

Abstract
The rapid progress in high-throughput sequencing has significantly enriched our capacity for studying the mitochondrial DNA (mtDNA). In addition to performing specific mitochondrial targeted sequencing, an increasingly popular alternative approach is using the off-target reads from exome sequencing to infer mtDNA variants, including single nucleotide polymorphisms (SNPs) and heteroplasmy. However, the effectiveness and practicality of this approach have not been tested. Recently, RNAseq data have also been suggested as a good source for alternative data mining, but whether mitochondrial variants can be detected from RNAseq data has not been validated. We designed a study to evaluate the practicability of mtDNA variant detection using exome and RNA sequencing data. Five breast cancer cell lines were sequenced through mitochondrial targeted, exome, and RNA sequencing. Mitochondrial targeted sequencing was used as the gold standard to compute the validation and false discovery rates of SNP and heteroplasmy detection in exome and RNAseq data. We found that exome and RNA sequencing can accurately detect mitochondrial SNPs. However, the lower false discovery rate makes exome sequencing a better choice for heteroplasmy detection than RNAseq. Furthermore, we examined three alignment strategies and found that aligning reads directly to the mitochondrial reference genome or aligning reads to the nuclear and mitochondrial references genomes simultaneously produced the best results, and that aligning to the nuclear genome first and afterwards to the mitochondrial genome performed poorly. In conclusion, our study provides important guidelines for future studies that intend to use either exome sequencing or RNAseq data to infer mitochondrial SNPs and heteroplasmy.
Keywords: mitochondria, data mining, SNP, heteroplasmy
Background
Mammalian cells each contain approximately 100 mitochondria, which themselves contain between 2 and 10 copies of mitochondrial DNA (mtDNA) [1]. Because of this, mutations to mtDNA often result in heteroplasmic cells, with both normal and mutant copies of mtDNA [2, 3]. While heteroplasmy of the mtDNA is common in normal individuals, varying in frequency between different tissue types [4, 5], heteroplasmy that results in mitochondrial dysfunction, affecting the production of ATP through oxidative phosphorylation, has been linked to many neurological diseases [6] and drug toxicities [7, 8].
Before the advent of high-throughput sequencing technology, the best options for complete mitochondrial genome sequencing were direct Sanger sequencing and Affymetrix’s MitoChip v.2.0, which contains a microarray of 25-mer probes complementary to the revised Cambridge Reference Sequence (rCRS) [9]. Quantifying mtDNA heteroplasmy has been accomplished via a number of different methods (i.e. real-time amplification refractory mutation system quantitative polymerase chain reaction (PCR) [10], PCR restriction fragment length polymorphism analysis [11], allele-specific oligonucleotide dot-blot analysis [12] and pyrosequencing [13]), but the small number of targets available to these methods limits their utility. With high-throughput sequencing technology having emerged as a reliable, cost-effective option, the mitochondrial genome, including mtDNA heteroplasmy, is available for study like never before, and while all three of the major sequencing platforms (Illumina HiSeq, Roche 454 and Applied Biosystems SOLid) are capable of sequencing mtDNA [14, 15], Illumina has dominated that market in terms of use. This study will focus on the Illumina platform.
Direct sequencing of the mitochondrial genome with high-throughput sequencing technology can generate incredibly high read depth, in the tens of thousands [5, 16–18], but this is not the only option. Information about the mitochondrial genome can be obtained through indirect methods as well by extracting the mtDNA sequences produced by exome and whole genome high-throughput sequencing data. Although mtDNA is not the target of these sequencing types, there is usually significant coverage of the mitochondrial genome, comprising about 1–5% of reads from exome sequencing data [19]. Because of the high copy number of mtDNA per cell, mtDNA coverage can exceed the coverage of even the targeted genomic regions with an average depth of around 100 [20, 21], and research has demonstrated the feasibility of extracting mtDNA sequences from exome sequencing data [22]. Additional possibilities have emerged as well, including the inference of mtDNA mutations from exome sequencing data, and in fact, The Cancer Genome Atlas project has inferred all of its mtDNA somatic mutations in this way [23, 24]. These methods have even facilitated the diagnosis of mitochondrial disorders from the mtDNA content in exome sequencing data [25].
Exome sequencing has been widely used for mtDNA studies [19, 20, 25–31]. However, to date, no study has evaluated the accuracy of this approach. We performed mitochondrial targeted sequencing and exome sequencing on five breast cancer cell line samples. This allowed us to use the targeted mitochondrial sequencing data as a gold standard to evaluate the true positive rate and false discovery rate (FDR) when using exome sequencing data to determine mitochondrial single nucleotide polymorphisms (SNPs) and heteroplasmy. Additionally, RNAseq data has been suggested as an alternative data source for mining [32, 33]. Thus, we performed RNAseq on these five cell lines, allowing us to evaluate the practicability of identifying mitochondrial SNPs and heteroplasmy using RNAseq data.
As previously suggested, mitochondrial alignment is sensitive to nuclear mitochondrial sequences (nuMTs) [26]. NuMTs are DNA sequences that are similar to mtDNA but have been copied into the nuclear genome in the distant past. Such nuMTs can cause ambiguity during alignment. Therefore, we compared three distinct alignment approaches to identify the best approach for mitochondrial alignment.
Method
We cultured five breast cell lines in this study (MDAMB157, SUM159, HS578T, CAL51 and MDAMB436). For all five cell lines, we performed mitochondrial targeted sequencing, exome sequencing and RNAseq. The sequencing of all samples was performed at Vanderbilt Technologies for Advanced Genomics.
MtDNA enrichment was done using the amplification kit from Affymetrix’s Genechip Human Mitochondria Resequencing Array 2.0 (Affymetrix, USA). The Affymetrix protocol specifically amplifies the entire mitochondrial genome from genomic DNA using overlapping primers to eliminate the bias that may be introduced from the PCR method. The enriched mtDNA were barcoded and sequenced using the Illumina MiSeq sequencing platform (Illumina, USA). MtDNA fragments with an average size of 120 nucleotides were sequenced from both ends. The average coverage of mitochondrial genome was 99.9%. For exome sequencing, the exomes were captured using Illumina’s TrueSeq capture kit. Seventy-five nucleotide paired-end sequencing runs were performed using Illumina’s HiSeq 2000 platform. For RNAseq, total RNA was isolated with the Aurum Total RNA Mini Kit. All samples were quantified on the QuBit RNA assay. RNA quality was checked using Agilent Bioanalyzer. RNA integrity number for both samples was 10. The ribosome RNA reduction was performed using the Ribo-Zero Magnetic Gold Kit (Human/Mouse/Rat) (Epicentre). The RNA libraries were sequenced on Illumina High HiSeq 2500 with paired-end 100 base pair long reads.
To study the effect of nuMTs on alignment, we examined three distinct alignment approaches: (1) align all reads to the nuclear reference genome plus the mitochondrial reference genome simultaneously, relying on the aligner to make the correct decision on where the mitochondrial reads should align; (2) align all reads, including potential nuMT reads, directly to the mitochondrial genome; and (3) first align all reads to the nuclear genome only, then align the unmapped reads to the mitochondria genome. The third approach is the most conservative approach, which would eliminate all nuMT reads along with some true mtDNA reads. The overall alignment approaches are illustrated in Figure 1.
Figure 1.
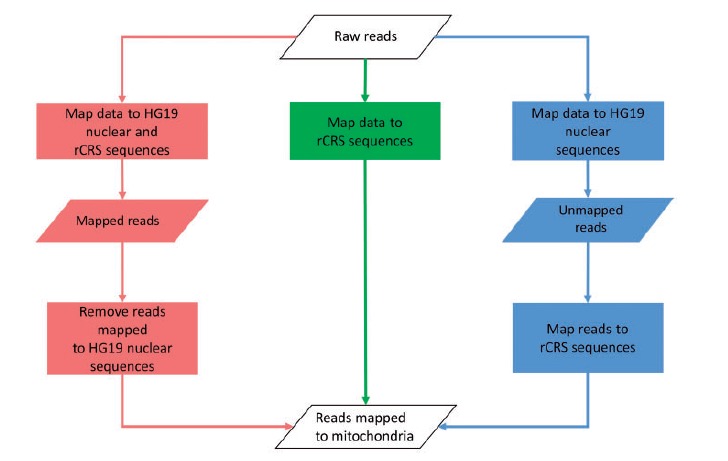
Workflow of the three mitochondrial mapping strategies we presented. Red = Method 1; green = Method 2; blue = Method 3. A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
Alignment was performed using Burrows–Wheeler Aligner (BWA) [34]. The nuclear genome we used is the human reference genome HG19, and the mitochondrial genome we used is the rCRS. We marked duplicates using Picard [35], and then performed local realignment and local recalibration using the Genome Analysis Toolkit [36] developed by the Broad Institute. SNPs and heteroplasmy were inferred using MitoSeek [27].
To evaluate the efficiency of conducting mitochondrial research using exome sequencing and RNAseq data, we computed two statistics for SNPs and heteroplasmy. The first one is the validation rate, and the second one is the FDR. If the number of SNPs or heteroplasmies identified by mitochondrial targeted sequencing is A (gold standard) and the subset of A that can be validated in another approach (either exome or RNA sequencing) is B, then the validation rate is B/A. If the number of SNPs or heteroplasmies identified by the alternative method (exome or RNA sequencing) is C, and the subset of C that is not validated by the gold standard mitochondrial targeted sequencing data is D, then the FDR is D/C. We also obtained haplogroup information (Supplementary Table S1) for each sample by checking the SNP results against phylotree.org’s mtDNA phylogeny tree [37].
Results
We achieved high quality sequencing data. For mitochondrial sequencing, we sequenced on average 2.3 million reads per sample. For exome sequencing, we sequenced on average 59 million reads. For RNAseq, we sequenced on average 34 million reads. We conducted thorough quality control on our sequencing data based on the multi-stage quality control protocol [38, 39] developed previously. No quality issues were detected after thorough quality control (Table 1). Haplogroup results showed consistent haplogroup determination across all three types of sequencing for each cell line (Supplementary Table S1). The ethnicities of the original contributor of three of five cell lines used in our study are known (MDAMB157—Black, HS578T—White, MDAMB436—White). The haplogroup results of these three cell lines matched the correct ethnicity groups (MDAMB157—L3f1b1a, HS578T—J2c1 and MDAMB436—H4a1a1).
Table 1.
Sample description and sequenced reads
| Sample name | Total reads | BQ | GC (%) |
|---|---|---|---|
| Mitochondria | |||
| MDAMB157 | 1 408 257 | 37 | 49 |
| SUM159 | 1 544 359 | 36 | 46 |
| HS578T | 2 024 445 | 36 | 46 |
| CAL51 | 1 588 780 | 36 | 46 |
| MDAMB436 | 5 846 096 | 37 | 47 |
| Exome | |||
| MDAMB157 | 41 111 584 | 33 | 52 |
| SUM159 | 60 845 870 | 30 | 51 |
| HS578T | 39 824 496 | 35 | 52 |
| CAL51 | 48 237 104 | 35 | 52 |
| MDAMB436 | 93 702 907 | 33 | 51 |
| RNAseq | |||
| MDAMB157 | |||
| SUM159 | 36 556 608 | 34 | 61 |
| HS578T | 36 147 448 | 35 | 59 |
| CAL51 | 38 752 295 | 34 | 61 |
| MDAMB436 | 30 976 269 | 31 | 76 |
For each of the three alignment approaches, we examined four mitochondrial quality control statistics: median depth, coverage, mapping quality and total mapped reads for exome sequencing and RNAseq data. RNAseq data achieved higher median depth for mitochondrial regions (Figure 2A). This is not a surprising result because RNAs in the mitochondria should be captured during the RNA library construction. However, mitochondria are not within the capture regions of the exome capture kit. Coverage was computed as the percentage of mitochondrial loci that have read depth >20. Exome sequencing achieved higher coverage than RNAseq data (Figure 2B). The median depth and coverage statistics together suggest that exome sequencing’s coverage of the mitochondria is more uniformly distributed than RNAseq’s coverage. As expected, RNAseq tends to have high coverage for the coding regions and leave other non-coding regions in mtDNA unsequenced. RNAseq and exome sequencing data achieved similar mapping quality scores (Figure 2C). RNAseq data managed to align more reads to the mitochondrial genome than did exome sequencing data even when the exome sequencing data had almost double the total number of reads of the RNAseq data (Figure 2D). For all four of these statistics, the rCRS-alone alignment approach achieved a slightly better value compared with the simultaneous HG19-rCRS alignment approach, while the sequential HG19 followed by rCRS alignment approach had the lowest values for these four quality control statistics.
Figure 2.
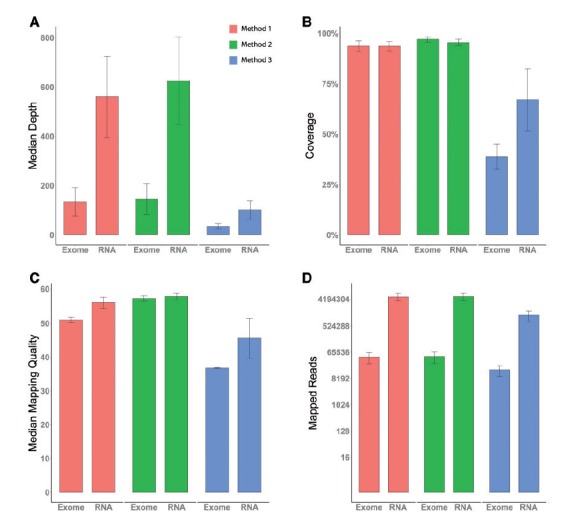
We examined four quality measurements across all sequencing types and alignment methods. (A) The median depth. (B) Coverage, defined as percentage of positions in rCRS that have depth >20. (C) Median mapping quality as reported by BWA. (D) Mapped reads. A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
Next, we examined the SNP validation rate (Figure 3A) and FDR (Figure 3B) using exome sequencing and RNAseq data by all three types of alignment approaches. The simultaneous HG19-rCRS and rCRS-alone alignments achieved a perfect SNP validation rate for both exome sequencing and RNAseq data, meaning that every SNP identified in our samples through mitochondrial targeted sequencing was also identified using exome sequencing as well as RNA sequencing data. The sequential HG19 followed by rCRS alignment approach identified the lowest number of true SNPs. The simultaneous HG19-rCRS and rCRS-alone alignment approaches on exome sequencing data achieved the lowest possible FDR at 0%. The same approaches on RNAseq data obtained 4.5% and 3.9% FDR, which were still tolerable. The sequential HG19 followed by rCRS alignment approach received the lowest validation rate and the highest FDR, which make it a less ideal alignment approach in comparison. Based on both validation rate and FDR, both the simultaneous HG19-rCRS and rCRS-alone alignment approaches offer excellent validation rates and sufficient FDR for SNP identification.
Figure 3.
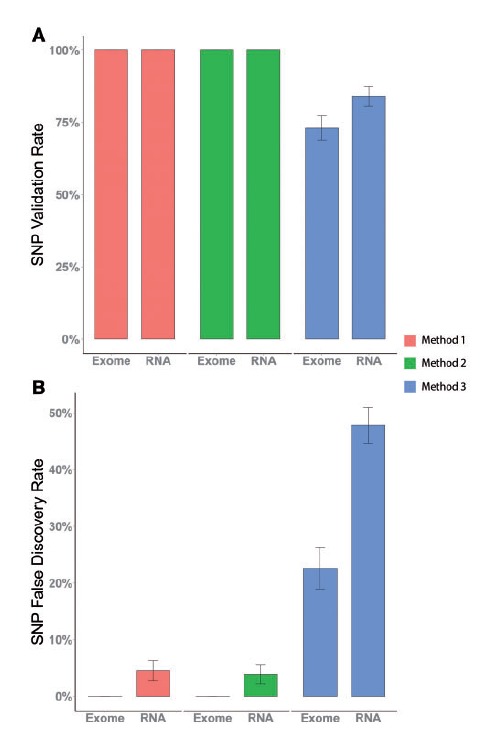
(A) Validation rate of SNPs identified from exome and RNAseq data from all three alignment strategies using mitochondrial targeted sequencing as the gold standard. (B) FDR of SNPs identified from exome and RNAseq data from all three alignment strategies using mitochondrial targeted sequencing as the gold standard. A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
Finally, we examined the effectiveness of detecting mitochondrial heteroplasmy using exome sequencing and RNAseq data for all three alignment approaches (Figure 4A). Heteroplasmy was tested at three different thresholds: 1%, 5% and 10%. One of the obvious trends is that as the heteroplasmy detection threshold increases, the validation rate increases as well. All three alignment approaches produced roughly the same validation rate for heteroplasmies, and RNAseq data produced a higher validation rate than did exome sequencing data. The overall validation rate for heteroplasmy is much lower than the SNP validation rate for both exome sequencing and RNAseq data.
Figure 4.
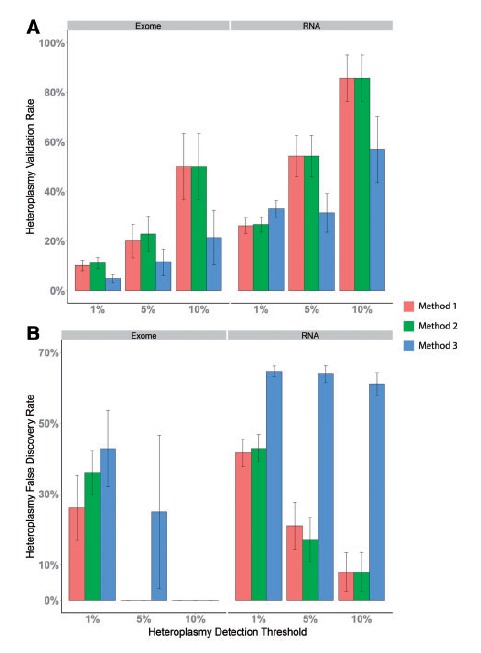
(A) Validation rate of heteroplasmies identified from exome and RNAseq data from all three alignment strategies using mitochondrial targeted sequencing as the gold standard. (B) FDR of heteroplasmies identified from exome and RNAseq data from all three alignment strategies using mitochondrial targeted sequencing as the gold standard. The x-axis denotes the heteroplasmy detection thresholds at alternative allele depth greater than 1%, 5% or 10%. A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
For FDR, the patterns were more complex (Figure 4B). Several conclusions can be drawn from the FDR analysis. First, a higher heteroplasmy detection threshold tends to produce lower false positives. Five scenarios produced zero FDR (exome sequencing with simultaneous HG19-rCRS and rCRS-alone alignment approaches at the 5% and 10% heteroplasmy detection thresholds, and the exome sequencing with sequential HG19 followed by rCRS alignment approach at the 10% heteroplasmy detection threshold). Second, RNAseq data generated substantially higher FDR as compared with exome sequencing data at the same detection thresholds. Even at the 10% heteroplasmy detection threshold, the simultaneous HG19-rCRS and rCRS-alone alignments still generated 8% FDR on RNA sequencing data. Third, the sequential HG19 followed by rCRS alignment approach generated the highest FDR, especially for RNA sequencing data (>60% across all three thresholds), which rendered it useless for heteroplasmy detection.
RNA editing is a rare molecular process through which some cells can make discrete changes to specific nucleotide sequences within an RNA molecule after it has been generated by RNA polymerase. A recent study examined these potential RNA editing sites within mitochondrial RNA and found three common RNA editing sites at positions C295T, G2129A and G6691A [40]. We compared the DNA–RNA difference in our data and found DNA–RNA differences at the three supposed RNA editing sites. Additionally, we found three additional RNA editing candidates at positions C296T, G5746A and T5878C. C296T occurred in four of five cell lines. G5746A and T5878C occurred in all five cell lines (Table 2).
Table 2.
RNA DNA position difference occurred in at least four of the five cell lines
| Sample | Position | DNA |
RNA |
Reported RNA editing | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Alternate | Reference reads | Alternate reads | Alternate frequency | Reference | Alternate | Reference reads | Alternate reads | Alternate frequency | |||
| CAL51 | 295 | C | C | 376 | 0 | 0 | C | T | 351 | 11 | 0.0304 | Y |
| CAL51 | 296 | C | C | 380 | 0 | 0 | C | T | 352 | 5 | 0.014 | N |
| CAL51 | 2129 | G | G | 679 | 0 | 0 | G | A | 7745 | 174 | 0.022 | Y |
| CAL51 | 5746 | G | G | 1363 | 3 | 0.0022 | G | A | 2453 | 117 | 0.0455 | N |
| CAL51 | 5878 | T | T | 1660 | 4 | 0.0024 | T | C | 1721 | 26 | 0.0149 | N |
| CAL51 | 6691 | G | G | 903 | 0 | 0 | G | A | 538 | 6 | 0.011 | Y |
| HS578T | 295 | C | T | 4 | 8 | 0.6667 | C | T | 4 | 44 | 0.9167 | Y |
| HS578T | 296 | C | C | 15 | 0 | 0 | C | C | 49 | 0 | 0 | N |
| HS578T | 2129 | G | G | 682 | 0 | 0 | G | A | 7719 | 180 | 0.0228 | Y |
| HS578T | 5746 | G | G | 1293 | 0 | 0 | G | A | 1725 | 58 | 0.0325 | N |
| HS578T | 5878 | T | T | 1611 | 0 | 0 | T | C | 2514 | 28 | 0.011 | N |
| HS578T | 6691 | G | G | 958 | 0 | 0 | G | A | 1807 | 14 | 0.0077 | Y |
| MDAMB157 | 295 | C | C | 111 | 0 | 0 | C | T | 302 | 11 | 0.0351 | Y |
| MDAMB157 | 296 | C | C | 109 | 0 | 0 | C | T | 305 | 1 | 0.0033 | N |
| MDAMB157 | 2129 | G | G | 393 | 0 | 0 | G | A | 7709 | 188 | 0.0238 | Y |
| MDAMB157 | 5746 | G | G | 935 | 0 | 0 | G | A | 5302 | 211 | 0.0383 | N |
| MDAMB157 | 5878 | T | T | 1370 | 1 | 0.0007 | T | C | 2494 | 3 | 0.0012 | N |
| MDAMB157 | 6691 | G | G | 665 | 0 | 0 | G | A | 5258 | 12 | 0.0023 | Y |
| MDAMB436 | 295 | C | C | 198 | 0 | 0 | C | T | 107 | 13 | 0.1083 | Y |
| MDAMB436 | 296 | C | C | 197 | 0 | 0 | C | T | 111 | 2 | 0.0177 | N |
| MDAMB436 | 2129 | G | G | 966 | 2 | 0.0021 | G | A | 7546 | 152 | 0.0197 | Y |
| MDAMB436 | 5746 | G | G | 1815 | 4 | 0.0022 | G | A | 2268 | 81 | 0.0345 | N |
| MDAMB436 | 5878 | T | T | 2384 | 4 | 0.0017 | T | C | 3840 | 25 | 0.0065 | N |
| MDAMB436 | 6691 | G | G | 1245 | 2 | 0.0016 | G | A | 4031 | 32 | 0.0079 | Y |
| SUM159 | 295 | C | C | 708 | 0 | 0 | C | T | 212 | 6 | 0.0275 | Y |
| SUM159 | 296 | C | C | 680 | 0 | 0 | C | T | 203 | 4 | 0.0193 | N |
| SUM159 | 2129 | G | G | 1416 | 2 | 0.0014 | G | A | 7791 | 150 | 0.0189 | Y |
| SUM159 | 5746 | G | G | 2883 | 0 | 0 | G | A | 2140 | 103 | 0.0459 | N |
| SUM159 | 5878 | T | T | 3656 | 3 | 0.0008 | T | C | 1536 | 19 | 0.0122 | N |
| SUM159 | 6691 | G | G | 2096 | 1 | 0.0005 | G | A | 888 | 4 | 0.0045 | Y |
One interesting phenomenon we observed during the study was related to orphan read alignment. Orphan reads by definition are the reads that have only one member of a pair mapped and the other member unmapped. When applying the sequential HG19 followed by rCRS alignment approach, the majority of the reads that aligned to the rCRS were orphan reads. But these same reads were not orphan reads when using the simultaneous HG19-rCRS alignment approach. The reason is rather elusive but can be explained by Figure 5. There are two reasons that orphan reads were mapped to the rCRS when using the sequential approach. In the first scenario, when mapping a paired-end read to the nuclear genome and the rCRS simultaneously, the first read of the pair can be mapped to nuMTs on the nuclear genome and the second read of the pair can be mapped to the rCRS. The paired-end read is mapped to different chromosomes and it is considered discordant but not orphan because both reads of the pair are mapped. When we try to align the same paired-end read to the nuclear genome first and the rCRS second, the first read of the pair is still mapped to nuMTs on the nuclear genome, and the second read of the pair is unmapped. That unmapped read is subsequently mapped to the rCRS as an orphan (Figure 5A). In the second scenario, when a paired-end read is mapped to the nuclear genome and the rCRS simultaneously, both reads of the pair are aligned to the rCRS. When mapping the same paired-end read to the nuclear genome first, the first read in the pair is aligned to a nuMT on the nuclear genome, even though it might not be the best match globally. The second read of the pair is unmapped and subsequently mapped to rCRS as an orphan read (Figure 5B). The two scenarios explain the reason behind the large quantity of orphan reads mapped to rCRS when using the sequential alignment approach. We conducted an additional analysis, in which we counted the orphan reads from the sequential alignment approach. The result did improve, but it still remained the worst of the three alignment strategies. Note that the scenarios of orphan reads described in our study are based on the alignment results produced by BWA aligner. Other aligner may assign orphan reads differently from BWA.
Figure 5.
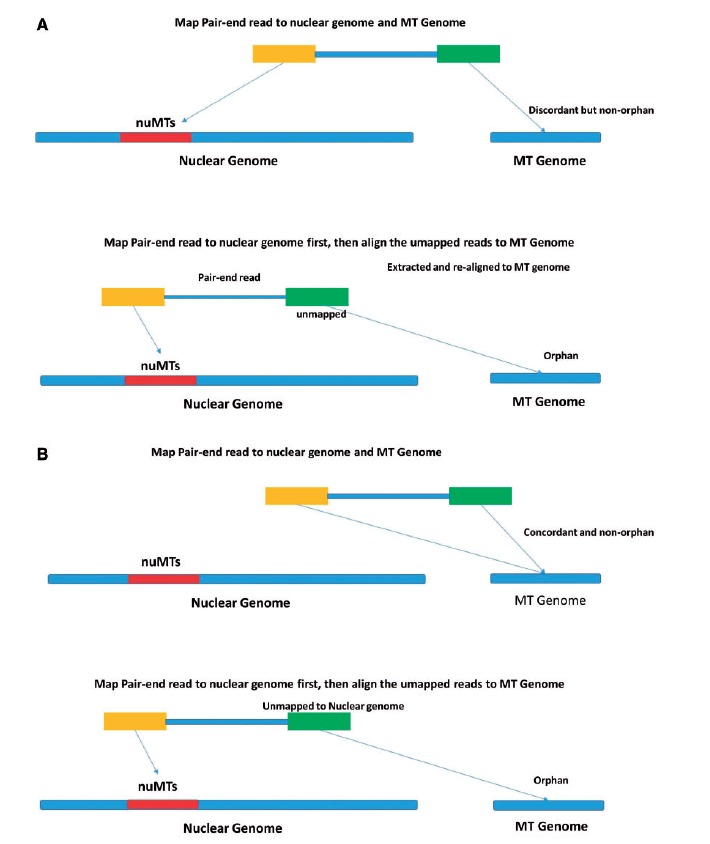
(A) Scenario 1 of orphan read on mitochondria: one read from the pair is originally mapped to nuMTs, leaving the other read from that pair as an orphan. (B) Scenario 2 of orphan read on mitochondria: one read from the pair is originally mapped to mitochondrial sequences, but when only nuclear genome is provided during alignment, this read is forced to map to nuMTs (although this alignment is globally sub-optimal), leaving the other read from that pair as an orphan. A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
Discussion
Based on our analysis results, several important conclusions can be drawn. For SNP calling, exome sequencing data can be used to detect mitochondrial SNPs with nearly perfect validation rates and low FDR when compared with the gold standard mitochondrial targeted sequencing. RNAseq data can also be used to detect mitochondrial SNPs, however, at a higher, yet still tolerable, FDR (<5%). This conclusion about RNAseq data is consistent with a previous finding of RNAseq data’s ability to identify nuclear genome SNPs, but at a higher false-positive rate [32]. By nature, accurate SNP calling from RNA sequencing data is much more challenging than that from DNA sequencing data. It is possible that the FDR of SNP calling from RNA sequencing data can be further improved by using more complex SNP calling tools. For heteroplasmy, exome sequencing data can identify a portion (10–50%) of all heteroplasmies depending on the detection threshold applied. However, exome sequencing also detected 0–36% false-positive heteroplasmies. Increasing the detection threshold to 5% decreased the false-positive rate to 0%.
RNAseq data are not ideal for detecting heteroplasmy compared with exome sequencing data based on our analysis results. This conclusion is within the expectation. To sequence RNA, RNA must first be reverse transcribed to complementary DNA and usually reverse transcriptase PCR is required to increase the quantity of the RNA. Both of these processes introduce errors that are not easily identifiable, thus increasing the FDR of SNP and heteroplasmy detections.
Of all three alignment approaches, simultaneous HG19-rCRS and rCRS-alone approaches performed similarly and produced trustworthy results. On one hand, when computation efficiency is a major concern, such as applications where a big sample size is involved, the rCRS-alone approach is recommended considering the additional computational effort needed for the simultaneous HG19-rCRS approach; on the other hand, the simultaneous HG19-rCRS approach can help filter out reads that come from nuMTs, lowering the possibility that these reads will map to mitochondria. In our study, we exclusively used the popular aligner BWA. If more computationally efficient aligners, such as STAR [41], are used, the extra computation time used to align to HG19-rCRS simultaneously may be negligible. By aligning all reads to the nuclear genome first (without the mitochondrial reference), many true mitochondrial reads were forced to align to the nuMTs on the nuclear reference. This had two consequences. First and foremost, there was a loss of depth of coverage on the mtDNA. Second, many of the mitochondrial reads that did not get mapped to the nuclear nuMTs were of lower quality, which caused the sequential alignment approach to not produce any reliable SNPs or heteroplasmy. Thus, the sequential alignment approach should not be used.
Key Points
Mitochondrial SNP can be accurately detected in exome and RNA sequencing data.
A portion of the mitochondria heteroplasmies can be detected in exome and RNA sequencing data.
RNAseq data has higher false discovery rate for mitochondrial variant detection as compared with exome sequencing data.
Alignment strategy plays an important role in mitochondrial variant detection accuracy.
RNA editing sites are identified in mitochondria.
Supplementary Material
Acknowledgement
We would like to thank Stephanie Page Hoskins and Margot Bjoring for editorial support.
Funding
Cancer Center Support Grant (P30 CA68485 to Y.G. and Y.S.); National Institute of Health (CA95131 and RC2CA148375 to J.P.); Komen for the Cure Foundation (KG262005 to B.L.); Vanderbilt Institute for Clinical and Translational Research (VR7875 towards sequencing of the two total RNA library samples).
References
- 1. Robin ED, Wong R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J Cell Physiol 1988;136:507–13. [DOI] [PubMed] [Google Scholar]
- 2. Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 2010;42:30–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Durbin RM, Altshuler DL, Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Samuels DC, Li C, Li B, et al. Recurrent tissue-specific mtDNA mutations are common in humans. Plos Genetics 2013;9:e1003929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. He Y, Wu J, Dressman DC, et al. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010;464:610–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fernandez-Vizarra E, Bugiani M, Goffrini P, et al. Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum Mol Genet 2007;16:1241–52. [DOI] [PubMed] [Google Scholar]
- 7. Lemasters JJ, Qian T, Bradham CA, et al. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J Bioenerg Biomembr 1999;31:305–19. [DOI] [PubMed] [Google Scholar]
- 8. Wallace KB, Starkov AA. Mitochondrial targets of drug toxicity. Annu Rev Pharmacol Toxicol 2000;40:353–88. [DOI] [PubMed] [Google Scholar]
- 9. Andrews RM, Kubacka I, Chinnery PF, et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 1999;23:147. [DOI] [PubMed] [Google Scholar]
- 10. Bai RK, Wong LJ. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: a single-step approach. Clin Chem 2004;50:996–1001. [DOI] [PubMed] [Google Scholar]
- 11. Holt IJ, Harding AE, Petty RK, et al. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet 1990;46:428–33. [PMC free article] [PubMed] [Google Scholar]
- 12. Liang MH, Johnson DR, Wong LJ. Preparation and validation of PCR-generated positive controls for diagnostic dot blotting. Clin Chem 1998;44:1578–9. [PubMed] [Google Scholar]
- 13. White HE, Durston VJ, Seller A, et al. Accurate detection and quantitation of heteroplasmic mitochondrial point mutations by pyrosequencing. Genet Test 2005;9:190–9. [DOI] [PubMed] [Google Scholar]
- 14. Payne BA, Wilson IJ, Yu-Wai-Man P, et al. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet 2013;22:384–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Craven L, Tuppen HA, Greggains GD, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010;465:82–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo Y, Cai Q, Samuels DC, et al. The use of next generation sequencing technology to study the effect of radiation therapy on mitochondrial DNA mutation. Mutat Res 2012;744:154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tang S, Huang T. Characterization of mitochondrial DNA heteroplasmy using a parallel sequencing system. Biotechniques 2010;48:287–96. [DOI] [PubMed] [Google Scholar]
- 18. Ameur A, Stewart JB, Freyer C, et al. Ultra-deep sequencing of mouse mitochondrial DNA: mutational patterns and their origins. Plos Genet 2011;7:e1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Samuels DC, Han L, Li J, et al. Finding the lost treasures in exome sequencing data. Trends Genet 2013;29:593–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Picardi E, Pesole G. Mitochondrial genomes gleaned from human whole-exome sequencing. Nat Methods 2012;9:523–4. [DOI] [PubMed] [Google Scholar]
- 21. Bogenhagen D, Clayton DA. The number of mitochondrial deoxyribonucleic acid genomes in mouse L and human HeLa cells. Quantitative isolation of mitochondrial deoxyribonucleic acid. J Biol Chem 1974;249:7991–5. [PubMed] [Google Scholar]
- 22. Larman TC, Depalma SR, Hadjipanayis AG, et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci USA 2012;109:14087–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dinwiddie DL, Smith LD, Miller NA, et al. Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics 2013;102:148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ye F, Samuels DC, Clark T, et al. High-throughput sequencing in mitochondrial DNA research. Mitochondrion 2014;17:157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo Y, Li J, Li CI, et al. MitoSeek: extracting mitochondria information and performing high-throughput mitochondria sequencing analysis. Bioinformatics 2013;29:1210–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Falk MJ, Pierce EA, Consugar M, et al. Mitochondrial disease genetic diagnostics: optimized whole-exome analysis for all mitocarta nuclear genes and the mitochondrial genome. Discov Med 2012;79:389–99. [PMC free article] [PubMed] [Google Scholar]
- 29. Nemeth AH, Kwasniewska AC, Lise S, et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 2013;136:3106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sevini F, Giuliani C, Vianello D, et al. mtDNA mutations in human aging and longevity: controversies and new perspectives opened by high-throughput technologies. Exp Gerontol 2014;56:234–44. [DOI] [PubMed] [Google Scholar]
- 31. McMahon S, LaFramboise T. Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 2014;35:1046–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han L, Vickers KC, Samuels DC, et al. Alternative applications for distinct RNA sequencing strategies. Brief Bioinform 2014;16:629–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vickers KC, Roteta LA, Hucheson-Dilks H, et al. Mining diverse small RNA species in the deep transcriptome. Trends Biochem Sci 2015;40:4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Katibah GE, Qin YD, Sidote DJ, et al. Broad and adaptable RNA structure recognition by the human interferon-induced tetratricopeptide repeat protein IFIT5. Proc Natl Acad Sci USA 2014;111:12025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 2009;30:E386–94. [DOI] [PubMed] [Google Scholar]
- 38. Guo Y, Zhao S, Sheng Q, et al. Multi-perspective quality control of Illumina exome sequencing data using QC3. Genomics 2014;103:323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guo Y, Ye F, Sheng Q, et al. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform 2014;15:879–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hodgkinson A, Idaghdour Y, Gbeha E, et al. High-resolution genomic analysis of human mitochondrial RNA sequence variation. Science 2014;344:413–15. [DOI] [PubMed] [Google Scholar]
- 41. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.