

Abstract
Chlamydia trachomatis is an obligate intracellular pathogen characterized by a unique biphasic developmental cycle that alternates between infectious and non-infectious organisms. Chlamydial ChxR is a transcriptional activator that has been implicated in the regulation of the development cycle. We used a reverse genetics approach to generate three chxR null mutants. All three mutants grew normally in cultured mammalian cells. Whole genome sequencing identified SNPs in other genes; however, none of the mutated genes were common to all three ChxR null mutants arguing against a genetic compensatory mechanism that would explain the non-essential in vitro growth phenotype. Comparative proteomics identified five proteins, CT005, CT214, CT565, CT694 and CT695, that were significantly downregulated in all ChxR null mutants. This group includes established inclusion membrane and type III secreted proteins. ChxR transcriptional regulation of these genes was confirmed by qRT-PCR. Importantly, while ChxR null mutants exhibited no growth deficiencies in in vitro, they did show significant differences in in vivo growth using a mouse genital tract model. Collectively, our findings demonstrated that ChxR is a transcriptional activator that regulates the expression of virulence genes whose functions are restricted to in vivo infection.
Keywords: Chlamydia, ChxR, transcription, in vivo infection
ChxR transcriptionally regulates chlamydial virulence genes that function in in vivo infection.
INTRODUCTION
Chlamydia trachomatis is a mucosotropic obligate intracellular pathogen that is the leading cause of ocular and sexually transmitted infections. Infections are typically self-limiting, though repeated infections and the pathogen's propensities to cause persistent infection and to suppress host immunity can result in serious long-term complications such as infertility, ectopic pregnancy and blindness (Schachter 1978; WHO 2001). The obligate intracellular nature of C. trachomatis and the historic lack of tractable genetic systems have presented significant challenges in the study of chlamydial biology and pathogenesis. However, some genetic tools for manipulating C. trachomatis have recently been described. Forward, reverse and transformation-based genetic methodologies are established for both lymphogranuloma venereum (LGV) and non LGV C. trachomatis (Kari et al.2011; Wang et al.2011; Nguyen and Valdivia 2012). However, to date targeted gene knock out has only been described for serovar L2 (Johnson and Fisher 2013; Mueller et al.2016). Thus, for non-LGV serovars random mutagenesis, using a TILLING-based reverse genetic method remains a useful approach for conducting targeted mutagenesis (Kari et al.2011).
Chlamydiae are characterized by a unique biphasic developmental cycle that alternates between an extracellular, metabolically inactive, infectious elementary body (EB) and an intracellular, metabolically active, non-infectious reticulate body (RB) (Moulder 1991). Many aspects of the developmental cycle, including its regulation, are not fully understood. ChxR (CT630) has been proposed to function in the chlamydial developmental cycle as a RB-to-EB conversion stage-specific transcriptional activator (Koo et al.2006). The potential role of ChxR in this basic biological process is supported by its highly conserved nature, uniform presence in all chlamydial species and developmentally regulated expression (Koo et al.2006). Furthermore, biochemical and structural characterization has demonstrated that ChxR has DNA-binding activity and sequence homology to known regulators (Koo et al.2006; Hickey et al.2009, 2011; Hickey, Weldon and Hefty 2011; Barta et al.2014). However, a direct role for ChxR in the developmental regulation of the chlamydial growth cycle has yet to be demonstrated.
Here we used a genetic approach to ask whether ChxR plays a role in the regulation of the chlamydial developmental cycle. We reasoned if ChxR functioned in this capacity it should be an essential gene. Unexpectedly, we generated multiple chxR null mutants using the TILLING reverse genetics method. Employing comparative proteomics, we identified several proteins that were substantially downregulated in all chxR mutants. Furthermore, quantitative RT-PCR analyses indicated that transcription of the corresponding genes was reduced in the mutant strains. Finally, while chxR null mutants exhibited no significant growth alterations in in vitro they did show significant growth deficiencies in murine model of female genital tract infection. Collectively, our findings indicate that ChxR is a transcriptional activator as originally reported but rather than controlling events essential for regulation of the chlamydial developmental cycle, it regulates the expression of genes that play an important role early during in vivo infection.
MATERIALS AND METHODS
Chlamydia trachomatis strains and cell culture
The parental C. trachomatis D-LC wild-type (WT) strain derived from the D/UW-3/Cx reference strain (Sturdevant et al.2010) and plaque cloned (Matsumoto et al.1998) chxR mutants were propagated in McCoy cells and purified as previously described (Caldwell et al.1981). In vitro experiments were carried out using McCoy or HeLa 229 cells obtained from ATCC. All in vitro infections were facilitated by centrifuging the culture plates at 545 × g for 1 h at room temperature. Following infection, the inoculum was removed, monolayers washed with Hanks balanced salt solution (HBSS) and fed with Dulbecco's Modified Eagle Medium (DMEM)-10 containing 1 μg/ml cycloheximide.
Chemical mutagenesis, library construction, mutation screen and whole genome sequencing
Highly mutagenized C. trachomatis organisms were generated by four rounds of consecutive mutagenesis using 5–7 mg/ml of ethyl methanesulfonate. Infected McCoy cells were treated with EMS at 19 h post-infection (PI) for 1 h, and EBs were harvested at 40 h PI. The library construction and mutation screen were performed as previously described (Kari et al.2011). Briefly, McCoy cells in multiple 96-well tissue-culture plates were infected with eight inclusion forming units (IFU) per well. After three rounds of passage, cultures were harvested, divided and frozen at −80°C. DNA extracted from one half of the third passage was used to PCR-amplify chxR. ChxR PCR amplicons were digested by CEL I, and digestion products were visualized by DNA agarose gel electrophoresis. Mutations were sequenced and mutants harboring nonsense mutations or an altered ATG in chxR were plaque cloned three times. Predicted alterations to chxR null mutant open reading frames were assessed using Gene Construction Kit software (Textco BioSoftware, Inc., Raleigh, NC). EBs were purified from each of the three plaque cloned chxR mutants by density gradient centrifugation. Genomic DNA was isolated and the DNA samples were used to prepare Illumina paired-end libraries according to the manufacturer's instructions (Illumina Inc., San Diego, CA). Libraries were subsequently subjected to cluster generation and paired-end sequencing (2×150 bp) by using the next-generation sequencing platform Illumina MiSeq (Illumina Inc., San Diego, CA, USA), according to the manufacturer's instructions. Reads were referenced mapped to C. trachomatis D LC chromosome (NCBI NC_017436.1) and plasmid (NCBI NC_017433.1). Gapped-read alignments were achieved with Bowtie 2 (Langmead and Salzberg 2012), and SNPs were called using Freebayes (Blankenberg et al.2014) and TRAMS version 1.0.0. In all cases, high-quality SNPs indicated were supported by a Phred quality score of ≥30. The genome sequences of ChxRG003, ChxRC457T and ChxRC490T have been deposited in the GenBank database under accession numbers SRR3240308, SRR3240309 and SRR3240310, respectively.
Antibodies and western blot analysis
Mouse monoclonal antibodies (mAb) were generated to recombinant chlamydial ChxR (αChxR 1F3 and 4H1) using protocols approved by the Animal Care and Use Committee (Rocky Mountain Laboratories, National Institute of Allergy and Infectious Disease). Two BALB/c mice were immunized three times by intraperitoneal injection with 10 μg recombinant ChxR protein adsorbed to aluminum hydroxide gel. Animals were euthanized 3 days following the last immunization, splenocytes were harvested and a fusion was performed with the mouse myeloma cell line Sp2/0 using polyethylene glycol 1500. Hybridomas were suspended in ClonaCell HY Medium D and cultured according to the manufacturer's recommendations (STEMCELL Technologies, Vancouver, British Columbia, Canada). Hybridomas were subcultured in Quantum Yield mAb Medium (Becton Dickinson) supplemented with 100 mM hypoxanthine, 16 mM thymidine, 5% J1774 macrophage conditioned media, 10% fetal bovine serum and 4 mM L-glutamine. Hybridoma supernatant was tested by enzyme-linked immunosorbent assay against the recombinant protein to determine reactivity and specificity. Hybridoma supernatant and immunoglobulin purified by protein A/G chromatography were used for downstream analysis. A mouse mAb to chlamydial HSP60 (A57-B9) was used as control (Yuan et al.1992). Western blot analysis of ChxR was performed as previously described (Kari et al.2011).
Proteomics
McCoy cells grown in 75 cm2 flasks (2 × 107 cells) were infected with WT C. trachomatis D LC or chxR mutant organisms at a multiplicity of infection (MOI) of 2. Cells were harvested by trypsinization at 36 h PI. Trypsinized cells were centrifuged at 500 × g for 5 min and suspended in 0.5 ml hot 2% SDS-50 mM HEPES (pH 8.2) and boiled for 10 min (Patton et al.2016). This method was sufficient to inactivate exogenous CPAF activity (Patton et al.2016). Protein extracts from whole-cell lysates were trypsin digested, labeled by isobaric mass tags and analyzed by shotgun proteomics using nanoflow liquid chromatography-tandem mass spectrometry (nLC-MS/MS) on a Q-Exactive Plus mass spectrometer (Thermo Fisher, Canoga Park, CA), as described previously (Patton et al.2016). The nLC-MS/MS data were searched against a mouse and a C. trachomatis D LC protein database (NC_017436.1). Proteomic statistics were performed on experimental groups (WT and chxR mutants) done in triplicate. If protein hits were not present in all three of the experimental groups they were discarded. These analyses resulted in a total of 7036 proteins identified with a 2-peptide 99% confidence threshold (1.0% false-discovery rate). Differentially expressed proteins were identified using the Linear Model for Micro Arrays (LIMMA) R package (Smyth 2004). The corresponding P values for each comparison were adjusted using the multiple testing procedure (Benjamini and Hochberg 1995). Protein hits with adjusted P values of <0.05 and fold changes of ≥2 were considered statistically significant. The mass spectrometry-based protein data sets are available at the following links: https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task = ecea003e34c54475b7effacae27a404a and ftp://massive.ucsd.edu/MSV000080635.
Quantitative RT-PCR
McCoy cells grown in six-well plate (1.2 × 106 cells per well) were infected with WT or chxR mutants at MOI = 1. Each infection group consisted of six replicates. The cells were harvested using TRIzol reagent (Life Technologies) at 36 h PI, and RNA was extracted in random order using TRIzol LS (Life Technologies) according to the manufacturer's recommendations. AgPath-ID One-Step reverse transcription polymerase chain reaction (RT-PCR) Kit (Invitrogen, Carlsbad, CA) was used to perform the qRT-PCR. Transcript copy numbers derived from six replicates were normalized to those of gyrA mRNA. The primer and probe sequences for the target transcripts are listed in the supplemental material (Table S1, Supporting Information).
Infection and growth analysis in vitro
McCoy cells and HeLa 229 cells were grown in 24-well plates at a cell density of 2 × 105 cells per well. Monolayers were inoculated with chlamydiae in SPG buffer at an MOI of 0.1. Chlamydiae were harvested at various times PI into SPG buffer for enumeration of recoverable IFUs in McCoy cells. Plaquing was used to clone the chxR null mutants and to assay plaquing efficiency and plaque forming kinetics. In all experiments, McCoy monolayers in six-well plates were infected with 20–100 IFU/well and processed for plaque assay as previously (Matsumoto et al.1998). For cloning, monolayers were visually examined and isolated plaques were picked for further propagation. Plaquing efficiency and plaque forming kinetics were assayed by staining monolayers with 0.03% neutral red at eight days PI to visualize and photograph plaques.
Infection and growth analysis in vivo
Ten-week-old female C3H/HeJ mice (Jackson Laboratories) were treated with 2.5 mg medroxyprogesterone acetate at 10 and 3 days prior to urogenital infection with WT C. trachomatis D LC and the three chxR null mutants. Groups of 10 mice per strain were inoculated intravaginally with 1×105 IFU in 5 μl SPG. The vaginal vault was swabbed prior to inoculation to remove mucous. Post-infection chlamydial shedding was monitored by swabbing the vaginal vault and culturing recovered chlamydiae on McCoy monolayers in 48-well plates to assay recoverable IFU. All handling procedures were approved by the RML Animal Care and Use Committee, and the research was conducted in full compliance with the guidelines in the Guide for the Care and Use of Laboratory Animals (NRC, 2011). The facilities are fully accredited by the American Association for Accreditation of Laboratory Animal Care.
RESULTS AND DISCUSSION
Isolation of chxR mutants
A TILLING-based reverse genetic approach was used to generate Chlamydia trachomatis D LC chxR mutants (Kari et al.2011). In this study, we used high-level EMS mutagenesis as our primary goal was to investigate whether ChxR is essential for chlamydial survival, as would be expected if it is a regulator of the developmental cycle. We screened ∼3000 mutants for chxR mutations using CEL I digestion and found 27 chxR mutants containing 7 synonymous and 20 non-synonymous mutations (Table S2, Supporting Information). Two of the 20 non-synonymous mutations at base pair 457 and 490 introduced premature stop codons (chxRC457T, chxRC490T), truncating the chxR open reading frame (ORF) by 225 and 192 base pairs, respectively (Table S2). One of the remaining 18 non-synonymous SNPs mutated the ATG initiation codon changing it to ATA (chxRG003A). Promoter regions were unchanged in all three mutants. The serovar D chxR mutants had no other initiation codons; therefore, all three mutants were predicted to lack at least the C-terminal portion of ChxR or the entire protein (Fig. 1A). The three mutants harboring the predicted inactivating mutations were isolated by multiple rounds of plaque cloning and propagated for further analysis.
Figure 1.

Protein expression of mutants with disrupted chxR. (A) Schematic depiction of the predicted ORFs of the WT serovar D and mutant strains containing SNPs disrupting chxR (polymorphisms are detailed in Table S2). (B) Temporal analysis of WT ChxR expression during the developmental cycle by immunoblotting. Infected McCoy cell lysates were harvested at various times PI. Equal amounts of protein were used for immunoblotting and probed with a αChxR mAb. ChxR is first detected by 24 h PI and expression increases late in the growth cycle as previously described (Koo et al.2006). (C) Specificity of αChxR mAbs. The truncated form of chxRC490T was recombinantly expressed and immunoblotted with αChxR 1F3 and αChxR 4H1 mAbs. αChxR 1F3 recognized the N-terminal truncated polypeptide while αChxR 4H1 did not. The bottom of panel A depicts the predicted epitope specificity range of each mAb. (D) Characterization of chxR mutants by immunoblotting infected cells lysates solubilizes using RIPA buffer and 8 M urea. Cell lysates infected with WT and the three chxR mutants were harvested at 36 h PI and probed with both αChxR mAbs, anti-chlamydial HSP60, anti-CPAF and anti-keratin18. ChxR is expressed only by parental WT strain. CPAF did not affect ChxR detection in WT organisms despite being proteolytically active as shown by the changes in protein mass of CPAF heterodimer, chlamydial HSP60 and host keratin following solubilization of infected cells in RIPA buffer but not 8 M urea.
chxR G003A, chxRC457T and chxRC490T are true chxR null mutants
To definitively show that a gene is not essential, it is critical to unambiguously demonstrate that the gene is not expressed or the gene product is dysfunctional. Hence, the WT and three chxRG003A, chxRC457T, chxRC490T strains were evaluated by western blot analysis for ChxR protein expression. We analyzed the temporal kinetics of ChxR and HSP60 in the WT strain (Fig. 1B). Western blot analysis using a αChxR mAb confirmed that ChxR is a mid-to-late cycle gene. Compared to HSP60, a known early gene which was first detected at 12 h PI, ChxR was not detected until 24 h PI and expression levels increased markedly at later time points PI (30-48 h). We selected the 36 h PI time point to evaluate ChxR expression in the mutants containing predicted ChxR inactivating mutations. Two mAbs specific to ChxR (1F3 and 4H1) were generated for this project but the epitopes they recognize are unknown. To address this question, the N-terminal 163 amino acids corresponding to the predicted truncated form of ChxR in chxRC490T (Fig. 1A) were expressed recombinantly and probed with αChxR mAbs. Figure 1C shows that αChxR 1F3 recognized the N-terminal polypeptide, while αChxR 4H1 was non-reactive, suggesting that αChxR 4H1 likely binds an epitope in the C-terminal portion of ChxR. By using both C- and N-terminal ChxR-specific antibodies, we found that neither full length nor truncated forms of ChxR were detected in any of the three mutants (Fig. 1D). Hence, we concluded that all three strains are true chxR null mutants. We excluded a role for the chlamydial protease activity factor (CPAF) in the degradation of chxR in our null mutants (Fig. 1D). CPAF was present in the WT and all three chxR mutants (Fig. 1D). However, CPAF did not affect ChxR expression in the WT strain regardless of whether infected cells were lysed using radioimmunoprecipitation (RIPA) buffer or 8M urea. CPAF proteolytic activity was evident as shown by the partial proteolytic cleavage of HSP60, dimeric CPAF and host keratin in cells solubilized in RIPA buffer but not 8M urea.
Whole genome sequencing of the chxR null mutants
While high-level EMS mutagenesis simplifies the mutant discovery process, it does complicate phenotype interpretation because other mutations are introduced in the genome. It is possible that a common compensatory mutation could exist in all three independent mutants, thus making chxR appear non-essential. To fully define the mutations present in each mutant, we sequenced their genomes and compared them to WT C. trachomatis serovar D. A summary of the genetic changes in the three-chxR mutants is shown in Table 1. As expected from a high-level EMS mutagenesis protocol, in addition to the chxR mutations already identified in our PCR-based screen, there were 40–50 additional mutations identified in each of the mutants (Table 1). Consistent with EMS mutagenesis, the majority of mutations were transitions of C/G to T/A that occurred randomly in the chromosome in both intervening and ORF sequences. Within ORFs, there was a mix of synonymous, non-synonymous and missense mutations. There were no mutations in the plasmid in any of the three mutants. Details of the genotype of the individual mutants are presented in the supplemental material (Tables S3 and S4, Supporting Information). Importantly, other than the mutations in chxR, there were no mutations present in a single locus common to all isolates that could have a potential compensatory function. Thus, we concluded that chxR is a non-essential gene for chlamydial survival in vitro and it does not directly function as a stage specific regulator of the chlamydial development cycle as initially hypothesized (Koo et al.2006).
Table 1.
Genetic characteristics of ChxR mutants.
| Mutants | |||
|---|---|---|---|
| Mutation type | chxR G003A | chxR C490T | chxR C457T |
| Total number of mutations | 44 | 50 | 41 |
| Non-synonymous SNPs | 34 | 32 | 26 |
| Synonymous SNPs | 6 | 11 | 9 |
| Nonsense SNPs | 0 | 4 | 3 |
| Intergenic | 4 | 3 | 3 |
| Mutation in ChxR | ATG > ATA (base 3) | CGA > TGA (base 490) | CGA > TGA (base 457) |
The chxR nulls exhibit no significant growth deficiency in vitro
Although our findings clearly showed that ChxR was not essential for chlamydial in vitro growth, it was important to more thoroughly characterize the mutant strains in assays that were capable of detecting subtle differences in the pathogenesis of in vitro infection. To this end, we studied the ability of the chxR nulls to infect and replicate in two eukaryotic cell lines. McCoy and HeLa cells were infected with the WT or chxR nulls, and the infectious progeny were enumerated from samples harvested at various time points PI (Fig. 2A and B). Comparison of the growth curves revealed no major differences between WT and mutant strains. The plaquing efficiencies and morphologies of WT and chxR null mutants were evaluated in McCoy cells (Fig. 2C). No differences in plaquing efficiency or plaque size were observed between the WT strain and chxR null mutants. These in vitro data clearly demonstrate that infection, growth and propagation of the null mutants were essentially unaffected by the loss ChxR.
Figure 2.

One step growth curves and plaquing efficiency of chxR null mutants. (A) McCoy and (B) HeLa 229 cells were infected at an MOI of 0.1. Cells were harvested at different times PI and the number of recoverable IFU determined. Mean and standard deviation are shown (n = 3). Similar growth kinetics were observed for the parental WT strain and all three chxR null mutants in McCoy and HeLa cells. (C) Plaquing efficiency and plaque size were similar between WT and ChxR null mutant strains. Monolayers of McCoy cells grown in six-well TC plates were infected with 100 IFU of WT and the three chxR null mutants. After 8 days incubation, plaques were identified following neutral red staining and evaluated by number and phenotypic characteristics. No differences in plaquing efficiencies or plaque size were observed between the WT strain and chxR null mutants.
Comparative proteomics of WT and the chxR mutants
Abundant structural and biochemical evidence exists indicating that ChxR is a DNA-binding protein (Hickey et al. 2011; Barta et al.2014) and that it regulates chlamydial gene expression (Koo et al.2006). Given this, we reasoned that ChxR null mutants would show alterations in their proteome, when compared to WT. Thus, we employed comparative proteomics as a way to discover genes that might be regulated by ChxR. Protein lysates were prepared from WT and chxR null infected McCoy cells at 36 h PI. The lysates were equivalently labeled with tandem mass tags (TMTs) and analyzed by nLC-MS/MS. A total of 734 chlamydial proteins were identified. Employing a >2-fold change in mean TMT intensity and an adjusted P value of <0.05 across three replicates with 99% confidence of 2-peptide identification resulted in a limited list of proteins altered in the various mutants compared to WT (Fig. 3) (Table S5, Supporting Information). The proteomics data corroborated our western blot and whole genome sequencing findings that ChxR is highly downregulated in all three chxR nulls (6.5-, 6.0- and 5.9-fold) downregulated in chxRG003AchxRC490TchxRC457T, respectively (Table S5). Furthermore, genes other than ChxR identified as having nonsense mutations are also highly downregulated in the proteome. For example, in the chxRC490T mutant, a nonsense mutation occurs in CT289 and the protein is highly downregulated in the proteome (6.3-fold). Of particular interest is a subset of genes (CT005, CT214, CT565, CT694 and CT695) in all three chxR nulls that are significantly downregulated but lack mutations, as determined by whole genome sequencing (Tables S3 and S4). These data implicate ChxR as transcriptional activator of the expression of these genes as ChxR is the only common locus mutated in each of the three nulls. All of the genes identified as potentially regulated by ChxR are expressed early in the developmental cycle (Belland et al.2003). CT005 and CT214 are predicted inclusion membrane proteins (Weber et al.2015), and CT694 and CT695 are type three secretion effectors (Hower et al.2009). CT005 is particularly interesting because it has been suggested to function in the interaction with multiple host proteins important for chlamydial intracellular survival and growth (Mirrashidi et al.2015).
Figure 3.
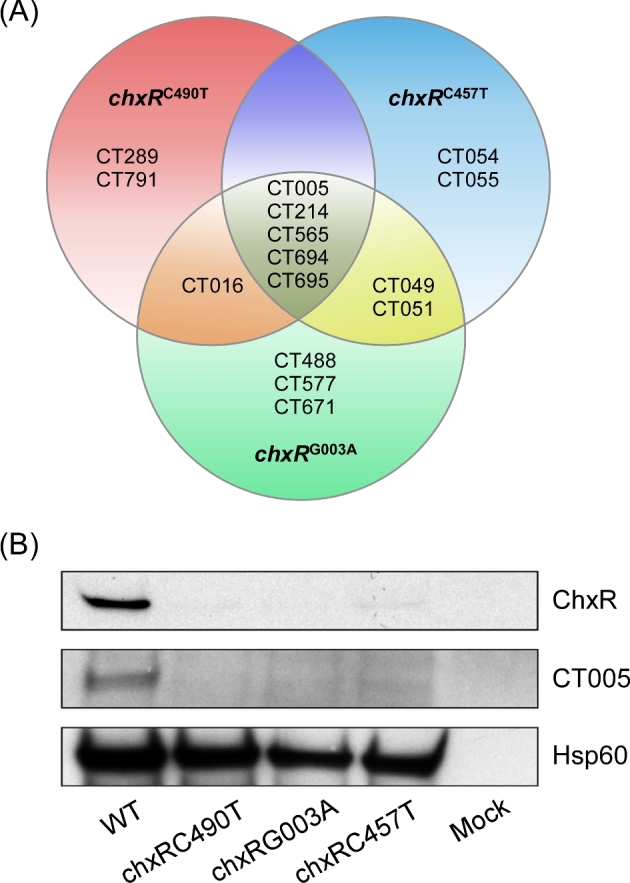
Comparative proteomics of chxR mutants. (A) A Venn diagram showing proteins downregulated in chxR mutant strains compared to WT organisms. Compared to WT, proteins CT005, CT214, CT565, CT694 and CT695 were downregulated in all three chxR mutants. (B) Western blot validation of proteome results. Lysates of WT and the three chxR null mutants were electrophoresed on SDS-PAGE gels and immunoblotted using ChxR, CT005 and chlamydial HSP60 antibodies. All three chxR mutants did not react with αChxR mAb or anti-CT005.
Regulation of gene expression by ChxR
The proteomics data implicated CT005, CT214 and CT694 as genes potentially being regulated by ChxR. To validate this possibility, we performed qRT-PCR analysis for these genes in McCoy cells infected with WT and chxR null mutants (Fig. 4). Total RNA was isolated from infected McCoy cells at 36 h PI and used for qRT-PCR analysis. The results demonstrate that the expression of CT005, CT214 and CT694 is significantly reduced in all three chxR nulls. No difference was observed between WT and null mutants using the negative control gene CT671. The reduction of CT005 expression was particularly strong (5- to 7-fold). This finding is consistent with the proteomics results and directly supports the conclusion that ChxR regulates these genes.
Figure 4.
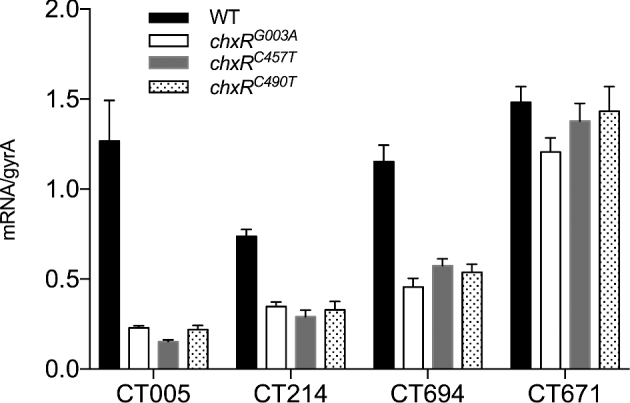
ChxR regulates the transcription of CT005, CT214 and CT694. qRT-PCR transcript copy numbers of CT005, CT214, CT694 and CT671 in WT and chxR null mutants normalized to chlamydial gyrA. Compared to WT, transcripts of CT005, CT214 and CT694 are reduced in the three chxR mutants. In contrast, no difference was observed between WT and null mutants using negative control gene CT671. Of note, CT005 transcripts were significantly downregulated (P < 0.05, two-tailed Student's t-test) in all three mutants, a finding that is consistent with the western blot results shown in Fig. 3.
chxR null mutants are partially attenuated for in vivo growth
Our findings clearly show that ChxR regulates expression of CT005, CT214, CT565, CT694 and CT695 but plays no substantial role in in vitro infection or growth. In a final series of experiments, we asked whether regulation of these genes might be important to in vivo infection. Female C3H/HeJ mice were infected intravaginally with the WT and three chxR nulls. Infections were monitored at various times postchallenge by culturing chlamydiae recovered from vaginal swabs on monolayers of McCoy cells. Notably, we observed significant differences between the WT strain and all three chxR nulls in infectious burdens recovered from vaginal cultures on days 3 and 10 PI (Fig. 5). Thus, we conclude that one or more of the ChxR activated genes (CT005, CT214, CT565, CT694 and CT695) are virulence factor(s) that function in chlamydial host–cell interactions that are important to in vivo infection.
Figure 5.
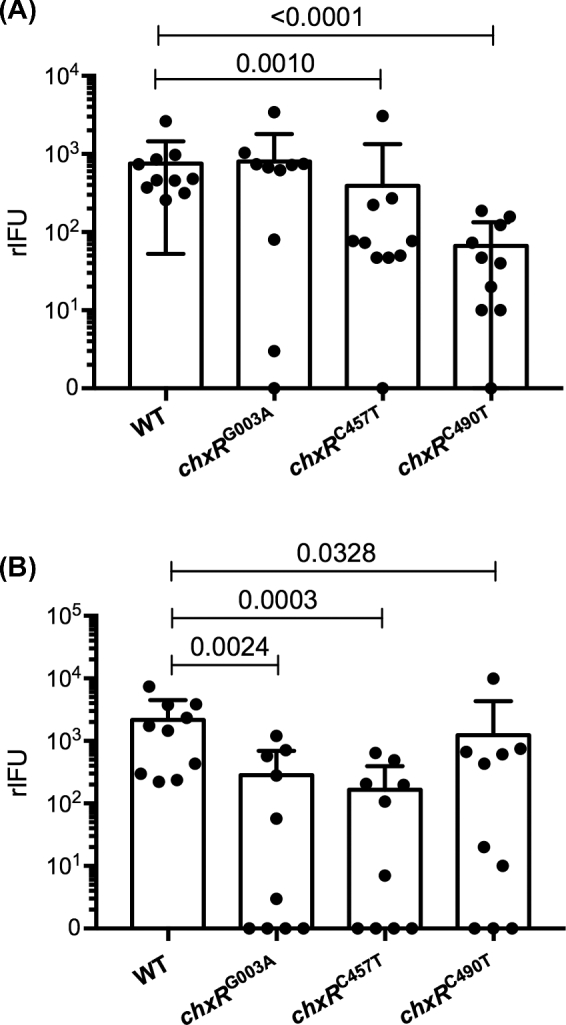
Infectivity of WT and chxR null mutants in C3H/HeJ mice. C3H/HeJ mice were intravaginally inoculated with 105 IFU of WT or chxR null mutant strains (10 mice per strain). The infectious burden and course of infection was monitored by culturing chlamydiae from vaginal swabs on monolayers of McCoy cells. Mean recoverable IFU with standard error of the mean are shown for each strain at days 3 (A) and 10 (B) PI. All three null mutants exhibited a statistically significant (P < 0.01, Mann-Whitney U test) reduction in rIFU at days 3 and 10 PI.
CONCLUDING REMARKS
Our experiments demonstrate that chxR is not an essential gene for chlamydial growth in cultured cells, a finding that does not support a role for ChxR as a global transcriptional regulator of the chlamydial developmental cycle. On the other hand, our findings support the conclusion that ChxR is a transcriptional regulator of CT005, CT214, CT565, CT694 and CT695 and the regulation of these genes is important to in vivo chlamydial infection. Interestingly, three of the genes (CT005, CT565 and CT695) are expressed early in the chlamydial developmental cycle (Belland et al.2003). CT005 and CT214 are inclusion membrane proteins, whereas CT694 and CT695 are type III secreted proteins. Since these proteins are expressed early in the chlamydial developmental cycle and play a role in the initial stages of in vivo infection intuitively suggests that they may function in the evasion of innate host immunity. Collectively, our data suggest that ChxR is expressed late in the developmental cycle, and specifically regulates virulence factors that are important to chlamydial host interaction. It is noteworthy that there is measurable expression of ChxR regulated genes in all three chxR mutants (Fig. 5). Thus, ChxR nulls may present an intermediate or partial in vivo phenotype. To definitively address the function of the ChxR regulated genes identified in this study, it will be necessary to isolate isogenic mutants of each gene by allelic exchange. Unfortunately, allelic exchange is currently restricted to LGV strains (Mueller et al.2016). When genetic allelic exchange becomes available for non-LGV strains, it will be essential to knockout the genes identified here and test their in vivo growth characteristics in animal model systems.
Supplementary Material
Supplementary data are available at FEMSPD online.
Acknowledgments
We thank the NIAID Rocky Mountain Laboratory core facility for qRT-PCR analysis, Dr. Raphael Valdivia Duke University for providing antibody specific to CT005, and Garrett Westmacott and Stuart McCorrister for proteomics support. The authors have no conflict of interest to declare.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSPD online.
FUNDING
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases and National Institutes of Health and the Public Health Agency of Canada.
Conflict of interest. None declared.
REFERENCES
- Barta ML, Hickey JM, Anbanandam A et al. . Atypical response regulator ChxR from Chlamydia trachomatis is structurally poised for DNA binding. PLoS One 2014;9:e91760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belland RJ, Zhong G, Crane DD et al. . Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. P Natl Acad Sci USA 2003;100:8478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B-Met 1995;57:289–300. [Google Scholar]
- Blankenberg D, Von Kuster G, Bouvier E et al. . Dissemination of scientific software with Galaxy ToolShed. Genome Biol 2014;15:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell HD, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun 1981;31:1161–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey JM, Hefty PS, Lamb AL. Expression, purification, crystallization and preliminary X-ray analysis of the DNA-binding domain of a Chlamydia trachomatis OmpR/PhoB-subfamily response regulator homolog, ChxR. Acta crystallogr F 2009;65:791–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey JM, Lovell S, Battaile KP et al. . The atypical response regulator protein ChxR has structural characteristics and dimer interface interactions that are unique within the OmpR/PhoB subfamily. J Biol Chem 2011;286:32606–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey JM, Weldon L, Hefty PS. The atypical OmpR/PhoB response regulator ChxR from Chlamydia trachomatis forms homodimers in vivo and binds a direct repeat of nucleotide sequences. J Bacteriol 2011;193:389–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hower S, Wolf K, Fields KA. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol Microbiol 2009;72:1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CM, Fisher DJ. Site-specific, insertional inactivation of incA in Chlamydia trachomatis using a group II intron. PLoS One 2013;8:e83989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari L, Goheen MM, Randall LB et al. . Generation of targeted Chlamydia trachomatis null mutants. P Natl Acad Sci USA 2011;108:7189–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo IC, Walthers D, Hefty PS. et al. ChxR is a transcriptional activator in Chlamydia. P Natl Acad Sci USA 2006;103:750–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Izutsu H, Miyashita N et al. . Plaque formation by and plaque cloning of Chlamydia trachomatis biovar trachoma. J Clin Microbiol 1998;36:3013–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirrashidi KM, Elwell CA, Verschueren E et al. . Global mapping of the Inc-human interactome reveals that retromer restricts Chlamydia infection. Cell Host Microbe 2015;18:109–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder JW. Interaction of chlamydiae and host cells in vitro. Microbiol Rev 1991;55:143–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller KE, Wolf K, Fields KA. Gene deletion by fluorescence-reported allelic exchange mutagenesis in Chlamydia trachomatis. MBio 2016;7:e01817–01815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen BD, Valdivia RH. Virulence determinants in the obligate intracellular pathogen Chlamydia trachomatis revealed by forward genetic approaches. P Natl Acad Sci USA 2012;109:1263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton MJ, McCorrister S, Grant C et al. . Chlamydial protease-like activity factor and type III secreted effectors cooperate in inhibition of p65 nuclear translocation. MBio 2016;7:e01427–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter J. Chlamydial infections (First of three parts). N Engl J Med 1978;298:428–34. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol 2004;3:Article3. [DOI] [PubMed] [Google Scholar]
- Sturdevant GL, Kari L, Gardner DJ et al. . Frameshift mutations in a single novel virulence factor alter the in vivo pathogenicity of Chlamydia trachomatis for the female murine genital tract. Infect Immun 2010;78:3660–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kahane S, Cutcliffe LT et al. . Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 2011;7:e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MM, Bauler LD, Lam J et al. . Expression and localization of predicted inclusion membrane proteins in Chlamydia trachomatis. Infect Immun 2015;83:4710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. World Health Organization. Global Prevalence and Incidence of Selected Curable Sexually Transmitted Infections: Overview and Estimates. Geneva: 2001, 1–43. [Google Scholar]
- Yuan Y, Lyng K, Zhang YX et al. . Monoclonal antibodies define genus-specific, species-specific, and cross-reactive epitopes of the chlamydial 60-kilodalton heat shock protein (hsp60): specific immunodetection and purification of chlamydial hsp60. Infect Immun 1992;60:2288–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data are available at FEMSPD online.