

Abstract
Essentials.
The antiphospholipid syndrome predisposes to thrombosis due to activation of endothelium and blood components.
The role of anti‐β2GPI antibodies in VWF release and ADAMTS13 function is not well understood.
Some anti‐β2GPI antibodies induce endothelial release of soluble VWF but not VWF strings.
An anti‐β2GPI antibody can decrease ADAMTS13 activity in vitro similar to ex vivo results.
Background
Antiphospholipid syndrome (APS) is characterized by recurrent thromboembolic events in the setting of pathologic autoantibodies, some of which are directed to β2‐Glycoprotein 1 (β2GPI). The mechanisms of thrombosis in APS appear to be multifactorial and likely include a component of endothelial activation. Among other things, activated endothelium secretes von Willebrand factor, a hemostatic protein that in excess can increase the risk of thrombosis.
Objective
We hypothesized that anti‐β2GPI antibodies could regulate the release and modulation of VWF from endothelial cells.
Patients/Methods
Isolated anti‐β2GPI antibodies from patients with APS were assayed for their ability to induced VWF release from HUVECs and modulate the effects of ADAMTS13 in a shear‐dependent assay.
Results
We observed that anti‐β2GPI antibodies from some patients with APS induced VWF release from human endothelial cells but did not induce formation of cell‐anchored VWF‐platelet strings. Finally, we also determined that one of the Anti‐β2GPI antibodies tested can inhibit the function of ADAMTS13, the main modulator of extracellular VWF.
Conclusions
These results suggest that VWF and ADAMTS13 may play a role in the prothrombotic phenotype of APS.
Keywords: ADAMTS13, antiphospholipid syndrome, endothelial cells, von Willebrand factor
1. INTRODUCTION
Antiphospholipid antibody syndrome (APS) is characterized by a predisposition to arterial and venous thromboebolism in the presence of antiphospholipid antibodies (APLA). APS afflicts people of all ages, including children and adolescents, and is characterized by up to a 40‐fold increased risk of arterial and venous thromboembolism in the setting of persistent APLA.1 APS is an autoimmune disorder with the pathological formation of antibodies against various host antigens. In addition to the thromboembolic events, APS has a significant effect on maternal/fetal health, as women with APLA are at a higher risk for miscarriages and thrombotic events during pregnancy.2 The most common thrombotic events associated with APS are ischemic stroke and deep venous thromboembolism. Additionally, the diagnosis of APS may commit a patient to indefinite anticoagulation, decreasing their quality of life and exposing them to risks of hemorrhage.3, 4 The underlying mechanism that predisposes these patients to thrombosis is not well understood but is thought to be the multifactorial activation of various components of the blood and vasculature.1, 5
Many of the pathophysiological mechanisms in APS have been attributed to anti‐β2GPI IgG antibodies.6 Clinically, patients with anti‐β2GPI antibodies are at a higher risk for thrombotic events as compared to other types of APLA.7, 8 Anti‐β2GPI antibodies activate leukocytes, platelets, endothelial cells and, more recently, have been shown to augment thrombus formation in vivo.6, 9, 10, 11 One important consequence of endothelial activation is the release of the hemostatic protein von Willebrand factor (VWF). We therefore sought to investigate the effects of anti‐β2GPI antibodies on the regulation of VWF in APS.
VWF has two main functions in hemostasis: (i) to bind to exposed subendothelial collagen at sites of vessel injury and capture platelets during primary hemostasis, and (ii) to stabilize coagulation factor VIII. In addition, in certain pathological states, endothelial‐released VWF remains attached to the endothelial surface, providing a surface on which platelets can accumulate.
While deficiency of VWF characterizes von Willebrand disease, elevations of VWF have also been implicated in thrombotic disorders, specifically cardiovascular disease, myocardial infarction, and arterial ischemic stroke.12, 13, 14, 15 The unique predisposition of APS to arterial stroke suggests that VWF, which has significant importance in arterial, platelet‐rich thrombi, may play a role in the predisposition to thromboembolism seen in APS. Several reports have demonstrated an increase in cell surface VWF on endothelial cells treated with APL‐Abs and higher levels of active VWF in the serum of patients with anti‐β2GPI antibodies.16, 17, 18, 19
VWF is modulated by ADAMTS13 (a‐disintegrin‐and‐metalloproteinase‐with‐a‐thrombospondin‐type‐motif, member 13), which cleaves highly hemostatic ultra large VWF (ULVWF) complexes into smaller units. Deficiency of ADAMTS13, either congenital (Upshaw‐Schulman syndrome) or acquired (usually due to acquired autoantibodies), leads to thrombocytopenia, microangiopathic hemolytic anemia, and microvascular thrombosis in the clinical syndrome of thrombotic thrombocytopenic pupura.20 In this syndrome, patients are at high risk of thrombosis due to the presence of circulating ultra large molecular weight VWF that is highly hemostatically active. Previously, there have been varied reports of ADAMTS13 levels, activity, and the presence of anti‐ADAMTS13 antibodies in APS.16, 17, 21 More recently, reduced ADAMTS13 activity was shown to correlate with thrombotic risk in a large cohort of patients with APS.22
We therefore sought to investigate the role of VWF and ADAMTS13 in thrombosis associated with APS. In this study, we specifically investigated the role of anti‐β2GPI in VWF release from endothelial cells and the role of anti‐β2GPI in the modulation of ADAMTS13 activity.
2. MATERIALS AND METHODS
2.1. Materials
Control human and rabbit IgG were purchased from Jackson ImmunoResearch (West Grove, PA, USA) or Zymed Laboratories (San Francisco, CA, USA). Rabbit anti‐human VWF polyclonal antibody was from DAKO (Carpinteria, CA, USA). HRP‐linked secondary antibody, bovine serum albumin (BSA) was from ThermoFisher Scientific, (Waltham, MA, USA). PBS, Tyrode's buffer, MgCl2, EGTA, Triton‐X, paraformaldehyde, p‐nitrophenylphosphate, (PNPP), phorbol‐12‐myristate‐13‐acetate (PMA) were all from Sigma Aldrich (St. Louis, MO, USA).
2.2. Patient samples
Patients were enrolled in a IRB‐approved study (University of Colorado COMIRB 09‐0816) and blood was collected into sodium citrate (BD Vacutainer, San Jose, CA, USA). Platelet‐poor plasma was isolated via centrifugation of whole blood, twice, at 2500 × g for 30 minutes at 4C prior to cryopreservation at −80°C. Four patient samples were used in this study (APS155, APS698, APS25, APS203). Patients were confirmed to have the clinical diagnosis of APS per the updated Sapporo/Sydney criteria (which requires a history of a thromboembolic event).23 See Table 1 for further descriptive information regarding clinical characteristics and APLA levels. Of note, patients were chosen to represent both individuals with and without high levels of anti‐β2GPI to represent the clinical heterogeneity seen in APS.
Table 1.
Clinical and laboratory characteristics of APS patients and Anti‐β2GPI samples
| Patient | Clinical characteristics | APL Assays at time of lab draw (Range) Normal range = 0.0–14.9 GPL |
|---|---|---|
| APS155 |
APS Lower extremity deep venous thrombosis (DVT) (2 years prior to blood draw) Non‐acute state at time of draw Medications wafarin |
dRVVT (+) Anti‐β2GPI IgA – 5.5 Anti‐β2GPI IgM – 21.4 Anti‐β2GPI IgG – 49.2 Anti‐cardiolipin IgA – n/a Anti‐cardiolipin IgG – >100 Anti‐cardiolipin IgM – 30.2 |
| APS698 |
APS Systemic Lupus Erythematosus Lower extremity DVT (1 year prior to blood draw) Non‐acute state at time of draw Medications warfarin |
dRVVT (+) Anti‐β2GPI IgA – <4 Anti‐β2GPI IgM – 13.2 Anti‐β2GPI IgG – 25 Anti‐cardiolipin IgA – n/a Anti‐cardiolipin IgG – 22.2 Anti‐cardiolipin IgM – 25.7 |
| APS25 |
APS Multiple PE (2 days prior to draw) S/p thromboendoarterectomy Acute presentation of thrombosis Medications Subtherapeutic on warfarin at the time of draw; on unfractionated heparin |
dRVVT (+) Anti‐β2GPI IgA – <4 Anti‐β2GPI IgM – <4 Anti‐β2GPI IgG – 86.5 Anti‐cardiolipin IgA – negative Anti‐cardiolipin IgG – >100 Anti‐cardiolipin IgM – n/a |
| APS203 |
Catastrophic antiphospholipid syndrome (sentinel episode three years prior to lab draw) Non‐acute state at time of draw Medications warfarin |
dRVVT (+) Anti‐β2GPI IgA – n/a Anti‐β2GPI IgM – n/a Anti‐β2GPI IgG – 0.19 Anti‐cardiolipin IgA – negative Anti‐cardiolipin IgG – negative Anti‐cardiolipin IgM – negative |
2.3. Anti‐β2GPI antibody isolation
Anti‐β2GPI antibodies from subjects and from a β2GPI‐immunized rabbit were affinity purified using an Affigel HZ column to which purified human β2GPI was coupled (Bio‐Rad Laboratories, Hercules, CA, USA), as previously described.24, 25, 26
2.4. Recombinant ADAMTS13
cDNA for human ADAMTS13 was cloned into pcDNA‐3 vector with N‐terminal Myc and multi‐His tags (ThermoFisher Scientific, Waltham, MA, USA) and delivered into human embryonic kidney cells (HEK293) using lipids as the transfection mediator cells.27 The cells were grown in Dulbecco Modified Eagle Medium (Invitrogen, Carlsbad, CA, USA) containing 500 μg/ml hygromycin (Sigma Aldrich, St. Louis, MO, USA) and 10% fetal bovine serum at 37°C. Those expressing high levels of ADAMTS13 were selected by single cell cloning using flow cytometric cell sorting. Selected cells were then incubated in Optipro serum free media (ThermoFisher Scientific, Waltham, MA, USA) for 72 hours and the conditioned media was tested for ADAMTS13 content and frozen at −80°C.
2.5. Endothelial cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (Basel, Switzerland). Endothelial cells were maintained using EBM2‐MV (Lonza, Basel, Switzerland). All experiments were performed using cells of passage 6 or lower.
2.6. VWF release assays
Confluent HUVECs in 96‐well microplates were incubated in serum free‐media for a minimum of two hours before incubation for one hour in EBM2 serum‐free media containing patient‐derived anti‐β2GPI antibodies (600 nmol L−1) and ± β2GPI (Haematologic Technologies Inc, Essex Junction, VT, USA). After incubation, conditioned serum‐free medium was removed and frozen at −80°C. VWF antigen release was measured by ELISA as described previously.28 In brief, monoclonal antibodies (Blood Center of Wisconsin) were coated on a 96‐well plate and after blocking, conditioned medium is incubated for one hour. Total VWF bound is detected using an anti‐VWF polyclonal antibody and an HRP‐linked secondary antibody and optical densities (OD) are compared to a standard curve of pooled normal plasma (PNP) where the VWF concentration is assumed to be 100 mU/mL.
2.7. VWF propeptide assays
Stored conditioned media (described above) was assessed for VWF propeptide levels. Two monoclonal antibodies, 239.2 and 239.3, which recognize only the propeptide portion of the VWF molecule, (BloodCenter of Wisconsin) were coated on a 96‐well plate in a calcium carbonate buffer. The plate was then blocked with 1% bovine serum albumin (Sigma‐Aldrich) and washed with PBS‐Tween. Conditioned media obtained from VWF release assays (see above) was incubated for one hour at room temperature before application of VWF propeptide specific biotinlyated monoclonal antibodies 242.2 and 242.6 (BloodCenter of Wisconsin). Bound VWF propeptide was detected using streptavidin‐bound HRP (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and developed using PNPP. OD results are compared to a standard curve of PNP where the VWF propetide concentration is assumed to be 100 mU/mL.
2.8. Weibel‐Palade body assay
Confluent HUVECs on gelatin‐coated Laboratory‐Tek II chambered slides (ThermoFisher Scientific, Waltham, MA, USA) were incubated for two hours in serum‐free M199 medium containing 1% BSA and then with 100 nmol L−1 β2GPI and 600 nmol L−1 control or anti‐β2GPI antibodies (Zymed Laboratories, San Francisco, CA) for 30 and 60 minutse. Cells were then fixed by incubation in PBS containing 2 mmol L−1 MgCl2, 2 mmol L−1 EGTA, and 4% paraformaldehyde, permeabilized using PBS containing 2 mmol L−1 MgCl2, 2 mmol L−1 EGTA, and 0.5% Triton X‐100, and blocked in PBS containing 0.1% Tween and 1% BSA before incubation with rabbit‐anti‐human VWF primary antibody antibodies After washing with PBS, cells were sequentially incubated with Alexa Fluor 488–conjugated anti‐rabbit antibodies, Alexa Fluor 568–conjugated phalloidin, and 4,6 diamidino‐2‐phenylindole prior to imaging using a Zeiss LSM510 confocal microscope using a 60X objective. Multiple images were collected and then Weibel‐Palade Bodies were manually counted using ImageJ.29
2.9. VWF‐platelet string assay
Confluent HUVECs passaged as above were seeded into a collagen‐coated flow chamber (Ibidi U‐Slide IV 0.1, Ibidi USA, Madison, WI, USA) and incubated in serum‐free EBM2‐MV for a minimum of two hours before incubation for one hour with 600 nmol L−1 control or patient‐derived anti‐β2GPI antibodies. After one hour, the chambers were perfused with fixed platelets (4 × 107/mL) (Bio/Data Corporation, Horsham, PA, USA) for two minutes at a shear stress of 5 dynes/cm2. Multiple non‐overlapping bright‐field images of the channel were captured using a high‐speed camera (Olympus IX‐81, Olympus America, Center Valley, PA, USA) and analyzed in a blinded fashion for VWF‐platelet string formation wherein 25 μmol L−1 of a visualized VWF platelet string equals 1 string‐unit.
2.10. ADAMTS13 activity assays
ADAMTS13 activity in patient plasma was measured as the enzyme's ability to cleave a biotinlyated VWF A2 peptide substrate conjugated to HRP.30 Briefly, Patient plasma was incubated with the peptide substrate in 10 mmol L−1 HEPES (pH 7.4), 2 mmol L−1 CaCl2, and 3.5 mg/mL BSA containing serine protease inhibitors for 15 minute at room temperature. The cleavage reaction was terminated by adding 10 mmol L−1 EDTA. The reaction mixture was then incubated with streptavidin‐agarose beads for 5 minute to remove uncleaved substrate. Cleavage product in the supernatant was quantified using tetramethylbenzidine as substrate. ADAMTS13 activity was determined by the cleavage rate and normalized to PNP, which was assigned an activity of 100%.
For recombinant ADAMTS13 concentration determination, the FRETS‐VWF73 kit (Peptides International, Louisville, KY, USA) was used according to manufacturer's instruction. In brief, 100 μL of plasma dilutions (0%, 1%, 2%, 3%, 4%, 5%, and 6% plasma) and test solutions (0.5%, 1%, and 1.5%) were combined with FRETS‐VWF73 substrate. Emission at 450 nm was monitored over 2 hours at 30°C. Slopes of unknowns were compared to the slopes of standard plasma dilutions where PNP was assumed to have 100% activity.
2.11. VWF string cleavage model
Confluent HUVECs were seeded into a collagen‐coated flow chamber (Ibidi U‐Slide IV 0.1, Ibidi USA, Madison, WI, USA). Cells were then incubated in serum free EBM2‐MV for two hours and then stimulated with 50 ng/mL PMA for 30 minutes. After activation, fixed platelets (4 × 107 cells/mL) labeled with with a Pacific Blue anti‐CD41a antibody (Biolegend, San Diego, CA, USA) were perfused over the HUVECs at a shear stress of 5 dynes/cm2 for two minutes. 200 μL of Tyrode's buffer was then perfused and non‐overlapping bright‐field and fluorescent images at 10 standardized locations along each flow chamber are captured using a high‐speed camera (Hamamatsu ORCA‐Flash 4.0 attached to an Olympus IX‐81, Olympus America, Center Valley, PA, USA). After image capture, ADAMTS13 (standardized to 8% PNP activity) was perfused over the formed VWF‐platelet strings for two minutes followed by 200 μL of Tyrode's buffer prior to image acquisition at the 10 standardized locations. Images of the 10 standardized locations were then paired before/after ADAMTS13 perfusion. A standardized ImageJ/FIJI analysis macro was used to analyze and count each platelet using the particle analyzer function of ImageJ (see supplemental document for specific ImageJ/FIJI macro file). Platelet numbers before and after ADAMTS13 perfusion were compared. The difference served as a proxy for ADAMTS13 activity.
2.12. Multimer analysis
The equivalent of 1 mU VWF (determined via ELISA) from patient plasma was run in each lane of a 2% HGT agarose (Lonza, Rockland, ME, USA) resolving gel containing 1% sodium dodecyl sulfate (SDS) for 16 hours at 40V followed by electroblot transfer for one hour at 100V to an Immobilon‐P (Millipore, Billerica, MA, USA) membrane in a 4°C buffer containing 25 mmol L−1 Tris, 200 mmol L−1 glycine, 20% methanol and 0.03% SDS. The membrane was blocked for 1 hour with 5% nonfat dry milk, then incubated with an rabbit‐anti‐human VWF primary antibody (DAKO, Carpinteria, CA, USA) for one hour followed by a one‐hour incubation with a HRP‐linked goat anti‐rabbit antibody. Membranes were then developed with Western Lightning‐ECL (Perkin Elmer, Waltham, MA, USA) and bands were visualized by exposure to Fujifilm Super RX (Edison, NJ, USA). Multimer densitometric analysis was done using ImageJ/FIJI (see supplemental methods).
2.13. Statistical analysis
All experiments were performed a minimum of three times and statistical analysis was conducted via 1‐one ANOVA or paired t‐test; significance was defined by a p‐value <.05. Data was analyzed using GraphPad Prism version 7.00 for Mac (GraphPad Software, La Jolla, CA, USA).
3. RESULTS
3.1. VWF and VWFpp secretion from endothelial cells in response to anti‐β2GPI antibodies
In previous reports, APS serum or APS IgG fractions have been shown to increase cell surface VWF from endothelial cells.31, 32, 33 To determine the amounts of VWF released by endothelial cells into the cellular supernatant, endothelial cells were exposed to four patient‐derived anti‐β2GPI antibodies (APS25, APS203, APS155, and APS698) and the conditioned media were assayed by ELISA. Exposure to APS25 and APS203 as well as to a polyclonal rabbit anti‐β2GPI antibody was associated with a statistically significant increase in VWF release when compared to control human IgG and control rabbit IgG (Figure 1A). Interestingly, there was a wide variation in the amount of VWF released among the patient‐derived anti‐β2GPI antibodies. While APS25 and APS203 showed significant increases, APS155 and APS698 showed increased VWF release but the differences did not reach statistical significance when compared to control IgG (Figure 1A). Similarly, to confirm the acute release of VWF we also assayed VWF propeptide (VWFpp) which is expected to be released in equimolar amounts with mature VWF in medium conditioned by HUVEC exposed to the same patient‐derived anti‐β2GPI antibodies (Figure 1B). In this assay, APS25 induced a statistically significant increase in VWF release while the three other Anti‐β2GPI antibodies induced increased release but they did not reach significance. Since β2GPI has been proposed as a hemostatic modulator and is thought to bind to cells and act as a “targeting” molecule for anti‐β2GPI antibodies, we performed the same experiments in the presence of β2GPI.24, 26, 34 The results were similar to those without β2GPI (Supplemental Figure 1) although they did not reach statistical significance.
Figure 1.
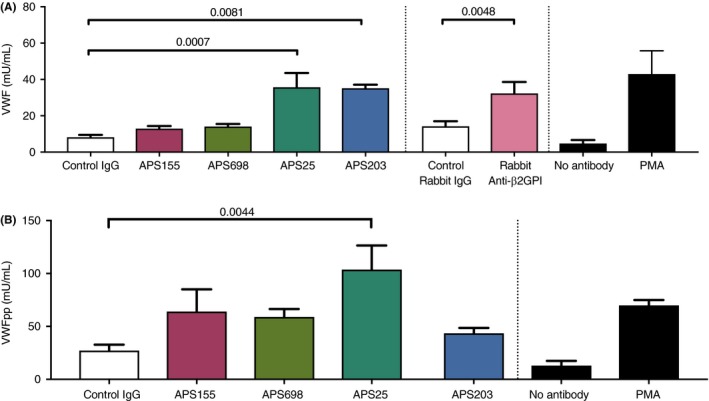
Anti‐β2GPI antibodies increase VWF and VWFpp release from endothelial cells. HUVECs were plated in a 96‐well plate and cultured until reaching 100% confluence. Cells were then washed in serum‐free EBM2 and incubated for 2 hours in serum‐free EBM2 prior to incubation in EBM2 plus the control or Anti‐β2GPI antibodies (600nM). Conditioned media is collected and frozen at ‐80ºC for future use. VWF and VWFpp quantities are assayed via VWF or VWFpp ELISAs and are reported in mU/mL of VWF. Control stimulation with serum free media alone (no control or anti‐β2GPI antibody) and PMA (50 ng/mL) are shown in black bars at the far right. N>3 and p‐values of significant relationships are shown as analyzed by a one‐way ANOVA with Holm‐Sidak’s multiple comparison test. Error bars represent the means SEM. P – values are shown as calculated
3.2. Anti‐β2GPI antibodies increase Weibel‐Palade formation in HUVECs
To further study the role of Anti‐β2GPI antibodies on the intracellular pathways of VWF expression and release, we assessed the formation of Weibel‐Palade bodies in HUVECs by immunofluorescence. An Anti‐β2GPI antibody in the presence of β2GPI induced increased Weibel‐Palade body formation as compared to control human Ig at both 30 and 60 minutes (Figure 2). These results are consistent with our early findings and suggest that anti‐β2GPI antibodies increase the intracellular formation and release of VWF from HUVECs.
Figure 2.
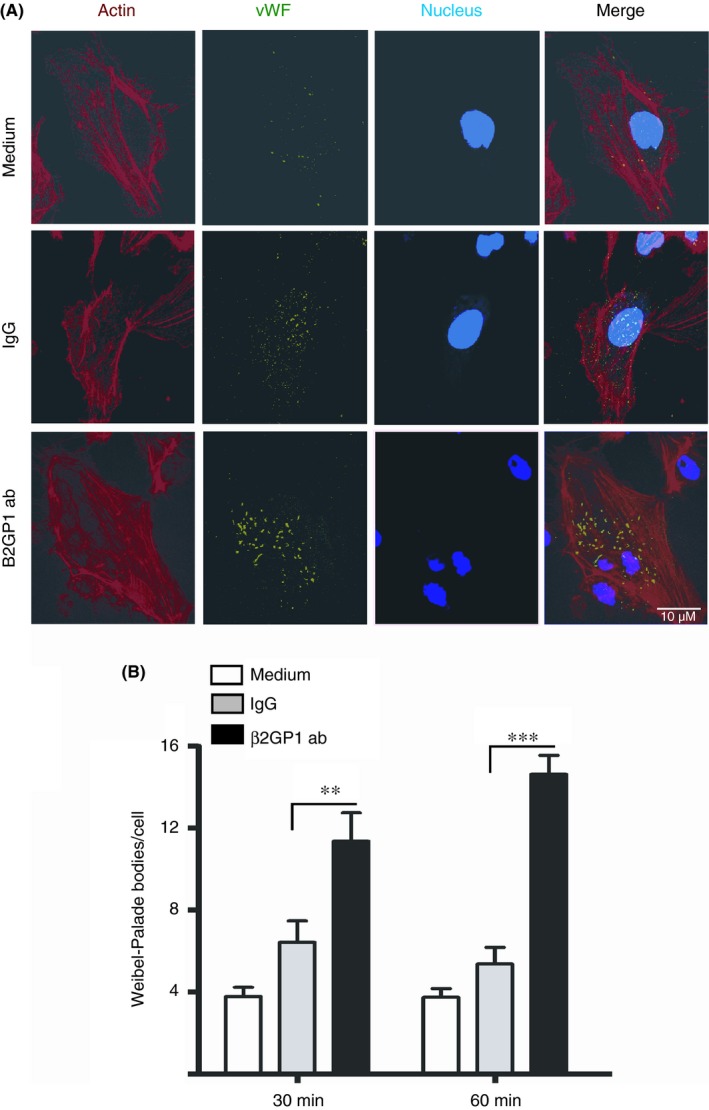
Anti‐β2GPI antibodies induce Weibel‐Palade body formation in HUVECs. Confluent monolayers of HUVEC on gelatin‐coated Laboratory‐Tek II chambered slides were incubated for 2 hours in serum‐free M199 medium containing 1% bovine serum albumin and then with 100 nmol L−1 Mβ2GPI and 600 nmol L−1 control or rabbit Anti‐β2GPI antibodies for 30 and 60 min in the tissue culture incubator. Cells were then fixed by incubation in phosphate‐buffered saline (PBS), permeabilized and then immunostained for Weibel‐Palade Bodies, actin filaments and nucleus using a 60X objective. Representative images of Weibel‐palades bodies at 60 minutes are shown in Figure 2A. (B) Quanitification of Weibel‐Palade bodies/cell. A minimum of 25 images (and therefore a minimum of 25 cells) were quantified from multiple independent experiments (n > 4) using ImageJ. Statistical significance is shown with capped lines (** P < .01, *** P < .001)
3.3. Anti‐β2GPI antibodies do not induce increase VWF‐platelet string formation from endothelial cells
VWF‐platelet strings have been shown to be released in response to agonists such as a histamine, thrombin, IL‐6 as well as chemical agonists such as PMA, and have been implicated in diseases such as TTP.19, 35 We sought to examine whether VWF‐platelet strings would be formed in response to anti‐β2GPI treatment similar to the VWF released into conditioned media demonstrated above. No statistically significant differences in the formation of VWF‐platelet strings in response to three different patient‐derived anti‐β2GPI antibodies (Figure S2) were observed when compared to control IgG.
3.4. Anti‐β2GPI antibodies cause dysfunction of ADAMTS13
We next investigated the effects of patient‐derived anti‐β2GPI antibodies on ADAMTS13 activity. Loss of ADAMTS13 activity, as measured in a VWF‐platelet string assay has been reported in TTP.19 We therefore tested the ability of ADAMTS13 to cleave VWF‐platelet strings in the presence of control or patient‐derived anti‐β2GPI antibodies. The anti‐β2GPI antibody APS155 significantly reduced ADAMTS13 activity as compared to control IgG. Differences attributable to APS203 and APS698 did not show statistical significance compared to control IgG but APS203 showed a trend towards a decrease in ADAMTS13 activity (Figure 3). These results were consistent with a FRET‐assay of ADAMTS13 activity in plasma isolated from APS155 and APS203 which demonstrated decreased ADAMTS13 activity when compared to control (Table 2), although within the normal rage for the enzyme values. Finally, a multimer gel with densitometric anaylsis of high molecular weight multimers demonstrates that plasma from APS155 may show increased high molecular weight multimers, further suggesting ADAMTS13 dysfunction (Figure 4). Interestingly, although within the normal range, ADAMTS13 activity in APS155 plasma showed decreased activity when compared to control plasma. Anti‐β2GPI derived from APS698 demonstrated a minimal effect on ADAMTS13 activity in our VWF‐platelet string cleaving assay and similarly demonstrated no inhibition but rather an increased function of ADAMTS13 in the FRET‐based assay.
Figure 3.
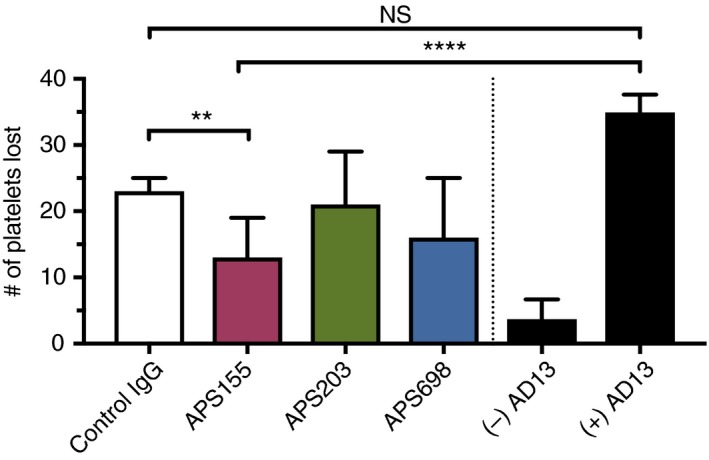
ADAMTS13 mediated VWF‐platelet string cleavage is inhibited by Anti‐β2GPI antibody APS155. HUVECs are seeded in a channel slide and are stimulated with 50 ng/mL PMA. After platelet perfusion, images are captured at standardized locations to represent the maximal formation of VWF‐platelet strings. After capture, ADAMTS13 (standardized to 8% of PNP), in the presence of control or patient‐derived Anti‐β2GPI antibodies, is perfused and images are again captured at the identical standardized locations. Loss of platelets after perfusion is thus the quantitative measure of ADAMTS13 activity. Control experiments in the presence and absence of ADAMTS13 are shown in black bars on the far right. Outliers were removed using Graphpad 7 ROUT function with Q=1% from all conditions. N3 and statistical significance is analyzed with a one‐way ANOVA with Holm‐Sidak’s multiple comparison test. Error bars represent the means SEM. Statistical significance is shown with capped lines (**P < .01, ****P < .0001)
Table 2.
ADAMTS13 activity results of plasma isolated from APS patients
| Samples | Assay 1 (%) | Assay 2 (%) | Average (%) | ± % |
|---|---|---|---|---|
| APS698 | 122.07 | 125.65 | 123.86 | 1.79 |
| APS155 | 75.59 | 83.24 | 79.42 | 3.83 |
| APS203 | 86.51 | 84.22 | 85.37 | 1.14 |
| Pooled normal plasma | 100.55 | 99.85 | 100.20 | 0.35 |
Using a FRET‐based assay, platelet poor plasma was assayed for ADAMTS13 activity in patients from the same draw as used for isolation of anti‐β2GPI antibodies. Results are shown in duplicate and a lab standard normal plasma as a standard control is also shown.
Figure 4.
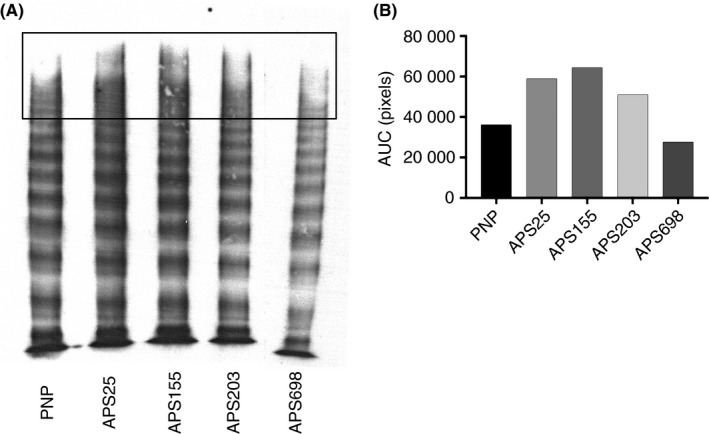
VWF Multimer Pattern from patients with APS. PPP from each APS patient was run on a VWF multimer gel to resolve multimer patterns via SDS agarose gel electrophoresis followed by western blotting. The same was standardized to 1 mU of total protein in each lane and the plasma sample loaded was the same draw as that used to isolated anti‐β2GPI antibodies as well as run the FRET‐based assay in Table 1. Two lanes between APS203 and APS698 were excluded from analysis and removed from this image. Densitometry analysis of high molecular weight bands was performed. The black box in Figure 4A demonstrates the approximate area upon which analysis was conducted, analysis of the band intensity as measured by ImageJ/FIJI is demonstrated in 4B.
4. DISCUSSION
In this report, we sought to further investigate the role of VWF in the prothrombotic nature of APS. We hypothesized that anti‐β2GPI antibodies would stimulate VWF release from endothelial cells and tested this hypothesis by assaying VWF release, both into the supernatant as well as cellular‐based VWF (VWF platelet strings). We also hypothesized that anti‐β2GPI antibodies would have an effect on ADAMTS13, the primary modulator of VWF function in vivo. We have demonstrated that some, but not all anti‐β2GPI antibodies induced the release of VWF and that this release is not associated with VWF‐platelet string formation. We have also demonstrated that the APS155 anti‐β2GPI antibody can induce dysfunction of ADAMTS13 represented by decreased cleavage of VWF strings. This finding was similar to decreased ADAMTS13 activity in a FRET‐based assay on APS155 plasma and this effect may be reflected in VWF multimer patterns.
In the VWF release assay, APS25 and APS203 induced significant increases in VWF release as compared to control human IgG. A similar finding was seen for a rabbit polyclonal anti‐β2GPI antibody. APS698 and APS155 demonstrated non‐statistically significant increases in VWF release. Similar findings were also seen in our VWFpp assay which largely confirmed the “acute” release of VWF from endothelial cells and minimized the concern of contaminating VWF from cellular media or other sources. Our Weibel‐Palade body findings suggest that anti‐β2GPI, in addition to inducing release of VWF may also affect VWF expression and packaging as represented by increased Weibel‐Palade body formation. The increase in VWF observed in our assay may mirror the in vivo effects seen in some of APS patients as they tend to have elevated levels of total and active VWF.15, 20 Elevations of VWF in turn can lead to thrombotic events.11, 12, 13, 14 Interestingly, the variation of VWF release induced by anti‐β2GPI antibodies from different patients suggests that elevations of VWF are not the sole driver of the prothrombotic state of APS and is consistent with the “multi‐factorial” activation hypothesis of APS. In contrast to our supernatant experiments, we did not detect an increase in VWF release in the form of VWF‐platelet strings on endothelial cells in response to anti‐β2GPI IgG. Although the presence of VWF‐platelet strings has been noted upon exposure of endothelial cells to agonists such as PMA, histamine, some pro inflammatory cytokines,35 and have been visualized in vivo, 36 the role of VWF‐platelet string formation in APS needs further evaluation.37 We also only investigated the formation at one shear rate, 5 dynes/cm2 and it is possible that a phenotype of increased VWF platelet‐string formation would be revealed at higher or lower shear rates.
Our results are consistent with previous reports that demonstrated VWF release from endothelial cells in the presence of APLA.31, 32, 33 However, all of these studies used either serum or a total IgG fraction from patients with APS and here we specifically focused on the effects of anti‐β2GPI antibodies derived from patients with APS to induce VWF release, as they have been shown to potentiate thrombosis in several in vivo models.7, 9, 10 In contrast to previous reports,24, 26 β2GPI was not strictly required for VWF release in our studies. Although it has been suggested that β2GPI is necessary for targeting of anti‐β2GPI antibodies, many of the previous studies specifically studying anti‐β2GPI induced VWF release31, 32 were conducted with unknown levels of β2GPI. Lai et al. (1996) incubated HUVECs with APLA antibodies in full endothelial cell medium that contained FBS; this likely contributed some unknown amount of bovine‐derived β2GPI. Lindsey et al. (1993) incubated HUVECs in Media 199 with 20% human serum initially that was later removed and replaced. It is unclear whether this second M199 contained human serum or other sources of β2GPI. We have not excluded the possibility that residual β2GPI from cell culture may have remained associated with the cells in our experimental assays since β2GPI has been shown to bind to annexin A2 on endothelial cells with high affinity.26 Furthermore, we did not test the effects of anti‐β2GPI antibodies on different endothelial cell types, it is possible that endothelial cells derived from varied tissue beds (ie, arterial vs. venous) may reveal further phenotypic differences. Further investigation into the mechanism of VWF release from endothelial cells mediated by anti‐β2GPI antibodies should expand on the role of β2GPI and assess varied endothelial cell types.
We also sought to investigate the effect of anti‐β2GPI antibodies on ADAMTS13 activity as ADAMTS13 dysfunction, which classically results in microvascular thrombi such as that seen in TTP, has been recently linked to thromboembolic events such as myocardial infarction, DVT, and stroke.38, 39, 40 In addition to thromboembolic events ADAMTS13 may have effects on early miscarriage; there are reports of higher miscarriage rates in TTP although a recent report noted no significant changes in ADAMTS13 levels in those with recurrent miscarriages.41, 42 We demonstrated that APS155 caused a mild decrease in ADAMTS13 activity by FRET assay and that anti‐β2GPI antibodies purified from that patient inhibited ADAMTS13's ability to cleave VWF‐platelet strings. This VWF‐platelet string model has been used previously to assess ADAMTS13 activity under conditions of shear stress which allows for the unraveling of VWF.20, 43 We also observed an increase of the higher molecular weight multimers in APS samples, as previously reported by Austin et al.21 We note that although decreased, the levels of ADAMTS13 in our samples were still within the normal range for ADAMTS13 activity, and thus anti‐β2GPI antibodies alone were unable to fully inhibit ADAMTS13 activity at the concentrations tested. However, a recent publication demonstrated that mildly decreased ADAMTS13 activity levels were associated with acute and chronic cerebrovascular disease patients suggestive that even mild decrease may represent a prothrombotic risk factor.40 These findings suggest that anti‐β2GPI antibodies may have heterogenous effects on VWF release/ADAMTS13 activity. This concept is further supported by Austin et al. who demonstrated heterogeneous levels of ADAMTS13 dysfunction in a large cohort of individuals with anti‐β2GPI antibodies, and that the IgG fraction of some of these patients inhibited ADAMTS13 activity. Contrary to our results, Hulstein et al. evaluated ADAMTS13 activity in APS plasma and found similar ADAMTS13 activity levels across all of their cohorts with and without anti‐β2GPI antibodies suggesting that anti‐β2GPI antibodies may not be responsible for decreased ADAMTS13 activity. One of the challenges in direct comparison of the various anti‐β2GPI (and APLA) studies is the inherent discordance that is seen in APLA assays amongst different laboratories.44, 45 Thus, while presence of anti‐β2GPI antibodies has been strongly associated with thrombosis,7, 8 different levels (based on lab sensitivities) or perhaps even different subsets of anti‐β2GPI may drive the prothrombotic phenotype. One advancement of our in vitro ADAMTS13 assay is that it specifically isolates anti‐β2GPI antibodies and tests the effects of anti‐β2GPI antibodies on ADAMTS13 activity using a method that directly assesses the activity of ADAMTS13 in cleaving VWF. Potential mechanisms of ADAMTS13 inhibition by anti‐β2GPI antibodies, such as binding to the A2 domain of VWF or the active site of ADAMTS13 due to cross‐reactive epitopes needs to be further elucidated. The effects of different shear rates is also worthy of future study.46
Global limitations of our study include a relatively small number of anti‐β2GPI antibodies tested in our VWF models and a single concentration tested for efficacy. We were primarily limited by the total amount of anti‐β2GPI antibodies available for all of our assays and thus chose to use a previously published concentration that demonstrated endothelial cell activation. Future investigations are needed to determine if alternative concentrations of anti‐β2GPI may result in different effects.
In summary, we demonstrated that some anti‐β2GPI antibodies can induce VWF release from human endothelial cells and inhibit ADAMTS13 activity. These findings have important implications for the role that VWF may play in the prothrombotic phenotype of APS and suggest that targeting VWF may ameliorate the predisposition to thrombosis seen in this disease.
AUTHOR CONTRIBUTIONS
CJ Ng contributed to concept and design, analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. KR McCrae contributed to concept and design, analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. K. Ashworth contributed to analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. LJ Sosa contributed to analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. V. Betapudi contributed to analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. MM Johnson contributed to analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. A Liu contributed to analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. J Dong contributed to analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. TC White‐Adams contributed to, analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. JA López contributed to concept and design, analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval. J Di Paola contributed to: concept and design, analysis and/or interpretation of data, critical writing or revising the intellectual content, and final approval.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Sandra Habericter for her generous gift of monoclonal anti‐VWF antibodies.
RELATIONSHIP DISCLOSURE
None of the authors have any disclosures relevant to this paper.
Supporting information
Ng CJ, McCrae KR, Ashworth K, et al. Effects of anti‐β2GPI antibodies on VWF release from human umbilical vein endothelial cells and ADAMTS13 activity. Res Pract Thromb Haemost. 2018;2:380–389. 10.1002/rth2.12090
Funding information
This work was supported by grants/support from the National Hemophilia Foundation (CJN), the Health Resources & Services Administration—Maternal & Child Health (5 H30 MC00008‐20‐00 to CJN/JDP), the National Institutes of Health (R01 HL084086 to JDP) and the Postle Family Chair for Pediatric Cancer and Blood Disorders (JDP).
REFERENCES
- 1. Urbanus RT, de Groot PG. Antiphospholipid antibodies—We are not quite there yet. Blood Rev. 2011;25:97–106. [DOI] [PubMed] [Google Scholar]
- 2. Lockshin MD. Pregnancy and antiphospholipid syndrome. Am J Reprod Immunol. 2013;69:585–7. [DOI] [PubMed] [Google Scholar]
- 3. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest. 2016;149:315–52. [DOI] [PubMed] [Google Scholar]
- 4. Giannakopoulos B, Krilis SA. How I treat the antiphospholipid syndrome. Blood. 2009;114:2020–30. [DOI] [PubMed] [Google Scholar]
- 5. Meroni PL, Borghi MO, Raschi E, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. 2011;7:330–9. [DOI] [PubMed] [Google Scholar]
- 6. de Groot PG, Urbanus RT. The significance of autoantibodies against β2‐glycoprotein I. Blood. 2012;120:266–74. [DOI] [PubMed] [Google Scholar]
- 7. Zoghlami‐Rintelen C, Vormittag R, Sailer T, et al. The presence of IgG antibodies against beta2‐glycoprotein I predicts the risk of thrombosis in patients with the lupus anticoagulant. J Thromb Haemost. 2005;3:1160–5. [DOI] [PubMed] [Google Scholar]
- 8. De Craemer A‐S, Musial J, Devreese KMJ. Role of anti‐domain 1‐β2 glycoprotein I antibodies in the diagnosis and risk stratification of antiphospholipid syndrome. J Thromb Haemost. 2016;14:1779–87. [DOI] [PubMed] [Google Scholar]
- 9. Arad A, Proulle V, Furie RA, Furie BC, Furie B. β₂‐Glycoprotein‐1 autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood. 2011;117:3453–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramesh S, Morrell CN, Tarango C, et al. Antiphospholipid antibodies promote leukocyte–endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via β2GPI and apoER2. J Clin Invest. 2011;121:120–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Romay‐Penabad Z, Aguilar‐Valenzuela R, Urbanus RT, et al. Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood. 2011;117:1408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Schie MC, Wieberdink RG, Koudstaal PJ, et al. Genetic determinants of von Willebrand factor plasma levels and the risk of stroke: the Rotterdam Study. J Thromb Haemost. 2012;10:550–6. [DOI] [PubMed] [Google Scholar]
- 13. Bongers TN, de Maat MPM, van Goor M‐LPJ, et al. High von Willebrand factor levels increase the risk of first ischemic stroke: influence of ADAMTS13, inflammation, and genetic variability. Stroke. 2006;37:2672–7. [DOI] [PubMed] [Google Scholar]
- 14. Rutten B, Maseri A, Cianflone D, et al. Plasma levels of active Von Willebrand factor are increased in patients with first ST‐segment elevation myocardial infarction: a multicenter and multiethnic study. Eur Heart J Acute Cardiovasc Care. 2015;4:64–74. [DOI] [PubMed] [Google Scholar]
- 15. Sakai H, Goto S, Kim JY, et al. Plasma concentration of von Willebrand factor in acute myocardial infarction. Thromb Haemost. 2000;84:204–9. [PubMed] [Google Scholar]
- 16. Hulstein JJJ, Lenting PJ, de Laat B, Derksen RHWM, Fijnheer R, de Groot PG. beta2‐Glycoprotein I inhibits von Willebrand factor dependent platelet adhesion and aggregation. Blood. 2007;110:1483–91. [DOI] [PubMed] [Google Scholar]
- 17. Habe K, Wada H, Matsumoto T, et al. Plasma ADAMTS13, von Willebrand Factor (VWF), and VWF propeptide profiles in patients with connective tissue diseases and antiphospholipid syndrome. Clin Appl Thromb Hemost. 2017;23:622–30. [DOI] [PubMed] [Google Scholar]
- 18. Cugno M, Borghi MO, Lonati LM, et al. Patients with antiphospholipid syndrome display endothelial perturbation. J Autoimmun. 2010;34:105–10. [DOI] [PubMed] [Google Scholar]
- 19. Bontadi A, Ruffatti A, Falcinelli E, et al. Platelet and endothelial activation in catastrophic and quiescent antiphospholipid syndrome. Thromb Haemost. 2013;109:901–8. [DOI] [PubMed] [Google Scholar]
- 20. Dong J‐F, Moake JL, Nolasco L, et al. ADAMTS‐13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–9. [DOI] [PubMed] [Google Scholar]
- 21. Austin SK, Austin SK, Starke RD, et al. The VWF/ADAMTS13 axis in the antiphospholipid syndrome: ADAMTS13 antibodies and ADAMTS13 dysfunction. Br J Haematol. 2008;141:536–44. [DOI] [PubMed] [Google Scholar]
- 22. Lee SJ, Kim J‐E, Han K‐S, Kim HK. Thrombotic risk of reduced ADAMTS13 activity in patients with antiphospholipid antibodies. Blood Coagul Fibrinolysis. 2016;27:907–12. [DOI] [PubMed] [Google Scholar]
- 23. Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306. [DOI] [PubMed] [Google Scholar]
- 24. Allen KL, Fonseca FV, Betapudi V, et al. A novel pathway for human endothelial cell activation by antiphospholipid/anti‐β2 glycoprotein I antibodies. Blood. 2012;119:884–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Allen KL, Hamik A, Jain MK, McCrae KR. Endothelial cell activation by antiphospholipid antibodies is modulated by Kruppel‐like transcription factors. Blood. 2011;117:6383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J, McCrae KR. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti‐beta2 glycoprotein I antibodies. Blood. 2005;105:1964–9. [DOI] [PubMed] [Google Scholar]
- 27. Yeh H‐C, Zhou Z, Choi H, et al. Disulfide bond reduction of von Willebrand factor by ADAMTS‐13. J Thromb Haemost. 2010;8:2778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. White‐Adams TC, Ng CJ, Jacobi PM, Haberichter SL, Di Paola JA. Mutations in the D'D3 region of VWF traditionally associated with type 1 VWD lead to quantitative and qualitative deficiencies of VWF. Thromb Res. 2016;145:112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu J‐J, Fujikawa K, Lian EC, McMullen BA, Kulman JD, Chung DW. A rapid enzyme‐linked assay for ADAMTS‐13. J Thromb Haemost. 2006;4:129–36. [DOI] [PubMed] [Google Scholar]
- 31. Lai KN, Leung JC, Lai KB, Lai FM, Wong KC. Increased release of von Willebrand factor antigen from endothelial cells by anti‐DNA autoantibodies. Ann Rheum Dis. 1996;55:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lindsey NJ, Dawson RA, Henderson FI, Greaves M, Hughes P. Stimulation of von Willebrand factor antigen release by immunoglobulin from thrombosis prone patients with systemic lupus erythematosus and the anti‐phospholipid syndrome. Br J Rheumatol. 1993;32:123–6. [DOI] [PubMed] [Google Scholar]
- 33. McCrae KR, DeMichele A, Samuels P, et al. Detection of endothelial cell‐reactive immunoglobulin in patients with anti‐phospholipid antibodies. Br J Haematol. 1991;79:595–605. [DOI] [PubMed] [Google Scholar]
- 34. Ma K, Simantov R, Zhang JC, Silverstein R, Hajjar KA, McCrae KR. High affinity binding of beta 2‐glycoprotein I to human endothelial cells is mediated by annexin II. J Biol Chem. 2000;275:15541–8. [DOI] [PubMed] [Google Scholar]
- 35. Bernardo A, Ball C, Nolasco L, Moake JF, Dong J‐F. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell‐derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104:100–6. [DOI] [PubMed] [Google Scholar]
- 36. Shim CY, Liu YN, Atkinson T, et al. Molecular imaging of platelet‐endothelial interactions and endothelial von Willebrand factor in early and mid‐stage atherosclerosis. Circ Cardiovasc Imaging. 2015;8:e002765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the von Willebrand factor strings puzzle. Blood. 2013;121:270–7. [DOI] [PubMed] [Google Scholar]
- 38. Maino A, Siegerink B, Lotta LA, et al. Plasma ADAMTS‐13 levels and the risk of myocardial infarction: an individual patient data meta‐analysis. J Thromb Haemost. 2015;13:1396–404. [DOI] [PubMed] [Google Scholar]
- 39. Llobet D, Tirado I, Vilalta N, et al. Low ADAMTS13 levels are associated with venous thrombosis risk in women. Thromb Res. 2017;157:38–40. [DOI] [PubMed] [Google Scholar]
- 40. Denorme F, Kraft P, Pareyn I, et al. Reduced ADAMTS13 levels in patients with acute and chronic cerebrovascular disease. PLoS ONE. 2017;12:e0179258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kentouche K, Voigt A, Schleussner E, et al. Pregnancy in Upshaw‐Schulman syndrome. Hamostaseologie. 2013;33:144–8. [DOI] [PubMed] [Google Scholar]
- 42. Pekcan MK, Sarıkaya E, Tokmak A, et al. ADAMTS‐3, ‐13, ‐16, and ‐19 levels in patients with habitual abortion. Kaohsiung J Med Sci. 2017;33:30–5. [DOI] [PubMed] [Google Scholar]
- 43. Nolasco LH, Turner NA, Bernardo A, et al. Hemolytic uremic syndrome‐associated Shiga toxins promote endothelial‐cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005;106:4199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Favaloro EJ. Variability and diagnostic utility of antiphospholipid antibodies including lupus anticoagulants. Int J Lab Hematol. 2013;35:269–74. [DOI] [PubMed] [Google Scholar]
- 45. Adams M. Measurement of lupus anticoagulants: an update on quality in laboratory testing. Semin Thromb Hemost. 2013;39:267–71. [DOI] [PubMed] [Google Scholar]
- 46. Han Y, Xiao J, Falls E, Zheng XL. A shear‐based assay for assessing plasma ADAMTS13 activity and inhibitors in patients with thrombotic thrombocytopenic purpura. Transfusion. 2011;51:1580–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials