiency in SLP76 or to combined pharmacological inhibition of the Src‐family kinases Syk and Bruton's tyrosine kinase (Btk) partially impaired the ability of platelets to prevent inflammatory bleeding.94, 102 Thus, GPVI‐mediated platelet responses likely also contribute to sealing of vascular breaches caused by neutrophils. In that regard, it was shown that even when not causing aggregation, collagen binding to GPVI can induce platelet shape change125 and release of soluble platelet factors such as angiopoietin‐1, serotonin, and ATP, all of which are regulators of endothelial cell barrier function or survival.126 How platelet secretion and aggregation can be mechanistically uncoupled remains to be determined but a first glimpse was provided recently by studies showing the existence of specific forms and pathways of platelet secretion in response to low‐dose agonists.127, 128 A recent study by Deppermann et al., showed that mice lacking platelet alpha‐ and/or dense‐granule secretion showed no signs of inflammatory bleeding in IC‐induced dermatitis or LPS‐induced lung inflammation.113 These findings suggest that platelet shape change and spreading might be more important than granule secretion for GPVI‐dependent inflammation‐associated hemostasis.
3. BACK TO THE PAST: DIRECT EVIDENCE THAT PLATELETS CAN SECURE INJURED VESSELS INDEPENDENTLY OF AGGREGATION
Recent data converge to suggest that non‐aggregated platelets adhering, spreading and/or releasing their granular content, in part through GPVI engagement, help to seal neutrophil‐induced vascular breaches, thereby providing hemostasis in inflamed tissues (Figure 1).98 But is there any direct evidence of platelets maintaining vascular integrity in the absence of aggregation? In a series of studies ranging from the 1960s to the 1970s, using electron microscopy, Hans Rudolf Baumgartner showed that platelets rapidly adhered and spread out to form a continuous monolayer that covered the subendothelium exposed following ballooning‐induced endothelial denudation in large arteries (Figure 1).129, 130, 131 In contrast to thrombi that disappeared rapidly, this “pseudo‐endothelium” made of non‐aggregated platelets persisted in time and rendered the vascular surface nonthrombogenic.129, 130 Collagenase treatment of the subendothelial surface prevented the formation of this platelet carpet, which in light of recent findings might be considered as a hint for a role of platelet/collagen interactions, and thus for GPVI, in this function. In a non‐traumatic model of endothelial denudation, Baumgartner showed that single platelets filled small endothelial gaps forming following reserpin‐induced capillary dilatation.132 Areas of wider basement membrane exposure were covered by several platelets, however, “platelet aggregates as seen during the formation of platelet thrombi were never observed”.132 Baumgartner concluded that “in certain physiological conditions platelets function as ‘bouche‐trou’ when for any reason the endothelial separate and the underlying basement lamina becomes exposed”. One could argue that Baumgartner's observations might not apply to inflamed vessels as endothelial denudation only partially models the vascular modifications and damage accompanying inflammation. In fact, inflammation is not only associated with exposure of the basement membrane through endothelial gap formation but also with leukocyte‐mediated damage to it.98, 133, 134 Nonetheless, Baumgartner's studies provided evidence that, even under flow conditions, platelet adhesion to the basement membrane does not necessarily cause thrombus formation.
Figure 1.
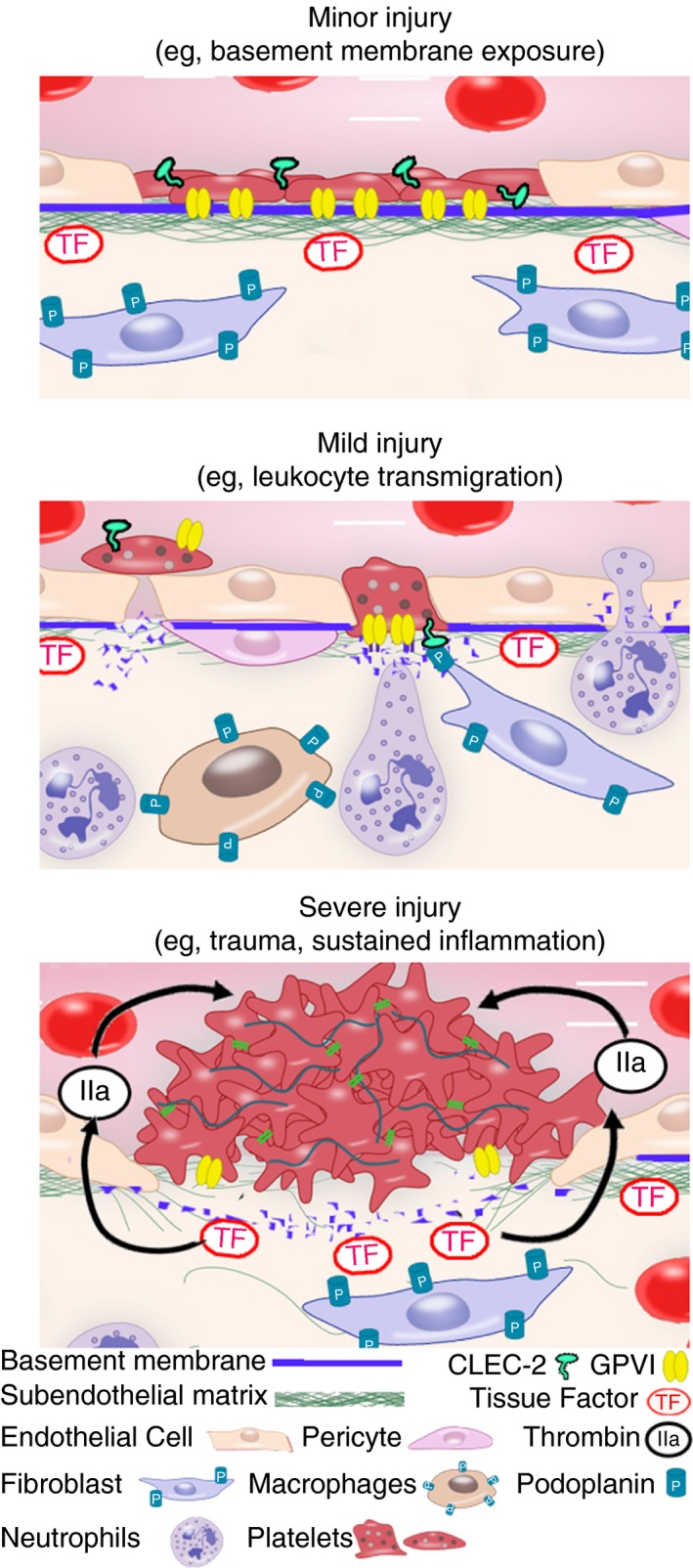
Platelets can maintain vascular integrity in multiple ways depending on the type and severity of vascular injury. Upper panel: The early work of Hans R. Baumgartner showed that in case of minor injury restricted to the endothelial layer (e.g, endothelial denudation, vasodilatation, and opening of endothelial junctions), platelets adhere and spread over the exposed basement membrane acting as “bouche‐trou”, either individually or through the formation of a nonthrombogenic “pseudo‐endothelium” in wider areas of denuded basement membrane. Notably, the adhesion of platelets to the exposed subendothelial matrix is collagenase‐sensitive, indicating a possible role for platelet/collagen interactions, and thus for GPVI, in this function. Middle panel: Recent and early studies indicate that, in case of mild injury where the basement is damaged (e.g, by neutrophil infiltration during acute inflammatory reactions), platelets seal vascular breaches by filling endothelial gaps and/or bridging endothelial junctions over areas of basement membrane disruption. Platelet GPVI contributes to the recruitment and adhesion of platelets to these sites. Engagement of platelets through CLEC‐2 by podoplanin‐expressing cells in direct vicinity of blood vessels (e.g, fibroblasts and inflammatory macrophages) provides a backup mechanism in the absence of GPVI. Lower panel: In a more classical view of hemostasis, in case of severe injury like in trauma or sustained inflammatory reactions, the exposure of the deeper layers of the vessel wall and/or of tissues surrounding vessels leads to thrombin generation and full platelet activation, thereby triggering platelet aggregation through integrin αIIbβ3
In studies contemporary to Baumgartner, Palade, Majno, Cotran, and other illustrious pathologists published electron microscopy images of the interactions between platelets and microvessels in the context of inflammation.98 Most of these studies show images of single platelets sealing endothelial gaps in microvessels at sites of increased permeability or neutrophil extravasation (Figure 1).98 Therefore, visual evidence that, besides fibrin deposits and platelet aggregates, nonaggregated platelets can repair small breaches in inflamed vessels has been available for over 50 years. Yet, it was only until recently that the importance of these interactions for the prevention of inflammatory bleeding, as well as their underlying mechanisms, including the role of GPVI engagement, were unraveled.
4. GPVI SECURING INFLAMED VESSELS: CLINICAL IMPLICATIONS AND APPLICATIONS
In preclinical studies, GPVI antagonists have proven to effectively inhibit thrombosis without significantly affecting primary hemostasis.42, 43 As a consequence, GPVI might appear as a new and ideal target for antithrombotic therapy that would combine efficacy with safety. In this context, the fact that GPVI deficiency or blockade in mice is associated with an increased risk of bleeding in the inflamed skin,94, 102 heart,103 and possibly lungs102 (Table 1), suggest that targeting GPVI might not be as safe as initially thought. Nevertheless, besides cutaneous petechiae which, as presented in the present review, can be caused by inflammation, inflammatory bleeding is not a common observation in GPVI‐deficient patients. There are several possible explanations for the lack of clinical reports of inflammation‐induced bleeding in GPVI‐deficient patients. First, we know from mouse studies that the role of GPVI in inflammation‐associated hemostasis can be partly compensated for through engagement of other platelet receptors like CLEC‐2.102, 122 Second, it is important to recall that platelets are extremely potent at preventing inflammatory bleeding, as inflammatory bleeding was observed only in mice with severe thrombocytopenia and could be rescued by restoring platelet counts to levels as low as 5‐10% of normal mouse platelet count.67, 68, 98 In a comparable manner, it was shown that even trace amounts of surface GPVI were sufficient to maintain residual function including adhesion to collagen under static conditions.9, 135 Moreover, consistent with the notion that even a low copy number of functional GPVI is sufficient for hemostasis, the parents of the first GPVI‐deficient patient identified as such had platelets who retained normal function despite containing approximately 50% the normal amount of GPVI.3 Last but not least, the protective role of GPVI in inflammation observed in the inflamed skin and lungs cannot be generalized to all inflammatory situations. In fact, not all organs are prone to inflammatory bleeding98 (Table 1). No bleeding was observed in mice with severe thrombocytopenia that were subjected to rheumatoid arthritis or to peritonitis.79, 80, 84, 94 Furthermore, even in organs that are prone to inflammatory bleeding, GPVI is not always required for inflammation‐associated hemostasis (Table 1). For example, GPVI was shown to be dispensable for the prevention of hemorrhagic transformation following cerebral ischemia‐reperfusion, which instead more classically relies on integrin αIIbβ3.113, 116, 136 Therefore, not all organs are at risk of inflammatory bleeding, and even less with respect to the use of GPVI inhibitors. Nonetheless, the bleeding risk associated with the use of GPVI antagonists might be worth investigating in models of inflammation. Apart from animal models of inflammation, the tourniquet test may provide a simple clinical test for this purpose. Historically, like the bleeding time, the tourniquet test was used to diagnose thrombocytopenia and hemostasis defects.137 Nowadays, it is part of the World Health Organization–recommended tests for the diagnosis of hemorrhagic fevers like dengue, a condition which incidentally nicely illustrates how the combination of infection and thrombocytopenia can cause bleeding. The tourniquet test is basically a model of mild cutaneous ischemia‐reperfusion injury induced by a sphygmomanometer cuff inflated around the upper arm to stop blood flow for several minutes, before being deflated to allow reperfusion in the forearm. The test is considered positive when more than 10‐20 petechial bleeding spots per square inch are observed. The formation of petechiae in the tourniquet test has long been interpreted as a consequence of capillary fragility, but recent studies have shown that it involved leukocyte‐mediated vascular damage.138, 139, 140 This test might thus turn useful to predict the anti‐GPVI drugs‐associated risk of inflammatory bleeding.
There is one situation where targeting the ability of platelets to prevent inflammatory bleeding may be beneficial. Experiments in tumor‐bearing mice have shown that, like in inflamed organs, platelets continuously prevent leukocyte‐induced bleeding in solid tumors independently of aggregation.119, 121, 141 Immunodepletion of platelets potentiated the effect of chemotherapy in mice by improving drug accumulation specifically to the tumor site through induction of tumor bleeding.120 Targeting platelet function was thus proposed as a way to improve chemotherapy.142 Yet, the identity of the platelet receptor(s) involved in the prevention of tumor bleeding remains unknown. If GPVI were to be involved in this function, this might open the way to new applications for anti‐GPVI drugs in the field of cancer.
5. CONCLUSION
Recent studies have shown that, in various inflammatory situations, and from the very onset of inflammation, platelets ensure hemostasis independently of aggregation. In some of these situations, exemplified by IC‐mediated dermatitis, platelets recruited early to inflammation sites partly through GPVI seal small vascular breaches caused by infiltrating leukocytes, thereby preventing bleeding. These recent data considered together with observations made in past electron microscopy studies indicate that sealing of those breaches involves platelets filling gaps in the endothelial lining and spreading over damaged areas of the exposed basement membrane. Both GPVI adhesion and signaling properties would thus be required for this function. The role of GPVI in this unconventional form of hemostasis shows that platelets can stop bleeding in multiple ways, depending on the type and severity of vascular injury, which likely determine the pathways and extent of platelet activation in a context‐dependent manner.
6. ISTH BERLIN REPORT
The duality of platelets, which exert both potentially damaging and protective actions in inflamed tissues,143 is reflected in the works presented at the ISTH 2017 in Berlin. The group of Michael Hickey extended its previous observations on the proinflammatory role of platelets in a mouse model of IC‐induced acute glomerulonephritis. After showing that GPVI and integrin αIIbβ3 mediate platelet recruitment to the inflamed glomerus, thus allowing subsequent platelet P‐selectin‐dependent recruitment of neutrophils.99, 110 they now provide evidence that platelet activation also contributes to stimulation of neutrophil intravascular activation. In particular, their new results show that the ADP/P2Y12 and thromboxane/TP receptor pathways stimulate neutrophil oxidative activity in inflamed glomerular capillaries.144
The regulatory action of platelets towards immune cell activities is not restricted to neutrophils.83 In her presentation, Julie Rayes from the group of Steve Watson in Birmingham showed that platelets modulate the proinflammatory phenotype of podoplanin‐expressing inflammatory macrophages during caecal ligation and puncture‐induced sepsis. In this model, deletion of platelet‐CLEC‐2 resulted in an increased severity of sepsis that was associated with a dysregulation of inflammatory cytokine production, thus indicating a protective role of platelet CLEC‐2.63 These findings showing a protective role of platelet CLEC‐2 in inflammation have since been published64 and are in line with those of a recent study demonstrating that platelet CLEC‐2 protects against LPS‐induced lung injury via its interaction with podoplanin on inflammatory alveolar macrophages.61 While it was previously shown that engagement of platelet CLEC‐2 by podoplanin on inflammatory macrophages triggers platelet activation,145 these new data indicate that this interaction induces reciprocal regulation between platelets and inflammatory macrophages. In a more general way, these results are consistent with a model where platelets and leukocytes regulate their recruitment and activation during inflammatory reactions in a mutual and reciprocal manner. It is worth mentioning that, in general, recent studies on the role of the podoplanin/CLEC‐2 axis in inflammation have used PF4 cre mice to generate platelet‐specific CLEC‐2‐deficient mice. Recent studies have shown that the PF4‐driven cre recombinase is expressed on a subset of immune cells and activated dentritic cells, thus suggesting the possible implication of cells other than platelets in the observed effects.146, 147
A series of studies converged to support the concept that platelets can ensure inflammation‐associated hemostasis independently of major platelet activation pathways, but also that the functional hierarchy between platelet receptors for this function varies with the cause and site of inflammation. The group of Tom van der Poll showed that, in contrast to the previously reported role of GPVI and CLEC‐2 in the prevention of bleeding in models of IC‐induced dermatitis and LPS‐induced lung injury, neither GPVI nor CLEC‐2 were needed for inflammation‐associated hemostasis in a model of sepsis induced by Klebsiella pneumoniae lung infection.148, 149 Notably, GPVI‐depleted mice showed increased bacterial growth in lungs, thus indicating that GPVI contributes to host defense during pneumonia‐derived sepsis. In a model of inflammatory bowel disease induced by oral administration of dextran sodium sulfate, the group of Cécile Oury showed that prevention of intestinal bleeding by platelets involved P2X1 but not P2Y12 or GPVI.150 In collaboration with the group of Steve Watson, we showed that GPVI played the major role in preventing bleeding in IC‐mediated dermatitis, with CLEC‐2 being involved only in the absence of GPVI.62 In addition, we showed that neither receptor plays a prominent part in preventing bleeding in LPS‐inflamed lungs, which partly relied on GPIb.
Collectively, these results demonstrate that the contribution of platelet adhesion receptors to vascular integrity varies between vascular beds and inflammatory challenges. Among the parameters that vary with the vascular bed and inflammatory stimulus, the type and expression levels of ligands of platelet activating and inhibitory receptors, the type of leukocytes involved, local hemodynamic factors (e.g, arterial vs. venous and systemic vs. pulmonary pressures and resistances), as well as the severity of the injury, likely contribute to determine how platelets are engaged for inflammation‐associated hemostasis.
RELATIONSHIP DISCLOSURE
The authors have no conflicts of interest to report.
Boulaftali Y, Mawhin M‐A, Jandrot‐Perrus M, Ho‐Tin‐Noé B. Glycoprotein VI in securing vascular integrity in inflamed vessels. Res Pract Thromb Haemost. 2018;2:228–239. 10.1002/rth2.12092
Funding Information
This work was supported by grants from Force Hémato, INCA, and La Fondation Arc (PJA 20151203107) to B. H., and by La Fondation pour la Recherche Medicale (FRM ARF20140129191), Marie Curie Action (Reintegration grant 708973), and DHU Fire 012 to Y.B.
REFERENCES
- 1. Jandrot‐Perrus M, Busfield S, Lagrue AH, et al. Cloning, characterization, and functional studies of human and mouse glycoprotein VI: a platelet‐specific collagen receptor from the immunoglobulin superfamily. Blood. 2000;96:1798–807. [PubMed] [Google Scholar]
- 2. Sugiyama T, Okuma M, Ushikubi F, Sensaki S, Kanaji K, Uchino H. A novel platelet aggregating factor found in a patient with defective collagen‐induced platelet aggregation and autoimmune thrombocytopenia. Blood. 1987;69:1712–20. [PubMed] [Google Scholar]
- 3. Moroi M, Jung SM, Okuma M, Shinmyozu K. A patient with platelets deficient in glycoprotein VI that lack both collagen‐induced aggregation and adhesion. J Clin Invest. 1989;84:1440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clemetson JM, Polgar J, Magnenat E, Wells TN, Clemetson KJ. The platelet collagen receptor glycoprotein VI is a member of the immunoglobulin superfamily closely related to FcalphaR and the natural killer receptors. J Biol Chem. 1999;274:29019–24. [DOI] [PubMed] [Google Scholar]
- 5. Ezumi Y, Uchiyama T, Takayama H. Molecular cloning, genomic structure, chromosomal localization, and alternative splice forms of the platelet collagen receptor glycoprotein VI. Biochem Biophys Res Commun. 2000;277:27–36. [DOI] [PubMed] [Google Scholar]
- 6. Nieswandt B, Brakebusch C, Bergmeier W, et al. Glycoprotein VI but not alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J. 2001;20:2120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuijpers MJ, Schulte V, Bergmeier W, et al. Complementary roles of glycoprotein VI and alpha2beta1 integrin in collagen‐induced thrombus formation in flowing whole blood ex vivo. FASEB J. 2003;17:685–7. [DOI] [PubMed] [Google Scholar]
- 8. Chen H, Kahn ML. Reciprocal signaling by integrin and nonintegrin receptors during collagen activation of platelets. Mol Cell Biol. 2003;23:4764–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nieswandt B, Watson SP. Platelet‐collagen interaction: is GPVI the central receptor? Blood. 2003;102:449–61. [DOI] [PubMed] [Google Scholar]
- 10. Jung SM, Moroi M, Soejima K, et al. Constitutive dimerization of glycoprotein VI (GPVI) in resting platelets is essential for binding to collagen and activation in flowing blood. J Biol Chem. 2012;287:30000–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jung SM, Tsuji K, Moroi M. Glycoprotein (GP) VI dimer as a major collagen‐binding site of native platelets: direct evidence obtained with dimeric GPVI‐specific Fabs. J Thromb Haemost. 2009;7:1347–55. [DOI] [PubMed] [Google Scholar]
- 12. Loyau S, Dumont B, Ollivier V, et al. Platelet glycoprotein VI dimerization, an active process inducing receptor competence, is an indicator of platelet reactivity. Arter. Thromb Vasc Biol. 2012;32:778–85. [DOI] [PubMed] [Google Scholar]
- 13. Poulter NS, Pollitt AY, Owen DM, et al. Clustering of glycoprotein VI (GPVI) dimers upon adhesion to collagen as a mechanism to regulate GPVI signaling in platelets. J Thromb Haemost. 2017;15:549–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arthur JF, Shen Y, Kahn ML, Berndt MC, Andrews RK, Gardiner EE. Ligand binding rapidly induces disulfide‐dependent dimerization of glycoprotein VI on the platelet plasma membrane. J Biol Chem. 2007;282:30434–41. [DOI] [PubMed] [Google Scholar]
- 15. Gibbins JM, Okuma M, Farndale R, Barnes M, Watson SP. Glycoprotein VI is the collagen receptor in platelets which underlies tyrosine phosphorylation of the Fc receptor gamma‐chain. FEBS Lett. 1997;413:255–9. [DOI] [PubMed] [Google Scholar]
- 16. Tsuji M, Ezumi Y, Arai M, Takayama H. A novel association of Fc receptor gamma‐chain with glycoprotein VI and their co‐expression as a collagen receptor in human platelets. J Biol Chem. 1997;272:23528–31. [DOI] [PubMed] [Google Scholar]
- 17. Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124:2013–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miura Y, Takahashi T, Jung SM, Moroi M. Analysis of the interaction of platelet collagen receptor glycoprotein VI (GPVI) with collagen. A dimeric form of GPVI, but not the monomeric form, shows affinity to fibrous collagen. J Biol Chem. 2002;277:46197–204. [DOI] [PubMed] [Google Scholar]
- 19. Auger JM, Kuijpers MJ, Senis YA, Watson SP, Heemskerk JW. Adhesion of human and mouse platelets to collagen under shear: a unifying model. FASEB J. 2005;19:825–7. [DOI] [PubMed] [Google Scholar]
- 20. Pugh N, Simpson AM, Smethurst PA, de Groot PG, Raynal N, Farndale RW. Synergism between platelet collagen receptors defined using receptor‐specific collagen‐mimetic peptide substrata in flowing blood. Blood. 2010;115:5069–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morton LF, Hargreaves PG, Farndale RW, Young RD, Barnes MJ. Integrin alpha 2 beta 1‐independent activation of platelets by simple collagen‐like peptides: collagen tertiary (triple‐helical) and quaternary (polymeric) structures are sufficient alone for alpha 2 beta 1‐independent platelet reactivity. Biochem. J. 1995;306(Pt 2):337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Asselin J, Gibbins JM, Achison M, et al. A collagen‐like peptide stimulates tyrosine phosphorylation of syk and phospholipase C gamma2 in platelets independent of the integrin alpha2beta1. Blood. 1997;89:1235–42. [PubMed] [Google Scholar]
- 23. Polanowska‐Grabowska R, Gibbins JM, Gear ARL. Platelet adhesion to collagen and collagen‐related peptide under flow: roles of the alpha2beta1 integrin, GPVI, and Src tyrosine kinases. Arterioscler Thromb Vasc Biol. 2003;23:1934–40. [DOI] [PubMed] [Google Scholar]
- 24. Chen H, Locke D, Liu Y, Liu C, Kahn ML. The platelet receptor GPVI mediates both adhesion and signaling responses to collagen in a receptor density‐dependent fashion. J Biol Chem. 2002;277:3011–9. [DOI] [PubMed] [Google Scholar]
- 25. Sarratt KL, Chen H, Kahn ML, Hammer DA. Platelet receptor glycoprotein VI‐mediated adhesion to type I collagen under hydrodynamic flow. Ann Biomed Eng. 2004;32:970–6. [DOI] [PubMed] [Google Scholar]
- 26. Schulz C, Penz S, Hoffmann C, et al. Platelet GPVI binds to collagenous structures in the core region of human atheromatous plaque and is critical for atheroprogression in vivo. Basic Res Cardiol. 2008;103:356–67. [DOI] [PubMed] [Google Scholar]
- 27. Kato K, Kanaji T, Russell S, et al. The contribution of glycoprotein VI to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood. 2003;102:1701–7. [DOI] [PubMed] [Google Scholar]
- 28. Ozaki Y, Suzuki‐Inoue K, Inoue O. Novel interactions in platelet biology: CLEC‐2/podoplanin and laminin/GPVI. J Thromb Haemost. 2009;7(Suppl 1):191–4. [DOI] [PubMed] [Google Scholar]
- 29. Inoue O, Suzuki‐Inoue K, McCarty OJ, et al. Laminin stimulates spreading of platelets through integrin alpha6beta1‐dependent activation of GPVI. Blood. 2006;107:1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mammadova‐Bach E, Ollivier V, Loyau S, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood. 2015;126:683–91. [DOI] [PubMed] [Google Scholar]
- 31. Alshehri OM, Hughes CE, Montague S, et al. Fibrin activates GPVI in human and mouse platelets. Blood. 2015;126:1601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maurer E, Schaff M, Receveur N, et al. Fibrillar cellular fibronectin supports efficient platelet aggregation and procoagulant activity. Thromb Haemost. 2015;114:1175–88. [DOI] [PubMed] [Google Scholar]
- 33. Bültmann A, Li Z, Wagner S, et al. Impact of glycoprotein VI and platelet adhesion on atherosclerosis‐A possible role of fibronectin. J Mol Cell Cardiol. 2010;49:532–42. [DOI] [PubMed] [Google Scholar]
- 34. Nieswandt B, Bergmeier W, Eckly A, et al. Evidence for cross‐talk between glycoprotein VI and Gi‐coupled receptors during collagen‐induced platelet aggregation. Blood. 2001;97:3829–35. [DOI] [PubMed] [Google Scholar]
- 35. Hughan SC, Hughes CE, McCarty OJT, et al. GPVI potentiation of platelet activation by thrombin and adhesion molecules independent of Src kinases and Syk. Arterioscler Thromb Vasc Biol. 2007;27:422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schulte V, Reusch HP, Pozgajová M, Varga‐Szabó D, Gachet C, Nieswandt B. Two‐phase antithrombotic protection after anti‐glycoprotein VI treatment in mice. Arterioscler Thromb Vasc Biol. 2006;26:1640–7. [DOI] [PubMed] [Google Scholar]
- 37. Boylan B, Chen H, Rathore V, et al. Anti‐GPVI‐associated ITP: an acquired platelet disorder caused by autoantibody‐mediated clearance of the GPVI/FcRγ‐chain complex from the human platelet surface. Blood. 2004;104:1350–5. [DOI] [PubMed] [Google Scholar]
- 38. Dumont B, Lasne D, Rothschild C, et al. Absence of collagen‐induced platelet activation caused by compound heterozygous GPVI mutations. Blood. 2009;114:1900–3. [DOI] [PubMed] [Google Scholar]
- 39. Matus V, Valenzuela JG, Saez CG, et al. Platelet glycoprotein VI: an adenine insertion in exon 6 generates a truncated form of the protein associated with bleeding disorder in 4 chilean unrelated patients. Blood. 2012;120:3302. [Google Scholar]
- 40. Matus V, Valenzuela G, Sáez CG, et al. An adenine insertion in exon 6 of human GP6 generates a truncated protein associated with a bleeding disorder in four Chilean families. J Thromb Haemost. 2013;11:1751–9. [DOI] [PubMed] [Google Scholar]
- 41. Hermans C, Wittevrongel C, Thys C, Smethurst PA, Van Geet C, Freson K. A compound heterozygous mutation in glycoprotein VI in a patient with a bleeding disorder. J Thromb Haemost. 2009;7:1356–63. [DOI] [PubMed] [Google Scholar]
- 42. Zahid M, Mangin P, Loyau S, et al. The future of glycoprotein VI as an antithrombotic target. J Thromb Haemost. 2012;10:2418–27. [DOI] [PubMed] [Google Scholar]
- 43. Dütting S, Bender M, Nieswandt B. Platelet GPVI: a target for antithrombotic therapy?!. Trends Pharmacol Sci. 2012;33:583–90. [DOI] [PubMed] [Google Scholar]
- 44. Li H, Lockyer S, Concepcion A, et al. The fab fragment of a novel anti‐GPVI monoclonal antibody, OM4, reduces in vivo thrombosis without bleeding risk in rats. Arterioscler Thromb Vasc Biol. 2007;27:1199–205. [DOI] [PubMed] [Google Scholar]
- 45. Nieswandt B, Schulte V, Bergmeier W, et al. Long‐term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med. 2001;193:459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mangin PH, Tang C, Bourdon C, et al. A humanized glycoprotein VI (GPVI) mouse model to assess the antithrombotic efficacies of Anti‐GPVI agents. J Pharmacol Exp Ther. 2012;341:156–63. [DOI] [PubMed] [Google Scholar]
- 47. Matsumoto Y, Takizawa H, Nakama K, et al. Ex vivo evaluation of anti‐GPVI antibody in Cynomolgus monkeys: dissociation between anti‐platelet aggregatory effect and bleeding time. Thromb Haemost. 2006;96:167–75. [PubMed] [Google Scholar]
- 48. Ohlmann P, Hechler B, Ravanat C, et al. Ex vivo inhibition of thrombus formation by an anti‐glycoprotein VI Fab fragment in non‐human primates without modification of glycoprotein VI expression. J Thromb Haemost. 2008;6:1003–11. [DOI] [PubMed] [Google Scholar]
- 49. Penz S. Human atheromatous plaques stimulate thrombus formation by activating platelet glycoprotein VI. FASEB J. 2005;19:898–909. [DOI] [PubMed] [Google Scholar]
- 50. Massberg S, Konrad I, Bültmann A, et al. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 2004;18:397–379. [DOI] [PubMed] [Google Scholar]
- 51. Lebozec K, Jandrot‐Perrus M, Avenard G, Favre‐Bulle O, Billiald P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet's glycoprotein VI function without causing bleeding risks. MAbs. 2017;9:945–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Massberg S, Gawaz M, Grüner S, et al. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197:41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hechler B, Gachet C. Comparison of two murine models of thrombosis induced by atherosclerotic plaque injury. Thromb Haemost. 2011;105:3–12. [DOI] [PubMed] [Google Scholar]
- 54. Kuijpers MJE, Gilio K, Reitsma S, et al. Complementary roles of platelets and coagulation in thrombus formation on plaques acutely ruptured by targeted ultrasound treatment: a novel intravital model. J Thromb Haemost. 2009;7:152–61. [DOI] [PubMed] [Google Scholar]
- 55. Lockyer S, Okuyama K, Begum S, et al. GPVI‐deficient mice lack collagen responses and are protected against experimentally induced pulmonary thromboembolism. Thromb Res. 2006;118:371–80. [DOI] [PubMed] [Google Scholar]
- 56. Lecut C, Feeney LA, Kingsbury G, et al. Human platelet glycoprotein VI function is antagonized by monoclonal antibody‐derived fab fragments. J Thromb Haemost. 2003;1:2653–62. [DOI] [PubMed] [Google Scholar]
- 57. Goebel S, Li Z, Vogelmann J, et al. The GPVI‐Fc fusion protein Revacept improves cerebral infarct volume and functional outcome in stroke. PLoS ONE. 2013;8:e66960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grothusen C, Umbreen S, Konrad I, et al. EXP3179 inhibits collagen‐dependent platelet activation via glycoprotein receptor‐VI independent of AT1‐receptor antagonism: potential impact on atherothrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1184–90. [DOI] [PubMed] [Google Scholar]
- 59. Bynagari‐Settipalli YS, Cornelissen I, Palmer D, et al. Redundancy and interaction of thrombin‐ and collagen‐mediated platelet activation in tail bleeding and carotid thrombosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:2563–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bender M, May F, Lorenz V, et al. Combined in vivo depletion of glycoprotein VI and C‐type lectin‐like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arter Thromb Vasc Biol. 2013;33:926–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lax S, Rayes J, Wichaiyo S, et al. Platelet CLEC‐2 protects against lung injury via effects of its ligand podoplanin on inflammatory alveolar macrophages in the mouse. Am J Physiol Lung Cell Mol Physiol. 2017;313:L1016–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rayes J, Lax S, Gros A, et al. The contribution of platelet adhesion receptors to vascular integrity during inflammation is stimulus and organ dependent. Res Pr Thromb Haemost. 2017;1 Suppl 1:234(OC 29.2). [Google Scholar]
- 63. Rayes J, Lax S, Zuidscherwoude SM, Watson B, Grygielska S, Watson SP. Proinflammatory phenotype of macrophages via the interaction of CLEC‐2 and podoplanin. Res Pract Thromb Haemost. 2017;1 Suppl 1:12. [Google Scholar]
- 64. Rayes J, Lax S, Wichaiyo S, et al. The podoplanin‐CLEC‐2 axis inhibits inflammation in sepsis. Nat Commun. 2017;8:2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Payne H, Ponomaryov T, Watson SP, Brill A. Mice with a deficiency in CLEC‐2 are protected against deep vein thrombosis. Blood. 2017;129:2013–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hitchcock JR, Cook CN, Bobat S, et al. Inflammation drives thrombosis after Salmonella infection via CLEC‐2 on platelets. J Clin Invest. 2015;125:4429–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goerge T, Ho‐Tin‐Noe B, Carbo C, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111:4958–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ho‐Tin‐Noe B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost. 2011;9(Suppl 1):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carbo C, del Conde I, Duerschmied D. Petechial bleeding after sunburn in a patient with mild thrombocytopenia. Am J Hematol. 2009;84:523. [DOI] [PubMed] [Google Scholar]
- 70. Hillgruber C, Poppelmann B, Weishaupt C, et al. Blocking neutrophil diapedesis prevents hemorrhage during thrombocytopenia. J Exp Med. 2015;212:1255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Garraud O. Editorial: platelets as immune cells in physiology and immunopathology. Front Immunol. 2015;6:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mancuso ME, Santagostino E. Platelets: much more than bricks in a breached wall. Br J Haematol. 2017;178:209–19. [DOI] [PubMed] [Google Scholar]
- 73. Nurden AT. Platelets, inflammation and tissue regeneration. Thromb Haemost. 2011;105:13–33. [DOI] [PubMed] [Google Scholar]
- 74. May AE, Seizer P, Gawaz M. Platelets: inflammatory firebugs of vascular walls. Arterioscler Thromb Vasc Biol. 2008;28:s5–10. [DOI] [PubMed] [Google Scholar]
- 75. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123:2759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Manne BK, Xiang SC, Rondina MT. Platelet secretion in inflammatory and infectious diseases. Platelets. 2017;28:155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost. 2015;114:449–58. [DOI] [PubMed] [Google Scholar]
- 78. Zarbock A, Singbartl K, Ley K. Complete reversal of acid‐induced acute lung injury by blocking of platelet‐neutrophil aggregation. J Clin Invest. 2006;116:3211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Petri B, Broermann A, Li H, et al. Von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116:4712–9. [DOI] [PubMed] [Google Scholar]
- 80. Cloutier N, Pare A, Farndale RW, et al. Platelets can enhance vascular permeability. Blood. 2012;120:1334–43. [DOI] [PubMed] [Google Scholar]
- 81. Wetterholm E, Linders J, Merza M, Regner S, Thorlacius H. Platelet‐derived CXCL4 regulates neutrophil infiltration and tissue damage in severe acute pancreatitis. Transl Res. 2016;176:105–18. [DOI] [PubMed] [Google Scholar]
- 82. Hara T, Shimizu K, Ogawa F, et al. Platelets control leukocyte recruitment in a murine model of cutaneous arthus reaction. Am J Pathol. 2010;176:259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gros A, Ollivier V, Ho‐Tin‐Noe B. Platelets in inflammation: regulation of leukocyte activities and vascular repair. Front Immunol. 2015;5:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen‐dependent microparticle production. Science. 2010;327:580–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Duerschmied D, Suidan GL, Demers M, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. 2012;121:1008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hwaiz R, Rahman M, Zhang E, Thorlacius H. Platelet secretion of CXCL4 is Rac1‐dependent and regulates neutrophil infiltration and tissue damage in septic lung damage. Br J Pharmacol. 2015;172:5347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Massberg S, Brand K, Grüner S, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schulz C, Schäfer A, Stolla M, et al. Chemokine fractalkine mediates leukocyte recruitment to inflammatory endothelial cells in flowing whole blood: a critical role for P‐selectin expressed on activated platelets. Circulation. 2007;116:764–73. [DOI] [PubMed] [Google Scholar]
- 89. Massberg S, Enders G, Leiderer R, et al. Platelet‐endothelial cell interactions during ischemia/reperfusion: the role of P‐selectin. Blood. 1998;92:507–15. [PubMed] [Google Scholar]
- 90. Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein. E Nat Med. 2002;9:61–7. [DOI] [PubMed] [Google Scholar]
- 91. Carvalho‐Tavares J, Hickey MJ, Hutchison J, Michaud J, Sutcliffe IT, Kubes P. A role for platelets and endothelial selectins in tumor necrosis factor‐alpha‐induced leukocyte recruitment in the brain microvasculature. Circ Res. 2000;87:1141–8. [DOI] [PubMed] [Google Scholar]
- 92. Asaduzzaman M, Lavasani S, Rahman M, et al. Platelets support pulmonary recruitment of neutrophils in abdominal sepsis. Crit Care Med. 2009;37:1389–96. [DOI] [PubMed] [Google Scholar]
- 93. Losche W, Dressel M, Krause S, Redlich H, Spangenberg P, Heptinstall S. Contact‐induced modulation of neutrophil elastase secretion and phagocytic activity by platelets. Blood Coagul Fibrinolysis. 1996;7:210–3. [DOI] [PubMed] [Google Scholar]
- 94. Gros A, Syvannarath V, Lamrani L, et al. Single platelets seal neutrophil‐induced vascular breaches via GPVI during immune‐complex‐mediated inflammation in mice. Blood. 2015;126:1017–26. [DOI] [PubMed] [Google Scholar]
- 95. Zalavary S, Grenegård M, Stendahl O, Bengtsson T. Platelets enhance Fc‐gamma receptor‐mediated phagocytosis and respiratory burst in neutrophils: the role of purinergic modulation and actin polymerization. J Leukoc Biol. 1996;60:58–68. [DOI] [PubMed] [Google Scholar]
- 96. Bengtsson T, Zalavary S, Stendahl O, Grenegard M. Release of oxygen metabolites from chemoattractant‐stimulated neutrophils is inhibited by resting platelets: role of extracellular adenosine and actin polymerization. Blood. 1996;87:4411–23. [PubMed] [Google Scholar]
- 97. Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–9. [DOI] [PubMed] [Google Scholar]
- 98. Ho‐Tin‐Noe B, Boulaftali Y, Camerer E. Platelets and vascular integrity: how platelets prevent bleeding in inflammation. Blood. 2018;131:277–88. [DOI] [PubMed] [Google Scholar]
- 99. Devi S, Kuligowski MP, Kwan RYQ, et al. Platelet Recruitment to the Inflamed glomerulus occurs via an αIIbβ3/GPVI‐dependent pathway. Am J Pathol. 2010;177:1131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pierre S, Linke B, Suo J, et al. GPVI and thromboxane receptor on platelets promote proinflammatory macrophage phenotypes during cutaneous inflammation. J Invest Dermatol. 2017;137:686–95. [DOI] [PubMed] [Google Scholar]
- 101. Schulz C, von Bruhl ML, Barocke V, et al. EMMPRIN (CD147/basigin) mediates platelet‐monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J Thromb Haemost. 2011;9:1007–19. [DOI] [PubMed] [Google Scholar]
- 102. Boulaftali Y, Hess PR, Getz TM, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123:908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Pachel C, Mathes D, Arias‐Loza AP, et al. Inhibition of platelet GPVI protects against myocardial ischemia‐reperfusion injury. Arter Thromb Vasc Biol. 2016;36:629–35. [DOI] [PubMed] [Google Scholar]
- 104. Takaya N, Katoh Y, Iwabuchi K, et al. Platelets activated by collagen through the immunoreceptor tyrosine‐based activation motif in the Fc receptor gamma‐chain play a pivotal role in the development of myocardial ischemia‐reperfusion injury. J Mol Cell Cardiol. 2005;39:856–64. [DOI] [PubMed] [Google Scholar]
- 105. Schönberger T, Ziegler M, Borst O, et al. The dimeric platelet collagen receptor GPVI‐Fc reduces platelet adhesion to activated endothelium and preserves myocardial function after transient ischemia in mice. Am J Physiol Cell Physiol. 2012;303:C757–66. [DOI] [PubMed] [Google Scholar]
- 106. Zoja C, Remuzzi G. Role of platelets in progressive glomerular diseases. Pediatr Nephrol. 1995;9:495–502. [DOI] [PubMed] [Google Scholar]
- 107. Barnes JL. Platelets in glomerular disease. Nephron. 1997;77:378–93. [DOI] [PubMed] [Google Scholar]
- 108. Cameron JS. Platelets in glomerular disease. Annu Rev Med. 1984;35:175–80. [DOI] [PubMed] [Google Scholar]
- 109. Mahan JD, Hebert LA, McAllister C, et al. Platelet involvement in experimental immune complex‐mediated glomerulonephritis in the nonhuman primate. Kidney Int. 1993;44:716–25. [DOI] [PubMed] [Google Scholar]
- 110. Kuligowski MP, Kitching AR, Hickey MJ. Leukocyte recruitment to the inflamed glomerulus: a critical role for platelet‐derived P‐selectin in the absence of rolling. J Immunol. 2006;176:6991–9. [DOI] [PubMed] [Google Scholar]
- 111. Gazit SL, Mariko B, Therond P, et al. Platelet and erythrocyte sources of S1P are redundant for vascular development and homeostasis, but both rendered essential after plasma S1P depletion in anaphylactic shock. Circ Res. 2016;119:e110–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lee RH, Piatt R, Conley PB, Bergmeier W. Effects of ibrutinib treatment on murine platelet function during inflammation and in primary hemostasis. Haematologica. 2017;102:e89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Deppermann C, Kraft P, Volz J, et al. Platelet secretion is crucial to prevent bleeding in the ischemic brain but not in the inflamed skin or lung in mice. Blood. 2017;129:1702–6. [DOI] [PubMed] [Google Scholar]
- 114. Hirahashi J, Hishikawa K, Kaname S, et al. Mac‐1 (CD11b/CD18) links inflammation and thrombosis after glomerular injury. Circulation. 2009;120:1255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. de Stoppelaar SF, van‘t Veer C, Claushuis TA, Albersen BJ, Roelofs JJ, van der Poll T. Thrombocytopenia impairs host defense in gram‐negative pneumonia‐derived sepsis in mice. Blood 2014;124:3781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115:2323–30. [DOI] [PubMed] [Google Scholar]
- 117. Iannacone M, Sitia G, Isogawa M, et al. Platelets prevent IFN‐alpha/beta‐induced lethal hemorrhage promoting CTL‐dependent clearance of lymphocytic choriomeningitis virus. Proc Natl Acad Sci U S A. 2008;105:629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Loria GD, Romagnoli PA, Moseley NB, Rucavado A, Altman JD. Platelets support a protective immune response to LCMV by preventing splenic necrosis. Blood. 2013;121:940–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ho‐Tin‐Noe B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008;68:6851–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Demers M, Ho‐Tin‐Noe B, Schatzberg D, Yang JJ, Wagner DD. Increased efficacy of breast cancer chemotherapy in thrombocytopenic mice. Cancer Res. 2011;71:1540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ho‐Tin‐Noe B, Carbo C, Demers M, Cifuni SM, Goerge T, Wagner DD. Innate immune cells induce hemorrhage in tumors during thrombocytopenia. Am J Pathol. 2009;175:1699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Boulaftali Y, Hess PR, Kahn ML, Bergmeier W. Platelet immunoreceptor tyrosine‐based activation motif (ITAM) signaling and vascular integrity. Circ Res. 2014;114:1174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Galt SW, Lindemann S, Medd D, et al. Differential regulation of matrix metalloproteinase‐9 by monocytes adherent to collagen and platelets. Circ Res. 2001;89:509–16. [DOI] [PubMed] [Google Scholar]
- 124. Seizer P, Borst O, Langer HF, et al. EMMPRIN (CD147) is a novel receptor for platelet GPVI and mediates platelet rolling via GPVI‐EMMPRIN interaction. Thromb Haemost. 2009;101:682–6. [DOI] [PubMed] [Google Scholar]
- 125. Drummond AH, Gordon JL. Proceedings: kinetics of the platelet release reaction induced by collagen. Br J Pharmacol. 1974;52:130P. [PMC free article] [PubMed] [Google Scholar]
- 126. Ollivier V, Syvannarath V, Gros A, et al. Collagen can selectively trigger a platelet secretory phenotype via glycoprotein VI. PLoS ONE. 2014;9:e104712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Elaïb Z, Adam F, Berrou E, et al. Full activation of mouse platelets requires ADP secretion regulated by SERCA3 ATPase‐dependent calcium stores. Blood. 2016;128:1129–38. [DOI] [PubMed] [Google Scholar]
- 128. Eckly A, Rinckel JY, Proamer F, et al. Respective contributions of single and compound granule fusion to secretion by activated platelets. Blood. 2016;128:2538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Baumgartner HR, Haudenschild C. Adhesion of platelets to subendothelium. Ann N Y Acad Sci. 1972;201:22–36. [DOI] [PubMed] [Google Scholar]
- 130. Baumgartner HR. The role of blood flow in platelet adhesion, fibrin deposition, and formation of mural thrombi. Microvasc Res. 1973;5:167–79. [DOI] [PubMed] [Google Scholar]
- 131. Baumgartner HR, Stemerman MB, Spaet TH. Adhesion of blood platelets to subendothelial surface: distinct from adhesion to collagen. Experientia. 1971;27:283–5. [DOI] [PubMed] [Google Scholar]
- 132. Tranzer J, Baumgartner HR. Filling gaps in the vascular endothelium with blood platelets. Nature. 1967;216:1126–8. [DOI] [PubMed] [Google Scholar]
- 133. Cochrane CG, Aikin BS. Polymorphonuclear leukocytes in immunologic reactions. The destruction of vascular basement membrane in vivo and in vitro. J Exp Med. 1966;124:733–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Voisin MB, Woodfin A, Nourshargh S. Monocytes and neutrophils exhibit both distinct and common mechanisms in penetrating the vascular basement membrane in vivo. Arter. Thromb Vasc Biol. 2009;29:1193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Snell DC, Schulte V, Jarvis GE, et al. Differential effects of reduced glycoprotein VI levels on activation of murine platelets by glycoprotein VI ligands. Biochem J. 2002;368:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kraft P, Schuhmann MK, Fluri F, et al. Efficacy and safety of platelet glycoprotein receptor blockade in aged and comorbid mice with acute experimental stroke. Stroke. 2015;46:3502–6. [DOI] [PubMed] [Google Scholar]
- 137. Quick AJ. The bleeding time as a test of hemostatic function. Am J Clin Pathol. 1975;64:87–94. [DOI] [PubMed] [Google Scholar]
- 138. Hughes SF, Hendricks BD, Edwards DR, Bastawrous SS, Roberts GE, Middleton JF. Mild episodes of tourniquet‐induced forearm ischaemia‐reperfusion injury results in leukocyte activation and changes in inflammatory and coagulation markers. J Inflamm (Lond). 2007;4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Hughes SF, Cotter MJ, Evans SA, Jones KP, Adams RA. Role of leucocytes in damage to the vascular endothelium during ischaemia‐reperfusion injury. Br J Biomed Sci. 2006;63:166–70. [DOI] [PubMed] [Google Scholar]
- 140. Hughes SF, Hendricks BD, Edwards DR, Middleton JF. Tourniquet‐applied upper limb orthopaedic surgery results in increased inflammation and changes to leukocyte, coagulation and endothelial markers. PLoS ONE. 2010;5:e11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ho‐Tin‐Noe B, Goerge T, Wagner DD. Platelets: guardians of tumor vasculature. Cancer Res. 2009;69:5623–6. [DOI] [PubMed] [Google Scholar]
- 142. Demers M, Wagner DD. Targeting platelet function to improve drug delivery. Oncoimmunology. 2012;1:100–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Rayes J, Watson SP. Platelet GPVI repairs its own damage. Blood. 2015;126:933–4. [DOI] [PubMed] [Google Scholar]
- 144. Finsterbusch M, Hickey MJ. Platelets interact with neutrophils and promote intravascular neutrophil activation in acute glomerulonephritis. Res Pract Thromb Haemost. 2017;1(Suppl 1):15. [Google Scholar]
- 145. Kerrigan AM, Navarro‐Nuñez L, Pyz E, et al. Podoplanin‐expressing inflammatory macrophages activate murine platelets via CLEC‐2. J Thromb Haemost. 2012;10:484–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Pertuy F, Aguilar A, Strassel C, et al. Broader expression of the mouse platelet factor 4‐cre transgene beyond the megakaryocyte lineage. J Thromb Haemost. 2015;13:115–25. [DOI] [PubMed] [Google Scholar]
- 147. Lowe KL, Navarro‐Núñez L, Bénézech C, et al. The expression of mouse CLEC‐2 on leucocyte subsets varies according to their anatomical location and inflammatory state. Eur J Immunol. 2015;45:2484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Claushuis AM, de Vos AF, Nieswandt B, et al. Different roles for platelet glycoprotein VI and CLEC2 during gram negative pneumonia derived sepsis. Res Pract Thromb Haemost. 2017;1(Suppl 1):114. [Google Scholar]
- 149. Claushuis AM, de Vos AF, Nieswandt B, et al. Platelet glycoprotein VI aids in local immunity during pneumonia‐derived sepsis caused by gram‐negative bacteria. Blood. 2018;131:864–76. [DOI] [PubMed] [Google Scholar]
- 150. Wéra O, Delierneux C, Servais L, et al. Interplay between platelets, neutrophils and coagulation in bleeding in a mouse model of inflammatory bowel disease. Res Pract Thromb Haemost. 2017;1(Suppl 1):462. [Google Scholar]